
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the reaction pathway of lithium borohydride-hydroxide-based multi-component systems for enhanced hydrogen storage†
Sweta
Munshi
 *a,
Gavin S.
Walker
a,
Kandavel
Manickam
a,
Thomas
Hansen
b,
Martin
Dornheim
a and
David M.
Grant
a
*a,
Gavin S.
Walker
a,
Kandavel
Manickam
a,
Thomas
Hansen
b,
Martin
Dornheim
a and
David M.
Grant
a
aAdvanced Materials Research Group, Faculty of Engineering, University of Nottingham, UK. E-mail: swetamunshi.sm@gmail.com
bInstitut Laue-Langevin, Grenoble, France
First published on 13th September 2024
Abstract
Complex hydride–metal hydroxide multicomponent hydrogen storage systems have high potential for hydrogen storage because their dehydrogenation thermodynamics can be tuned while maintaining a high hydrogen storage capacity. Out of all the ratios explored using lithium borohydride and lithium hydroxide (LiBH4–xLiOH, x = 1, 3, 4), a particularly promising system is LiBH4–3LiOH with a maximum storage capacity of 7.47 wt%. Thermal and diffraction studies along with in situ neutron diffraction reveal new insights into the intermediate phases involved in the reaction pathway, enabling the identification of a detailed reaction schematic. The onset decomposition temperature was reduced to 220 °C for the hand-milled 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 system, releasing 6 wt% of H2 by 370 °C. Li3BO3 was the main decomposition product. Other than a small trace of water, no toxic gas release was detected along with the H2 release. Ball-milling showed improved reaction kinetics by releasing around 6 wt% between 200 and 260 °C in one step. The destabilization was achieved through the coupling reaction between Hδ− in [BH4]− and Hδ+ in [OH]−. Among all the catalysts investigated, the addition of 5 wt% NiCl2 led to further improvement in reaction kinetics. This resulted in a decrease in the onset decomposition temperature to 80 °C and released 6 wt% of H2 below 300 °C. The systems have exhibited improvements in kinetics and operational temperature, showing potential as a single use hydrogen storage material.
:3 system, releasing 6 wt% of H2 by 370 °C. Li3BO3 was the main decomposition product. Other than a small trace of water, no toxic gas release was detected along with the H2 release. Ball-milling showed improved reaction kinetics by releasing around 6 wt% between 200 and 260 °C in one step. The destabilization was achieved through the coupling reaction between Hδ− in [BH4]− and Hδ+ in [OH]−. Among all the catalysts investigated, the addition of 5 wt% NiCl2 led to further improvement in reaction kinetics. This resulted in a decrease in the onset decomposition temperature to 80 °C and released 6 wt% of H2 below 300 °C. The systems have exhibited improvements in kinetics and operational temperature, showing potential as a single use hydrogen storage material.
1. Introduction
The development of a cost-effective and energy-dense hydrogen storage system with a moderate operating condition is one of the key challenges in the hydrogen economy. Extensive research has explored various materials for hydrogen storage including complex hydrides such as borohydrides [BH4]−,1–4 alanates [AlH4]−5,6 and amides [NH2],−7–9 demonstrating promising gravimetric densities. Lithium-based complex hydrides, particularly due to the lightweight of lithium, gathered interest as potential hydrogen storage materials. Lithium borohydride has gained much interest in this field due to its high gravimetric and volumetric hydrogen capacities of 18.5 wt% and 121 kg m−3, respectively.2 However, they suffer from high thermodynamic stability and slow dehydrogenation kinetics. This necessitates high temperatures, exceeding 400–600°C, for major hydrogen release.1,2In recent studies, several effective methods such as milling,10–12 catalyst addition13–15 and nano-confinement16,17 have been studied to improve the hydrogen storage properties of LiBH4. Among these, the additive-based method is a simple way to tune LiBH4 thermodynamic properties (i.e. destabilization).18 Vajo et al.19 demonstrated the destabilization of LiBH4 by incorporating MgH2 as an additive, leading to a 2LiBH4–MgH2 composite with a reversible hydrogen capacity of 8–10 wt% and rapid reaction kinetics at moderate temperatures (300–400 °C). Subsequent research led to the development of destabilized LiBH4 composites with enhanced hydrogen properties, including LiBH4–Al/Ni/Ti,20–22 LiBH4–MgH2/CaH2/SrH2/CeH2,22–26 LiBH4–SiO2/TiO2/SnO2,27–29 and LiBH4–CaNi5.30 While the additives demonstrably enhance both thermodynamic and kinetic properties of LiBH4, most systems still require decomposition temperatures exceeding 300 °C for major hydrogen release. Therefore, the development of novel lightweight destabilizers capable of catalysing the dehydrogenation of LiBH4 at substantially lower temperatures remains an essential area of investigation.
The Hδ+/Hδ− coupling mechanism proposed by Chen31 and the development of complexes incorporating both H+ and H− were investigated through the reaction of LiNH2 and LiH. In their study, hydrogen release was observed from the mixture [LiNH2–2LiH] at 150 °C, attributed to the reaction between Hδ+ in [NH2]− and Hδ− in LiH. Since then, numerous research studies have been published based on the H+/H− coupling reaction.32–34 This suggests that alkali metal hydroxides (such as LiOH, NaOH, and KOH) are likely to efficiently destabilize LiBH4 due to the interaction between Hδ+ in [OH]− and Hδ− in [BH4]−. The absence of B2H6 or other toxic gases in the released products, as reported in the literature,35 along with lithium's lightweight nature and the high hydrogen content of 11 wt% in the LiBH4–LiOH system, makes LiOH a promising additive for LiBH4. Although many studies have focused on LiBH4 hydrolysis in the LiBH4–H2O system36–38 which has some similarities with hydroxide systems, the solid–liquid reaction for hydrolysed systems raises significant safety concerns due to the unpredictable generation of hydrogen, as well as issues related to hydrogen efficiency, cost of borohydrides and poor reversibility. Investigating the destabilization of LiBH4 using alkali metal hydroxides in a solid–solid reaction could address many of these challenges, making it an area of considerable interest.
Cai et al.35 identified LiBH4–xLiOH as the most favourable system among various hydroxide-based options. The degree of LiBH4 destabilization by alkali metal hydroxides followed the trend LiOH > NaOH > KOH, attributed to the superior acidity of LiOH, due to lithium's highest electronegativity, releasing H2 at 200 °C for the LiBH4–LiOH system. Vajo et al.39 investigated similar systems and found that LiOH greatly destabilises LiBH4 by releasing hydrogen at 270 °C for the LiBH4–4LiOH composite. Due to magnesium's highest electronegativity, among all the hydroxides explored, LiBH4–xMg(OH)2-based systems showed an onset decomposition at 150 °C.40 However, the majority of the stored hydrogen was released above 300 °C, making this system unattractive.
The destabilised system LiBH4–xLiOH reported by researchers,35,39 was principally in the stoichiometric ratio of LiBH4–4LiOH, though discrepancies have been noted in the reported results. The variety of reported products resulting from the same additions emphasizes the crucial role of understanding the reaction pathway to identify the behaviour of the system and how to optimise it. Experiments on a 1:3 stoichiometry which may offer both improved kinetics and an alternative reaction pathway, have not been investigated. The literature on reported systems illustrates the lack of understanding of the system and the process involved, its behaviour under varying stoichiometry and reaction conditions. For this work, a detailed investigation on LiBH4–xLiOH systems was performed, and the stoichiometric ratio and milling treatment were investigated, and the study focused on the LiBH4–3LiOH system which was found to be the optimum stoichiometry. Nickel chloride as a catalyst precursor was investigated to accelerate the dehydrogenation kinetics and in situ powder neutron diffraction (PND) was used to investigate the reaction pathway.
