
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a crosslinked ether-based polymer for high-performance semi-solid lithium metal batteries via in situ integration†
Dezhi
Yang
ab,
Yanan
Yang
ab,
Yeying
Cui
ab,
Yiyang
Sun
 ab and
Tao
Zhang
*abc
ab and
Tao
Zhang
*abc
aState Key Lab. of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, PR China. E-mail: taozhang@mail.sic.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, PR China
cChinese Acad. Sci., Shanghai Inst. Ceram., CAS Key Lab. Mat. Energy Convers., Shanghai 201899, PR China
First published on 9th September 2024
Abstract
In situ ring-opening polymerization of ether-based monomers has shown promising application in solid-state lithium metal batteries owing to their positive lithium compatibility, mild reaction conditions, and facile preparation. However, typical poly(1,3-dioxolane) (PDOL) based electrolyte still struggles with low ionic conductivity, narrow electrochemical window, and poor thermal stability, which greatly retard its further progress. Herein, we constructed a crosslinked PDOL semi-solid electrolyte initiated by a three-armed crosslinker and high voltage resistant fluorinated solvents, which shows favourable stability toward lithium metal (lithium plating/stripping stably operating for 2700 h at 0.2 mA cm−2 with no significant polarization growth) and a high electrochemical oxidation window of up to 4.6 V. A lithium metal coin cell using this crosslinked semi-solid electrolyte equipped with LFP expresses excellent cycling stability with a capacity retention of 88% for 1000 cycles at 1C. A 50 mA h pouch cell with an NCM811 cathode exhibited favourable working ability. Meanwhile, the crosslinked electrolyte demonstrated better solid-state characteristics and thermal stability compared with a long-chain electrolyte. This work provides a prospective reference for the application of ether-based electrolyte in high safety and high energy density lithium metal batteries.
Introduction
Lithium-ion batteries have been widely used in various consumer electronics fields since their creation in the 1990s, bringing great convenience to our lives. However, with the increasing demand for energy storage devices, such as electric vehicles and energy storage power stations, state-of-the-art lithium-ion batteries encounter increasing difficulty in meeting the requirements for high energy density. Additionally, a higher energy density usually comes with a higher security risk due to unstable liquid electrolytes. Lithium metal is widely recognized as an ideal candidate for next-generation anode materials owing to its extremely high theoretical specific capacity (3860 mA h g−1) and low chemical potential (−3.04 V vs. SHE).1,2 Unfortunately, lithium metal is incompatible with the widely used liquid ester electrolytes because of the continuous side reactions between them.3 Therefore, replacing liquid electrolytes with solid electrolytes is considered the ultimate solution for lithium metal batteries.4,5 Among the various solid electrolytes, organic materials are superior to inorganic materials in terms of interface contact, processability, and cost.6–8 However, all-solid-state organic polymer electrolytes, such as polyethylene oxide (PEO) and polyvinylidene fluoride (PVDF), often suffer from poor room-temperature ionic conductivity (≤10−6 S cm−1), which limits their further development.9,10 Combining polymers and solvent molecules to form semi-solid gel electrolytes can significantly improve ionic conductivity and ameliorate interface contact while maintaining solid characteristics and has shown brilliant benefits in lithium metal batteries.11The in situ polymerization strategy represents significant superiority in constructing high-performance lithium metal batteries in recent years.12–14 By injecting a precursor composed of monomers, plasticizers, lithium salts, and initiators into the batteries, the formed gel electrolyte can sufficiently fill all the pores in the batteries after solidification. Compared with an ex situ process, an in situ process has better conformal contact and a simpler preparation procedure. In situ polymerization of cyclic ether monomers represented by poly(1,3-dioxolane) (PDOL) has attracted widespread attention in recent decades due to their satisfying alkali metal compatibility, mild polymerization conditions, and moderate ionic conductivity.15,16 However, it is still confronted with some serious challenges, such as insufficient room-temperature ionic conductivity, poor oxidation stability, and low thermal stability.17 Numerous strategies have been proposed to tackle the issues mentioned above, including molecular design,18 compounding with inorganic particles,19 and functional initiators.20 Among them, crosslinking copolymerization of DOL with a proper crosslinker can significantly improve ionic conductivity by inhibiting crystallization of the molecular chain, and the dense network structure is beneficial for improving the electrochemical stability and thermal stability.21 That means the selection of and search for appropriate crosslinkers are of great significance. Due to the suitable crosslinking degree, electrolytes with crosslinking polymerization based on DOL and three-armed crosslinkers can exhibit decent ionic conductivity and an improved electrochemical window, which have exhibited acceptable enhancement effects in all-solid-state batteries.22,23 But, limited by inadequate ionic conductivity, they still exhibit unsatisfactory electrochemical performance. Additionally, a crosslinker with more arms may lead to excessive crosslinking and result in the need to reduce solid content in the electrolyte to retain electrochemical performance, which may deviate from the original intention of solid state to some extent.24,25 In addition, extensive studies have shown that fluorinated solvents generally display excellent oxidation stability; those represented by hydrofluoroether (HFE) and fluoroethylene carbonate (FEC) have been widely applied in the research of high-voltage lithium metal batteries and showed desirable improvements.26–28
Taking inspiration from the above representations, we prepared a semi-solid electrolyte composed of crosslinked PDOL initiated by glycerol triglycidyl ether and high voltage resistance fluorinated solvents. Compared with long-chain PDOL semi-solid electrolytes, the crosslinked semi-solid electrolyte demonstrates higher ionic conductivity at room temperature (0.36 mS cm−1), a superior oxidation window (4.6 V), and better solid-state characteristic and thermal stability. The fabricated crosslinked electrolyte exhibits brilliant stability towards lithium metal with lithium plating/stripping stably operating for 2700 h at 0.2 mA cm−2 with no significant polarization growth, and the batteries using this electrolyte assembled with LFP keep working for 1000 cycles with a capacity retention of 88% at 1C. Even when assembled with NCM811, the battery can still exhibit favourable cycling stability. We believe this work will provide a promising reference for the development of ether-based electrolytes in high safety and high energy density lithium metal batteries.
