
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fusion of capsules to produce liquid-filled monoliths for carbon capture†
Chia-Min
Hsieh
a,
Luma
Al-Mahbobi
b,
Smita S.
Dasari
c,
Mohd
Avais
b,
Huaixuan
Cao
 c,
Peiran
Wei
d,
Yifei
Wang
b,
Micah J.
Green
c and
Emily B.
Pentzer
*ab
c,
Peiran
Wei
d,
Yifei
Wang
b,
Micah J.
Green
c and
Emily B.
Pentzer
*ab
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: emilypentzer@tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, College Station, TX 77840, USA
cArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA
dSoft Matter Facility, Texas A&M University, 1313 Research Pkwy, College Station, TX 77845, USA
First published on 9th October 2024
Abstract
Solid–liquid composites (SLCs) combine the properties of solids and liquids, enhancing functionalities and expanding potential applications. Traditional methods for creating SLCs often face challenges such as low mass transfer efficiency, difficulty in controlling separation behavior, and substantial waste production. Herein, we report a new approach to solve these challenges by using disulfide-based responsive polymeric capsule shells to make liquid-filled monoliths for carbon capture. The capsules are prepared through interfacial polymerization and contain either non-polar poly(α-olefin)432 or highly polar 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([HMIM][TFSI]) at 74–82 wt%. Upon gentle heating, the dynamic disulfide bonds of the isolated capsules undergo bond exchange, leading to the fusion of capsule shells into free-standing monoliths that retain >89 wt% of their liquid core and remain stable for at least two weeks. These monoliths demonstrate CO2 absorption rates and capacities comparable to their capsule counterparts; further, in response to radiofrequency (RF), they reach the CO2 desorption temperature in only ∼31 s. This innovative system addresses the limitations of conventional SLC fabrication techniques, offering a versatile and practical approach to fusing polymer capsule shells for applications across separation, energy storage, and carbon capture applications.
1 Introduction
Solid–liquid composites (SLCs) are an emerging class of materials that combine a liquid phase within a solid matrix (Scheme 1). They can expand the landscape of materials science by combining the complementary properties of solid, which provides mechanical support and ease of handling, and liquid, which provides a distinct solvating environment, ionic conductivity, or magnetic responsiveness.1–3 These heterogeneous structures can take various forms, including discrete liquid droplets in a solid, co-continuous solid–liquid networks, patterned liquid channels embedded in solids, or bulk liquids encapsulated by solids.1 SLCs provide several advantages over traditional single-phase materials. The combination of liquid and solid phases allows for enhanced mechanical properties, such as flexibility and toughness, while maintaining functional properties intrinsic to the liquid phase, like electrical or thermal conductivity.4–6 Moreover, the liquids within SLCs can endow the material with autonomous healing capabilities, as seen in systems containing healing agent capsules that self-repair upon mechanical damage.7 Properties can be fine-tuned by adjusting the liquid loading and droplet size, offering defined performance of the composite materials. The unique properties of SLCs make them promising candidates for a wide range of applications, from energy storage to environmental remediation and advanced electronics.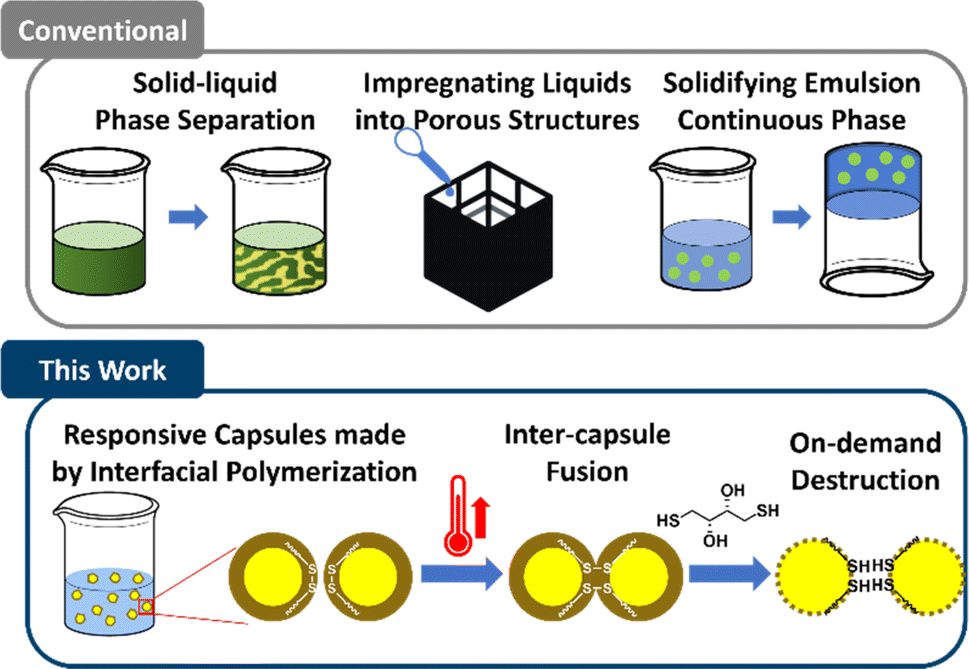 | ||
| Scheme 1 Overview of conventional syntheses of solid–liquid composites (SLCs, top) and the work presented herein using liquid-filled capsule shells of dynamic covalent bonds as feedstocks to produce SLCs (bottom). | ||
Recent developments in SLCs have demonstrated their broad potential. For instance, Ding et al. reported cold-compressed and sintered micro-structured composites that amalgamate ceramics and graphite with a eutectic mixture of lithium and sodium carbonate, effectively addressing the dual challenges of chemical compatibility and thermal efficiency—crucial for advancing energy storage solutions.8 Luo and Huang et al. developed a method to encapsulate ionic liquids (ILs) in polyurea capsules, simplifying the handling of the viscous ILs as solid powder and enabling use for the removal of contaminants from organic solvents or the capture of CO2 without additional energy input (e.g., stirring), thereby opening new avenues for environmental remediation.9–11 Extending this line of innovation, Majidi et al. incorporated liquid metal (LM) droplets within silicone elastomers.6 This combination not only enhanced flexibility and electrical conductivity but also imparted self-healing properties, significantly boosting the durability and adaptability of devices in soft electronics and robotics.
Methods to produce SLCs primarily include solid–liquid phase separation, impregnating liquids into porous structures, and solidifying the continuous phase of emulsions.1 Each approach presents unique limitations: phase separation methods often lack modularity and exhibit poor control over separation behavior, where small parameter changes can lead to process failure.12 Creating porous scaffolds followed by vacuum-assisted liquid infiltration frequently results in substantial liquid waste and increased energy consumption.3 Although solidifying the continuous phase of emulsions offers a promising route due to well-established emulsion platforms, this method requires predetermined geometries and mold fabrication, leading to inefficiencies in liquid feedstock transportation and limiting flexibility in SLC production.9,13 Thus, despite the advantages of SLCs, traditional methods for their fabrication have many limitations.
To address these limitations, we previously developed an innovative approach to encapsulate liquids in capsules with shells containing hindered urea bonds (HUBs), a type of dynamic covalent bond,14 which enabled fusion of capsule shells into monoliths upon compaction and mild heating.15,16 This approach offers several advantages over traditional methods to produce SLCs, including enhanced modularity due to emulsion-based preparation, minimal waste through theoretically complete liquid encapsulation, and on-demand production. While these benefits and the tunable transformation temperature based on HUB steric demands were noteworthy, the choice of dynamic covalent bond chemistry limited what types of liquid could be encapsulated. Specifically, cores of pure IL, of interest in carbon capture,11,17–22 could not be realized because to the core liquid plasticized the polymer shell, stabilizing an intermediate of bond exchange and thereby lowering the transformation temperature and compromising shell stability under ambient conditions. Moreover, an isocyanate is an intermediate of bond exchange with HUBs, which is highly reactive with nucleophiles, including water,14,23,24 and thus limits the practicality of this chemistry for broader SLC applications. Therefore, to significantly expand the possibilities of transforming liquid-filled capsules into SLCs, the dynamic covalent chemistry must be agnostic to the identity of the liquid core and produce capsules that are stable until triggered by an external stimulus, thus fusing on demand.
