
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the effect of Fe–N5 configuration in the oxygen reduction reaction using N-heterocycle functionalized carbon nanotubes†
Qi
Li
ab,
Qi
Zhao
 a,
Angus
Pedersen
bc,
Mi
Zhang
a,
Zhipeng
Lin
d,
Yue
Xu
ab,
Patrick L.
Cullen
d,
Andrei
Sapelkin
e,
Devis
Di Tommaso
af,
Maria-Magdalena
Titirici
*b and
Christopher R.
Jones
*a
a,
Angus
Pedersen
bc,
Mi
Zhang
a,
Zhipeng
Lin
d,
Yue
Xu
ab,
Patrick L.
Cullen
d,
Andrei
Sapelkin
e,
Devis
Di Tommaso
af,
Maria-Magdalena
Titirici
*b and
Christopher R.
Jones
*a
aDepartment of Chemistry, Queen Mary University of London, London, E1 4NS, UK. E-mail: c.jones@qmul.ac.uk
bDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
cDepartment of Materials, Royal School of Mines, Imperial College London, London, SW7 2AZ, UK
dSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK
eDepartment of Physics, Queen Mary University of London, London, E1 4NS, UK
fDigital Environment Research Institute, Queen Mary University of London, Empire House, 67-75 New Road, London E1 1HH, UK
First published on 26th September 2024
Abstract
Atomic iron embedded in N-doped carbon materials (Fe–N–C) are regarded as the most promising alternative to platinum-based catalysts in anion exchange membrane fuel cells, due to their high oxygen reduction reaction (ORR) activity. The molecular catalyst iron phthalocyanine (FePc) is often employed to explore the correlation between a model single Fe structure and ORR activity. Recent advancements have introduced a penta-coordinated Fe–N5 structure achieved by binding FePc onto pyridine-functionalized carbon surfaces, demonstrating outstanding ORR activities. To investigate the effect of the axial pyridine ligand, in this work, four different N-functionalized carbon nanotubes (N-CNTs) were prepared, each adorned with a unique N-heterocycle (pyridine, quinoline, isoquinoline, or acridine). Subsequent binding of FePc (FePc/N-CNTs) afforded penta-coordinated complexes in all cases. The characterization results demonstrated that the penta-coordination Fe–N5 configuration could be effectively constructed by this method. The ORR activities of these FePc/N-CNTs suggest that the local surface structure around the axial ligand on the functionalized CNTs influences catalytic performance.
Introduction
Anion exchange membrane fuel cells (AEMFCs) have received increasing attention as a viable alternative to proton exchange membrane fuel cells (PEMFCs), particularly due to the emergence of low-cost atomically dispersed Fe–N–C catalysts that have shown high activity in the alkaline oxygen reduction reaction (ORR) as compared to Pt-based catalysts.1,2 These Fe–N–C materials are typically synthesized through the pyrolysis of metal, carbon, and nitrogen precursors, forming an iron center coordinated with nitrogen atoms (Fe-Nx, x = 2–5) via lone pair electrons within a large carbon matrix.3,4 This N–C structure ideally provides excellent electronic conductivity, hierarchical pores, and stability for catalytic activity.5,6 In contrast to these pyrolyzed Fe–N–C catalysts, molecular catalysts with well-defined structures are often employed as model catalysts to investigate reaction mechanisms and structure–activity relationships.7 Among the various heterogeneous molecular iron-based electrocatalysts, iron-phthalocyanine (FePc) has been known as a highly promising catalyst for ORR in alkaline environments for a long time.8,9Recently, researchers have explored carbon surface engineering and molecular structure refinement strategies to develop an axial-ligand-coordinated Fe-based molecular catalyst to further enhance the ORR activity and durability of Fe macrocycles through the “push effect” or “pull effect”.10–13 Specifically, electron-withdrawing or electron-donating groups are axially coordinated with Fe macrocycles to modulate the electron density at the Fe center respectively. Studies have highlighted that this effect could be achieved by complexing FePc onto a matrix surface doped with oxygen atoms or by covalently attaching pyridine groups.14–17 However, most of these oxygen-doping and functionalization techniques on carbon are typically conducted under strongly acidic conditions (e.g. HCl, HNO3, and HBF4), which can introduce additional defects, damage to the carbon surface, and further influence the ORR turnover frequency (TOF) of Fe–N–C catalysts.18–20 The complexity of these modified carbon surfaces poses challenges when investigating the reaction mechanisms of FePc and understanding the relationship between its structure and ORR activity, especially given the sensitivity of Fe centers to their local electronic environment.20 As a result, a key study by Cao et al. used a milder aryl diazonium method to achieve covalent attachment of pyridine molecules to the surface of CNTs and found that a reduced distance between FePc and the Py-CNTs was associated with improved ORR performance.21 It is noteworthy that throughout these significant advances, only a few different functionalization methods have been employed and most studies focussed on grafting pyridine as the axial ligand. Further research is therefore needed to investigate the effects of related N-heterocyclic coordinating groups on the catalytic performance of FePc-based catalysts.
Given our interests in the covalent functionalization of CNTs,22,23 in this study we sought to prepare a group of structurally related N-CNTs and compare how different pyridine-type axial ligands impact catalytic performance. To this end, covalent surface immobilization of four different nitrogen heterocycles (pyridine, quinoline, acridine, and isoquinoline) was achieved by exploiting the aryl diazonium method with the corresponding commercially available amino-substituted heterocycles. Subsequently, these N-CNTs were utilized as the carbon matrix for the binding of FePc. The morphology, distribution, as well as state of iron in the final catalysts (FePc/N-CNTs), were characterized using techniques such as transmission electron microscopy (TEM), X-ray diffraction (XRD), high angle annular dark field (HAADF) scanning transmission electron microscopy – energy dispersive X-ray spectroscopy (STEM-EDXS), X-ray photoelectron spectroscopy (XPS), and X-ray absorption spectroscopy. The ORR activity of these catalysts was evaluated and compared through rotating disk electrode (RDE) testing in alkaline media. Density functional theory (DFT) was also conducted to compute the free energy profiles of the ORR on FePc/N-CNTs. Our results revealed significant differences among the four FePc/N-CNTs, highlighting the influence of the heterocycle structure and the potential irregularities in the surface introduced by the functionalization reaction.
Experiment
Material synthetic procedures
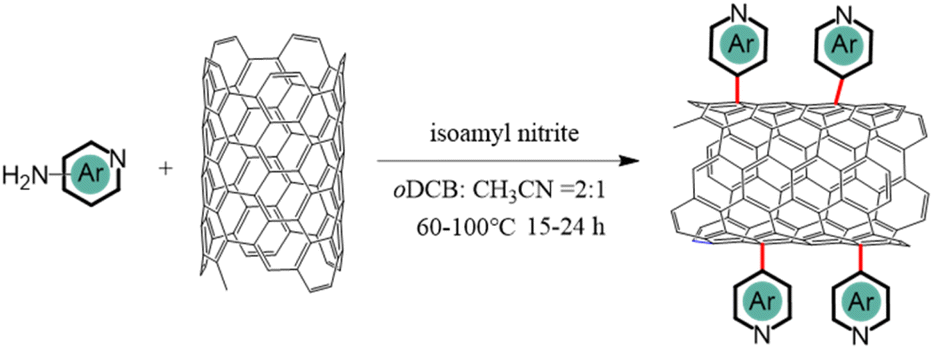 | ||
| Scheme 1 Synthesis of N-CNTs. | ||
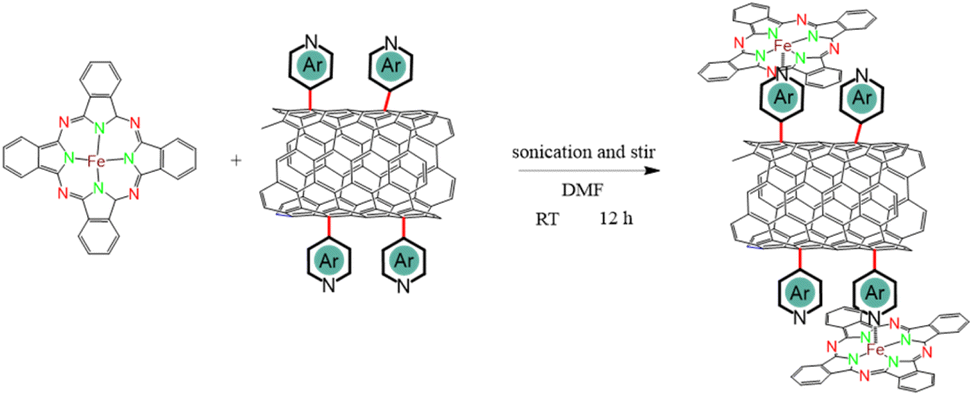 | ||
| Scheme 2 Synthesis of FePc/N-CNTs. | ||
Inks containing the different catalysts were prepared for RDE testing to evaluate their performance for the catalysis of ORR. 8 mg of catalyst powder was weighed and mixed with 1400 μL of deionized (DI) water, 528 μL of EtOH, and 72 μL of 5 wt% NafionD-521 dispersion in water and 1-propanol (Alfa Aesar). The Nafion dispersion was used as a binder for the components of the ink. The inks were then placed in a bath sonicator (132 kHz ultrasonic cleaner, VWR) for 40 min before undergoing ultrasonic liquid processing in a probe sonicator (Fisherbrand model 705, USA) for 20 min per sample, under a 5 s on and 5 s off regime. The working electrodes were then prepared by pipetting 14 μL of the inks onto a polished glassy carbon electrode of diameter 5 mm, resulting in a catalyst loading of 0.28 mg cm−2. The catalyst-modified electrodes were left rotating at 700 rpm for 1 h to dry at room temperature before electrochemical testing. A commercial 20 wt% Pt/C (JM HiSPEC 9100) working electrode was prepared in the same way. During the calibration, a 3 mm Pt RDE tip (Metrohm) works as the working electrode rotated at 1600 rpm for five cyclic voltammograms at 10 mV s−1 between −0.8 and −0.9 VHg/HgO with a Pt rod (Metrohm) and Hg/HgOsat as a counter and reference electrode, respectively. An average value for the conversion between Hg/HgOsat and RHE was obtained from the forward and backward scans at zero current.24
Cyclic voltammograms (CV) were obtained after purging the electrolyte for at least 15 min and acquired at 100 mV s−1 at 0 rpm for both N2 and O2 saturated electrolytes (measurements at the tenth cycle displayed). Linear sweep voltammograms (LSV) were obtained at 1600 rpm with a scan rate of 10 mV s−1. The pseudocapacitance was corrected by subtracting N2 saturated measurements at 1600 rpm from the O2 saturated results. The electrochemical measurements were repeated at least three times. The iR correction (90%) was applied post-measurement, with R = 36–38 Ω, determined by the intercept of the real axis in electrochemical impedance spectroscopy.