Experimental
Synthesis of electrolytes
1,3-Dioxolane (DOL, 99.8%, Aladdin), glycerol triglycidyl ether (GTE, Aladdin), 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (HFE, 99.8%, DoDoChem), fluoroethylene carbonate (FEC, 99.9%, DoDoChem), ethyl methyl carbonate (EMC, 99.98%, DoDoChem), lithium bis(trifluoromethane)sulfonimide (LiTFSI, 99%, Aladdin), lithium tetrafluoroborate (LiBF4, 99.9%, Aladdin), and lithium difluoro(oxalato)borate (LiDFOB, 99%, Aladdin) were used without further treatment, except that GTE was dried with 4 Å molecular sieves for one week.First, DOL, GTE, HFE, FEC and EMC were mixed at a volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:2:1, in which DOL and GTE were used to synthesize the crosslinked polymer while HFE, FEC and EMC served as liquid plasticizers. Next, 1 M LiTFSI, 0.5 M LiBF4 and 0.1 M LiDFOB were dissolved in the mixture above to form the electrolyte precursor. In the precursor, LiBF4 acted as initiator to complete the ring-opening polymerization at room temperature while LiDFOB served as an additive to protect the electrode materials. Then, the precursor was injected into coin cells or pouch cells and the crosslinked poly(1,3-dioxolane) based gel polymer electrolyte (CPGPE) was obtained in situ after resting for about 30 h at 30 °C. For comparison, poly(1,3-dioxolane) based gel polymer electrolyte (PGPE) was prepared by dissolving the same lithium salts in a mixture of DOL, HFE, FEC, EMC at a volume ratio of 5:2:2:1 with no GTE. The precursor slowly transformed from liquid to solid after 3 days. All the preparation processes were performed in an Ar filled glove box with H2O and O2 both below 0.1 ppm.
:1:2:2:1, in which DOL and GTE were used to synthesize the crosslinked polymer while HFE, FEC and EMC served as liquid plasticizers. Next, 1 M LiTFSI, 0.5 M LiBF4 and 0.1 M LiDFOB were dissolved in the mixture above to form the electrolyte precursor. In the precursor, LiBF4 acted as initiator to complete the ring-opening polymerization at room temperature while LiDFOB served as an additive to protect the electrode materials. Then, the precursor was injected into coin cells or pouch cells and the crosslinked poly(1,3-dioxolane) based gel polymer electrolyte (CPGPE) was obtained in situ after resting for about 30 h at 30 °C. For comparison, poly(1,3-dioxolane) based gel polymer electrolyte (PGPE) was prepared by dissolving the same lithium salts in a mixture of DOL, HFE, FEC, EMC at a volume ratio of 5:2:2:1 with no GTE. The precursor slowly transformed from liquid to solid after 3 days. All the preparation processes were performed in an Ar filled glove box with H2O and O2 both below 0.1 ppm.
Characterization
CPGPE and PGPE were cracked and soaked in deionized water and ethanol sequentially for ultrasonic cleaning several times and then dried for structural characterization. The obtained purified polymers were named CPDOL and PDOL, respectively. DOL and PDOL were dissolved in deuterated DMSO-d6 for 1H NMR and 13C NMR measurements (Bruker 400M). CPDOL was insoluble in deuterated DMSO-d6, so solid 1H NMR and 13C NMR were conducted after it was ground and crushed (Agilent 600M). FTIR spectra were measured to investigate the molecular structures of monomer, crosslinker, and polymers (IRAffinity-1). Raman spectra were also used to analyze the structural information of the molecules above using an excitation wavelength of 785 nm (inVia). Crystallization information of the polymers was analyzed by XRD characterization (D8 Advance). TG-DSC was conducted to analyze the thermal stability of the electrolytes from room temperature to 350 °C (STA409PC). The morphologies of the separator and cycled electrodes were identified using a field-emission scanning electron microscope (ZEISS Sigma300). Surface species of the cycled cathode and anode were detected by XPS measurement (Thermo Scientific K-Alpha).Battery assembly and testing
CR2025 type coin cells were assembled to evaluate the main electrochemical performances. Celgard 2500 separator was used as the supporting skeleton for CPGPE and PGPE. For the measurement of ionic conductivity, different electrolytes were sandwiched between two stainless steel electrodes (SS|electrolyte|SS), and the electrochemical impedance spectra (EIS) of SS|electrolyte|SS cells were measured on a Solartron 1260A workstation with a bias voltage of 5 mV in the range from 106 to 1 Hz. Ionic conductivity (σ) was obtained from eqn (1),| σ = L/(R × S), | (1) |
| σ = Aexp(−Ea/RT), | (2) |
| tLi+ = (Iss(ΔV − I0R0))/(I0(ΔV − IssRss)), | (3) |
LFP or NCM811, SP and PVDF were mixed in NMP at a mass ratio of 8:1:1. The mixture was scraped onto 16 µm aluminum foil and dried at 80 °C for 2 h in air. The LFP cathode (2 or 5 mg cm−2) or NCM811 cathode (2.5 mg cm−2) was obtained after further drying of 12 h at 80 °C in a vacuum oven. To assemble the semi-solid lithium metal batteries, the LFP or NCM811 cathode, lithium metal, and Celgard 2500 separator were integrated into coin cells, the CPGPE or PGPE electrolyte precursor was injected into the cells, and the sealed cells were kept at 30 °C for 30 h or 3 days to form semi-solid lithium metal batteries. Also, Li|electrolyte|Li and Li|electrolyte|Cu cells were assembled to evaluate their stability with lithium metal using the same curing process mentioned above. In addition, commercial liquid electrolyte composed of 1 M LiPF6 in EC:DMC:DEC (1:1:1, vol%) was used for comparison. To evaluate the performance of batteries using CPGPE under extremely harsh conditions, 5 × 4 cm lithium metal pouch cells with NCM811 cathode were assembled for shearing and bending tests. Finally, a 50 mA h lithium metal pouch cell using NCM811 cathode with high loading of 8 mg cm−2 was prepared to check the potential for practical application.