Herein, we introduce responsive polyurea capsule shells containing disulfide bonds and use thermally induced bond exchange to produce SLCs and demonstrate the versatility of this method with two different core materials: the hydrocarbon lubricant poly(α-olefin)432 (PAO-432) and IL 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([HMIM][TFSI]), the utility of the latter was demonstrated in carbon capture.11,25,26 As depicted in Scheme 1, capsules are prepared via interfacial polymerization in emulsions by reaction of a diisocyanate with cystamine, a diamine containing a disulfide bond, which is chosen for its dynamic nature and is widely used in responsive synthetic polymers.27–42 These capsules contain 74–82 wt% core of either nonpolar PAO-432 or highly polar [HMIM][TFSI], demonstrating the broad applicability of the approach to different core liquids. Upon gentle heating (100 to 160 °C), the shells undergo bond exchange, leading to fusion and formation of a free-standing monolith that retains >89 wt% of the liquid core and is stable under ambient conditions for at least two weeks. Thus, the integration of this dynamic covalent chemistry enables the transformation of a powdery feedstock into free-standing SLCs of nearly any desired shape, giving access to systems that can address not only how composition but also how macroscopic structures, such as shape and surface area, influence performance-related properties. We demonstrate that the IL-containing SLCs absorb CO2 at comparable rates and capacity to the capsule feedstock and that the temperature required for CO2 desorption can be rapidly achieved by applying radiofrequency (RF), thereby enabling efficient and uniform heating for regeneration compared to conventional convection heating approaches.43 Notably, the presence of disulfide bonds also means these materials can be destroyed on demand using reducing agents. The work reported herein showcases the versatility and practicality of this capsule system for producing SLCs, with potential applications across separations,10,13 energy storage,44–50 carbon capture,11,17–21 and more.
2 Results and discussion
2.1 Synthesis of capsules
Liquid-filled SLC capsules were synthesized employing aqueous or non-aqueous emulsions as templates, stabilized by graphene oxide (GO) or 1-octadecylamine-functionalized graphene oxide (C18–GO), respectively. Notably, these particle surfactants were selected with the goal to use them to enhance heating in response to the application of RF.51,52 Adapted from our prior reports,10,13 capsule synthesis involved a two-step process: one monomer (diamine or diisocyanate) was incorporated into the dispersed phase, and then this was introduced to a dispersion of GO in water or C18–GO in octane followed by shear mixing to form an emulsion. Then, a complementary monomer (diisocyanate or diamine) was added to the continuous phase, which initiated step-growth polymerization between the diamine and diisocyanate at the fluid–fluid interface, thereby forming a robust polyurea shell, as depicted in Scheme 2. Two diisocyanates, isophorone diisocyanate (IPDI) and 1,3-bis(2-isocyanato-2-propyl)benzene (BZDI), were used, and the diamine used was cystamine (Cys). For control experiments, i.e., capsules with shells that did not contain disulfide bonds, IPDI and ethylene diamine (EDA) were used as monomers following the same method. Detailed experimental procedures can be found in the Experimental methods. | ||
| Scheme 2 Top: Schematic of PAO and IL capsule synthesis via nanosheet-stabilized emulsions. For PAO capsules, monomer A is diisocyanate, monomer B is diamine, and GO stabilized the emulsion; for IL capsules, monomer A is diamine, monomer B is diisocyanate, and C18–GO stabilized the emulsion. Bottom: Chemical structures of the monomers and core liquids, along with representative polymer chemistry of the shells. | ||
To assess the impact of core polarity on the formation of SLCs and disulfide bond exchange, capsules with core liquids of two distinct polarities, PAO-432 (a non-polar hydrocarbon) and [HMIM][TFSI] (a liquid salt at room temperature), were prepared. Sample nomenclature is established based on the shell and core material, formatted as [diisocyanate]×[diamine]_[core]. For example, IPDI × Cys_PAO denotes a capsule with a polyurea shell composed of IPDI and cystamine, encapsulating a core of PAO. Fig. S1† shows optical microscopy images of all emulsions, with either PAO-432 or [HMIM][TFSI] as the dispersed phase. After interfacial polymerization, 1-propylamine was added to quench residual isocyanates, then capsules were isolated by gravity filtration, and their formation was initially verified by optical microscopy, as shown in Fig. S2.†
2.2 Characterization of PAO capsules
The structural integrity of the synthesized capsules was first established, as any compromises in capsule integrity may lead to leakage of the encapsulated liquid, potentially undermining performance in intended applications. The scanning electron microscopy (SEM) images provided in Fig. 1a–c show the successful synthesis of the three PAO capsules and their morphologies, with insets showing individual capsules. IPDI × Cys_PAO and IPDI × EDA_PAO capsules are spherical, whereas BZDI × Cys_PAO capsules are more ellipsoidal. The non-spherical morphology of the BZDI × Cys_PAO capsules may originate from the hydrolysis of BZDI by water during shear-mixing, which produces an amine that can react with equivalents of BZDI, thereby maintaining the ellipsoidal shape of the emulsion droplets, as seen in Fig. S1b.† A significant difference in size distribution between the capsules and their precursor emulsion droplets is observed. For instance, the diameters of the IPDI × Cys_PAO capsules ranged from 15 to 100 μm, whereas the corresponding emulsion droplets varied from 5 to 40 μm (compare Fig. 1a and S1a†). For BZDI + PAO, both capsules and droplets maintained ellipsoidal shapes, with the long axes of the droplets extending up to 50 μm (Fig. S1b†) and those of the capsules reaching up to 75 μm (Fig. 1b). The larger size of the capsules compared to the emulsion droplets may be attributed to the accumulation of polymer at the droplet interface during interfacial polymerization, which thickens the polymer shell and increases the overall size of the capsules,53 and/or coalescence of smaller droplets with larger ones, which enlarges droplet size concurrent with capsules formation. | ||
| Fig. 1 SEM images of capsules: (a) IPDI × Cys_PAO, (b) BZDI × Cys_PAO, and (c) IPDI × EDA_PAO, with insets showing individual capsules. (d) Particle size distribution of the PAO capsules from laser diffraction. (e) Offset FTIR spectra of the PAO capsules. (f) TGA weight loss profiles of the PAO capsules. | ||
To quantify the distribution of capsule size, the PAO-containing capsules were dispersed in water and characterized by laser diffraction. As depicted in Fig. 1d and detailed in Table 1, the average volume-equivalent diameter was 54.6 ± 50.0 μm for IPDI × Cys_PAO, 51.9 ± 31.0 μm for BZDI × Cys_PAO, and 15.3 ± 8.6 μm for IPDI × EDA_PAO. The disulfide-containing capsules had a broad and bimodal size distribution, whereas the control capsules had a narrower distribution. This irregularity in the disulfide-containing capsules suggests the possibility of inter-capsule fusion during preparation, potentially triggered by addition of 1-propylamine, which can facilitate bond exchange by breaking the disulfide bonds through nucleophilic attack and forming thiolate anions.40,54–57 Residual amine groups may also contribute to unwanted bond exchange at room temperature.