The double layer capacitance (Cdl) values based on the CVs within the potential of non-faradic range (0.785–0.985 VRHE) at various scan rates (20, 40, 60, 80 and 100 mV s−1) in N2 saturated 0.1 M KOH solution.4 The Cdl was obtained by plotting ΔJ (anodic current density − cathodic current density) at 0.9 VRHE against the scan rates. The slope of the above profiles was assigned to 2Cdl.
Site density and turnover frequency. The total Fe site density of the bulk catalyst could be found from the Fe content based on ICP-MS measurement, SDICP (sites cm−2), according to
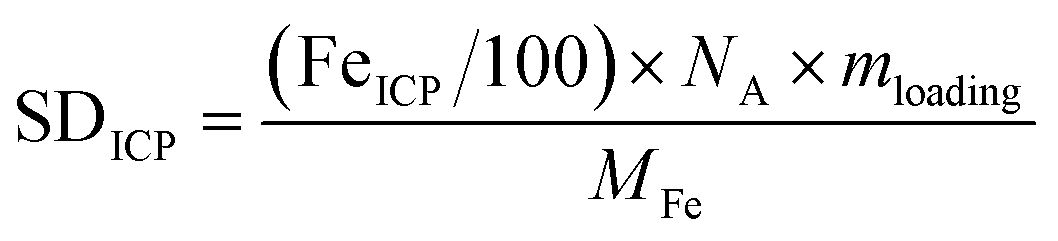 | (1) |
The electrochemically active Fe sites of the catalyst could be calculated based on the Fe III/II peak integration in CV, which could be converted to the site density through the following equation:
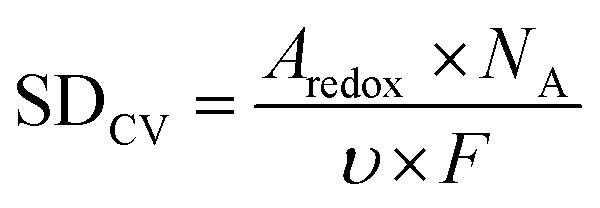 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 [s A mol−1].
485 [s A mol−1].
The kinetic current density (jkin) was determined from the geometric disk current (jd) and geometric diffusion-limited current density (jlim) at 0.2 VRHE according to the Koutecky–Levich equation:
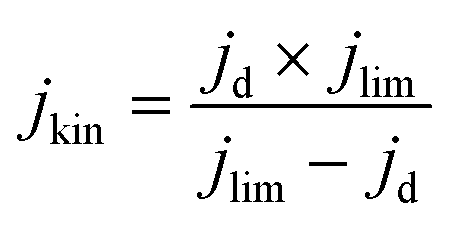 | (3) |
Subsequently, the kinetic mass activity could be found:
 | (4) |
TOF values were determined in the range of 0.8–0.6 VRHE based on the following equation:
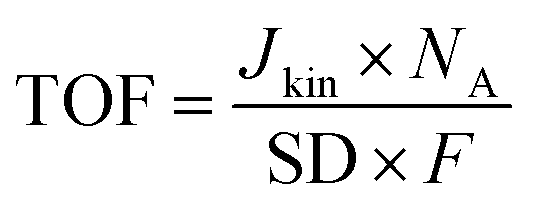 | (5) |
To calculate the number of electrons transferred (n), electrochemical measurements were carried out in an RRDE employing the same ink formulation, loading, and procedures as stated previously for the RDE. The Pt ring was set to 1.27 VRHE. The number of electrons transferred (n) was calculated from the disk and the ring current using the equation:
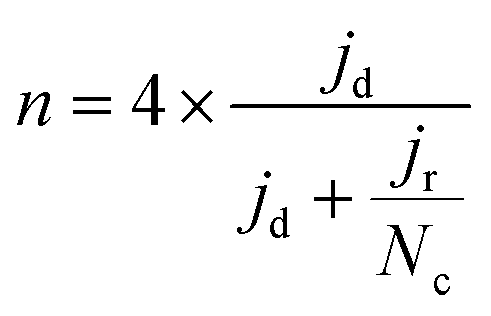 | (6) |
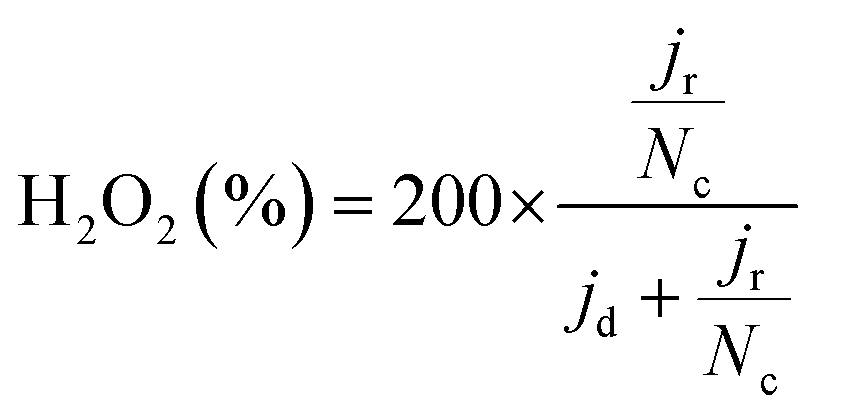 | (7) |
The distribution of relaxation times (DRT) analysis was performed using the MatlabDRTTools software developed by Ciucci and Chen.25 A Gaussian discretization method was selected with the inductive data excluded. This is because the DRT method inherently does not handle inductive processes well. A 2nd order regularization derivative was selected for the analysis to reduce the noise in the solution of the DRT. The regularization parameter (lambda) was selected as 10−3.26 The resistance of the processes was calculated by integrating the area under the corresponding peaks. The impedance can be described by eqn (8);
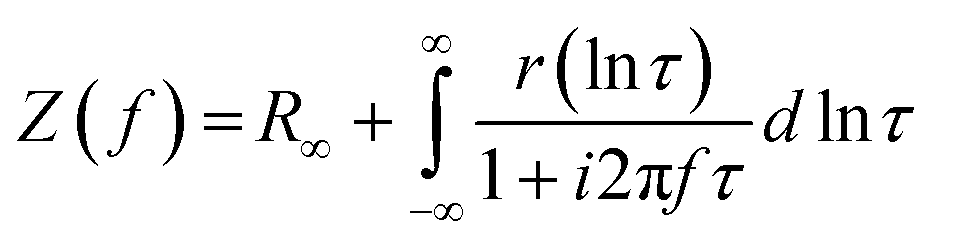 | (8) |
Results and discussion
Materials synthesis and characterization
Three related nitrogen-containing heterocycles – namely, pyridine, quinoline, and acridine – were initially selected as potential candidates for axial coordination. This choice was informed by several key considerations. Firstly, all three heterocycles possess a single pyridine-type nitrogen atom carrying a lone pair of electrons, likely facilitating axial coordination with iron atoms. Secondly, the introduction of additional aromatic rings in different positions around the pyridine core would provide a simple model to investigate the impact of stereoelectronic modifications to axial ligands on the final ORR catalytic activities. Lastly, the corresponding primary amine-substituted heterocycle starting materials were all commercially available and could therefore be attached to the surface of CNTs through an aryl diazonium strategy.As illustrated in Fig. 1a, the initial step involved anchoring each different heterocycle onto the surface of CNTs, to facilitate subsequent axial coordination with FePc through the pyridine-type nitrogen atoms. The functionalization of CNT surfaces was accomplished via radical aryl-diazonium reactions employing isoamyl nitrite in ortho-dichlorobenzene (oDCB) solution, as illustrated in Scheme 1. Notably, an alternative approach employing aryl derivatives, HBF4, and NaNO2 in a hydrochloric acid solution was avoided in order to mitigate against potential surface damage and oxidation associated with acid exposure.20 As such, three related N-CNTs were prepared through the utilization of distinct precursors: pyridine-CNT (Py-CNT), quinoline-CNT (Q-CNT), and acridine-CNT (A-CNT).