2. Experimental
2.1 Materials
LiBH4 (≥95%, Sigma-Aldrich), anhydrous LiOH (≥98%, Sigma-Aldrich), NiCl2 (98%, Sigma-Aldrich), Li11BD4 (11B > 99, D > 98, Katchem), D2O (99.9%, Sigma-Aldrich) and Li2O (97%, Sigma-Aldrich) were used as received. All samples, except D2O, were stored and handled in an MBRAUN Unilab argon-filled glove box (oxygen and water levels below 0.1 ppm).:1. Ball milling was conducted using a Fritsch Rotary P5 under an argon atmosphere. As-received LiOH was pre-milled at 400 rpm for 2 hours to obtain a fine powder. For LiBH4–xLiOH (x = 1, 3, 4) composites, samples were milled at 200 rpm for 30 minutes.
For the powder neutron diffraction (PND) study, anhydrous litihum deuteroxide (LiOD) was prepared by reacting Li2O with excess D2O according to eqn (1) and (2). About 500 mg of Li2O was slowly added to 1 mL of D2O in a Schlenk flask under argon flow while continuously stirring. This formed hydrous material, LiOD·D2O, which was then heated at 175 °C for 4 h under vacuum to get rid of all the D2O.
| Li2O + 3D2O → 2LiOD·D2O | (1) |
 | (2) |
LiBD4–xLiOD (x = 1, 3, 4) composites were prepared by initially ball milling prepared LiOD separately for 2 hours to reduce the particle size. The LiBD4-xLiOD mixtures were then hand-milled using a mortar and pestle for 15 minutes. This approach was chosen to avoid hydrogen evolution during ball milling and to accommodate the large quantity required for the PND experiment. To facilitate a more accurate comparison with PND data, separate thermal gravimetric analysis (TGA) and temperature programmed desorption – mass spectrometry (TPD-MS) (ESI, ES3†) experiments were conducted using hydride samples prepared under similar milling conditions to the PND samples.
2.2 Characterisation
Thermal gravimetric analysis (TGA) was conducted on a Netzsch 209 F1 instrument under a constant 100 mL min−1 argon flow in the temperature range of 30–570 °C and at a heating ramp of 10 °C min−1. The desired amount of sample was placed in alumina crucibles and sealed within an aluminium pan and a lid. The lid was pierced shortly before loading the pan into the instrument.Temperature programmed desorption – mass spectrometry (TPD-MS) was performed on a Hiden CATLAB microreactor coupled with a mass spectrometer (MS, QIC 20). Around 10 mg of samples were loaded into the sample cell and studied between 30 and –570 °C with a heating ramp of 10 °C min−1 under an argon flow of 100 mL min−1.
Ex situ powder X-ray diffraction (XRD) was performed using a DaVinci Bruker D8 Advance with a CuKα source (λ = 1.5418 Å) in the 2θ range of 10–70°, a step size of 0.02° and a time/step of 0.4 s. Samples were placed on a Si crystal wafer and covered with plastic tape to protect the materials from oxidation.
In situ powder neutron diffraction (PND) measurements were carried out at the Institute Laue–Langevin (ILL) in Grenoble, France. A D1B neutron diffractometer was used for the LiBH4–3LiOH system (λ = 2.52 Å). Around 1.5 g of samples were loaded into a 316 L stainless steel vessel under argon. The experiment was performed under a self-generated D2 atmosphere (initially under a static vacuum). Around 2 g of sample was heated up to 570 °C, starting at 50 °C with a heating ramp of 1 °C min−1 and data were collected in 5 min time intervals. Data analysis was performed on the Large Array Manipulation Program (LAMP), designed by ILL.
Diffuse reflectance infra-red Fourier transform spectroscopy (DRIFTS) was carried out on a Bruker IFS 66/S using a sealed Pike environment cell (HC-900). Samples were mixed with potassium bromide in 1:10 ratio using a mortar and pestle in an argon-filled glove box. The sample was then loaded into a crucible and sealed inside the environment cell. The samples were heated internally with a temperature controller. The spectra were recorded with a resolution of 8 cm−1 after a KBr background scan had been recorded for the same resolution.
3. Results and discussion
3.1 The effect of stoichiometry on the interaction between [BH4]− and [OH]−
The decomposition behaviour of LiBH4–xLiOH (x = 1, 3, 4) composites is illustrated in Fig. 1. Upon heating as-received LiBH4, material decomposition starts at 270 °C, with main decomposition starting above 380 °C and reaching up to 12 wt% by 570 °C, consistent with previous findings.1,2,19 Conversely, the thermal decomposition of LiOH starts at 450 °C, releasing H2O, and reaches about 11 wt% by 570 °C.41 For LiBH4–LiOH, the incorporation of LiOH notably reduces the operating temperature required for hydrogen desorption from LiBH4. It should be noted that hydrogen desorption from LiBH4–LiOH composite exhibits multistep reaction characteristics within the temperature range of 175–450 °C, which is obviously different from the pristine LiBH4. This phenomenon suggests that the presence of LiOH changes the dehydrogenation reaction mechanism of LiBH4, viz., there is a chemical reaction between LiBH4 and LiOH. Initially, as the temperature increases, approximately 4 wt% of hydrogen is released between 170 to 270 °C. Subsequently, between 350 and –450 °C, an additional H2 release of 2.6 wt% was observed, followed by a further 0.9 wt% liberated upon heating beyond 500 °C. By 570 °C, a total mass loss of approximately 7.5 wt% is observed for the 1:1 system out of a theoretical hydrogen storage capacity of 10.9 wt%, indicating that the observed mass loss may primarily result from hydrogen loss. Considering all weight loss corresponds solely to hydrogen, the weight loss corresponds to 68.6% of the total hydrogen, equivalent to 3.4H. To influence the reaction pathway further by increasing the amount of LiOH from 1 mol to 3 mol in LiBH4–xLiOH composites, a gradual high-temperature shift in the dehydrogenation curves is observed, accompanied by a significant increase in the dehydrogenation amount at lower temperatures. In the non-isothermal experiment, a total mass loss of slightly more than 6 wt% is observed within the temperature range of 220 to 260 °C, out of a theoretical hydrogen storage capacity of 7.47 wt%, corresponding to 80.8% of the total hydrogen, i.e., 5.6H. Increase of the added amount of LiOH to the ratio of 1:4, resulted in a notable decrease in the dehydrogenation quantity in the low temperature regime when compared to the 1:3 system. A significant increase in the dehydrogenation temperature was observed. The LiBH4–4LiOH system showed three-step decomposition starting at 230–280 °C, 320–400 °C and a large mass loss step at temperature above 430 °C. The major decomposition for this system was observed at 230 °C, losing around 4.2 wt% by 300 °C. Another small weight loss was observed above 330 °C, losing around 0.7 wt% by 380 °C. Post 440 °C, a big drop in weight could be seen of around 5.6 wt%, continued till 570 °C. Interestingly, a total of around 11.1 wt% mass loss was observed for the 1:4 system out of 6.85 wt% theoretical hydrogen storage capacity which indicates the release of side products along with H2.