| Capsules | Emulsion size (μm) | Capsule size (μm) | T d,5% (°C) | Fusion temperature (°C) | Capsule liquid component (wt%) | Monolith liquid component (wt%) |
|---|---|---|---|---|---|---|
| a Length of the long axes of the droplets. b Monolith analyzed immediately after preparation. c Monolith left at ambient conditions on the benchtop for 14 days. | ||||||
| IPDI × Cys_PAO | 5–40 | 54.6 ± 50.0 | 201 | 140 | 76.6 ± 0.5 | 68.2 ± 4.7b (60.2 ± 3.9)c |
| BZDI × Cys_PAO | 5–50a | 51.9 ± 31.0 | 214 | n/a | 65.1 ± 0.6 | n/a |
| IPDI × EDA_PAO | 5–40 | 15.3 ± 8.6 | 202 | n/a | 59.5 ± 2.6 | n/a |
The chemical composition of the PAO-containing capsules was investigated using Fourier-transform infrared (FTIR) spectroscopy (Fig. 1e), and the thermal stability of these capsules was assessed by thermogravimetric analysis (TGA, Fig. 1f). The FTIR spectra all displayed characteristic stretching frequencies associated with polyurea, including N–H stretching and bending vibrations at approximately 3320 cm−1 and 1560 cm−1, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1635 cm−1. Notably, no peak due to isocyanate (∼2250 cm−1) was observed, indicating complete consumption of the isocyanate groups. Furthermore, the capsules exhibited pronounced sp3 C–H stretching vibrations between 2850 and 2950 cm−1, supporting the presence of the saturated hydrocarbon (i.e., PAO) in the core.
O stretching at 1635 cm−1. Notably, no peak due to isocyanate (∼2250 cm−1) was observed, indicating complete consumption of the isocyanate groups. Furthermore, the capsules exhibited pronounced sp3 C–H stretching vibrations between 2850 and 2950 cm−1, supporting the presence of the saturated hydrocarbon (i.e., PAO) in the core.
From TGA data, all PAO capsules showed 5% weight loss temperatures (Td,5%) at ∼200 °C (201, 214, and 202 °C for IPDI × Cys_PAO, BZDI × Cys_PAO, and IPDI × EDA_PAO, respectively), as summarized in Table 1. Evaluation of the first derivative of the TGA curves reveals two main degradation events: the first event at around 260 °C corresponds to the loss of the PAO core, and the second peak at higher temperatures (at 304 and 329 °C for IPDI × Cys_PAO and IPDI × EDA_PAO, respectively) is attributed to the decomposition of the polyurea shell. Notably, in the case of BZDI × Cys_PAO, the shell degradation peak overlaps with that of PAO, thus manifesting a single broad peak. The TGA profiles of the shells and cores are summarized in the ESI (Fig. S3, S4 and Table S1†). Despite general concerns regarding the thermal stability of many dynamic covalent bonds,58,59 the responsive capsules showed similar thermal stability to the control capsules, highlighting that integration of dynamic disulfide bonds into the polyurea shell does not compromise the thermal stability of the capsules.
2.3 Fusion and destruction of PAO capsules
The dynamic behavior of the PAO capsule shells, particularly their ability to fuse into cohesive structures or degrade in response to reducing agents, is pivotal for their application in smart materials, including structural composites and controlled release systems.60 We evaluated the fusion of these capsule shells by loading them into a silicone mold, lightly tapping to compact, and then subjecting the sample to incremental heating in an oven, at temperatures between 70 to 160 °C (see Experimental methods for details). SEM images in Fig. 2a–c show that IPDI × Cys_PAO capsules successfully fused into a monolithic structure at 140 °C; in contrast, BZDI × Cys_PAO capsules and the control IPDI × EDA_PAO capsules did not fuse into monoliths but retained their original capsule morphology. | ||
| Fig. 2 (a) Cross-sectional SEM image of IPDI × Cys_PAO monolith, (b) SEM image of BZDI × Cys_PAO capsules, and (c) SEM image of IPDI × EDA_PAO post heat treatment; insets showing photos of the materials after heat treatment. Optical images of fused IPDI × Cys_PAO monoliths shaped into (d) a star, (e) the state of Texas, and (f) a heart. (g) Stress–strain curve from the compression testing of IPDI × Cys_PAO compact capsules and fused monoliths with the shaded area representing the standard deviation (n = 3). Optical microscope image of (h) IPDI × Cys_PAO monolith and (i) IPDI × EDA_PAO capsules after exposure to DTT. | ||
Characterization of the fused IPDI × Cys_PAO structure by FTIR spectroscopy confirmed the presence of the PAO core and no distinctive change to the polymers, as shown in Fig. S5a.† The retention of the liquid core upon fusion was verified and quantified by extracting PAO with hexanes, evaporating hexanes, and weighing the residue. As highlighted in Table 1, the capsules were 76.6 ± 0.5 wt% PAO (i.e., pre-fusion), and the monoliths were 68.2 ± 4.7 wt% PAO (i.e., after thermally induced shell fusion). Thus, the core retention after fusion was as high as 89%. To assess the long-term stability of the liquid monolith, we left the IPDI × Cys_PAO monoliths on the benchtop for two weeks. After this period, they still contained 60.2 ± 3.9 wt% PAO, corresponding to a 78.6% retention rate compared to the pre-fusion PAO content. This result demonstrates the efficacy of our method in creating liquid-containing monoliths with substantial and prolonged core retention. The versatility of the IPDI × Cys_PAO monoliths was demonstrated by molding them into various geometries, such as stars, the state of Texas, or hearts (Fig. 2d–f), highlighting the potential for custom-shaped applications in filtration and adsorption systems with enhanced purification efficiency.61 Compression tests, as illustrated in Fig. 2g, highlighted a significant enhancement in the mechanical properties of the fused monolith compared to the capsules. The average compressive modulus of IPDI × Cys_PAO monoliths reached 1.76 MPa, a substantial increase from the 0.29 MPa measured in compact capsules without heating. Thus, fusion not only transforms the physical form but also significantly improves the mechanical properties.
The disulfide bonds within the polymer shell not only give the ability to fuse the capsules into a monolith, but also enable shell degradation. Specifically, exposure of the capsules/monoliths to the reducing agent dithiothreitol (DTT) initiated the reduction of the disulfide bonds and, thus, the degradation of the polyurea and release of the PAO. When the IPDI × Cys_PAO monoliths are soaked in water containing DTT, an oil-in-water emulsion stabilized by GO and fragmented oligomers is formed (Fig. 2h).15 In contrast, control capsules (IPDI × EDA_PAO) lacking disulfide bonds maintained their integrity under the same conditions (Fig. 2i). The ability to degrade the shells under specific conditions underscores the chemical responsivity of these materials and their potential utility in applications requiring controlled release and degradation.
The observation that the disulfide-containing shells of BZDI × Cys_PAO did not form a monolith, despite the application of elevated temperatures, is surprising. We attribute this anomaly to the aqueous emulsion preparation process, which could lead to partial hydrolysis of the isocyanate of BZDI to an amine that competed with cystamine during shell formation, thus reducing the number of disulfide bonds within the shell that can undergo exchange. This phenomenon was not observed with IPDI, which is less susceptible to hydrolysis due to its aliphatic nature, in contrast to the aromatic BZDI.62 Solid-state electron paramagnetic resonance (EPR) analysis performed at room temperature supported this hypothesis; the spectra in Fig. 3a reveal a strong signal at 3339 G for IPDI × Cys_PAO, indicative of the presence of unpaired electrons (e.g., formed upon thermally induced homolytic cleavage of the disulfide bonds). In contrast, BZDI × Cys_PAO capsules had a weaker signal at the same position, suggesting a lower concentration of radicals which we attribute to fewer disulfide bonds, which likely contributed to the insufficient fusion at elevated temperatures. The absence of any signal in IPDI × EDA_PAO further highlights the lack of dynamic disulfide bonds in these capsule shells. The g-value, a dimensionless parameter, indicates the interaction strength between unpaired electrons and the magnetic field. The calculated g-values were 2.002 for both IPDI × Cys_PAO and BZDI × Cys_PAO, aligning with prior reports of disulfide-containing polymers41,63 and emphasizing the critical role of disulfides in facilitating inter-capsule fusion and monolith formation (see ESI eqn (S1) for details†). This g-value indicates an environment similar to that of a free electron, and suggests the presence of sulfur-based radicals formed from the homolytic cleavage of disulfide bonds.41,64,65 X-ray photoelectron spectroscopy (XPS) analysis corroborated these findings, as the peak due to the S 2p binding energy (163 eV) was more prominent in the IPDI × Cys_PAO shell, but barely detectable in BZDI × Cys_PAO, consistent with our hypothesis regarding disulfide bond incorporation (Fig. 3b and c). These analyses also suggest that aqueous systems provide limited control over the precise composition, which is crucial for tailoring properties and functionalities.