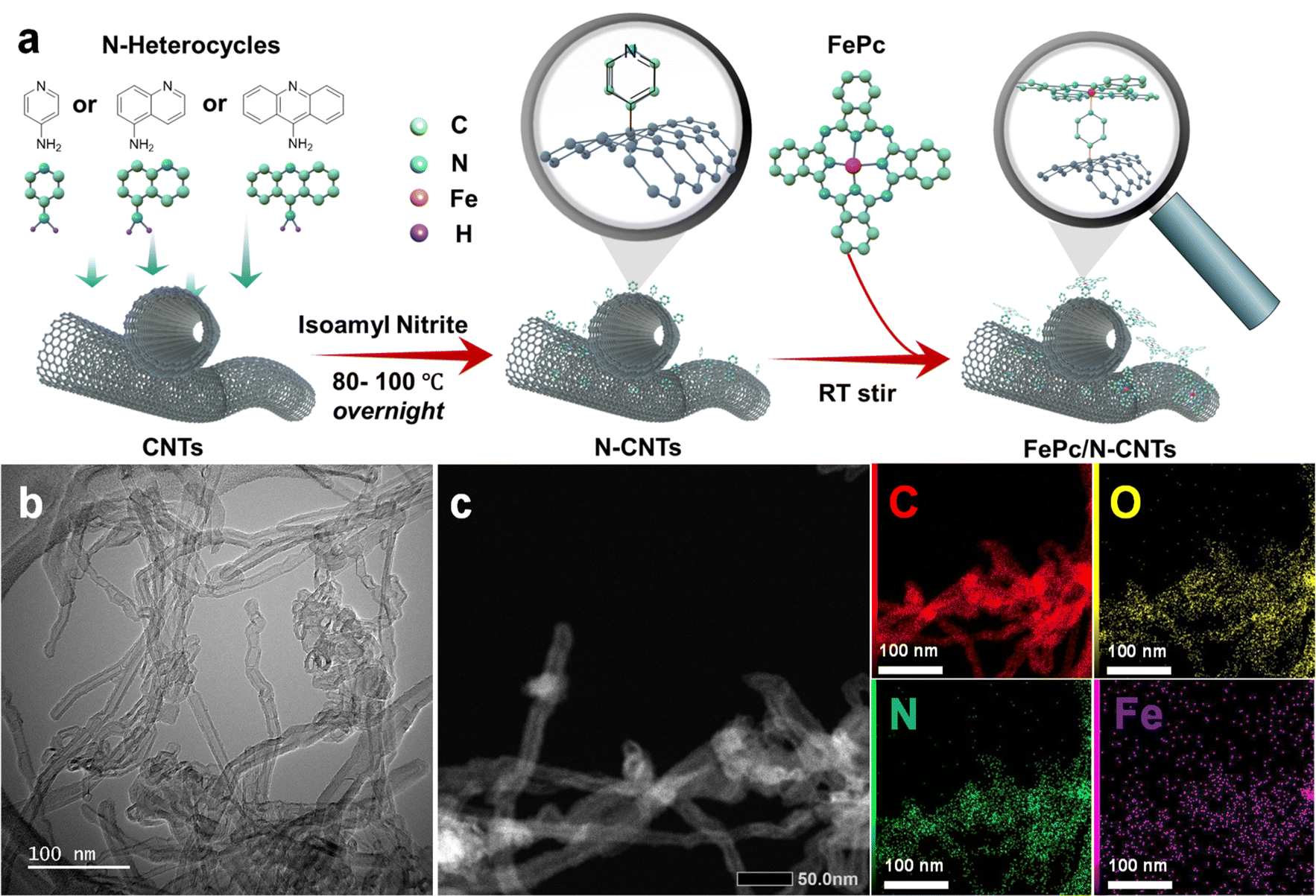 | ||
| Fig. 1 (a) Schematic illustration of the overall synthetic procedure for FePc/N-CNTs; (b) TEM image of FePc/Py-CNT; (c) HAADF-STEM of FePc/Py-CNT and EDS elemental mapping. | ||
The successful covalent functionalization of these molecules was verified through Raman spectroscopy and XPS. In Fig. S1a,† the Raman spectra of all N-CNT samples exhibited two distinctive bands at 1360 and 1590 cm−1, corresponding to the disordered carbon (D band) and graphitic carbon (G band), respectively.27 Additional peaks were observed at ∼1620 cm−1, attributed to the D′ band of CNT.28 Following the aryl diazonium reaction, a slight increase in the ID/IG ratio was noted, likely stemming from alterations in the surface lattice as a result of covalent grafting.29,30 XPS analysis was employed to assess the nitrogen content. Notably, the XPS survey spectra revealed the presence of a nitrogen peak in all functionalized CNT samples compared to pristine CNT, indicating the successful anchoring of N-heterocycles on the CNTs (Fig. S1b†). The calculated coherence length La decreases from 128 nm (CNTs) to approximately 110 nm (N-CNTs), suggesting alterations in the structural integrity of the CNT surface and decreased crystallinity due to the functionalization process.31–33 By adjusting the reaction conditions (Table S1†), the nitrogen content in each functionalized N-CNTs was 1–2 at% based on XPS measurements (Fig. S2e and Table S2†), with the oxygen content both before and after the aryl-diazonium reaction maintained at approximately 2–3 at% (Table S2†). This suggests that surface defects primarily result from the diazonium reaction rather than the oxidation of the CNT during synthesis. Additionally, high-resolution N 1s spectra and C 1s spectra were fitted and analyzed (Fig S2 and S3†), revealing that the signal at approximately 399.3 eV corresponds to the pyridine nitrogen atom, while the signal at approximately 401.2 eV, similar to graphite N in pyrolyzed materials,34 is attributed to the pyridinium nitrogen atom, likely stemming from the protonation of the pyridine N atom during the synthesis process.35
The N-heterocycle-functionalized CNTs were used as a platform to coordinate FePc macrocycles in order to achieve the desired penta-coordinated structure, as illustrated in Scheme 2. To ensure successful axial functionalization and comprehend the impact of the axial group, it is crucial to uniformly disperse FePc onto the surface and to minimize aggregation. Considering the previously reported low solubility of FePc (131.62 mg L−1 ± 65.81 mg L−1 in DMF),36 FePc solutions with a concentration of 50 mg L−1 were prepared and then sonicated to avoid aggregation of the planar conjugate structures. The XRD findings presented in Fig. S4† indicate that following FePc adsorption, the (002) peak at approximately 26° in these hybrid materials remains. Notably, no distinct diffraction peaks associated with powdered FePc were observed, indicating no remaining aggregated FePc. The morphology and microstructure of the hybrid materials were comprehensively examined through SEM and TEM. The SEM images in Fig. S5,† along with the low-magnification TEM micrographs in Fig. 1b and S6,† reveal that the hybrid materials maintain the fibrous structure of CNTs, indicative of a low aggregation of FePc and/or polymerization of the N-heterocyclic structures following the two-step reaction. Additional TEM images and corresponding selected area electron diffraction (SAED) of FePc/N-CNTs were provided in Fig. S7.† The diffusive diffraction rings are the characteristic feature of polycrystalline carbon, and the d-space of diffraction rings was measured to be approximately 0.34 nm corresponding to CNT (002) peaks. HAADF-STEM and EDS mapping further confirmed the homogeneous distribution of carbon, nitrogen, oxygen, and iron elements on the CNT surface, as depicted in Fig. 1c and S8.†
XPS was utilized to study the electronic structures of these hybrid materials. The high-resolution Fe 2p spectra in Fig. S9b† displayed FePc powder peaks decomposed into Fe 2p3/2 at 709.3 eV and Fe 2p1/2 at 722.5 eV, respectively. The spectra of FePc/N-CNTs were not fitted due to the strong noise caused by the low loading, while a broader Fe 2p3/2 peak was observed in the hybrid FePc/N-CNT samples compared to the FePc powder. Additionally, a potential positive shift in the Fe 2p3/2 peak in the FePc/N-CNT samples indicated a potential rise in the Fe oxidation state, which is attributed to the change of Fe electron density caused by the functional groups and CNT substrate.37,38 The high-resolution N 1s spectra were studied and shown in Fig. S9a.† The N 1s peak of the FePc powder was deconvoluted into two peaks corresponding to the N species within the FePc structure. Based on the literature,39–41 the peak at 398.7 eV was assigned to the Fe–N signal, while a smaller peak at 400.5 eV was attributed to bridge N (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) at the meso position of the macrocycles. In the hybrid samples, two additional peaks at approximately 399.3 eV and 401.3 eV were allocated to pyridinic N and pyridinium N resulting from the molecular functionalization of the CNTs.