Calculations
Results and discussion
Preparation and characterization
Density functional theory (DFT) calculations were conducted to provide guidelines for electrolyte design. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of several molecules are listed in Fig. 1a and divided by HFE, which is known to be inert in ion transport. On the left side are ether molecules with generally higher LUMO energies than the ester molecules in the right area, which shows the better stability to lithium metal of ethers. On the right side are ester molecules, usually with lower HOMO energies than ether molecules, which shows better resistance to oxidation of esters. It is worth noting that the highly fluorinated hydrofluoroether (HFE) has the lowest HOMO energy of −9.119 eV, showing excellent oxidation stability. In addition, DOL and PDOL (dimers) both display a slightly high HOMO energy, indicating poor oxidation stability. GTE exhibits a lower HOMO energy (−6.899 eV) than DOL (−6.692 eV) and PDOL (−6.827 eV), indicating that crosslinking polymerization of DOL and GTE may help improve the oxidation stability of PDOL-based electrolyte. Based on the above analysis, we designed a semi-solid gel polymer electrolyte consisting of crosslinked PDOL and fluorinated solvents with the expectation of simultaneously achieving favourable stability to high voltage and lithium metal. Due to the weakness to lithium salt dissociation of the fluorinated solvents, a small amount of EMC was added to promote the dissolution of lithium salts and improve ionic conductivity. In general, the volume ratio of monomers and solvents is controlled to be 1:1 in the precursor solution, which is commonly seen in previous reports.32–34 The effect of the ratio of DOL and GTE on the curing state was studied. In the set gradient, complete solidification can only be achieved in an appropriate time when the volume ratio of GTE to DOL is no less than 1:4. However, more crosslinkers reduce the ionic conductivity due to excessive crosslinking (Fig. S1†). In addition, it is found that the crosslinked polymerization of DOL and GTE expresses strong selectivity towards initiators, and the widely used long-chain polymerization initiators, such as LiPF6 (ref. 16) and LiFSI,35 have difficulty triggering this reaction even at high concentrations for a long time (Fig. S2†). Based on the optimized formula composition, DOL and GTE form a crosslinked network structure via in situ polymerization, and the network adsorbed the other solvents and converted to solid state (Fig. 1b).
 | ||
| Fig. 1 (a) The calculated HOMO and LUMO energies of DOL, PDOL, GTE, HFE, FEC, and EMC. (b) A schematic diagram of the formation process of crosslinked gel polymer electrolyte. (c) Crosslinking polymerization mechanism between DOL and GTE. (d) FTIR spectra of DOL, GTE, PDOL, and CPDOL. (e) 1H NMR spectra and (f) 13C NMR spectra of DOL, PDOL, CPDOL elution, and CPDOL. | ||
The mechanism of crosslinked polymerization is shown in Fig. 1c. LiBF4 partially decomposes and produces Lewis acidic BF3. After being protonated by trace amounts of water, H+(BF3OH)− triggers the ring-opening crosslinked polymerization between DOL and GTE.22 Due to the randomness of the crosslinking reaction, it is difficult to define a specific molecular structure after the reaction, and it may even form an extremely irregular network structure (Fig. S3†). To explore the structures of the polymerization products, CPGPE and PGPE were carefully washed to remove the influences of lithium salts and solvents (Fig. S4†). FTIR, Raman, NMR and XRD were used to analyze the structural changes before and after polymerization. As shown in the FTIR spectra (Fig. 1d), the long-chain vibrational peak at 847 cm−1 was observed for both PDOL and CPDOL, indicating the successful occurrence of ring-opening polymerization. The C–O–C vibrational peak slightly red shifts from 1118 cm−1 for PDOL to 1102 cm−1 for CPDOL, implying a special chain structure in the crosslinked network.22,23 In the Raman spectra (Fig. S5†), CPDOL exhibits characteristic peaks of both PDOL and GTE, suggesting the occurrence of copolymerization between DOL and GTE. Further, liquid and solid NMR were measured to determine the changes in the chemical environments of elements before and after reaction (Fig. 1e and f). Two 1H spectral characteristic peaks at 4.78 ppm and 3.77 ppm assigned to the H in O–CH2–O and O–CH2–CH2–O are observed in DOL, and they shift to 4.63 ppm and 3.59 ppm in PDOL, which suggests the occurrence of ring-opening polymerization.35,36 However, CPDOL was found difficult to be dissolved in DMSO-d6 like DOL or PDOL (Fig. S6†), and no PDOL signal was detected in the DMSO-d6 elution immersed with CPDOL for a long time, which indirectly indicates the difference between PDOL and CPDOL. CPDOL was crushed for solid NMR measurement, and the characteristic peaks at 4.63 ppm and 3.59 ppm assigned to the H in O–CH2–O and O–CH2–CH2–O further shift to lower values of 4.16 ppm and 3.14 ppm, which directly confirms the formation of a crosslinked network. Also, some other weak characteristic peaks (4.30 ppm, 3.28 ppm, and 2.78 ppm) were also observed, which may be contributed by various H from GTE. The same results also appear in the 13C spectra. Two 13C spectral characteristic peaks assigned to the C in O–CH2–O and O–CH2–CH2–O shift from 94.54 ppm and 64.29 ppm for DOL to 95.18 ppm and 66.86 ppm for PDOL, and they further shift to 98.36 ppm and 69.88 ppm for CPDOL. The XRD patterns of different polymers are displayed in Fig. S7;† the pure PDOL shows an obvious crystallization peak like polyethylene oxide (PEO),37,38 which is well-known to be detrimental to ion transport. In comparison, the pure CPDOL shows a completely amorphous structure. Although PDOL or PEO exhibits amorphous characteristics after being composited with lithium salt, it should be believed that an intrinsic polymer with an amorphous state will be more conducive to ion transport.