 | ||
| Fig. 3 (a) Offset solid-state EPR spectra of IPDI × Cys_PAO, BZDI × Cys_PAO, and IPDI × EDA_PAO. (b) Offset XPS survey spectra of isolated shells of IPDI × Cys_PAO, BZDI × Cys_PAO, and IPDI × EDA_PAO. (c) High-resolution S 2p peak of IPDI × Cys_PAO and BZDI × Cys_PAO isolated shells. | ||
2.4 Characterization of IL capsules
In complement to the PAO capsules, IL-containing capsules could be prepared under water-free conditions and provide a distinct core environment due to the high polarity of the IL. Moreover, contrasting the PAO capsules to the IL capsules gives insight into the influence of core polarity on capsule morphology and stability, as previously reported for capsule shells containing hindered urea bonds.15,16 SEM imaging reveals the morphology of the IL capsules (Fig. 4a–c). BZDI × Cys_IL capsules, prepared from a non-aqueous emulsion, maintain a spherical shape (Fig. 4b), whereas the BZDI × Cys_PAO capsules produced from an aqueous emulsion were ellipsoidal (Fig. 1b). A comparative analysis with their precursor emulsions (Fig. S1c and d†) shows a markedly larger size of the capsules, with droplet sizes ranging from 10–20 μm and capsules ∼45 μm, similar to that observed for the PAO capsules prepared in water. Laser diffraction analysis quantified the average diameters of the capsules: 39.4 ± 40.2 μm for IPDI × Cys_IL, 73.6 ± 116.5 μm for BZDI × Cys_IL, and 33.1 ± 20.3 μm for IPDI × EDA_IL, as shown in Fig. 4d and summarized in Table 2. Similar to the PAO capsules discussed above, disulfide bonds in the capsule shells led to broader and bimodal size distributions, possibly suggesting that fusion was triggered by 1-propylamine. Additionally, even the non-dynamic IL capsule (IPDI × EDA_IL) showed slight fusion and loss of spherical shapes (Fig. 4c). This could be due to the IPDI monomer being soluble in both the dispersed and continuous phases, which destabilizes the emulsion and causes irregular shapes. | ||
| Fig. 4 SEM images of capsules: (a) IPDI × Cys_IL, (b) BZDI × Cys_IL, and (c) IPDI × EDA_IL, with insets showing details of individual capsules. (d) Particle size distribution of the IL capsules determined by laser diffraction. (e) Offset FTIR spectra of the IL capsules. (f) TGA weight loss profiles of the IL capsules. | ||
| Capsules | Emulsion size (μm) | Capsule size (μm) | T d,5% (°C) | Fusion temperature (°C) | Capsule liquid component (wt%) | Monolith liquid component (wt%) |
|---|---|---|---|---|---|---|
| a Monoliths analyzed immediately after preparation. b Monoliths left at room temperature on the benchtop for 14 days. | ||||||
| IPDI × Cys_IL | 10–20 | 39.4 ± 40.2 | 247 | 100 | 82.1 ± 0.3 | 80.1 ± 1.9a (79.9 ± 0.7)b |
| BZDI × Cys_IL | 10–20 | 73.6 ± 116.5 | 221 | 160 | 74.3 ± 1.5 | 71.3 ± 2.8 (75.2 ± 2.2) |
| IPDI × EDA_IL | 10–45 | 33.1 ± 20.3 | 246 | n/a | 80.9 ± 0.8 | n/a |
The IL-containing capsules were characterized by FTIR spectroscopy and TGA weight loss profiles. FTIR spectra verified the successful consumption of isocyanate groups, indicated by the absence of peaks at ∼2250 cm−1, and the presence of polyurea, with peaks similar to those observed in the spectra of PAO capsules (Fig. 4e and 1e). Notably, the IL capsules also exhibited a peak at 3150 cm−1, attributed to the sp2 C–H stretching from the imidazolium cation and supporting encapsulation of IL. The IL capsules all had Td,5% exceeding 220 °C, as shown in Fig. 4f and Table 2. Given the major weight loss of [HMIM][TFSI] occurred at 432 °C, we ascribed the major weight loss near and after 400 °C to the loss of the IL and weight loss at lower temperatures to be due to decomposition of polyurea shells (e.g., 269 °C for IPDI × Cys_IL, 223 and 272 °C for BZDI × Cys_IL, and 311 °C for IPDI × EDA_IL). These data support the structural integrity and stability of IL capsules under conditions relevant for many applications.
2.5 Fusion of IL capsules
Based on our prior work to use capsules of IL for carbon capture,11,17–21 we were inspired to evaluate the fusion of IL capsules into a monolith and its use as a free-standing CO2 absorbent, that could perhaps be used as filters for direct air capture. Employing the temperature-based fusion method outlined above, the IL capsules were compacted and heated to induce capsule shell fusion. SEM imaging showed that both IPDI × Cys_IL and BZDI × Cys_IL capsules successfully fused into monoliths (Fig. 5a and b), which occurred at 100 °C and 160 °C, respectively. In stark contrast, the control IPDI × EDA_IL capsules showed no fusion and maintained the original individual (Fig. 5c), spherical shape, as expected due to the lack of disulfide bonds in the capsule shell. The compressive moduli of these monoliths were enhanced compared to the capsule feedstock, as illustrated in Fig. S6.† The compressive modulus of IPDI × Cys_IL increased from 0.29 to 0.37 MPa, while that of BZDI × Cys_IL rose from 0.24 to 0.33 MPa. We attribute the less significant increase in compressive moduli to the plasticization of the polyurea by the encapsulated IL. | ||
| Fig. 5 SEM images of (a) IPDI × Cys_IL and (b) BZDI × Cys_IL cross-section, and (c) IPDI × EDA_IL capsules after heat treatment with insets showing the photos of the materials after heat treatment. | ||
To visually capture the fusion process, IPDI × Cys_IL capsules were incrementally heated from 70 °C to 100 °C on a glass slide under an optical microscope equipped with a heating stage and camera. The distinct thermal responsivity of the capsules is documented in Fig. S7a and b† and shown at 10× speed in Videos S1a–d.† When IPDI × Cys_IL capsules were heated at 70 °C and 80 °C for 10 minutes (Video S1a and b†), no obvious fusion was observed. However, at 90 °C, liquid expulsion from the shells was noticeable after 2 minutes (Video S1c†). Upon reaching 100 °C, pronounced fusion occurred within 3 minutes, with the capsules losing their original spherical morphology and merging into one structure (Video S1d†). In contrast, the IPDI × EDA_IL capsules show no change in shape (Fig. S5c and d†), underscoring the fusion behavior is dependent on shell composition.