) at the meso position of the macrocycles. In the hybrid samples, two additional peaks at approximately 399.3 eV and 401.3 eV were allocated to pyridinic N and pyridinium N resulting from the molecular functionalization of the CNTs.
The Fe K-edge X-ray absorption near-edge structure (XANES) analysis was utilized to further probe the electronic structure of the Fe atoms in a series of FePc/N-CNT samples. As depicted in Fig. 2a, the FePc powder sample displays a pre-edge peak at ≈7118 eV, attributable to the 1 s to 4pz electronic transition in the symmetrical FeN4 moiety.42,43 However, all the FePc/N-CNT samples show a pre-edge peak at approximately 7114 eV, instead of the peak at 7118 eV, akin to the signature of FePcCl, suggesting the distorted square-planar geometry caused by the axially coordinated groups (pyridine, quinoline, acridine) on the CNTs.16,37 In the Fourier-transformed extended X-ray absorption fine structure (FT EXAFS) analysis shown in Fig. 2b, all the samples exhibited a peak at approximately 1.9 Å, corresponding to the Fe–N coordination path, and an extra peak at 2.9 Å attributed to the second scattering path of the Fe–C coordination, indicating the preservation of the FePc structure. Additionally, no peak around 2.6 Å was observed for any FePc/N-CNTs, demonstrating the absence of Fe–Fe interactions. The specific structural information was confirmed by EXAFS fitting curves of both R space and k space results (Table S3 and Fig. S10†). The simulated Fe–N path coordination number of FePc/N-CNTs is approximately 5, indicating that the FePc molecule is likely axially coordinated by N-heterocycle ligands. Although we cannot entirely exclude the possibility of unanchored FePc within the FePc/N-CNTs, the results suggest that the majority of FePc molecules are probably coordinated to or influenced by the axial ligand. This implied that the FePc macrocycles in the hybrid materials were altered, by the additional nitrogen atom from the functionalized CNTs. The iron content in a series of the hybrid materials was studied by both ICP-MS and XPS and found to be approximately 0.22–0.35 wt% and 0.33–0.52 at% respectively (Table S4†), indicating a consistent Fe content. We conducted N2 adsorption and desorption tests for FePc/N-CNTs to investigate their specific Brunauer–Emmett–Teller (BET) surface area and pore size distribution. As shown in Fig. S11,† all these CNT samples demonstrate limited surface area and the specific BET surface area of FePc/N-CNTs exhibits similar values overall, showing a slight decrease compared to that of pristine CNTs (35 m2 g−1).
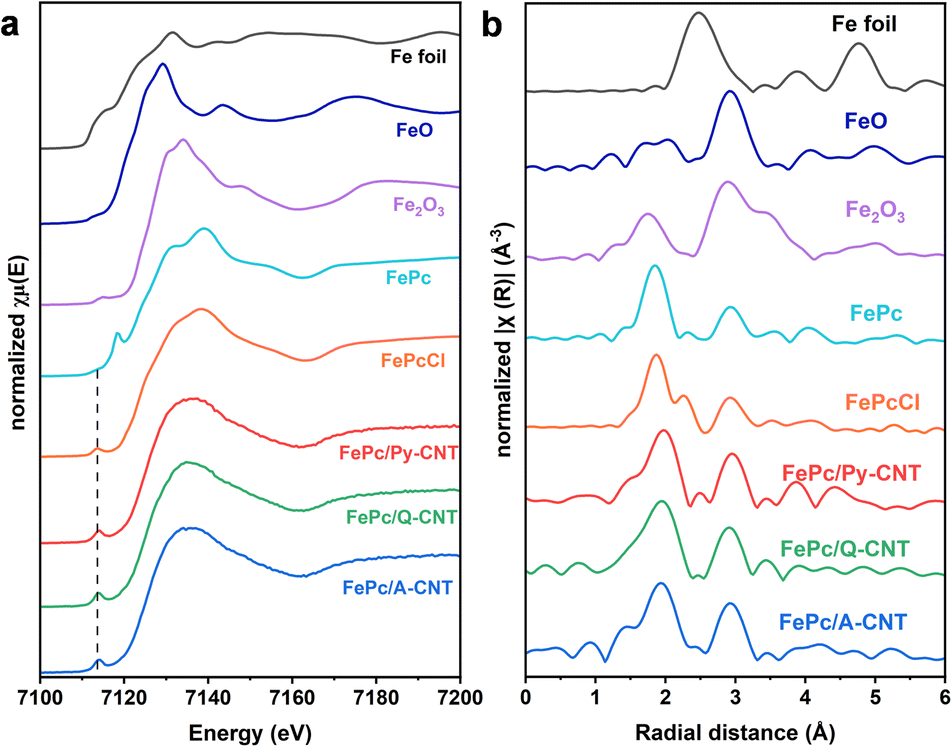 | ||
| Fig. 2 (a) Fe K-edge XANES spectra of as prepared FePc/N-CNTs in comparison with Fe foil, FeO, Fe2O3, Fe(II)Pc, and Fe(III)PcCl; (b) Fourier transform (phase-corrected) of FePc/N-CNTs, Fe foil, FeO, Fe2O3, Fe(II)Pc, and Fe(III)PcCl. | ||
Electrocatalytic ORR performance study
The electrochemical performance of the series of N-molecule functionalized CNTs (in the absence of coordinated Fe macrocycle) and the three FePc/N-CNTs was assessed in a three-electrode system with an RDE in N2 and O2-saturated KOH (0.1 M). A commercially available 20 wt% Pt/C catalyst was chosen as a benchmark for comparison with the synthesized FePc/N-CNTs catalysts. Initial investigations of the N-CNTs revealed low oxygen reduction performance (Fig. S12†), which will not be further discussed.FePc/CNT was prepared by sonicating and stirring FePc solutions with unfunctionalized pristine CNT (see ESI† for details). As observed in Fig. 3a, the catalyst derived from FePc/Py-CNTs displayed significantly improved ORR performance compared to FePc/CNT – highlighting the impact of the axially-coordinated pyridine, consistent with previous literature.13,16,44 While some unanchored FePc may be present in FePc/N-CNTs, the enhanced ORR activity is more likely attributed to the penta-coordinated structure. Interestingly, although all FePc/N-CNT samples exhibited axial coordination as evidenced by XANES, relative ORR kinetic performance varied according to the following sequence: FePc/Q-CNT < FePc/A-CNT < FePc/Py-CNT (Fig. 3b).