Intrinsic chemical and electrochemical properties of CPGPE and PGPE
The ability to fully infiltrate pores inside a battery is the innate advantage of in situ polymerization over an ex situ solution. The morphology of a Celgard 2500 separator before and after infiltration of CPGPE is shown in Fig. 2a. The average pore diameter of the Celgard 2500 separator is at the level of 0.1 µm. After the precursor infiltrates and solidifies, the pores of the separator are fully filled, suggesting intimate interfacial contact can be formed readily. Thermal stability is crucial for the safety of batteries. As the TG-DSC curve shows in Fig. 2b, CPGPE exhibits better thermal stability than PGPE and can maintain intact solid-state characteristics for 5 minutes at 110 °C while PGPE changes from solid to liquid under the same conditions (Fig. S8a†). CPGPE supported by a crosslinked network exhibits the solid-state characteristic of hard elasticity, while PGPE supported by a long chain is a highly viscous fluid (Fig. S8b†). It is the dense network structure that endows CPGPE with better thermal stability. | ||
| Fig. 2 (a) SEM images of the separator before and after infiltration of CPGPE. (b) TG-DSC curves of CPGPE and PGPE. (c) Plot of ionic conductivity with curing time for CPGPE. (d) Comparison of changes in ionic conductivity after three days for CPGPE and PGPE. (e) LSV curves of Li|CPGPE|SS and Li|PGPE|SS cells. (f) Temperature dependent ionic conductivity curve. Constant voltage polarization curves of (g) CPGPE and (h) PGPE. | ||
Ionic conductivity is the most critical index to support the electrochemical performance of electrolytes. After being left for about 30 h at 30 °C, the ionic conductivity of CPGPE is relatively stable at 0.36 mS cm−1 (based on Celgard 2500 separator, 25 µm) (Fig. 2c and S9†). In comparison, it takes three days to finish the transition from liquid state to solid state at 30 °C for PGPE. Because GTE, with large volume and high viscosity, shows weaker dissociation and solvation ability towards lithium salts than DOL, the ionic conductivity of the initial precursor for CPGPE is lower than that of CGPE. However, after the same curing time of three days, CPGPE exhibits a higher ionic conductivity of 0.33 mS cm−1 compared to that of PGPE of 0.15 mS cm−1 (Fig. 2d and S10†), suggesting the amorphous network structure is more beneficial to Li+ transport. DFT calculations were also used to analyse the mechanism behind the enhanced ionic conductivity of CPGPE. It can be affirmed that the biggest difference between CPDOL and PDOL is the additional three-dimensional nodes brought by GTE in CPDOL. As shown in Fig. S11,† the binding energy of Li+–GTE (−4.239 eV) is higher than that of Li+–PDOL (−2.653 eV), which demonstrates that the three-dimensional structure is more conducive to the dissociation of lithium salts and generates more active Li+, resulting in higher ionic conductivity.32
In addition to the advantage in ionic conductivity, CPGPE also shows a higher electrochemical window of 4.6 V than that of 4.4 V for PGPE (Fig. 2e), which is consistent with results of the electrochemical floating test (Fig. S12†). This could be partly contributed by the stable crosslinked network structure that possesses the higher HOMO energy of GTE than PDOL and fewer terminal hydroxyl groups (Fig. S13†)37 and partly to the high oxidation resistance from fluorinated solvents (Fig. 1a). Activation energy is calculated from the fitting curve of the conductivity–temperature relationship (Fig. 2f and S14†). CPGPE exhibits a lower activation energy than PGPE, which means the crosslinked network structure can promote