 | ||
| Fig. 1 Summarised TGA data for as-received reactants (LiBH4 and LiOH) and LiBH4–xLiOH (x = 1, 3, 4) composites studied on heating to 570 °C at 10 °C min−1 under argon flow. | ||
Fig. 2 shows the TPD-MS curves of the LiBH4–xLiOH (x = 1, 3, 4) composites. It is evident from the curves that mostly the H2 signal was observed during heating from room temperature to 570 °C for all composites, with only a minimal presence of H2O. Thus, it can be inferred that the gas released from the LiBH4–xLiOH (x = 1, 3, 4) composites upon heating is predominantly hydrogen. In the TPD-MS curve for the 1:1 system, five distinct dehydrogenation peaks are observed, providing compelling evidence for a stepwise dehydrogenation process. The peak temperatures for hydrogen desorption from the LiBH4–LiOH composite are measured to be 170, 200, 235, 422, and 540 °C. These temperatures are in good agreement with the results depicted in Fig. 1. Furthermore, an increase in the LiOH content in LiBH4–xLiOH composites causes the dehydrogenation peak to shift from 170 to 220 °C. Interestingly, for 1:3 composites, most of the stored hydrogen was liberated below 300 °C, a temperature below the melting point of LiBH4. No further weight loss or hydrogen release was discernible for the 1:3 system in Fig. 1 and 2. Conversely, for the 1:4 composite, a three-step hydrogen release profile was evident, consistent with observations in Fig. 1. Notably, a prominent H2O peak emerged for the 1:4 composite above 450 °C, indicating that the weight loss at this stage was attributable to the release of H2O from the system which justifies 11.1 wt% loss from the system.
 | ||
| Fig. 2 Summarised TPD-MS data for LiBH4–xLiOH (x = 1, 3, 4) composites studied on heating to 570 °C at 10 °C min−1 under argon flow. | ||
Further insights into the decomposition process are provided by the FTIR profile of LiBH4–xLiOH (x = 1, 3, 4) composites (Fig. 3). A notable decrease in O–H stretching band (3500–3700 cm−1) intensity is noted for the 1:1 composite, as the system undergoes heating from 25 °C to 300 °C,35,42 while there is minimal alteration in bands corresponding to B–H bending (1100 cm−1) and B–H stretching (2200–2500 cm−1).42 This implies that the weight loss observed during the initial dehydrogenation step below 300 °C is attributed to interactions between [BH4]− and [OH]− units, with most of the LiOH being consumed in this process, while the system remains rich in LiBH4. As the temperature increased up to 450 °C, the intensity of the B–H band gradually decreased, indicating that the second phase of weight loss observed in the TGA profile for the 1:1 composite post 350 °C arises from the decomposition of excess LiBH4 after this temperature threshold, thereby signifying the LiBH4 rich nature of this system. The FTIR profile for the 1:3 composite shows a significant reduction in the intensity of O–H and B–H stretching bands when the system is heated from 25 °C to 300 °C, which demonstrates that the liberation of H2 was due to the occurrence of the reaction between Hδ+ in [OH]− and Hδ− in [BH4]− at this stage.
 | ||
| Fig. 3 FTIR data for LiBH4–xLiOH (x = 1, 3, 4) composites upon heating up to 450 °C. | ||
The FTIR profile for the LiBH4–4LiOH system shows a significant reduction in the B–H band compared to O–H when heated to 300 °C. The IR data showed a weak B–H band at 300 °C which was no longer visible at 450 °C but was replaced by two small peaks at similar positions. The appearance of these two small peaks at 2200 and 2500 cm−1 is attributed to different B–H stretching in a closo-dodecaborane Li–B–H cluster coming from LiBH4 decomposition intermediate LixByHz post 320 °C,43,44 which supports the weight loss occurring at the second stage (320–400 °C) in Fig. 1. The third step after 430 °C showed a major weight loss of around 5.6 wt% and was due to H2O release. This is supported by the FTIR spectrum at 450 °C showing the continued presence of the O–H band. Hence, the third step is the thermal decomposition of the remaining LiOH following eqn (3), i.e. for the 1:4 system the LiOH was in excess.41
 | (3) |
Considering both the dehydrogenation temperature and hydrogen capacity, the optimal system is determined to be LiBH4–3LiOH in this case. To further investigate the reaction mechanism of the LiBH4–xLiOH systems, XRD was employed. Fig. 4 (i) shows the XRD patterns for the LiBH4–xLiOH system at room temperature after milling and (ii) after dehydrogenation. The post-milled XRD pattern displayed prominent peaks corresponding to the starting materials, LiBH4 [ICDD pdf card no. 01-070-9017] and LiOH [ICDD card no. 00-032-0564]. XRD analysis of the post-milled sample at room temperature revealed peaks characteristic of a new unknown phase, termed the 1st intermediate phase. When the XRD profile was examined at 570 °C, distinct and strong peaks of the final product, Li3BO3 [ICDD card no. 00-018-0718], were observed, while no peaks from LiBH4 or the 1st intermediate were visible. The presence of the B–O stretching bands at 1260 cm−1 and 1460 cm−1, indicative of the BO3 unit,45,46 became evident in the FTIR profile after reaching 300 °C (Fig. 3), providing additional confirmation for the formation of Li3BO3. Moreover, the presence of the LiH phase was also identified in dehydrogenated samples. For the 1:4 system, strong presence of Li2O was detected which supports LiOH decomposition post 450 °C, following eqn (3). The appearance of LiOH peaks in the dehydrogenated 1:1 and 1:3 samples might result from the substantial presence of LiH in these dehydrogenated samples.47 Exposure to moisture during the transfer process for XRD measurement allows LiH to react with the moisture, forming LiOH. Based on TG/IR/XRD results, the overall reaction for 1:1, 1:3 and 1:4 systems is as given below.