FTIR analysis of the fused monoliths verified the presence of IL and the polyurea (Fig. S5b and c†). The IL content was quantified by extraction using dimethyl sulfoxide-d6 (DMSO-d6) with mesitylene as an internal standard and characterization by 1H NMR spectroscopy (see Fig. S8† and Experimental methods for details). As summarized in Table 2, the IPDI × Cys_IL and BZDI × Cys_IL capsules contain 82.1 ± 0.3 and 74.3 ± 1.5 wt% IL. After fusion, most of the core was retained (80.1 ± 1.9 and 71.3 ± 2.8 wt%), with impressive retention rates of 97 wt% and 96 wt% for IPDI × Cys_IL and BZDI × Cys_IL, respectively, with negligible changes even after 14 days under ambient conditions. This notable retention may be attributed to strong interactions between the IL and the polyurea shell (e.g., van der Waals forces and hydrogen bonding), which contrasts with the gradual loss observed in the PAO monolith due to weaker interactions between hydrocarbon solvent and the polar polyurea shell.
Surprisingly, the temperature required for capsule shell fusion into a monolith differed in capsules with the same core liquid. The higher fusion temperature required to fuse BZDI × Cys_IL capsules may be attributed to π–π interactions between the aromatic moieties of the polymer backbone, which could limit efficient chain mobility and prevent the formation of bonds between neighboring capsule shells,66 thus necessitating higher temperatures for comparable levels of fusion. SEM images of the monoliths with the IL removed support this hypothesis; IPDI × Cys_IL capsules fused completely, losing their original texture and showing no artifacts of the capsule feedstocks (Fig. S9a†), whereas BZDI × Cys_IL capsules gave monoliths that had obvious spherical shapes (Fig. S9b†). Also notable is the lower temperature required for fusion of the IL capsules compared to the PAO capsules; this may be attributed to: (i) the use of a non-aqueous emulsion to prepare IL capsule gave higher loading of dynamic bonds; and/or (ii) IL core plasticizes the polyurea shell, stabilizing the transition state of the disulfide bond exchange and reducing the energy barrier for bond breaking and reformation. Regardless, even though polymer shells containing disulfide bonds can be used to produce SLCs with varied polarity of the liquid, subtle differences are observed in shell fusion temperature and stability.
2.6 CO2 capture and RF heating
SLCs containing ILs are employed in a wide array of applications, from energy storage44,47–50,67 to environmental management,9,10,13,48 with their ability to absorb CO2 particularly notable.11,17–19,21,25,68–73 Our previous studies established that IL capsules are highly effective CO2 absorbents due to their increased surface area and enhanced mass transfer rates compared to bulk, agitated IL.11 Building on these findings, we compared the CO2 uptake of the IPDI × Cys_IL and BZDI × Cys_IL capsules and their corresponding monoliths to establish if the latter can be used as free-standing CO2 absorbents (the capsules must be held in a vessel). The gravimetric CO2 uptake performance was measured at 1 bar of pure CO2 at 25 °C, and the results are displayed in Fig. S10.† Notably, IPDI × Cys_IL exhibited superior CO2 uptake capacity and rate compared to BZDI × Cys_IL samples, which likely stems from the lower CO2 permeability of the BZDI-based polymer due to π–π stacking, as previously reported.17 This empirical evidence highlights the critical impact of small changes in the polymer shell on properties for CO2 absorption; thus, we focused on the performance of IPDI × Cys_IL capsules and the corresponding monolith on CO2 absorption and thermal desorption.The CO2 uptake and release by IPDI × Cys_IL capsules and monoliths were evaluated by exposing the sample to a pure CO2 stream (1 bar) at 25 °C until weight gain plateaued (absorption), then heating the sample to 75 °C under a pure N2 stream until weight loss plateaued (desorption). Notably, both the capsules and monoliths demonstrated CO2 uptake capacity surpassing that of the bulk IL (Fig. 6a). The enhanced capacity can be attributed to the affinity of the polyurea shell to CO2, due to the abundance of secondary amine functionalities on the polymer.18 The monolith and capsules exhibited comparable CO2 uptake capacity, both reaching 0.12 mol CO2 per kg of sample (i.e., polymer and IL combined), although the monolith required more time to reach full capacity. The extended time required for monolith to reach saturation compared to the capsules presumedly results from reduced surface area. Despite the slower absorption rate, we constantly observed that the CO2 capacity of the monolith slightly exceeded that of the corresponding capsules (Fig. 6a and S10†). We hypothesize that this enhanced capacity results from the heat treatment during its formation which not only disrupts disulfide bonds but also hydrogen bond crosslinking between the polyurea chains, facilitating IL infusion into the shell and expanding the volume available for CO2 absorption. This heat-induced structural change might compensate for the lower IL content in the monolith, resulting in a capacity slightly higher than that of the capsules. The mechanism of CO2 absorption–desorption in these monoliths is currently being investigated, but it is clear that the comparable capacity and rate of uptake underscore the effectiveness of the monolithic structure in capturing CO2—a significant advancement for potential application scalability. Notably, the monoliths have slightly lower CO2 capacity compared to previously reported IL capsules, as shown in Table S2,† it is important to note that this system has not yet been optimized for CO2 uptake. Ongoing efforts to enhance the shell's permeability to CO2 are expected to improve its performance.
 | ||
| Fig. 6 (a) Three cycles of gravimetric CO2 absorption and desorption of IPDI × Cys_IL capsules (dark green), monolith (light green), and the bulk IL (orange). (b) RF heating of the IPDI × Cys_IL monolith (138 MHz at 2.5 W) with inset showing IR image of the heated sample (temperature given of the yellow spot). (c) Cyclic heating test of the IPDI × Cys_IL monolith by RF of 138 MHz at 3.2 W. | ||
In addition to efficient CO2 capturing, a critical challenge for such systems is the energy-efficient release of CO2.74 This is commonly achieved by bulk convection heating, which requires infrastructure and is not typically energy efficient.43,75 Motivated by this need, we envisioned that RF heating of the monolith could occur due to the integrated GO-based nanosheets. C18–GO nanosheets were used as surfactants in the emulsion templates during preparation and then reduced to reduced graphene oxide (rGO) during the subsequent heat treatment to form the monolith. Thus, nanosheets serve dual roles as surfactants and conductive particles. When subjected to an RF field of 138 MHz at 2.5 W, the IPDI × Cys_IL monolith reached the temperature necessary for CO2 desorption (75 °C) in just 31 seconds (Fig. 6b). When the application of RF was stopped, the monolith quickly cooled to room temperature and was thus ready for another CO2 absorption cycle (e.g., within 105 seconds). Importantly, RF heating to a higher temperature of 85 °C with a power of 3.2 W over six cycles was demonstrated, without any discernible detriment to the structure or heating efficiency, as demonstrated in Fig. 6c. Thus, the SLC monoliths enable RF to achieve rapid temperature swing, which is crucial for efficient CO2 desorption and setting the stage for further development of these composite materials.