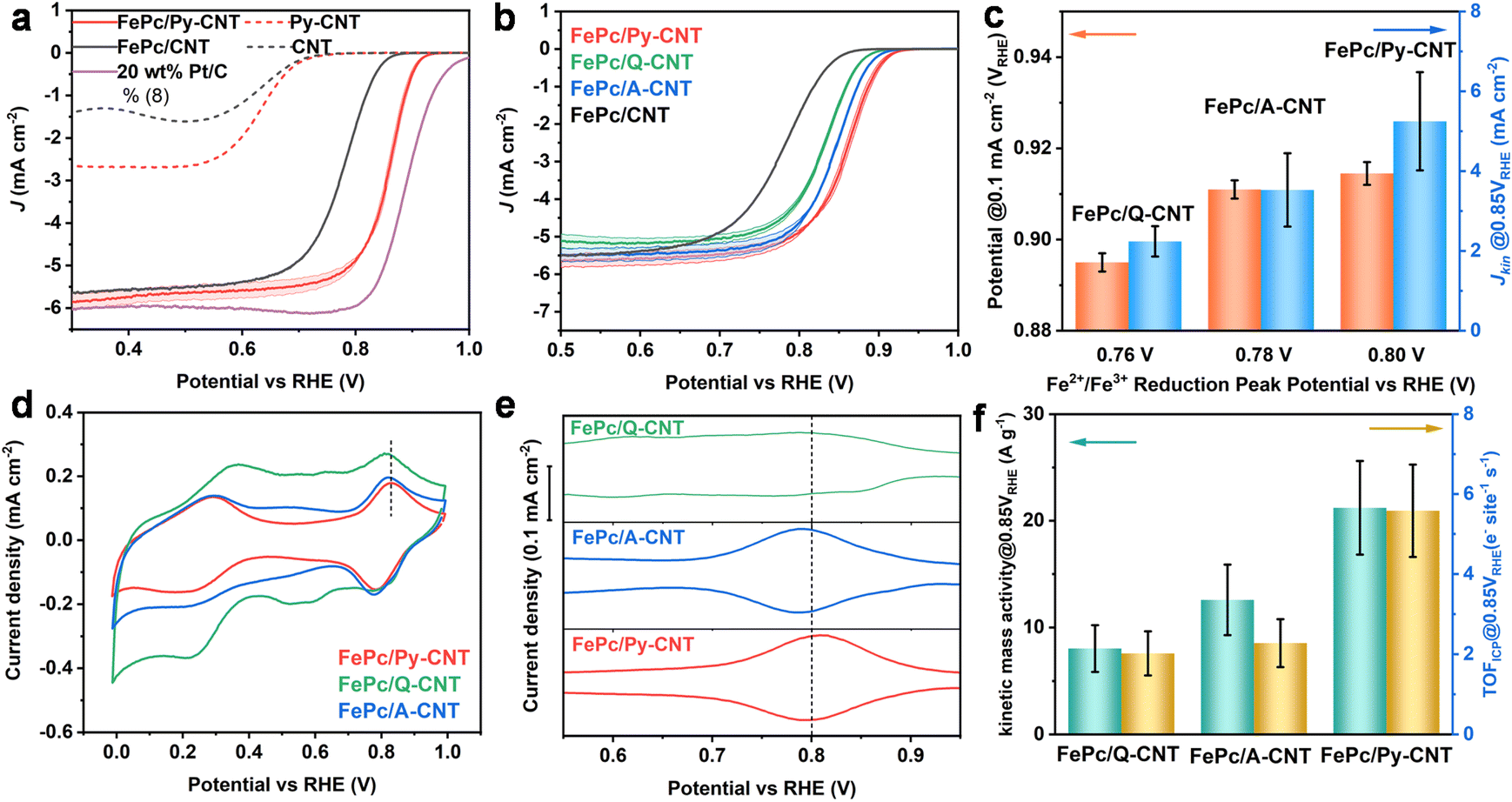 | ||
| Fig. 3 All the RDE data of FePc/N-CNT, CNT, N-CNT, and 20 wt% Pt/C was collected at room temperature (25 °C) with a Hg/HgO reference electrode and graphite rod counter electrode in 0.1 M KOH. (a) and (b) Cathodic LSV curves obtained by subtracting N2-saturated LSV from the O2-saturated result (with a rotation rate of 1600 rpm recorded at 10 mV s−1). Error bars represent the standard deviation from three separate measurements. (c) Comparison of onset potential (potential@0.1 mA cm−2) and kinetic current density@0.85 VRHE. (d) CV N2-saturated, in static conditions with a scan rate of 100 mV s−1 (e) SWV was obtained in N2-saturated, in static conditions, with a frequency of 2 Hz, with 4 mV steps, and a modulation amplitude of 20 mV. (f) Comparison of kinetic mass activity at 0.85 VRHE and turnover frequency at 0.85 VRHE (active site density was calculated based on ICP results). | ||
We hypothesized that the observed kinetic disparity in LSV was caused by variations in the binding energy of different oxygen intermediates during the ORR process. To investigate this, CV and square wave voltammograms (SWV) were performed to monitor the potential ORR intermediates by tracking the position of the high-potential peak (Fe2+/Fe3+ redox peak). This peak is commonly associated with the desorption of *OH intermediates from the Fe center of FePc and can be utilized to characterize the *OH binding energy via tracking the peak position.45 In Fig. 3d and e FePc/Py-CNT displays the highest peak potential, indicating a weak *OH bond and superior ORR kinetics. Conversely, FePc/A-CNT displayed discernible Fe2+/Fe3+ redox peaks with slightly lower potential shifts. In contrast, FePc/Q-CNT exhibited the lowest peak potential and smallest peak size, reflecting relatively sluggish ORR kinetics. Furthermore, we observed that the onset potential (potential at 0.1 mA cm−2) and kinetic current density at 0.85 VRHE increases with higher Fe2+/Fe3+ redox peak potential (Fig. 3c), implying a potential link between this kinetic disparity in LSV and *OH bond energy. In addition, the kinetic mass activity and TOF (based on ICP results) were calculated as shown in Fig. 3f. Given there are some inactive Fe sites in the material caused by stacking of FePc and burying in the material, the electrochemically active Fe sites were calculated based on the Fe2+/Fe3+ peak integration in CV (SDCV) and TOFCV were further calculated, as shown in Table S5.† They show similar SDCV, as a result, their TOF CV exhibits the same trends as TOFICP. All kinetic parameters exhibited a positive correlation with Fe2+/Fe3+ redox peaks, thereby supporting our initial hypothesis. The specific kinetic, as well as other related details, are presented in Table S5.†
In a further investigation on the stability of the best-performing sample (FePc/Py-CNT) by CV scan in N2-saturated 0.1 M KOH at 100 mV s−1 scan rate from 0.5 VRHE to 0.9 VRHE (Fig. S13†), it was observed that the LSV curves before and after cycles nearly overlap, indicating good stability. However, the Fe2+/Fe3+ redox peak area in the CV during 5000 cycles shows a significant decrease, suggesting a loss or deactivation of Fe active sites over the cycling process. More details regarding the test conditions are provided in the ESI.† Combining these results with the ICP findings that all samples exhibited similar Fe content, it is assumed that the kinetic performance of these types of catalysts is more reliant on their structure.
To probe the relationship between the structure and electrochemical activity of these catalysts, the effect of the axial ligand on the FePc environment was investigated further through UV-vis experimental analysis. Comparing the UV-vis spectra of mixed FePc/N-heterocycle samples, in the absence of CNTs, with a pristine FePc solution, FePc/Py displayed a 5–6 nm blue shift in the FePc Q band around 659 nm, as shown in Fig. 4a (photograph and details shown in Fig. S14†). Additionally, the acridine solution showed a slight blue shift (1 nm), while the quinoline solution did not exhibit a pronounced peak shift (pyridine > acridine > quinoline). This blue shift in the FePc Q band indicates that higher energy (lower wavelength) is required for the π → π* transitions of the macrocycles, signifying an electron density change of the macrocycle.