desolvation of Li+ and homogenize lithium plating.24 In addition, CPGPE shows a higher Li+ transfer number compared to PGPE, and this contributes to both reduction of polarization and uniform lithium plating (Fig. 2g and h). LiTFSI, which is easy to dissociate, is the dominating lithium salt in the semi-solid electrolyte, ensuring high ionic conductivity. The binding energy between TFSI− and GTE (−1.201 eV) is higher than that between TFSI− and PDOL (−1.152 eV) (Fig. S15†), which means the three-dimensional structure can more effectively restrict the movement of anions and further confirms the higher Li+ transfer number of CPGPE.23 These results discussed above fully demonstrate that CPGPE has significant advantages over PGPE in Li+ transport, oxidation resistance, thermal stability, etc., which provide favorable support for the construction of high-performance and safe lithium metal batteries.
Evaluation of stability to lithium anode by lithium symmetrical cells
Lithium symmetric cells were assembled to evaluate the stability of electrolytes to lithium metal. The cell with CPGPE can stably operate for 2700 h at a current density of 0.2 mA cm−2 with no significant polarization growth, while the PGPE system shows obvious polarization growth (Fig. 3a–c). Additionally, CPGPE and PGPE both show clear advantages over commercial ester-based liquid electrolyte (LE) in terms of stability to lithium metal. EIS measurements were conducted on the initial and cycled Li‖Li cells (Fig. S16†). Compared to the initial state, the cycled cells all show decreased impedance values and another semicircle (Rx2) appears in the mid-frequency region because of the reaction activation.39 It can be seen that CPGPE shows the smallest impedance value after cycling. In addition, the later semicircle (Rx2) in the mid-frequency region after cycling is often defined as interface impedance,40,41 which is closely related to interface reactions. The ratio of Rx2 in the total impedance of the mid-frequency region for the three systems is compared, and the result shows that CPGPE displays the smallest ratio of interface impedance, which corresponds well to its smallest polarization voltage and most uniform lithium anode after cycling.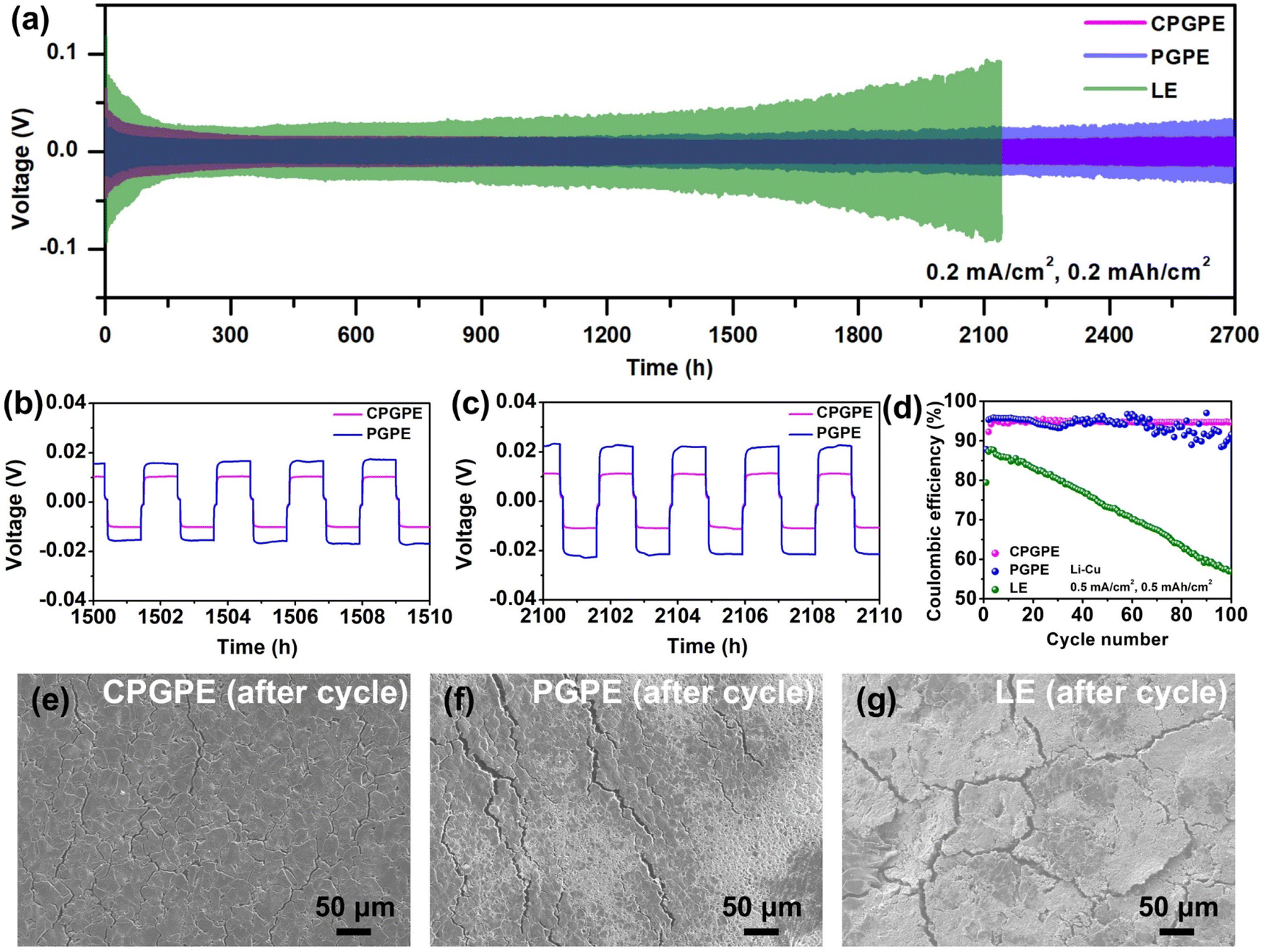 | ||
| Fig. 3 (a–c) Cycling performances of lithium symmetrical cells assembled with CPGPE, PGPE, and LE cycled at 0.2 mA cm−2. (d) Cycling performances of Li‖Cu cells assembled with different electrolytes. SEM images of the cycled lithium metal for (e) CPGPE, (f) PGPE, and (g) LE. | ||
Also, Li‖Cu cells were assembled to further evaluate the stability to lithium metal of different electrolytes. As shown in Fig. 3d and S17,† the Li|CPGPE|Cu cell exhibits excellent cycling stability at 0.5 mA cm−2, and a high coulombic efficiency of 94.7% remains after 100 cycles. The Li|PGPE|Cu cell shows a decreased cycling stability at the same current density and obvious fluctuations in Coulomb efficiency can be seen after 60 cycles. The Li|LE|Cu cell expresses a continuously declining coulombic efficiency, with a poor value of only 53.9% left after 100 cycles, which may be caused by the serious interface side reactions.
Furthermore, the cycled Li‖Li cells were disassembled to observe the surface morphology of lithium metal by SEM (Fig. 3e–g). The surface of the lithium metal is relatively flat and dense for CPGPE and shows obvious cracks and by-products for PGPE. In contrast, the surface of the lithium metal appears to have significant loose side reaction products and cracks with a tendency to fall off.34,42 The results above sufficiently illustrate that the ether-based crosslinked network polymer can effectively guide uniform deposition of Li+ and reduce interface side effects.