3 Conclusion
This study introduces an innovative responsive capsule system that incorporates disulfide bonds into polyurea shells and their temperature-induced fusion into monoliths for carbon capture, marking a significant development of high stability, high liquid content SLCs. Demonstrating the ability to encapsulate liquid cores ranging in polarity from nonpolar PAO to highly polar IL, these capsules exhibit a unique capability for inter-shell fusion, resulting in free-standing, structurally intact, and mechanically robust liquid monoliths that maintain >89 wt% of the encapsulated liquid. This presents a novel pathway for crafting SLCs with high loading comparable to polyHIPEs (polymers synthesized from high internal phase emulsions).76 Furthermore, IL-containing monoliths, prepared from the IDPI × Cys_IL capsule feedstock, demonstrated effectiveness in carbon capture, achieving CO2 absorption capacity surpassing the unencapsulated IL and comparable to the capsules. The rapid efficiency of RF heating brings the monolith to the CO2 desorption temperature within only 31 seconds, enhancing the practicality of the system. The adaptability of this system is further illustrated through the successful employment of an IR heating lamp to achieve capsule fusion, as depicted in Fig. S11,† indicating potential compatibility with selective laser sintering (SLS) 3D printing techniques. Taken together, this work not only advances SLC development by elucidating the complex interplay between core substances, shell compositions, and manufacturing processes but also opens avenues for applying responsive materials in making SLCs to address environmental challenges and beyond.4 Experimental methods
4.1 Materials
Graphite flakes, sulfuric acid (H2SO4), potassium permanganate (KMnO4, ≥99%), 1-propylamine (98.0%), isophorone diisocyanate (mixture of isomers, 98%), cystamine dihydrochloride (96%), dichloromethane (DCM), ethylenediamine (≥99%), and hexanes (≥98.5%) were purchased from Sigma-Aldrich. N,N-Dimethylformamide (DMF, anhydrous, ≥99.8%), hydrogen peroxide (H2O2, 30% w/w in water), isopropanol (≥99.5%), and toluene (≥99.5%), were from Fisher Scientific. 1,3-Bis(2-isocyanato-2-propyl)benzene (BZDI, ≥97.0%) was purchased from VWR. Potassium hydroxide (KOH), octane (99.0%), and dithiothreitol (99%) were from Oakwood Chemical. 1-Octadecylamine was obtained from Alfa Aesar. 1-Hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([HMIM][TFSI], 99.0%) was purchased from Iolitec. PAO was received from ExxonMobil and Chevron Phillips Chemical Company. All reagents except cystamine dihydrochloride were used as received without further purification.4.2 Instrumentation
Fisher brand ultrasonic bath (model CPX 3800) and vortex (model 9454FIALUS) were used along with a Thermo Scientific centrifuge, model 75005703. A waring commercial blender (model 7010S) was used to blend GO flakes. Emulsification was performed by a hand-held tissue-tearor from Biospec Product Inc. (model 985370). Fourier transform infrared (FTIR) spectroscopy was performed using a JASCO FTIR spectrometer, model FTIR-4600LE MidIR with a diamond-coated ZnSe crystal in ATR mode. A Cressington 108 Sputter Coater was used for sputter coating. SEM and EDS analysis was performed on a TESCAN VEGA SEM equipped with an Oxford Instruments EDS system. Particle size analysis was conducted on a Horiba Partica LA-960 particle sizer on samples of capsules dispersed in water. TGA was run using a TGA 5500 instrument from TA Instruments under N2, and samples were heated to 100 °C (20 °C min−1), held for 5 minutes, and heated to 600 °C (10 °C min−1). Compression tests were achieved using a DMA 850 instrument with a compression clamp. 1H NMR spectroscopy (for IL percentage measurement) was done using Avance NEO 400 instruments with mesitylene as the internal reference and DMSO-d6 as the solvent. Solid-state EPR on capsules was done using a Bruker Elexsys E500 console with a standard resonator. Optical microscopy images were taken using a microscope from Amscope equipped with an A35180U3 camera from Amazon, and the PE120 heating stage apparatus was coupled to a T96 Linkpad Controller and a water circulation pump from Linkam Scientific. The RF fields were generated using a co-planar stationary RF applicator and a signal generator (RIGOL DSG800). The field was amplified using an RF power amplifier (Minicircuits HL-100W-GAN+). A cylindrical monolith with a diameter of ∼5 mm and a height of ∼2 mm was used during all experiments, and the real-time temperature was recorded using a thermal camera (FLIR Systems, A655sc). The sample was exposed to 138 MHz at 2.5 W for a regular heating test. The same IL monolith was exposed to an on-and-off RF heating cycle at 138 MHz and 3.2 W for the cyclic heating test, repeated across six cycles. XPS was performed using an Omicron X-ray photoelectron spectrometer employing an Mg/Al sourced X-ray beam.4.3 Synthesis of GO and C18–GO
The synthesis of GO followed a modified Hummer's method.77 Graphite flakes (3.0 g) were dispersed in sulfuric acid (400 mL). Then, potassium permanganate (KMnO4, 3.0 g) was added to oxidize the graphene sheets. The potassium permanganate (3.0 g) was added every 24 hours, four times in total. The whole process was performed at room temperature. After four days, the reaction was poured into an ice bath, and excess KMnO4 was quenched by adding aqueous hydrogen peroxide dropwise until the solution turned translucent, with stirring, to prevent overheating. The product was isolated by centrifugation and then washed with isopropanol until litmus paper showed a neutral pH. Finally, the GO flakes were dried under reduced pressure at room temperature overnight and blended into powder before being stored at 4 °C.To prepare C18–GO, GO (100 mg) was dispersed in DMF (50 mL). The GO dispersion was then added to a 1-octadecylamine solution in DMF (50 mL, 8 mg mL−1), and the mixture was heated to 50 °C overnight. The mixture was subsequently washed with toluene until 1-octadecylamine was no longer detectable in the supernatant by FTIR analysis. Finally, the dispersion medium was changed to octane, and the concentration was adjusted based on the original amount of GO used.
4.4 Synthesis of cystamine
Cystamine was recovered from its dihydrochloride salt using a procedure reported in the literature.39 Cystamine dihydrochloride (6.0 g, 26.6 mmol) was dissolved in distilled water (75 mL). Potassium hydroxide (KOH, 4.5 g) was then added to the solution and stirred at room temperature for 15 minutes. The mixture was extracted with dichloromethane (DCM) three times, and the combined organic layers were dried over sodium sulfate (NaSO4). A pale-yellow oil was obtained (yield: 96%) after drying under vacuum. 1H NMR (400 MHz, CDCl3) δ 1.31 (br s, 4H), 2.76 (t, 4H), 3.02 (t, 4H).4.5 Synthesis of the disulfide responsive capsules in aqueous emulsions
For the synthesis of IPDI × Cys_PAO capsules, PAO (dispersed phase, 1.0 mL) was combined with isophorone diisocyanate (IPDI, 273 μL, 1.29 mmol) in a 20 mL vial. Graphene oxide (GO) in water dispersion (5 mL, 2 mg mL−1) and saturated brine (50 μL) were added and homogenized using a hand-held tissue tearor for three consecutive cycles of 20 seconds with 15 second intervals to form an oil-in-water (o/w) emulsion. Deionized water (0.5 mL) was used to dilute the emulsion, and cystamine (253.2 mg, 1.66 mmol) in water (1.25 mL) was added to the continuous phase, followed by hand swirling. After capped, the reaction mixture was left undisturbed for three days. Then, 7 vol% aqueous solution of 1-propylamine (40 mL) was added, and the mixture was allowed to react for an additional 24 hours. The capsules were then isolated by gravity filtration, washed with water until neutral filtrate was obtained, and dried under reduced pressure at room temperature for 120 hours. For BZDI × Cys_PAO capsules, 1,3-bis(1-isocyanato-1-methylethyl)benzene (BZDI, 300 μL, 1.30 mmol) was used as the diisocyanate monomer; for IPDI × EDA_PAO capsules, ethylene diamine (111 μL, 1.66 mmol) was used as the diamine monomer.4.6 Synthesis of the disulfide responsive capsules in non-aqueous emulsions