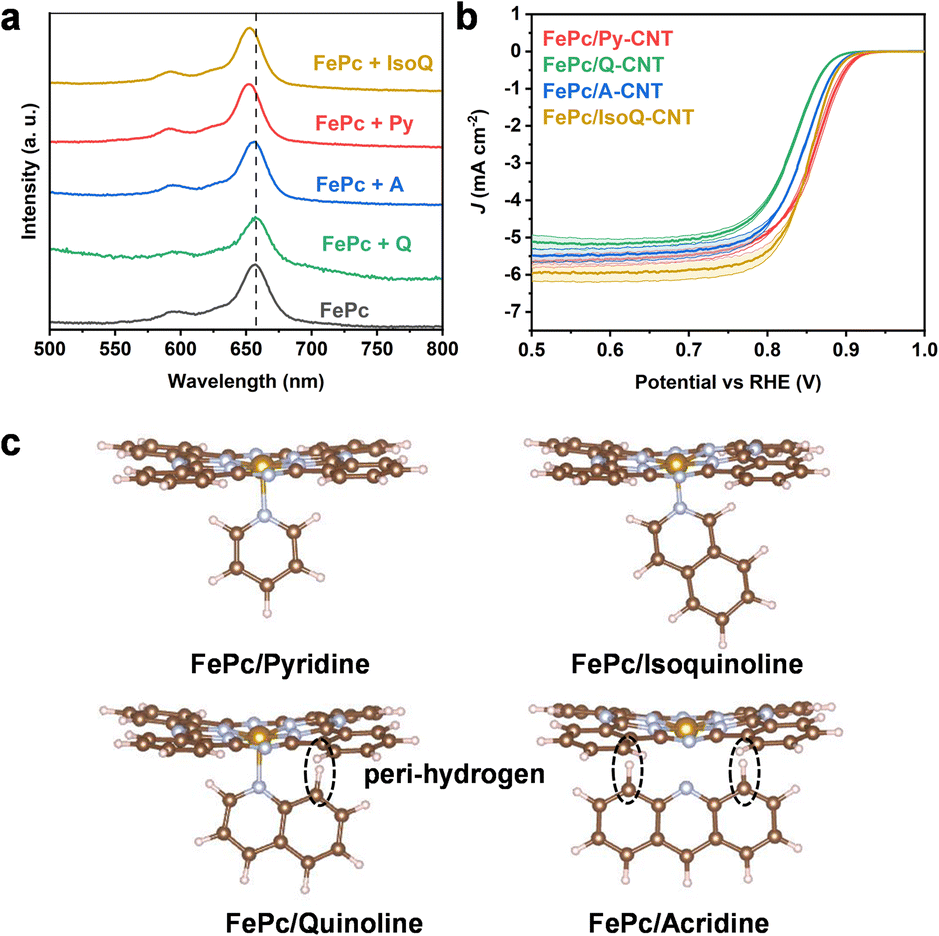 | ||
| Fig. 4 (a) UV-vis of FePc with different molecules (pyridine, quinoline, acridine, isoquinoline) in DMF at room temperature. (b) Cathodic LSV curves obtained by subtracting N2-saturated LSV from the O2-saturated result (in 0.1 M KOH with a rotation rate of 1600 rpm recorded at 10 mV s−1), error bars represent the standard deviation from three separate measurements. (c) The scheme of axial coordination and the steric hindrance caused by peri hydrogens. | ||
One potential explanation for the observed trend in peak shifts could be attributed to the steric hindrance caused by peri hydrogens in Fig. 4c. Previous research has indicated that peri steric hindrance is more prominent in quinoline and acridine in comparison to pyridine, leading to lower nitrogen atom basicity (low pKa).46 Furthermore, acridine consistently exhibits higher basicity than quinoline due to the increased stabilizing effect of the aromatic rings on both sides of the acridinium cation.46,47 This peri-hydrogen steric hindrance may also impact the coordination of Fe and could potentially account for the trends in ORR performance.
In light of this, the use of isoquinoline was proposed as an axial ligand on the surface of CNTs to coordinate with FePc, thereby circumventing the proposed peri-hydrogen steric hindrance seen in quinoline. The feasibility of employing isoquinoline was supported by UV analysis (4–5 nm blue shift, Fig. 4a). The XANES results of FePc/IsoQ-CNT also indicate axial coordination, showing the pre-edge peak at approximately 7114 eV (Fig. S15†). Furthermore, the EXAFS fitting results suggest that FePc/Py-CNT and FePc/IsoQ-CNT show higher coordination numbers in the first shell (Table S3†), which matches the results observed in UV spectroscopy. Interestingly, the FePc/IsoQ-CNT sample also exhibited improved ORR performance compared to FePc/Q-CNT, providing tentative support for the steric hindrance hypothesis; however, it falls short of the catalytic activity of FePc/Py-CNT (as shown in Fig. 4b) and its CV and SWV was shown in Fig. S16.†
Given that the UV experiment mentioned above was conducted using an ideal model, it is important to note that the actual surface structure of functionalized CNTs is more intricate. The diazonium reaction of various molecules may lead to the formation of distinct surface structures, including irregular surfaces that could result from side reactions (more details regarding possible side reactions are discussed in the ESI and Scheme S1†). Additionally, oxygen reduction is a multifaceted process influenced by numerous factors. In this study, the electrochemically active surface area (ECSA) of N-CNTs and electrochemical impedance spectroscopy (EIS) of FePc/N-CNTs have been further investigated.
The ECSA can be deduced by the double-layer capacitance (Cdl) of the N-CNTs, which was estimated via different CV scans at the non-faradaic region, as shown in Fig. S17.† The results showed that A-CNT displayed the highest Cdl while the Py-CNT exhibited the lowest, suggesting that more aromatic rings in the immobilized molecule led to higher Cdl observed.
EIS was performed for FePc/N-CNTs at the mixed kinetic-diffusion region (0.85 VRHE) and the results were analyzed via equivalent circuit fitting and the distribution of relaxation times (DRT) to estimate the resistances. The fitting equivalent circuit used here was shown as an insert figure in Fig. 5a according to previous research, even though the appropriate interpretation of RDE EIS results is still under debate (details are provided in ESI and Fig. S18†).48–50 As shown in Fig. 5a, all these samples show a higher-frequency semicircle regarding charge-transfer resistance and a lower-frequency semicircle associated with O2 transport. Their polarization resistance is similar for all these four samples while significant differences can be observed in the lower-frequency semicircles, suggesting their different resistance to oxygen transport. DRT is an emerging model-free approach to analyzing EIS spectra to provide a clearer representation of the electrochemical process. It has been recently used for calculating and monitoring the evolution of H+ transport, O2 diffusion, and ORR kinetics of Fe–N–C-based PEMFCs.26,51,52 Here, DRT analysis was performed for RDE-based EIS spectra. As can be observed in Fig. 5b, the two main specific peaks could be assigned to oxygen transport resistance (P1 in lower frequency region 0.1–2 Hz) and ORR resistance (P2 in higher frequency region 2–100 Hz), which supports the equivalent circuit fitting of two semicircles' in Nyquist plot. In addition, acid fuel cells in previous works also exhibited these two resistances at the same frequency.26,52 The peak (P1) shifting towards higher intensities and lower frequencies of FePc/N-CNTs could be observed, suggesting their O2 transport is slower and more challenging for FePc/Q-CNT. Fig. 5c shows the resistance calculated from DRT (integrating the area under each peak) and equivalent circuit fitting (Table S6†). The resistance calculated from both methods is similar and the improved oxygen transportation correlates with the enhancement in ORR performance, especially with respect to the kinetic parameters observed. We hypothesize that the variation in oxygen transport could be attributed to the complex surface characteristics of N-CNTs, and the distribution of FePc; however, further investigations are necessary to elucidate the functional properties of the CNT surface and their impact on charge and oxygen transport.
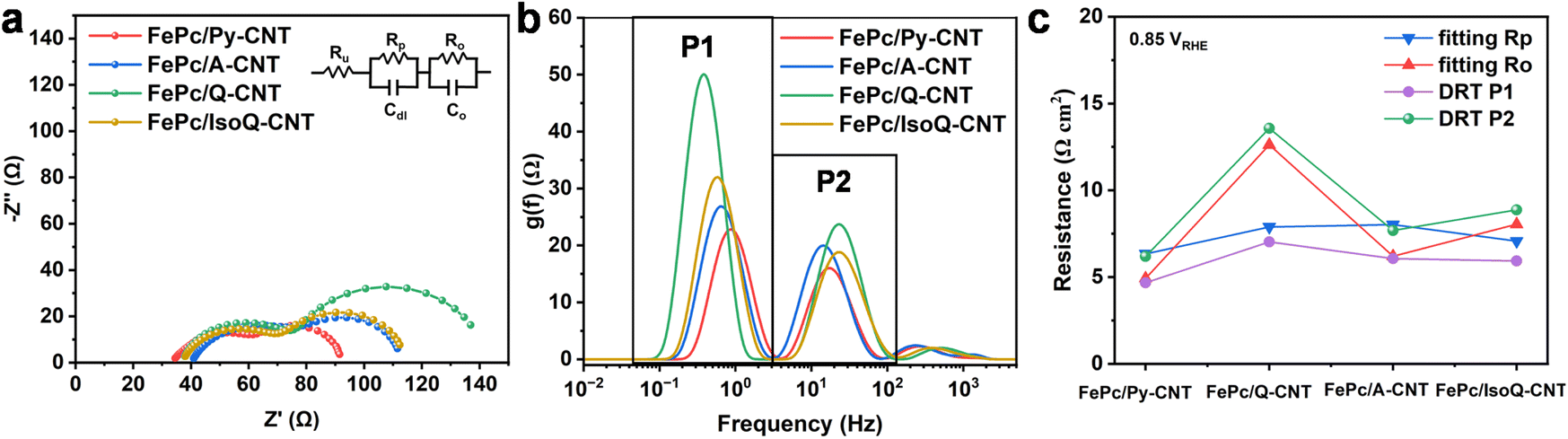 | ||
| Fig. 5 (a) Electrochemical impedance of FePc/N-CNTs was collected at 0.85 VRHE in a frequency range of 0.1 Hz to 1 M Hz, in oxygen-saturated 0.1 M KOH at a rotational rate of 1600 rpm. The results of the electrochemical impedance spectroscopy fitting with the equivalent circuit are detailed in the ESI.†Rp: Polarization resistance and Ro: the oxygen transport resistance. (b) DRT results from EIS spectra. (c) Corresponding calculated resistances from the equivalent circuit fitting and DRT analysis. | ||
Due to the high aryl-diazonium reaction efficiency of amino-isoquinoline, we endeavored to fabricate an IsoQ-CNT with a high functional content. This was to explore the impact of surface N content, which could potentially render the surface more irregular. This was achieved by performing the CNT functionalization with an increased amount of amino-isoquinoline. The synthesized FePc/isoQ-CNT2 exhibited a similar Fe content to FePc/isoQ-CNT (Table S4†), but showed higher functionalization proportions (3 at% N of IsoQ-CNT2, 1.75 at%N of IsoQ-CNT, Table S2†), suggesting that increased loading of isoquinoline led to more aryl radical-mediated surface aggregation, thereby increasing overall N content without increasing available coordination sites for FePc macrocycles. In comparison to FePc/isoQ-CNT, FePc/isoQ-CNT2 exhibited a slight decline in ORR performance (Fig. S19†), implying that attempts to overload N-heterocycles onto the CNT surface may adversely affect the ORR performance.