Performance of lithium metal batteries
Lithium metal batteries were assembled to evaluate the practical electrochemical performances of CPGPE and PGPE. As shown in Fig. 4a and b, the LFP‖Li battery assembled with CPGPE exhibits an initial discharge capacity of 132.2 mA h g−1 and decays to 117 mA h g−1 after 1000 cycles at 1C with a capacity retention of 88% and a high coulombic efficiency of over 99.9%, while the LFP‖Li battery assembled with PGPE exhibits an initial capacity of 124.6 mA h g−1 and decays to 100.6 mA h g−1 after 600 cycles with a lower capacity retention of 81%. The LFP‖Li battery assembled with LE shows a sudden collapse after 150 cycles accompanied by abnormal fluctuation of coulombic efficiency, which is a common phenomenon in ester liquid lithium metal batteries. Even at a higher rate of 2C, the LFP‖Li battery assembled with CPGPE still exhibits a satisfactory capacity retention of 91.7% and a coulombic efficiency of over 99.9% after 500 cycles (Fig. S18†). Also, the LFP|CPGPE|Li semi-solid battery with higher active materials loading of 5 mg cm−2 exhibits a favourable capacity retention of 97.7% after 150 cycles at 1C, which shows promisingly practical application prospects (Fig. S19†). The results also show clear advantages over the widely studied PDOL based lithium metal batteries in recent years (Table S1†).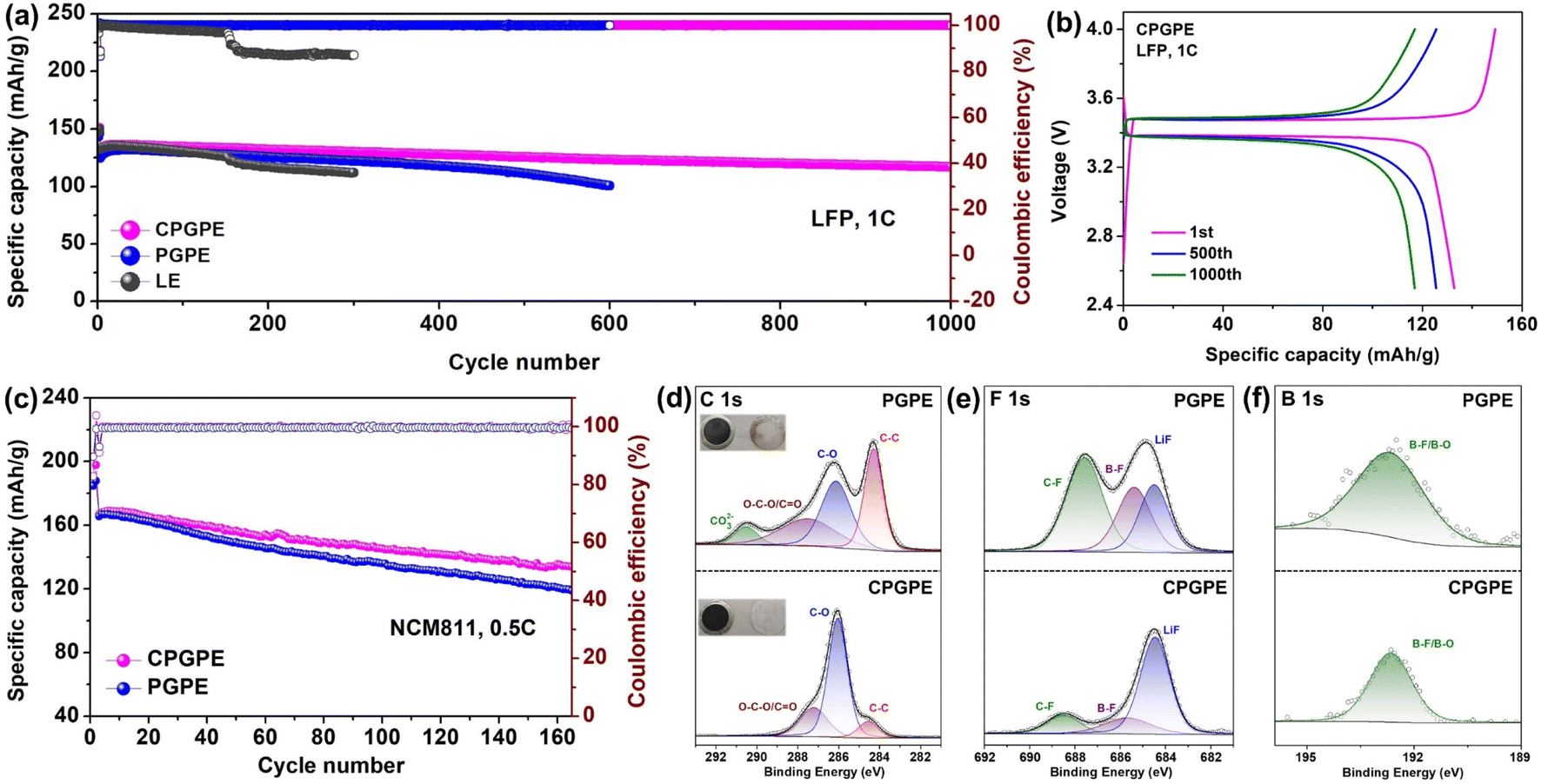 | ||
| Fig. 4 (a) Cycling performances of the LFP‖Li batteries assembled with CPGPE, PGPE, and LE at 1C. (b) Charge/discharge curves of the LFP|CPGPE|Li battery at different cycles. (c) Cycling performances of the NCM811‖Li batteries assembled with CPGPE and PGPE. XPS (d) C 1s spectra, (e) F 1s spectra, and (f) B 1s spectra of the washed cathodes from the cycled NCM811‖Li batteries assembled with CPGPE and PGPE. | ||
NCM811‖Li batteries were assembled to evaluate the high voltage stabilities of CPGPE and PGPE (Fig. 4c). The NCM811|CPGPE|Li battery has an initial capacity of 167 mA h g−1 at 0.5C and a capacity retention of 80.2% after 165 cycles, while the battery assembled with PGPE shows a lower capacity retention of 71.7% in the same conditions. The advantage of CPGPE over PGPE is more apparent at 0.2C because it experiences a longer time in the high voltage region at a smaller cycling rate (Fig. S20†). To study the mechanism of improvement in the performance of batteries from CPGPE, XPS measurements were conducted to detect the chemical compositions of the cycled NCM811 cathode. The NCM811‖Li batteries using PGPE and CPGPE were disassembled after 120 cycles at 0.5C. In the C 1s spectra of the washed NCM811 cathode (Fig. 4d), the O–C–O peak and C–O peak correspond to the O–CH2–O–CH2–CH2 unit in PDOL or CPDOL.43 The larger proportion of peaks representing the polymer in the CPGPE system may be due to the CPDOL inside the cathode being more difficult to wash away compared to the PDOL. An obvious CO32− peak was observed in the PGPE system which represents the decomposition of solvents and PDOL, while there was no obvious CO32− peak in the CPGPE system, indicating suppressed decomposition reactions.24,44 Similarly, in the F 1s spectra (Fig. 4e), the CPGPE system exhibits a smaller proportion of C–F and B–F peaks compared to the PGPE system, indicating the weaker decomposition of fluorinated solvents and borates. It must be said that reasonable decomposition of borates will help form a stable CEI on the cathode and protect it from continuous side reactions.45,46 The same situation was also observed in the B 1s spectra (Fig. 4f).