Following the procedure outlined for aqueous emulsions, the non-aqueous synthesis of IPDI × Cys capsules commenced with mixing the [HMIM][TFSI] (dispersed phase, 1.0 mL) with cystamine (198 mg, 1.30 mmol) in a 20 mL vial. C18–GO dispersed in octane (5 mL, 2 mg mL−1) was added and homogenized using a hand-held tissue tearor, following the same cycle timings to create an IL/o emulsion. After dilution with octane (0.5 mL), IPDI (369 μL, 1.66 mmol) in octane (1.25 mL) was added, and the mixture was left to stand for three days, mirroring the aqueous capsule synthesis timing. Then, the mixture was quenched using 7 vol% octane solution of 1-propylamine (40 mL), filtered, and washed with hexanes until neutral. The capsules were then vacuum-dried at room temperature for 12 hours. For BZDI × Cys_IL capsules, 1,3-bis(1-isocyanato-1-methylethyl)benzene (BZDI, 383 μL, 1.66 mmol) was used as the diisocyanate monomer; for IPDI × EDA_IL capsules, ethylene diamine (87 μL, 1.30 mmol) was used as the diamine monomer.4.7 Capsule fusion experiments via an oven or IR irradiation
For all liquid-core capsules, the capsule powders were tapped into a silicone mold and heated in an oven for 10 minutes at 70 °C. If no fusion occurred, the temperature was increased by 10 °C increments, with each increment followed by an additional 10 minutes of heating. The fusion temperature was defined as the temperature at which the capsules fused after 10 minutes of heating. This process was continued up to a maximum temperature of 160 °C. A 250 W IR lamp was used as the heat source for the irradiation-induced fusion experiments. The experiment setup was depicted in Fig. S11a.†4.8 Determining wt% IL via1H NMR spectroscopy
A known mass (15–50 mg) of ionic liquid (IL) capsules or monoliths was added to 1 mL of mesitylene in DMSO-d6 solution (approximately 0.05 M). The IL was extracted from the capsules by vortexing for 10 seconds, followed by sonication for 10 seconds, repeated alternately 3 times. A PTFE syringe filter (0.22 μm pore size) was then used to remove capsule shells. From the 1H NMR spectrum, the integral ratio of the N-methyl of [HMIM][TFSI] (3.84 ppm) to the methyl of mesitylene (2.21 ppm) was obtained to calculate IL wt%. Each measurement was repeated three times.4.9 Determining wt% of PAO via mass difference
A known mass (15–50 mg) of PAO capsules and monoliths was dispersed in hexanes. Then, the same extraction and filtration were performed as described above. The resulting solution was subjected to rotary evaporation, followed by vacuum drying overnight. The mass of the core material was subsequently determined. Each measurement was repeated three times.4.10 Degradation of monolith and capsules
Approximately 30 mg of monolith or capsules and 20 mg of dithiothreitol were placed in a 4 mL vial containing 2 mL of water. The entire mixture was heated at 60 °C for 30 minutes without stirring.4.11 SEM sample preparation
A 5 mm × 5 mm piece of conductive carbon double-sided tape was attached to a stub (12.7 mm in diameter and 8 mm pin length), which was used as the sample substrate. The samples were placed onto the conductive carbon double-sided tape and subsequently sputter-coated with a 10 nm layer of gold (Au).Data availability
All data generated or analyzed during this study are included in this article and its ESI.† The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.Author contributions
E. B. P. conceptualized and supervised the project. C.-M. H. designed the experiments and carried out all laboratory experiments and data analyses under the guidance of E. B. P. L. A.-M. conducted CO2 absorption experiments. S. S. D., H. C., and M. J. G. performed and helped with the RF heating experiments. M. A. assisted with EPR experiments. P. W. provided suggestions on the irradiation-induced fusion experiments. Y. W. provided suggestions on the synthesis and analysis of responsivity. C.-M. H. wrote the initial manuscript. E. B. P., L. A.-M., S. S. D., M. A., and P. W. contributed to manuscript revision.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors gratefully acknowledge financial support from: the National Science Foundation (Award 2103182), Department of Energy (Grant No. DE-SC0022214), and Texas A&M University. Special thanks are extended to the TAMU Soft Matter Facility (RRID: SCR_022482), the Microscopy and Imaging Center (RRID: SCR_022128), and the Materials Characterization Facility (RRID: SCR_022202) at Texas A&M University for their invaluable assistance and use of their facilities.References
- R. W. Style, R. Tutika, J. Y. Kim and M. D. Bartlett, Adv. Funct. Mater., 2021, 31, 2005804 CrossRef CAS.
- M. D. Bartlett and R. W. Style, Soft Matter, 2020, 16, 5799–5800 RSC.
- C.-M. Hsieh, C. E. Cipriani and E. B. Pentzer, Polym. Int., 2024, 73, 793–802 CrossRef.
- Z. Wang, R. Bo, H. Bai, S. Cao, S. Wang, J. Chang, Y. Lan, Y. Li and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 22553–22562 CrossRef CAS PubMed.
- Y. Wu, Z. Fang, W. Wu, K. Li, M. Lin and Z. Dang, Adv. Eng. Mater., 2021, 23, 2100372 CrossRef CAS.
- C. Majidi, K. Alizadeh, Y. Ohm, A. Silva and M. Tavakoli, Flexible Printed Electron., 2022, 7, 013002 CrossRef.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- Z. Ge, F. Ye and Y. Ding, ChemSusChem, 2014, 7, 1318–1325 CrossRef CAS.
- Q. Luo and E. Pentzer, ACS Appl. Mater. Interfaces, 2020, 12, 5169–5176 CrossRef CAS.
- Q. Luo, Y. Wang, Z. Chen, P. Wei, E. Yoo and E. Pentzer, ACS Appl. Mater. Interfaces, 2019, 11, 9612–9620 CrossRef CAS PubMed.
- Q. Huang, Q. Luo, Y. Wang, E. Pentzer and B. Gurkan, Ind. Eng. Chem. Res., 2019, 58, 10503–10509 CrossRef CAS.
- Y. Yahata, K. Kimura, Y. Nakanishi, S. Marukane, T. Sato, Y. Tsujii and K. Ohno, Langmuir, 2019, 35, 3733–3747 CrossRef CAS.
- K. J. Edgehouse, N. Rosenfeld, D. E. Bergbreiter and E. B. Pentzer, Ind. Eng. Chem. Res., 2021, 60, 14455–14463 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Y. Wang, P. Wei, Q. Zhou, C. Cipriani, M. Qi, S. Sukhishvili and E. Pentzer, Chem. Mater., 2022, 34, 5821–5831 CrossRef CAS.
- Y. Wang, K. Quevedo and E. Pentzer, Polym. Chem., 2021, 12, 2695–2700 RSC.
- S. S. Gaur, K. J. Edgehouse, A. Klemm, P. Wei, B. Gurkan and E. B. Pentzer, J. Polym. Sci., 2021, 59, 2980–2989 CrossRef CAS.
- L. Al-Mahbobi, A. Klemm, C. Taylor, B. Gurkan and E. Pentzer, ACS Appl. Eng. Mater., 2024, 2, 1298–1305 CrossRef CAS.
- C. D. L. Taylor, A. Klemm, L. Al-Mahbobi, B. J. Bradford, B. Gurkan and E. B. Pentzer, ACS Sustain. Chem. Eng., 2024, 12, 7882–7893 CrossRef CAS.
- M. Zeeshan, M. K. Kidder, E. Pentzer, R. B. Getman and B. Gurkan, Front. Sustain., 2023, 41, 1167713, DOI:10.3389/frsus.2023.1167713.
- Y.-Y. Lee, K. Edgehouse, A. Klemm, H. Mao, E. Pentzer and B. Gurkan, ACS Appl. Mater. Interfaces, 2020, 12, 19184–19193 CrossRef CAS.
- G. P. Dennis, K. E. O'Harra, X. Liu, E. M. Jackson, C. H. Turner and J. E. Bara, RSC Appl. Polym., 2023, 1, 111–122 RSC.
- M. Hutchby, C. E. Houlden, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 8721–8724 CrossRef CAS PubMed.
- H. Ying and J. Cheng, J. Am. Chem. Soc., 2014, 136, 16974–16977 CrossRef CAS PubMed.