DFT calculation of ORR
DFT calculations were conducted to investigate the variation in the ORR process between FePc/CNT and FePc/N-CNTs. The DFT model was built based on the proposed ideal axial coordinated FePc. Bader charge analysis reveals that Fe centers in N-heterocycle coordinated FePc (q = 1.12–1.15) exhibit more positive charge than that of FePc (q = 1.11), indicating that more electrons are transferred between the Fe centers and the axial coordinated groups (Fig. S21†). Additionally, the projected density of states (PDOS) (Fig. S22†) showed a downward shift of the D-band center of Fe sites of N-heterocycle coordinated FePc in comparison to FePc, suggesting a reduced binding energy of O2. Notably, a correlation was found between the D-band center and O2 adsorption energy, as shown in Fig. 6a. Given the previous study by Zagal and Koper reported that FePc showed a strong O2 adsorption,45 this downward shift of the Fe D-band center potentially facilitates the ORR process of N-heterocycle coordinated FePc. Interestingly, after geometry optimization, the quinoline ligand in the FePc/Q model appears to rotate slightly to mitigate steric hindrance from the peri-hydrogens (Fig. S23†), which likely accounts for its calculated activity being similar to that of FePc/Py. However, it is important to note that the DFT calculations are based on an idealized model, while the actual CNT composite presents a more complex surface environment. This complexity makes it more challenging for quinoline to overcome steric hindrance by adjusting its position.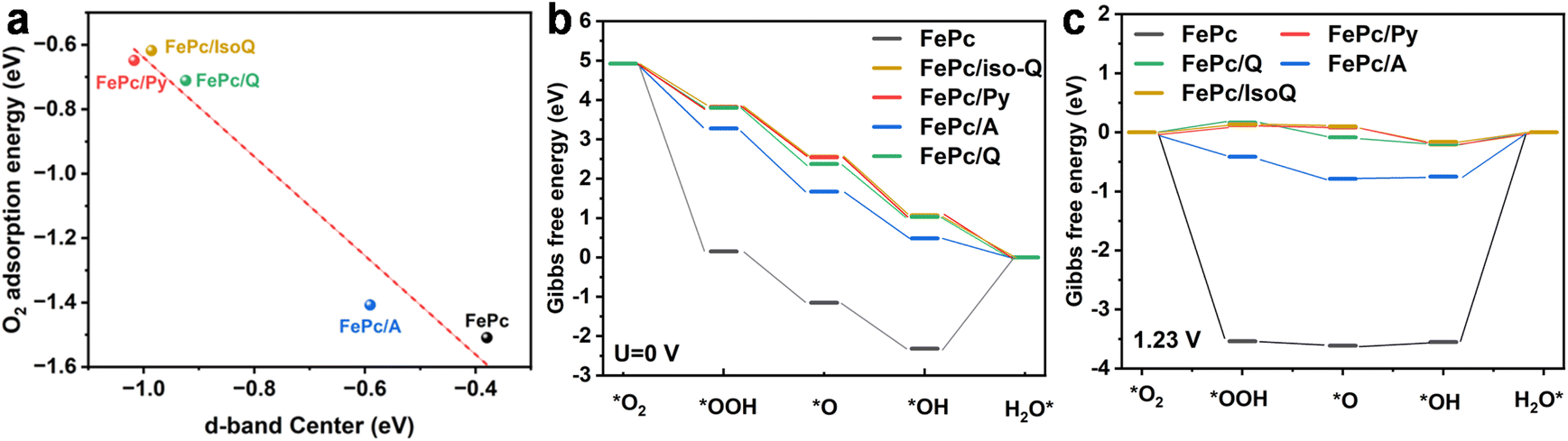 | ||
| Fig. 6 (a) The correlation between the D-band center and the oxygen adsorption energy (b) free energy diagram at 0 V for ORR of FePc and FePc/N-heterocycles, with a single-site mechanism. (c) Free energy diagram at 1.23 V for ORR of FePc and FePc/N-heterocycles. | ||
We assumed the FePc and penta-coordinated FePc proceed via a four-step ORR mechanism (Fig. S20†) through *O2, *OOH, and *OH as intermediates. The model structure of catalysts with intermediates is shown in Fig. S23.† FePc exhibits a substantial Gibbs energy change during the initial step (*O2 to *OOH) (Fig. 6b), suggesting overly strong O2 adsorption. One crucial parameter for evaluating catalytic activity is the limiting reaction barrier, determined from the free energy of the rate-determining step (RDS). To illustrate the thermodynamic limiting step more clearly, Fig. 6c provides a free energy diagram at potentials of 1.23 V for FePc and FePc/N-heterocycles. The RDS of FePc and FePc/N-heterocycles is the oxidation of *OH to H2O* and those axially coordinated FePc/N-heterocycles catalysts show a much lower limiting barrier of approximately 0.20 eV to 0.75 eV compared to that of FePc (limiting barrier of 3.55 eV), suggesting that the axial coordinating FePc/N catalysts can relieve high O2 adsorption. The O2 adsorption energy was investigated as a descriptor of the catalysts' activity, as illustrated in Fig. S24.† It was discovered that a decrease in adsorption energy corresponded to a lower theoretical overpotential. However, we do not observe any apparent correlations between the experimental TOFICP and the O2 binding energy. This observation suggests that there are many other factors influencing the catalytic activity that merit careful consideration.
Conclusions
Overall, this study focused on synthesizing several structurally related FePc/N-CNTs and investigating the effect of these Fe–N axially coordinating materials in ORR catalysis. This has extended the method of covalent functionalization of carbon nanomaterials via the aryl diazonium reaction to afford a wider range of N-heterocyclic ligands that all adsorb FePc onto N-CNTs to achieve penta-coordination. Given that ORR performance is dependent on the catalyst activity and O2 diffusion, it was found that the local structure around the axial ligand does affect the overall ORR activity, with three main factors: (i) O2 transport resistance, (ii) steric bulk around the axial coordination groups (hindrance generally affecting the environment around the Fe center) and (iii) O2 adsorption energy. It was found that FePc/Py-CNTs exhibited the best ORR performance and the variation of axial ligands via the addition of aromatic rings fused to the pyridine may introduce steric hindrance and/or cause potential irregular local surface structures on the functionalized CNTs. This results in sluggish O2 transportation and the observed reduction in catalytic activity. Finally, these findings mean that the local surface structure on modified carbon substrates also needs to be considered when designing and assessing new catalyst activity.Data availability
Data for this article, including XAS, UV, DFT and electrochemistry, are available at figshare at https://doi.org/10.6084/m9.figshare.26499562.Author contributions
Q. L. conceived the work, carried out the synthesis, characterization, and electrochemical testing, and wrote the initial draft. Q. Z. carried out the DFT calculation and wrote the related part. A. P. performed the ICP measurement, assisted with the electrochemical analysis, and revised the manuscript. M. Z. assisted with the sample preparation and XPS characterization. Z. L. recorded the STEM images and performed N2 adsorption. Y. X. co-designed the work and assisted with the sample preparation. P. L. C. provided supervision and revised the manuscript. A. S. aided in the XAS analysis, provided supervision, and revised the manuscript. D. D. T. provided the supervision and revised the final manuscript. M.-M. T. provided funding, co-supervised the work, and revised and edited the final manuscript. C. R. J. provided funding, supervised the work, and revised and edited the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Q. Li, Q. Zhao, Z. Lin, Y. Xu, and M. Zhang thank the Chinese government for the CSC scholarship award. We would also like to thank: the UK Catalysis Hub block allocation group for XAFS beamtime (SP34632-2), M. M. Tititici thanks RAEng CiET 819\2\60 for the financial support. A. Pedersen thanks the EPSRC Centre for Doctoral Training in the Advanced Characterisation of Materials (grant no. EP/L015277/1) and EPSRC Doctoral Prize Fellowship (EP/W524323/1).References
- J. Hyun and H.-T. Kim, Energy Environ. Sci., 2023, 16, 5633–5662 RSC.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- A. Pedersen, A. Bagger, J. Barrio, F. Maillard, I. E. L. Stephens and M. M. Titirici, J. Mater. Chem. A, 2023, 11, 23211–23222 RSC.