 | (4) |
 | (5) |
 | (6) |
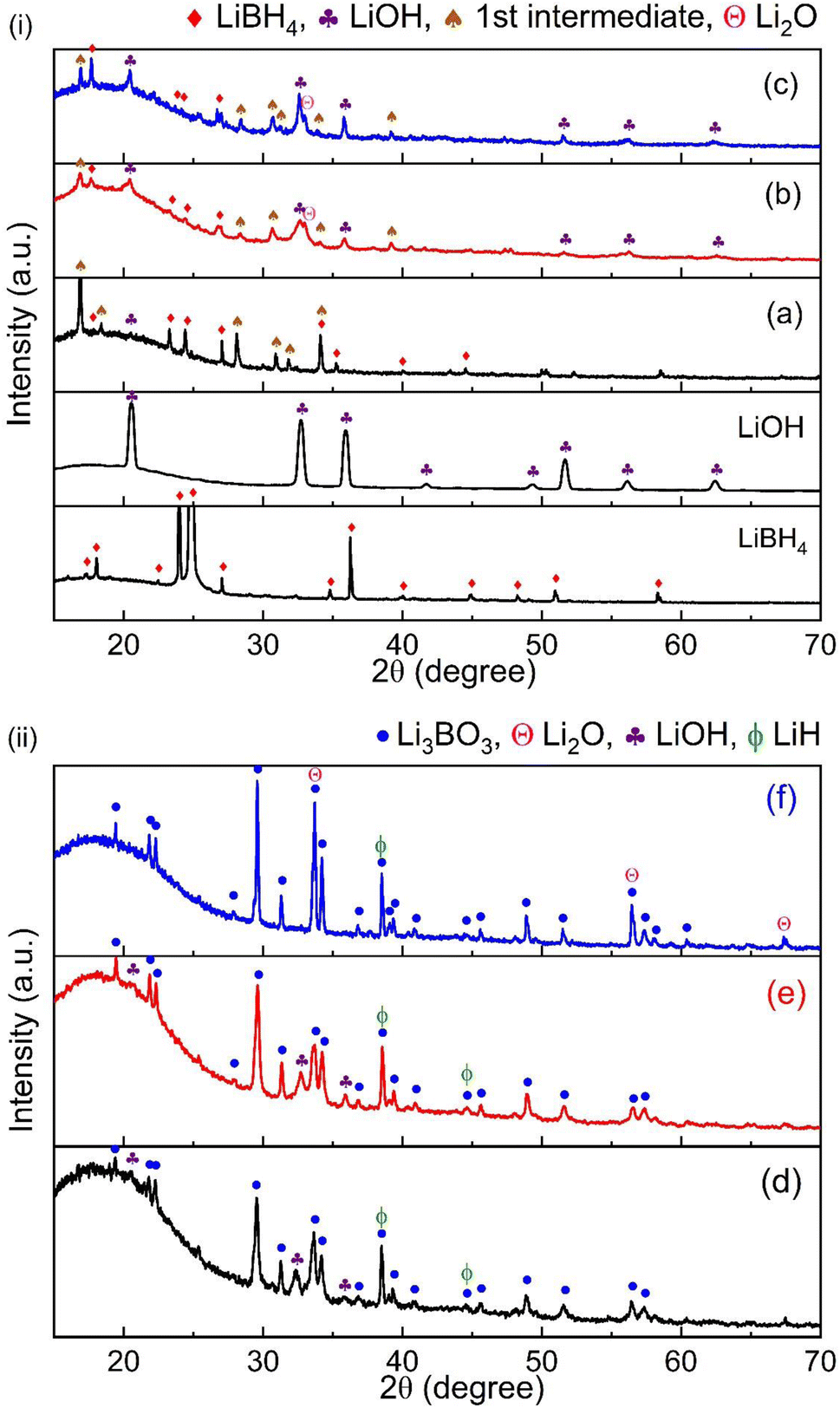 | ||
| Fig. 4 (i) XRD patterns of post-milled room temperature LiBH4–xLiOH (x = 1, 3, 4) composites – (a) 1:1, (b) 1:3, (c) 1:4, and (ii) post decomposition pattern at 570 °C – (d) 1:1, (e) 1:3, and (f) 1:4. | ||
3.2 Inclusion of nickel chloride in the LiBH4–3LiOH composite
The optimal stoichiometry from the work above was the LiBH4–3LiOH system. The effect of a catalyst on this system was studied by incorporating NiCl2 to enhance reaction kinetics. TGA/TPD-MS decomposition data for 5 wt% NiCl2 added LiBH4:3LiOH system are given in Fig. 5, showing a reduction in the decomposition temperature. Starting at 80 °C, a total of 6 wt% mass loss was observed by 300 °C with most of it released at T < 200 °C for the NiCl2 catalysed composite (out of a 7.1 wt% theoretical storage capacity). Total weight loss corresponds to 84.5% of the total hydrogen, i.e. 5.9H for the NiCl2 catalysed system. A small weight loss of 0.7 wt% above 400 °C was also observed by the end of the experiment. The reason behind the catalytic activity from the addition of NiCl2 could be due to forming Ni/Ni–B compounds after reacting with LiBH4, starting at 80 °C (ESI, ES3†). The process involves, LiBH4 reacting with NiCl2 to form Ni/Ni–B, LiCl and release H2.48,49 These compounds likely aid in activating the B–H bond within the material, thereby reducing the reaction kinetic barrier. The TPD-MS profile reveals two H2 peaks at 80 °C and 150 °C. The first peak results from the LiBH4–NiCl2 reaction (Fig. 5), while the second peak is due to the formed Ni/Ni–B acting as a catalyst to facilitate the H+…H− reaction in [BH4]− – [OH]−.
 | ||
| Fig. 5 TGA-TPD-MS data for LiBH4–3LiOH-5 wt% NiCl2 doped systems heated to 570 °C at 10 °C min−1 under Ar flow. | ||
3.3 Reaction mechanism for the LiBH4–xLiOH composite
Given the excellent results obtained with the LiBH4–xLiOH composite, additional investigations into these composites were necessary to fully understand the reaction mechanism. Fig. 6 shows the in situ PND profile for the LiBD4–3LiOD system. The presence of Li2O contamination in the prepared LiOD sample resulted in the observed Li2O peaks in the starting material. | ||
| Fig. 6 PND profile for LiBD4–3LiOD on heating to 570 °C at 1 °C min−1 under a self-generated D2 atmosphere (λ = 2.52 Å).50 | ||
Due to the complexity of the overall 3D plot, it was challenging to understand the occurrence of phase changes and the products generated along with changes in temperature. Therefore, separate intensity profiles were created for each phase to understand the progress of the reaction throughout the decomposition process (Fig. 7).
 | ||
| Fig. 7 Peak intensity profile for different species observed during the PND experiment for the LiBD4–3LiOD system upon heating to 570 °C. | ||
A reminder that the PND samples were hand-milled, separate TGA and TPD-MS results of these samples are given in ESI, ES3.† At the start of the NPD experiment, intense peaks of starting materials, LiBD4 and LiOD were observed. Two peaks from Li2O were also observed at room temperature owing to unreacted oxide in the prepared LiOD. At above 120 °C, the intensity of the reactants LiBD4 and LiOD started to decrease, and the peaks completely disappeared at 220 °C. Besides the reactant phases, the presence of a weak peak at 28° (2θ) at room temperature was attributed to the formation of the 1st intermediate, also evident in the XRD profile (Fig. 4). This peak continued to grow until 180 °C, accompanied by other peaks associated with the 1st intermediate at 30.1°, 47.5° and 57.7° in 2θ, which began to appear after 150 °C. The diffraction pattern for the 1st intermediate matches closely with lithium tetrahydroxyborate, LiB(OH)4 [ICDD: 00-044-0419], but will be the deuterated analogue, LiB(OD)4. The formation of the LiB(OH)4 phase can be evidenced from the peak at 1427 cm−1 assigned to B–OH bonds appearing below 100 °C in the FTIR spectrum of LiBH4–xLiOH composites (Fig. 3).51Fig. 7 displays the PND profile for LiB(OH)4 (decomposed at 180 °C), which is in agreement with the observations by Betourne et al.52 The formation of LiB(OD)4 occurred due to reaction between [BH4]− and [OH]− where [OH]− replaced H in [BH4]−via a nucleophilic reaction following short-living intermediates in the order of BH3OH− > BH2(OH)2− > BH(OH)3− > B(OH)4−.53 A plausible reaction for the formation of LiB(OD)4 is presented in eqn (7) and the mechanism presented in Fig. 8. This indicates the occurrence of a reaction at a temperature below 200 °C.
| 0.05LiBH4 + 0.2LiOH → 0.05LiB(OH)4 + 0.2LiH | (7) |
 | ||
| Fig. 8 Nucleophilic substitution reaction between [BH4]− and [OH]−. | ||
In addition, the appearance of the LiD peak in the same temperature range (Fig. 6 and 7) as to the formation of LiB(OD)4 further supports the proposed reaction in eqn (7). A similar intermediate was also reported by DEMİRCİ et al.36 and Goudon et al.37 during a borohydride hydrolysis reaction, which follows a similar reaction mechanism. The appearance of the LiB(OD)4/LiB(OH)4 phase in our post-milled sample suggests that the formation of this intermediate may have started during the milling process.