In terms of lithium anodes, the cycled lithium anode from the NCM811|CPGPE|Li battery exhibits a flat and dense surface, while that from the NCM811|PGPE|Li battery shows more cracks (Fig. S21†), which is consistent with the results in the Li symmetric batteries. XPS depth profiling was applied to analyse the surface composition of the cycled lithium anodes (Fig. 5a and b). In the C 1s spectra of different depths of etching, the lithium anode from the NCM811|CPGPE|Li battery exhibits lower O–C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–O peak proportions than that the lithium anode from the NCM811|PGPE|Li battery, suggesting a weaker decomposition of polymers in the CPGPE system than in the PGPE system.23 In the F 1s spectra, the cycled lithium anode from the NCM811|PGPE|Li battery shows a continuously enhanced LiF peak with increasing etching depth, indicating excessive consumption of fluorinated solvents and lithium salts.47,48 In comparison, the CPGPE system shows relatively stable LiF content at different depths, suggesting that the crosslinked polymer effectively alleviated the fluorinated solvent and anion involved side reactions in lithium anode. Also, the O 1s and B 1s spectra in Fig. S22† show a similar trend to the results above. As shown in the O 1s spectra, the proportion of C–O and O–CO peaks corresponding to organic components for the cycled CPGPE system are significantly less than those for the cycled PGPE system at different depths, indicating the suppressed decomposition reactions between organic molecules and the lithium anode due to the better compatibility to lithium metal of CPGPE.37,49 In the B 1s spectra, the cycled PGPE system shows a continuously increasing signal strength of B–F/B–O with increasing etching depth, indicating excessive lithium salt decomposition. In contrast, the cycled CPGPE system exhibits a relatively stable signal strength of B–F/B–O at different etching depths, suggesting the formation of a stable SEI film. In regard to the better electrochemical performance of a crosslinked network polymer than a long-chain polymer, a reasonable explanation can be provided by the following points: (i) the completely amorphous network structure with a larger free volume is more beneficial to the desolvation and transport of Li+, (ii) a more stable molecular structure and fewer terminal hydroxyl groups enhance high voltage stability, and (iii) a crosslinked polymer limits the movement of anions to some extent (higher migration numbers), promoting uniform deposition of Li+ on the lithium anode.
O, and C–O peak proportions than that the lithium anode from the NCM811|PGPE|Li battery, suggesting a weaker decomposition of polymers in the CPGPE system than in the PGPE system.23 In the F 1s spectra, the cycled lithium anode from the NCM811|PGPE|Li battery shows a continuously enhanced LiF peak with increasing etching depth, indicating excessive consumption of fluorinated solvents and lithium salts.47,48 In comparison, the CPGPE system shows relatively stable LiF content at different depths, suggesting that the crosslinked polymer effectively alleviated the fluorinated solvent and anion involved side reactions in lithium anode. Also, the O 1s and B 1s spectra in Fig. S22† show a similar