- F. L. Bernard, E. A. Duarte, B. B. Polesso, R. B. Duczinski and S. Einloft, Environ. Challenges, 2021, 4, 100109 CrossRef CAS.
- S. Kaviani, S. Kolahchyan, K. L. Hickenbottom, A. M. Lopez and S. Nejati, J. Chem. Eng., 2018, 354, 753–757 CrossRef CAS.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS.
- R. Zhang, T. Nie, Y. Fang, H. Huang and J. Wu, Biomacromolecules, 2022, 23, 1–19 CrossRef.
- P. Ju, J. Hu, F. Li, Y. Cao, L. Li, D. Shi, Y. Hao, M. Zhang, J. He and P. Ni, J. Mater. Chem. B, 2018, 6, 7263–7273 RSC.
- P.-H. Hsu and A. Almutairi, J. Mater. Chem. B, 2021, 9, 2179–2188 RSC.
- D. Yue, G. Cheng, Y. He, Y. Nie, Q. Jiang, X. Cai and Z. Gu, J. Mater. Chem. B, 2014, 2, 7210–7221 RSC.
- R. Bej and S. Ghosh, Bioconjugate Chem., 2019, 30, 101–110 CrossRef CAS PubMed.
- Y. Tachibana, T. Baba and K. Kasuya, Polym. Degrad. Stab., 2017, 137, 67–74 CrossRef CAS.
- Y. Li, W. Guo, W. Li, X. Liu, H. Zhu, J. Zhang, X. Liu, L. Wei and A. Sun, Chem. Eng. J., 2020, 393, 124583 CrossRef CAS.
- S. Guggari, F. Magliozzi, S. Malburet, A. Graillot, M. Destarac and M. Guerre, ACS Sustain. Chem. Eng., 2023, 11, 6021–6031 CrossRef CAS.
- H. Wang, Y. Huang, Z. Shi, X. Zhou and Z. Xue, ACS Macro Lett., 2022, 11, 991–998 CrossRef CAS.
- H. Liu, R. Sun, S. Feng, D. Wang and H. Liu, Chem. Eng. J., 2019, 359, 436–445 CrossRef CAS.
- S. Yang, J. Bai, X. Sun and J. Zhang, Chem. Eng. J., 2023, 461, 142066 CrossRef CAS.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- N. Patil, A. C. Camacho, N. K. Mishra, P. Singhla, C. B. Sweeney, M. A. Saed, M. Radovic and M. J. Green, Adv. Eng. Mater., 2019, 21, 1900276 CrossRef.
- Q. Luo, P. Wei, Q. Huang, B. Gurkan and E. B. Pentzer, ACS Appl. Mater. Interfaces, 2018, 10, 16707–16714 CrossRef CAS PubMed.
- S. N. Lak, C.-M. Hsieh, L. AlMahbobi, Y. Wang, A. Chakraborty, C. Yu and E. B. Pentzer, ACS Appl. Eng. Mater., 2023, 1, 2279–2287 CrossRef CAS.
- P. Wei, C. E. Cipriani and E. B. Pentzer, Matter, 2021, 4, 1975–1989 CrossRef CAS.
- Y. Zhang, G. Zhu, B. Dong, F. Wang, J. Tang, F. J. Stadler, G. Yang, S. Hong and F. Xing, Nat. Commun., 2021, 12, 111 CrossRef.
- F. N. Ajjan, M. Ambrogi, G. A. Tiruye, D. Cordella, A. M. Fernandes, K. Grygiel, M. Isik, N. Patil, L. Porcarelli, G. Rocasalbas, G. Vendramientto, E. Zeglio, M. Antonietti, C. Detrembleur, O. Inganäs, C. Jérôme, R. Marcilla, D. Mecerreyes, M. Moreno, D. Taton, N. Solin and J. Yuan, Polym. Int., 2017, 66, 1119–1128 CrossRef CAS.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- C. V. Manohar, T. C. Mendes, M. Kar, D. wang, C. Xiao, M. Forsyth, S. Mitra and D. R. MacFarlane, Chem. Commun., 2018, 54, 3500–3503 RSC.
- D. Debnath, X. Zhao, M. Anas, D. L. Kulhanek, J. H. Oh and M. J. Green, Carbon, 2020, 169, 475–481 CrossRef CAS.
- A. Vashisth, S. T. Upama, M. Anas, J.-H. Oh, N. Patil and M. J. Green, Nanoscale Adv., 2021, 3, 5255–5264 RSC.
- J. Cui, Y. Wang, A. Postma, J. Hao, L. Hosta-Rigau and F. Caruso, Adv. Funct. Mater., 2010, 20, 1625–1631 CrossRef CAS.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- K. Yamawake and M. Hayashi, Polym. Chem., 2023, 14, 680–686 RSC.
- A. Durand-Silva, K. P. Cortés-Guzmán, R. M. Johnson, S. D. Perera, S. D. Diwakara and R. A. Smaldone, ACS Macro Lett., 2021, 10, 486–491 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS.
- Y. Wang, N. Starvaggi and E. Pentzer, Polym. Chem., 2023, 14, 4033–4047 RSC.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS.
- Q. Zhang, Y. Shi, X. Zhan and F. Chen, Colloids Surf., A, 2012, 393, 17–26 CrossRef CAS.
- L. Bonazzola, J. P. Michaut and J. Roncin, J. Chem. Phys., 1985, 83, 2727–2732 CrossRef CAS.
- E. Sagstuen, J.-P. Jørgensen and T. Henriksen, Radiat. Res., 1982, 89, 453–467 CrossRef CAS.
- E. Sagstuen, J. Chem. Phys., 1978, 69, 3206–3213 CrossRef CAS.
- M. Irigoyen, J. M. Matxain and F. Ruipérez, Front. Mater., 2022, 9, 859482, DOI:10.3389/fmats.2022.859482.
- C. Dong, X. Zhang, Y. Yu, L. Huang, J. Li, Y. Wu and Z. Liu, Chem. Commun., 2020, 56, 11993–11996 RSC.
- M. Nisar, F. L. Bernard, E. Duarte, V. V. Chaban and S. Einloft, J. Environ. Chem. Eng., 2021, 9, 104781 CrossRef CAS.
- B. B. Polesso, R. Duczinski, F. L. Bernard, D. J. Faria, L. M. dos Santos and S. Einloft, Heliyon, 2023, 9, e13298 CrossRef CAS.
- H. Wang, J. Zhu, L. Tan, M. Zhou and S. Zhang, Mater. Chem. Phys., 2020, 251, 122982 CrossRef CAS.
- R. Duczinski, B. B. Polesso, E. Duarte, F. L. Bernard, V. V. Chaban and S. Einloft, J. Mol. Liq., 2023, 385, 122394 CrossRef CAS.
- J. K. Stolaroff, C. Ye, D. T. Nguyen, J. Oakdale, J. M. Knipe and S. E. Baker, Energy Procedia, 2017, 114, 860–865 CrossRef CAS.
- J. K. Stolaroff, C. Ye, J. S. Oakdale, S. E. Baker, W. L. Smith, D. T. Nguyen, C. M. Spadaccini and R. D. Aines, Faraday Discuss., 2016, 192, 271–281 RSC.
- U. I. Premadasa, V. Bocharova, A. R. Miles, D. Stamberga, S. Belony, V. S. Bryantsev, A. Elgattar, Y. Liao, J. T. Damron, M. K. Kidder, B. Doughty, R. Custelcean and Y.-Z. Ma, Angew. Chem., Int. Ed., 2023, 62, e202304957 CrossRef CAS PubMed.
- C. Ellison, J. Hoffman and D. Shekhawat, Int. J. Greenhouse Gas Control, 2021, 107, 103311 CrossRef CAS.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04906c |
| This journal is © The Royal Society of Chemistry 2024 |