Post 180 °C, with decreasing intensity of LiB(OD)4 along with the reactants, a new pattern emerged, identified as the 2nd intermediate, in the PND pattern (Fig. 6 and 7). The PND pattern for this phase closely matches with the reported pattern of hydrated lithium borate (i.e. LiBO2·0.3H2O) by Koga et al.,54 that is LiBO2·0.3D2O for this PND experiment. Above 180 °C, LiB(OD)4 decomposes to form hydrated LiBO2 and releases H2O. A plausible reaction for LiBO2·0.3D2O formation is proposed in eqn (8).38,52
| 0.05LiB(OH)4 → 0.05LiBO2·0.3H2O + 0.08H2O | (8) |
The released H2O can react with LiD to form Li2O and release H2 as evident with an increase and decrease in the peak intensities of Li2O and LiD after 180 °C, respectively (Fig. 7); the reaction mechanism is presented in eqn (9). It is established that when a sub-stoichiometric amount of water is present only to interact with a single surface layer of LiH, Li2O forms.47,55 This reaction involves the release of H2 from the system, supported by a small increase in pressure observed in the PND pressure profile (Fig. 7). This also shows that only a small portion of reactants participate in this reaction mechanism, which did not significantly contribute to the increase in pressure, indicating that it is a side reaction. To further strengthen this argument, TGA and TPD-MS results for hand-milled LiBH4–3LiOH were analysed, ES3 (ESI).† At above 180 °C, the TGA results indicate a minor mass loss of around 0.2 wt% with corresponding results from TPD-MS, showing a broad H2 peak within the same temperature region. These findings provide further support to LiB(OH)4 decomposition above 180 °C. The stoichiometry of these side reactions, derived from the TG weight loss data, is presented below.
| 0.17LiH + 0.08H2O → 0.08Li2O + 0.17H2 | (9) |
At temperatures >220 °C, the peak intensities of the LiBD4/LiOD/LiBO2·0.3H2O phases were drastically reduced and subsequently disappeared (Fig. 6 and 7). This stage is also accompanied by a small pressure rise of up to 1.5 bar in the PND pressure profile. These results are aligned with the TGA results, showing a mass loss of around 0.7 wt% by 270 °C, starting at 220 °C (ESI, ES3†). The TPD-MS profile showed two small peaks of H2 in the same temperature region, confirming that the weight loss was due to H2 release from the system and occurred in two steps. This could be due to some of the activated LiBH4 and LiOH reacting via the Hδ+–Hδ− coupling reaction in 1:3 ratio to release H2. The stoichiometric reaction based on TGA weight loss is stated in eqn (10) and described in Fig. 9. Another small step of weight loss observed above 255 °C was due to LiBO2·0.3D2O decomposition following eqn (11) (ref. 56) forming dehydrated amorphous LiBO2. The presence of a shoulder in the TGA data above 250 °C, accompanied by the emergence of a small H2 peak in the TPD-MS profile, provides additional support for the argument. No appearance of LiBO2 post 250 °C along with the disappearance of Li2O suggested that Li2O reacted with LiBO2 to form the high temperature stable borate, Li3BO3, following eqn (12). This can be also explained with the Li2O·B2O3 binary phase diagram.57 Within the Li2O–B2O3 system, multiple distinct phases can exist at different temperatures based on reactant stoichiometry, such as LiBO2, Li3BO3, Li2B4O7, Li4B2O5 and several others. Given the reaction conditions and dominance of Li3BO3 as the main reaction product, support LiBO2 takes up Li2O to form Li3BO3.
| 0.1LiBH4 + 0.3LiOH → 0.1Li3BO3 + 0.1LiH + 0.3H2 | (10) |
| 0.05LiBO2·0.3H2O → 0.05LiBO2 + 0.01H2O | (11) |
| 0.05LiBO2 + 0.05Li2O → 0.05Li3BO3 | (12) |
 | ||
| Fig. 9 Coupling reaction mechanism between Hδ+–Hδ− in the [BH4]−/[OH]− unit. | ||
Upon heating above 270 °C, a noticeable rise in pressure data was observed from Fig. 7 along with increasing intensity of Li3BO3 and LiD while all the other phases disappeared. The growth of these two phases persisted until the completion of the reaction, indicating their status as primary products. This conclusion is further supported by post-decomposition XRD data for the LiBH4–3LiOH system (Fig. 4). To understand the reaction better, the TGA and TPD-MS data (ESI, ES3†) for hand-milled hydride system were examined. The TGA showed, starting at 270 °C, a large mass loss of around 5.3 wt% by 400 °C, also supported by TPD-MS data, showing an intense peak of H2 over that temperature range. The H2 release was driven by the remaining LiBH4 and LiOH reacting via the Hδ+ – Hδ− coupling reaction in 1:3 ratio to release H2, following eqn (13) and the schematic shown in Fig. 9, also identified as the main decomposition step. The weight loss observed from TGA ( ESI, ES3†) data in the main dehydrogenation step corresponds to 70.9% of the total hydrogen i.e., 5H.
| 0.84LiBH4 + 2.5LiOH → 0.84Li3BO3 + 0.84LiH + 2.5H2 | (13) |
No significant changes were observed above 400 °C, except for a minor peak of H2 detected in the TPD-MS data.
Based on observations, the postulated overall reaction is given below which agrees with the observed results.
| LiBH4 + 3LiOH → 0.99Li3BO3 + 0.96LiH + 0.04Li2O + 3.02H2 | (14) |
A point to note is that a significant change in reaction kinetics was observed for the ball-milled (Fig. 1) and hand-milled (ESI, ES3†) LiBH4–3LiOH systems. Starting at 220 °C, most of the stored hydrogen was released by 260 °C when the ball-milled sample was examined. Based on the results observed it can be concluded that milling effectively enhanced the homogeneity of mixing, which influenced the Hδ+–Hδ− reaction in [BH4]−/[OH]− by bringing them to closer proximity and promoting a single-step hydrogen desorption reaction, while also limiting side reactions temperature below 200 °C.
According to the reaction for the LiBH4–3LiOH system, the reaction pathway for other LiBH4–xLiOH composites becomes evident. In the LiBH4–LiOH system, an abundance of LiBH4 is observed, with surplus LiBH4 undergoing decomposition above 350 °C, as indicated in Fig. 1. The multistep hydrogen release below 300 °C for this system can be attributed to the occurrence of side reactions at lower temperatures. These include the decomposition of LiB(OH)4 after 170 °C, the reaction of excess LiBH4 with released H2O (200 °C), and the subsequent coupling reaction between LiBH4 and LiOH (230 °C) in Fig. 2. Conversely, in the LiBH4–4LiOH system, an excess of LiOH is present, with additional LiOH decomposing beyond 450 °C, also illustrated in Fig. 1. This proposition finds further support in the TPD-MS/FTIR (Fig. 2 and 3) profile. Moreover, it can be inferred that systems with a higher ratio of LiOH in LiBH4–xLiOH (x = 3, 4) effectively suppressed the occurrence of the reaction leading to LiB(OH)4 formation. Conversely, in the LiBH4–LiOH system, the limited availability of LiOH influenced the reaction of LiB(OH)4 formation, evidenced by the strong appearance of LiB(OH)4 in the post-milled XRD profile for the LiBH4–LiOH system (Fig. 4).