- T. Asset and P. Atanassov, Joule, 2020, 4, 33–44 CrossRef CAS.
- A. B. Jorge, R. Jervis, A. P. Periasamy, M. Qiao, J. Feng, L. N. Tran and M. M. Titirici, Adv. Energy Mater., 2019, 10, 1902494 CrossRef.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- J. H. Zagal, S. Specchia and P. Atanassov, Curr. Opin. Electrochem., 2021, 27, 100683 CrossRef CAS.
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Chem. Soc. Rev., 2021, 50, 12985–13011 RSC.
- J. S. Park and D. W. Chang, Energies, 2020, 13, 4073 CrossRef CAS.
- A. Pizarro, G. Abarca, C. Gutiérrez-Cerón, D. Cortés-Arriagada, F. Bernardi, C. Berrios, J. F. Silva, M. C. Rezende, J. H. Zagal, R. Oñate and I. Ponce, ACS Catal., 2018, 8, 8406–8419 CrossRef CAS.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- M. P. Oyarzún, N. Silva, D. Cortés-Arriagada, J. F. Silva, I. O. Ponce, M. Flores, K. Tammeveski, D. Bélanger, A. Zitolo, F. Jaouen and J. H. Zagal, Electrochim. Acta, 2021, 398, 139263 CrossRef.
- C. Z. Loyola, G. Abarca, S. Ureta-Zañartu, C. Aliaga, J. H. Zagal, M. T. Sougrati, F. Jaouen, W. Orellana and F. Tasca, J. Mater. Chem. A, 2021, 9, 23802–23816 RSC.
- Y. Yang, H. Wang, X. Tan, K. Jiang, S. Zhai, Y. Li, X. Xie, N. Chen, H. Zhang and Z. Li, Adv. Funct. Mater., 2024, 34, 2403535 CrossRef CAS.
- H. Meng, X. Liu, X. Chen, Y. Han, C. Zhou, Q. Jiang, T. Tan and R. Zhang, J. Energy Chem., 2022, 71, 528–538 CrossRef CAS.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef PubMed.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y. R. Lu, T. S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- A. A. Mohamed, Z. Salmi, S. A. Dahoumane, A. Mekki, B. Carbonnier and M. M. Chehimi, Adv. Colloid Interface Sci., 2015, 225, 16–36 CrossRef CAS PubMed.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- C. H. Choi, H.-K. Lim, M. W. Chung, G. Chon, N. Ranjbar Sahraie, A. Altin, M.-T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Dražić, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- X. Li, H. Qin, J. Han, X. Jin, Y. Xu, S. Yang, W. Zhang and R. Cao, Adv. Funct. Mater., 2024, 34, 2310820 CrossRef CAS.
- Q. Li, Y. Xu, A. Pedersen, M. Wang, M. Zhang, J. Feng, H. Luo, M. M. Titirici and C. R. Jones, Adv. Funct. Mater., 2023, 34, 2311086 CrossRef.
- Y. Xu, R. Xie, Q. Li, J. Feng, H. Luo, Q. Ye, Z. Guo, Y. Cao, M. Palma, G. Chai, M. M. Titirici and C. R. Jones, Small, 2023, 19, 2302795 CrossRef CAS PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- F. Ciucci and C. Chen, Electrochim. Acta, 2015, 167, 439–454 CrossRef CAS.
- A. Pedersen, R. Z. Snitkoff-Sol, Y. Presman, J. Barrio, R. Cai, T. Suter, G. Yang, S. J. Haigh, D. Brett, R. Jervis, M.-M. Titirici, I. E. L. Stephens and L. Elbaz, J. Power Sources, 2024, 609, 234683 CrossRef CAS.
- A. Jorio and R. Saito, J. Appl. Phys., 2021, 129, 021102 CrossRef CAS.
- S. Osswald, M. Havel and Y. Gogotsi, J. Raman Spectrosc., 2007, 38, 728–736 CrossRef CAS.
- S. L. Rebelo, A. Guedes, M. E. Szefczyk, A. M. Pereira, J. P. Araujo and C. Freire, Phys. Chem. Chem. Phys., 2016, 18, 12784–12796 RSC.
- P. Salice, E. Fabris, C. Sartorio, D. Fenaroli, V. Figà, M. P. Casaletto, S. Cataldo, B. Pignataro and E. Menna, Carbon, 2014, 74, 73–82 CrossRef CAS.
- L. Cheng, C. Ma, W. Lu, X. Wang, H. Yue, D. Zhang and Z. Xing, Chem. Eng. J., 2022, 433, 133527 CrossRef CAS.
- W. Li, D. Peng, W. Huang, X. Zhang, Z. Hou, W. Zhang, B. Lin and Z. Xing, Carbon, 2023, 204, 315–324 CrossRef CAS.
- Z. Li, L. Deng, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2023, 135, 101089 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- Y. Yamada, H. Tanaka, S. Kubo and S. Sato, Carbon, 2021, 185, 342–367 CrossRef CAS.
- F. Ghani, J. Kristen and H. Riegler, J. Chem. Eng. Data, 2012, 57, 439–449 CrossRef CAS.
- X. Li, T. Chen, B. Yang and Z. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202215441 CrossRef CAS PubMed.
- Y. Li, H. Cheng, M. Wang, J. Xu and L. Guan, Mater. Horiz., 2024, 11, 2517–2527 RSC.
- E. V. Basiuk, L. Huerta and V. A. Basiuk, Appl. Surf. Sci., 2019, 470, 622–630 CrossRef CAS.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. V. Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- Z. Mukadam, S. Liu, A. Pedersen, J. Barrio, S. Fearn, S. C. Sarma, M.-M. Titirici, S. B. Scott, I. E. L. Stephens, K. Chan and S. Mezzavilla, Energy Environ. Sci., 2023, 16, 2934–2944 RSC.
- S. C. Sarma, J. Barrio, A. Bagger, A. Pedersen, M. Gong, H. Luo, M. Wang, S. Favero, C. X. Zhao, Q. Zhang, A. Kucernak, M. M. Titirici and I. E. L. Stephens, Adv. Funct. Mater., 2023, 33, 2302468 CrossRef CAS.
- J. Barrio, A. Pedersen, S. C. Sarma, A. Bagger, M. Gong, S. Favero, C. X. Zhao, R. Garcia-Serres, A. Y. Li, Q. Zhang, F. Jaouen, F. Maillard, A. Kucernak, I. E. L. Stephens and M. M. Titirici, Adv. Mater., 2023, 35, 2211022 CrossRef CAS PubMed.
- J. Huang, Q. Lu, X. Ma and X. Yang, J. Mater. Chem. A, 2018, 6, 18488–18497 RSC.
- J. H. Zagal and M. T. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- R. S. Hosmane and J. F. Liebman, Struct. Chem., 2009, 20, 693–697 CrossRef CAS.
- M. Lõkov, S. Tshepelevitsh, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org Chem., 2017, 2017, 4475–4489 CrossRef.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- B. Ruiz-Camacho, J. C. Baltazar Vera, A. Medina-Ramírez, R. Fuentes-Ramírez and G. Carreño-Aguilera, Int. J. Hydrogen Energy, 2017, 42, 30364–30373 CrossRef CAS.
- R. K. Singh, R. Devivaraprasad, T. Kar, A. Chakraborty and M. Neergat, J. Electrochem. Soc., 2015, 162, F489–F498 CrossRef CAS.
- M. Heinzmann, A. Weber and E. Ivers-Tiffée, J. Power Sources, 2018, 402, 24–33 CrossRef CAS.
- Q. Meyer, S. Liu, Y. Li and C. Zhao, J. Power Sources, 2022, 533, 231058 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05483k |
| This journal is © The Royal Society of Chemistry 2024 |