4. Summary
The outcomes have exhibited improvements in kinetics and operational temperature, which holds potential for further development in technologies such as stationary hydrogen storage or portable hydrogen storage devices, in applications such as military use. The only by-product generated is a minimal amount of water, enabling the direct utilization of the produced gas in PEMFCs without the need for complex purification processes. Unfortunately, due to the exothermic nature (enthalpy of reaction −30 kJ mol−1 of H2) of the LiBH4–3LiOH system (ES1, ESI†), which shows poor reversibility, efforts are underway to improve its performance. Table 1 below summarises optimally configured systems investigated in this study, compared with some reported studies that demonstrate superior decomposition characteristics compared to most reported LiBH4-binary hydride systems.| System parameter | Gravimetric capacity (wt%) | Gravimetric capacity (wt%) in solid state (<300 °C) | Decomposition temperature (°C) | Decomposition enthalpy (KJ mol−1 H2) | Hydrogenation condition | Ref. |
|---|---|---|---|---|---|---|
| LiBH4–3LiOH | 7.47 | 6 | 220–250 | −30 | — | This work |
| 2LiBH4–MgH2 | 10 | 9.2 | 270–585 | +49 | 450 °C/24 bar | 58 and 59 |
| 6LiBH4–CeH2 | 7.4 | 6.1 | 180–430 | +58 | 350 °C/10 bar | 23 and 60 |
| 6LiBH4–CaH2 | 11.7 | 9 | 400–500 | +60 | 450 °C/80 bar | 61 and 62 |
| 6LiBH4–SrH2 | 9.1 | 8.7 | 217–500 | +48 | 450 °C/80 bar | 24 |
5. Conclusion
These results present the first detailed investigation of the LiBH4:xLiOH systems, exploring phase progression, stoichiometry, reaction environment and effect of doping. The robust affinity between Hδ− in LiBH4 and Hδ+ in LiOH serves as the driving force for hydrogen desorption from this system. The presence of the hydroxide group, [OH]−, demonstrated a more effective destabilization effect compared to numerous LiBH4-metal hydride systems. The dehydrogenation temperature for LiBH4–LiOH, LiBH4–3LiOH, and LiBH4–4LiOH decreased to 170 °C, 220 °C, and 230 °C, respectively and showed faster kinetics compared to pristine LiBH4. The results suggested the reaction was dependent on the [BH4]−:[OH]− ratio. As the [OH]− content increases, the dehydrogenation temperature also increases incrementally. This suggests a varying interaction between [BH4]− and [OH]− based on their stoichiometry. In the case of LiBH4–LiOH, one [OH]− reacts with [BH4]− which facilitates the liberation of H2 at a lower temperature. The limited availability of LiOH in the LiBH4–LiOH system indeed favours a nucleophilic reaction mechanism where the oxygen atom in the hydroxide (OH−) group attacks the boron–hydrogen (B–H) bond, resulting in the formation of the tetrahydroxyborate ion, B(OH)4−. This process follows a series of short-lived intermediates in the order: BH3OH− → BH2(OH)2− → BH(OH)3− → B(OH)4−. However, in systems with higher LiOH ratios, such as LiBH4–xLiOH (x = 3, 4), the interaction between [BH4]− and [OH]− stabilizes [BH4]−, leading to an observed increase in the dehydrogenation temperature. The higher concentration of LiOH in the milling pot likely alters the chemical environment, facilitating a different mechanism. To explore this further, the reaction mechanism has been studied and is discussed below. Specifically, the excess LiOH may reduce the distance between hydrogen cations (H+) and hydride anions (H−), stabilizing the system and favouring the coupling of H+ and H− to form hydrogen gas. This H+–H− coupling reaction becomes more prominent at temperatures above 200 °C, as observed in the LiBH4–xLiOH (x = 3, 4) systems. The primary decomposition product for all systems was Li3BO3. By altering the stoichiometry of LiOH, the reaction pathway was modified. Among all, the LiBH4–3LiOH system demonstrated by far the most favourable attributes. The addition of NiCl2 catalysed the system, resulting in a further significant decrease in the hydrogen desorption temperature from 220 °C to 80 °C. The LiBH4–3LiOH and LiBH4–3LiOH – 5 wt% NiCl2 systems stand out not only for their impressive gravimetric capacity but also for their solid-state reactions at temperatures below 300 °C (below the melting temperature of reactants).
6. Future work
Reversibility of these systems is challenging due to the exothermic nature of the reactions. However, recent studies have explored the possibility of regenerating borohydride from borate end products. Zhu et al.63 reported a cost-effective method to synthesize Mg(BH4)2 by converting B–O bonds to B–H bonds, achieved by milling MgH2 with borates such as B2O3/Mg(BO2)2 to produce Mg(BH4)2. This process does not require high-pressure hydrogen but does necessitate the use of the expensive reducing agent MgH2. Given that Li3BO3 and LiH are the primary reaction products in the LiBH4–3LiOH system, exploring the potential to regenerate LiBH4 by milling these end products without the need for an external reducing agent like MgH2 or additional magnesium remains a significant and promising area for future research.Also, Li3BO3 has several other important applications, such as being used as a CO2 absorbent64 or as an electrode material65,66 in the battery industry. Investigating the potential to reuse this product or regenerate it could be an interesting avenue for future work to enhance the overall efficiency of the system.
Data availability
XRD data can be accessed from here: https://figshare.com/s/a5919b638257530eac28. Neutron data: neutron data id officially published in the ILL website and can be downloaded from: https://doi.ill.fr/10.5291/ILL-DATA.1-04-185.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the United Kingdom CDT programme – Fuel Cell & Their Fuels, hosted by University of Nottingham and funded by Engineering and Physical Sciences Research Council (EPSRC) (grant ref. EP/L015749/1) and Leverhulme Trust International Professorship Program (grant ref. LIP-2021-018). The authors extend their gratitude to Andrew Weilhard and David Furniss for their assistance with TPD-MS and FTIR studies, and to Saad Salman for reviewing the manuscript. We would also like to thank ILL France (exp no: doi.ill.fr/10.5291/ILL-DATA.1-04-185) for hosting us and helping us with the neutron experiment.References
- A. Züttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, LiBH4 a new hydrogen storage material, J. Power Sources, 2003, 118(1), 1–7, DOI:10.1016/S0378-7753(03)00054-5.
- A. Züttel, S. Rentsch and P. Fischer, et al., Hydrogen storage properties of LiBH4, J. Alloys Compd., 2003, 356–357, 515–520, DOI:10.1016/S0925-8388(02)01253-7.
- M. Paskevicius, L. H. Jepsen and P. Schouwink, et al., Metal borohydrides and derivatives – synthesis, structure and properties, Chem. Soc. Rev., 2017, 46(5), 1565–1634, 10.1039/C6CS00705H.
- M. B. Ley, L. H. Jepsen and Y. S. Lee, et al., Complex hydrides for hydrogen storage – new perspectives, Mater. Today, 2014, 17(3), 122–128, DOI:10.1016/j.mattod.2014.02.013.
- C. Milanese, S. Garroni and F. Gennari, et al., Solid State Hydrogen Storage in Alanates and Alanate-Based Compounds: A Review, Metals., 2018, 8(8), 567, DOI:10.3390/met8080567.
- K. Suárez-Alcántara, J. R. Tena-Garcia and R. Guerrero-Ortiz, Alanates, a Comprehensive Review, Materials, 2019, 12, 2724, DOI:10.3390/ma12172724.
- J. B. Eymery, S. Cahen, J. M. Tarascon and R. Janot, Hydrogen storage by reaction between metallic amides and imides, 2007, https://www.osti.gov/etdeweb/biblio/21100402 Search PubMed.
- H. Cao, Y. Zhang, J. Wang, Z. Xiong, G. Wu and P. Chen, Materials design and modification on amide-based composites for hydrogen storage, Prog. Nat. Sci.: Mater. Int., 2012, 22(6), 550–560, DOI:10.1016/j.pnsc.2012.11.013.
- S. Qiu, H. Chu, Y. Zou, C. Xiang, F. Xu and L. Sun, Light metal borohydrides/amides combined hydrogen storage systems: composition, structure and properties, J. Mater. Chem. A, 2017, 5(48), 25112–25130, 10.1039/C7TA09113C.
- C. C. Koch, Synthesis of nanostructured materials by mechanical milling: problems and opportunities, Nanostruct. Mater., 1997, 9(1), 13–22, DOI:10.1016/S0965-9773(97)00014-7.
- L. Zaluski, A. Zaluska and J. O. Ström-Olsen, Nanocrystalline metal hydrides, J. Alloys Compd., 1997, 253–254, 70–79, DOI:10.1016/S0925-8388(96)02985-4.
- J. Huot, G. Liang, S. Boily, A. Van Neste and R. Schulz, Structural study and hydrogen sorption kinetics of ball-milled magnesium hydride, J. Alloys Compd., 1999, 293–295, 495–500, DOI:10.1016/S0925-8388(99)00474-0.
- T. Das, S. Banerjee, K. Dasgupta, J. B. Joshi and V. Sudarsan, Nature of the Pd–CNT interaction in Pd nanoparticles dispersed on multi-walled carbon nanotubes and its implications in hydrogen storage properties, RSC Adv., 2015, 5(52), 41468–41474 RSC.
- J. Mao, Z. Guo, X. Yu and H. Liu, Improved reversible dehydrogenation of 2LiBH4 + MgH2 system by introducing Ni nanoparticles, J. Mater. Res., 2011, 26(9), 1143–1150 CrossRef CAS.
- H. Kou, X. Xiao and J. Li, et al., Effects of fluoride additives on dehydrogenation behaviours of 2LiBH4 + MgH2 system, Int. J. Hydrogen Energy, 2012, 37(1), 1021–1026, DOI:10.1016/j.ijhydene.2011.03.027.
- J. J. Vajo and G. L. Olson, Hydrogen storage in destabilized chemical systems, Scr. Mater., 2007, 56(10), 829–834, DOI:10.1016/j.scriptamat.2007.01.002.
- A. F. Gross, J. J. Vajo, S. L. Van Atta and G. L. Olson, Enhanced Hydrogen Storage Kinetics of LiBH4 in Nanoporous Carbon Scaffolds, J. Phys. Chem. C, 2008, 112(14), 5651–5657, DOI:10.1021/jp711066t.
- Solid-state hydrogen storage: materials and chemistry, ed. G. Walker, Elsevier, 2008 Search PubMed.
- J. J. Vajo, S. L. Skeith and F. Mertens, Reversible Storage of Hydrogen in Destabilized LiBH4, J. Phys. Chem. B, 2005, 109(9), 3719–3722, DOI:10.1021/jp040769o.
- X. D. Kang, P. Wang, L. P. Ma and H. M. Cheng, Reversible hydrogen storage in LiBH4 destabilized by milling with Al, Appl. Phys. A: Solids Surf., 2007, 89(4), 963–966, DOI:10.1007/s00339-007-4198-z.
- G. L. Xia, Y. H. Guo, Z. Wu and X. B. Yu, Enhanced hydrogen storage performance of LiBH4–Ni composite, J. Alloys Compd., 2009, 479(1), 545–548, DOI:10.1016/j.jallcom.2008.12.128.
- J. Yang, A. Sudik and C. Wolverton, Destabilizing LiBH4 with a Metal (M = Mg, Al, Ti, V, Cr, or Sc) or Metal Hydride (MH2 = MgH2, TiH2, or CaH2), J. Phys. Chem. C, 2007, 111(51), 19134–19140, DOI:10.1021/jp076434z.
- J. H. Shim, J. H. Lim and R. S. ullah, et al., Effect of Hydrogen Back Pressure on Dehydrogenation Behaviour of LiBH4-Based Reactive Hydride Composites, J. Phys. Chem. Lett., 2010, 1(1), 59–63, DOI:10.1021/jz900012n.
- D. M. Liu, W. J. Huang, T. Z. Si and Q. A. Zhang, Hydrogen storage properties of LiBH4 destabilized by SrH2, J. Alloys Compd., 2013, 551, 8–11, DOI:10.1016/j.jallcom.2012.09.138.
- T. E. C. Price, D. M. Grant, V. Legrand and G. S. Walker, Enhanced kinetics for the LiBH4:MgH2 multi-component hydrogen storage system – The effects of stoichiometry and decomposition environment on cycling behaviour, Int. J. Hydrogen Energy, 2010, 35(9), 4154–4161, DOI:10.1016/j.ijhydene.2010.02.082.
- G. Barkhordarian, T. Klassen, M. Dornheim and R. Bormann, Unexpected kinetic effect of MgB2 in reactive hydride composites containing complex borohydrides, J. Alloys Compd., 2007, 440(1), L18–L21, DOI:10.1016/j.jallcom.2006.09.048.
- M. Au, W. Spencer, A. Jurgensen and C. Zeigler, Hydrogen storage properties of modified lithium borohydrides, J. Alloys Compd., 2008, 462(1), 303–309, DOI:10.1016/j.jallcom.2007.08.044.
- Y. Zhang, W. S. Zhang and M. Q. Fan, et al., Enhanced Hydrogen Storage Performance of LiBH4–SiO2–TiF3 Composite, J. Phys. Chem. C, 2008, 112(10), 4005–4010, DOI:10.1021/jp709814b.
- X. B. Yu, D. M. Grant and G. S. Walker, Low-Temperature Dehydrogenation of LiBH4 through Destabilization with TiO2, J. Phys. Chem. C, 2008, 112(29), 11059–11062, DOI:10.1021/jp800602d.
- M. Meggouh, D. M. Grant, O. Deavin, M. Brunelli, T. C. Hansen and G. S. Walker, Investigation of the dehydrogenation behavior of the 2LiBH4:CaNi5 multicomponent hydride system, Int. J. Hydrogen Energy, 2015, 40(7), 2989–2996, DOI:10.1016/j.ijhydene.2014.12.059.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Interaction between Lithium Amide and Lithium Hydride, J. Phys. Chem. B, 2003, 107(39), 10967–10970, DOI:10.1021/jp034149j.
- Y. Guo, G. Xia, Y. Zhu, L. Gao and X. Yu, Hydrogen release from ammine lithium borohydride, LiBH4·NH3, Chem. Commun., 2010, 46(15), 2599–2601 RSC.
- J. Luo, H. Wu, W. Zhou, X. Kang, Z. Fang and P. Wang, LiBH4·NH3BH3: a new lithium borohydride ammonia borane compound with a novel structure and favourable hydrogen storage properties, Int. J. Hydrogen Energy, 2012, 37(14), 10750–10757, DOI:10.1016/j.ijhydene.2012.04.049.
- T. He, H. Wu and G. Wu, et al., Borohydride hydrazinates: high hydrogen content materials for hydrogen storage, Energy Environ. Sci., 2012, 5(2), 5686–5689 RSC.
- W. Cai, H. Wang, D. Sun, Q. Zhang, X. Yao and M. Zhu, Destabilization of LiBH4 dehydrogenation through H+–H− interactions by cooperating with alkali metal hydroxides, RSC Adv., 2014, 4(6), 3082–3089, 10.1039/C3RA45847D.
- Ü. I. B. I. DEMIRCI, Sodium borohydride for the near-future energy: a “rough diamond” for Turkey, Turk. J. Chem., 2018, 42(2), 193–220 CrossRef.
- J. P. Goudon, F. Bernard, J. Renouard and P. Yvart, Experimental investigation on lithium borohydride hydrolysis, Int. J. Hydrogen Energy, 2010, 35(20), 11071–11076, DOI:10.1016/j.ijhydene.2010.07.023.
- L. Laversenne, C. Goutaudier, R. Chiriac, C. Sigala and B. Bonnetot, Hydrogen storage in borohydrides comparison of hydrolysis conditions of LiBH4, NaBH4 and KBH4, J. Therm. Anal. Calorim., 2008, 94, 785–790 CrossRef CAS.
- J. J. Vajo, S. L. Skeith, F. Mertens and S. W. Jorgensen, Hydrogen-generating solid-state hydride/hydroxide reactions, J. Alloys Compd., 2005, 390(1), 55–61, DOI:10.1016/j.jallcom.2004.08.042.
- Y. Liu, Y. Zhang, H. Zhou, Y. Zhang, M. Gao and H. Pan, Reversible hydrogen storage behaviour of LiBH4–Mg(OH)2 composites, Int. J. Hydrogen Energy, 2014, 39(15), 7868–7875, DOI:10.1016/j.ijhydene.2014.03.137.
- J. M. Kiat, G. Boemare, B. Rieu and D. Aymes, Structural evolution of LiOH: evidence of a solid–solid transformation toward Li2O close to the melting temperature, Solid State Commun., 1998, 108(4), 241–245, DOI:10.1016/S0038-1098(98)00346-9.
- H. Kou, G. Sang and Y. Zhou, et al., Enhanced hydrogen storage properties of LiBH4 modified by NbF5, Int. J. Hydrogen Energy, 2014, 39(22), 11675–11682, DOI:10.1016/j.ijhydene.2014.05.179.
- C. Zhou, J. B. Grinderslev and L. N. Skov, et al., Polymorphism, ionic conductivity and electrochemical properties of lithium closo-deca-and dodeca-borates and their composites, Li2B10H10–Li2B12H12, J. Mater. Chem. A, 2022, 10(30), 16137–16151 RSC.
- M. P. Pitt, M. Paskevicius, D. H. Brown, D. A. Sheppard and C. E. Buckley, Thermal stability of Li2B12H12 and its role in the decomposition of LiBH4, J. Am. Chem. Soc., 2013, 135(18), 6930–6941 CrossRef CAS PubMed.
- J. Zhu, Y. Mao, H. Wang, J. Liu, L. Ouyang and M. Zhu, Reaction Route Optimized LiBH4 for High Reversible Capacity Hydrogen Storage by Tunable Surface-Modified AlN, ACS Appl. Energy Mater., 2020, 3(12), 11964–11973, DOI:10.1021/acsaem.0c02140.
- C. C. Zhang, X. Gao and B. Yilmaz, Development of FTIR Spectroscopy Methodology for Characterization of Boron Species in FCC Catalysts, Catalysts, 2020, 10(11), 1327, DOI:10.3390/catal10111327.
- R. Ren, A. L. Ortiz, T. Markmaitree, W. Osborn and L. L. Shaw, Stability of Lithium Hydride in Argon and Air, J. Phys. Chem. B, 2006, 110(21), 10567–10575, DOI:10.1021/jp060068m.
- B. J. Zhang and B. H. Liu, Hydrogen desorption from LiBH4 destabilized by chlorides of transition metal Fe, Co, and Ni, Int. J. Hydrogen Energy, 2010, 35(14), 7288–7294, DOI:10.1016/j.ijhydene.2010.04.165.
- W. S. Tang, G. Wu and T. Liu, et al., Cobalt-catalyzed hydrogen desorption from the LiNH2–LiBH4 system, Dalton Trans., 2008, 18, 2395–2399, 10.1039/B719420J.
- M. Sweta, G. David and T. Hansen, Neutron Diffraction Study for LiBH4–LiOH System as a Hydrogen Storage, Materials, 2021 DOI:10.5291/ILL-DATA.1-04-185.
- S. Mondal and A. K. Banthia, Low-temperature synthetic route for boron carbide, J. Eur. Ceram. Soc., 2005, 25(2), 287–291, DOI:10.1016/j.jeurceramsoc.2004.08.011.
- E. Betourne and M. Touboul, Crystallographic data about hydrated and anhydrous lithium monoborates, Powder Diffr., 1997, 12(3), 155–159 CrossRef CAS.
- E. Petit, F. Salles, D. Alligier and U. B. Demirci, Hydrolysis of the Borohydride Anion BH4−: A 11B NMR Study Showing the Formation of Short-Living Reaction Intermediates including BH3OH−, Molecules, 2022, 27(6), 1975 CrossRef CAS PubMed.
- N. Koga and T. Utsuoka, Thermal dehydration of lithium metaborate dihydrate and phase transitions of anhydrous product, Thermochim. Acta, 2006, 443(2), 197–205 CrossRef CAS.
- R. P. Awbery, D. A. Broughton and S. C. Tsang, In situ observation of lithium hydride hydrolysis by DRIFT spectroscopy, J. Nucl. Mater., 2008, 373(1), 94–102, DOI:10.1016/j.jnucmat.2007.05.039.
- K. Chen, L. Ouyang, H. Wang, J. Liu, H. Shao and M. Zhu, A high-performance hydrogen generation system: Hydrolysis of LiBH4-based materials catalysed by transition metal chlorides, Renewable Energy, 2020, 156, 655–664, DOI:10.1016/j.renene.2020.04.030.
- G. Rousse, B. Baptiste and G. Lelong, Crystal structures of Li6B4O9 and Li3B11O18 and application of the dimensional reduction formalism to lithium borates, Inorg. Chem., 2014, 53(12), 6034–6041 CrossRef CAS PubMed.
- X. B. Yu, D. M. Grant and G. S. Walker, A new dehydrogenation mechanism for reversible multicomponent borohydride systems—the role of Li–Mg alloys, Chem. Commun., 2006,(37), 3906–3908 RSC.
- J. J. Vajo, S. L. Skeith and F. Mertens, Reversible storage of hydrogen in destabilized LiBH4, J. Phys. Chem. B, 2005, 109(9), 3719–3722 CrossRef CAS PubMed.
- P. Mauron, M. Bielmann, A. Remhof, A. Züttel, J. H. Shim and Y. W. Cho, Stability of the LiBH4/CeH2 composite system determined by dynamic PCT measurements, J. Phys. Chem. C, 2010, 114(39), 16801–16805 CrossRef CAS.
- A. Ibikunle, A. J. Goudy and H. Yang, Hydrogen storage in a CaH2/LiBH4 destabilized metal hydride system, J. Alloys Compd., 2009, 475(1), 110–115, DOI:10.1016/j.jallcom.2008.08.010.
- Y. Li, P. Li and X. Qu, Investigation on LiBH4–CaH2 composite and its potential for thermal energy storage, Sci. Rep., 2017, 7(1), 41754, DOI:10.1038/srep41754.
- Y. Zhu, S. Shen and L. Ouyang, et al., Effective synthesis of magnesium borohydride via B–O to B–H bond conversion, Chem. Eng. J., 2022, 432, 134322, DOI:10.1016/j.cej.2021.134322.
- T. Harada and T. A. Hatton, Tri-lithium borate (Li3BO3); a new highly regenerable high capacity CO2 adsorbent at intermediate temperature, J. Mater. Chem. A, 2017, 5(42), 22224–22233, 10.1039/C7TA06167F.
- E. Il’ina, S. Pershina, B. Antonov and A. Pankratov, Impact of Li3BO3 addition on solid electrode-solid electrolyte interface in all-solid-state batteries, Materials, 2021, 14(22), 7099, DOI:10.3390/ma14227099.
- M. Zhang, M. Zhu and W. Dai, et al., Surface coating with Li3BO3 protection layer to enhance the electrochemical performance and safety properties of Ni-rich LiNi0.85Co0.05Mn0.10O2 cathode material, Powder Technol., 2021, 394, 448–458, DOI:10.1016/j.powtec.2021.08.083.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05368k |
| This journal is © The Royal Society of Chemistry 2024 |