trend to the results above. As shown in the O 1s spectra, the proportion of C–O and O–CO peaks corresponding to organic components for the cycled CPGPE system are significantly less than those for the cycled PGPE system at different depths, indicating the suppressed decomposition reactions between organic molecules and the lithium anode due to the better compatibility to lithium metal of CPGPE.37,49 In the B 1s spectra, the cycled PGPE system shows a continuously increasing signal strength of B–F/B–O with increasing etching depth, indicating excessive lithium salt decomposition. In contrast, the cycled CPGPE system exhibits a relatively stable signal strength of B–F/B–O at different etching depths, suggesting the formation of a stable SEI film. In regard to the better electrochemical performance of a crosslinked network polymer than a long-chain polymer, a reasonable explanation can be provided by the following points: (i) the completely amorphous network structure with a larger free volume is more beneficial to the desolvation and transport of Li+, (ii) a more stable molecular structure and fewer terminal hydroxyl groups enhance high voltage stability, and (iii) a crosslinked polymer limits the movement of anions to some extent (higher migration numbers), promoting uniform deposition of Li+ on the lithium anode.
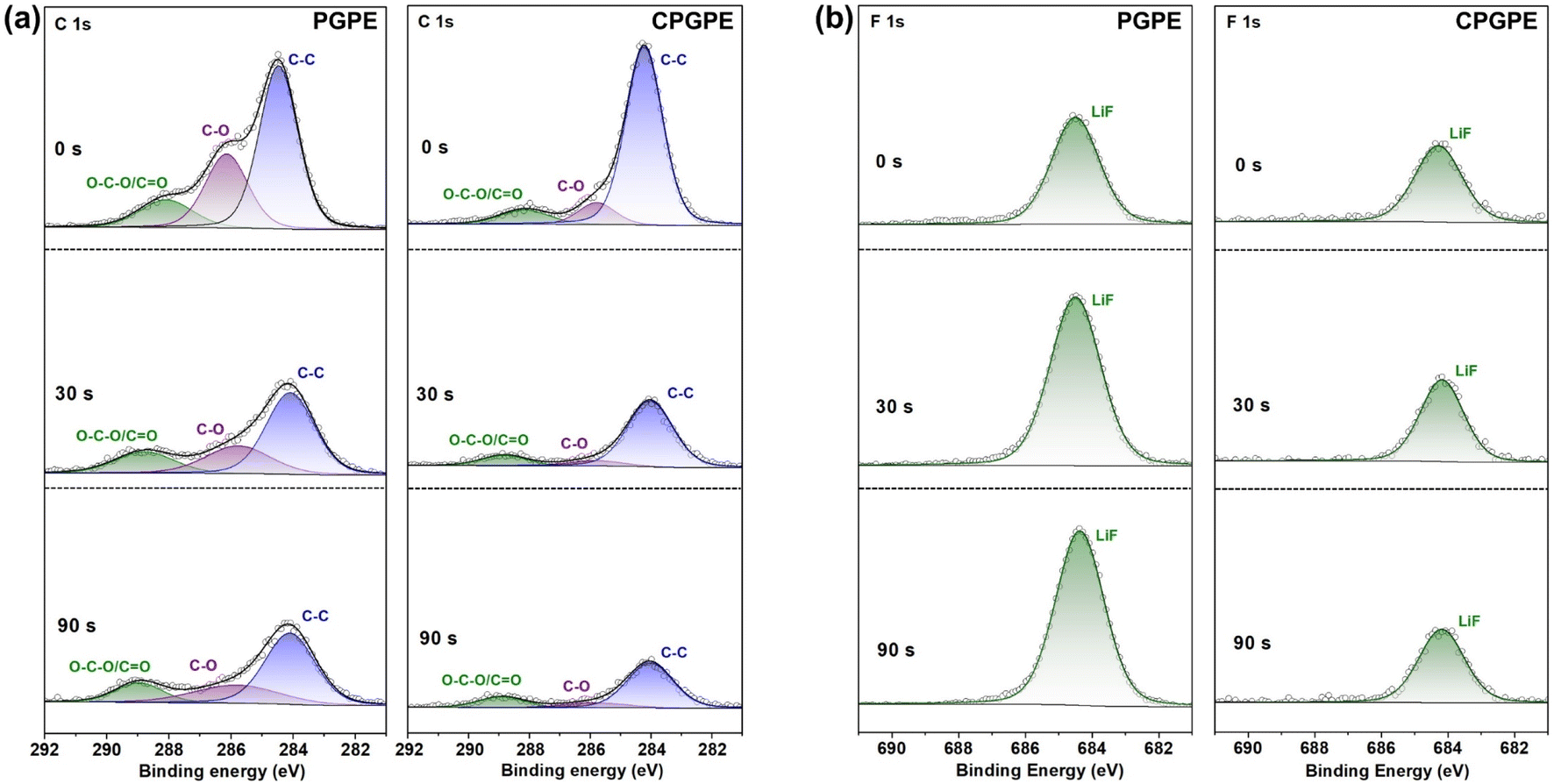 | ||
| Fig. 5 XPS (a) C 1s spectra and (b) F 1s spectra of the washed lithium anodes from the cycled NCM811‖Li batteries assembled with CPGPE and PGPE. | ||
Safety and work tolerance under harsh conditions
High safety is crucial for the application of lithium metal batteries. NCM811‖Li pouch cells with CPGPE and LE were assembled to evaluate the operational reliability under bending and shearing conditions. The NCM811‖Li pouch cell assembled with CPGPE can still maintain normal operation even after complete bending and multiple shearings (Fig. 6a–c). This ability of CPGPE can be attributed to the internal integration resulting from in situ solidification which prevents the interface from losing contact due to external deformation, and the hard elastic crosslinked electrolyte can effectively provide support to prevent hard short circuits between the cathode and anode. In comparison, the lamp bulb lighted by the NCM811‖Li pouch cell with LE shows a weakened brightness after bending and stops working after shearing (Fig. 6d–f), which may be caused by local contact loss and hard short circuits, respectively. Finally, a 50 mA h NCM811|CPGPE|Li battery with an active material loading of 8 mg cm−2 was fabricated to evaluate the practical working potential. As shown in Fig. 6g and h, the lithium metal battery can stably operate for 10 cycles at 0.1C, and it exhibits a capacity retention of 88% even in a highly bent state of over 90°, demonstrating its promising application prospects.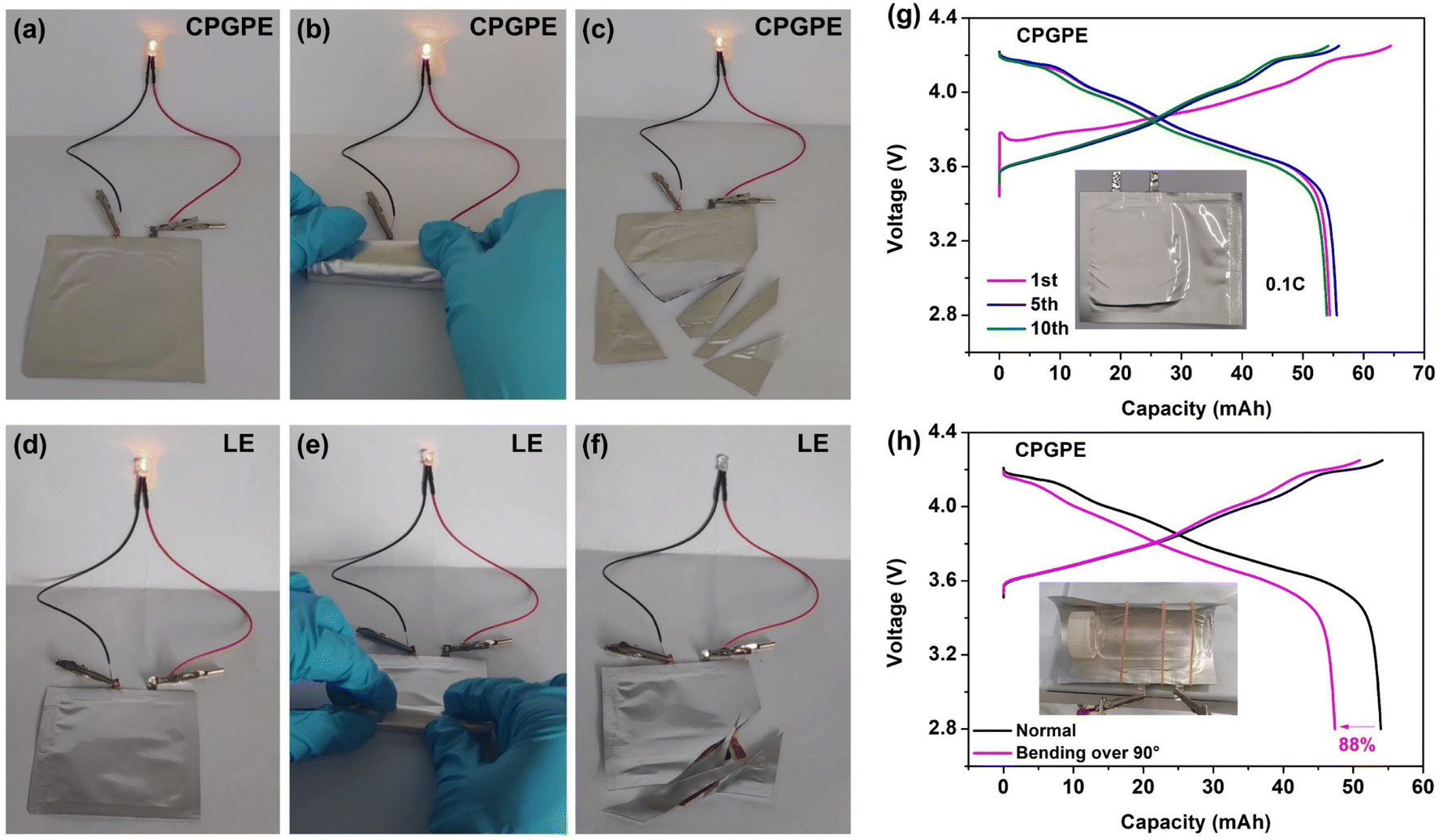 | ||
| Fig. 6 Evaluation of (a–c) the NCM811|CPGPE|Li battery and (d–f) NCM811|LE|Li battery operating in normal, folding, and shearing states. (g) Charge/discharge curve of the 50 mA h NCM811|CPGPE|Li pouch cell at 0.1C and (h) its operating ability at the bending state. | ||
Conclusion
In summary, an in situ fabricated semi-solid electrolyte composed of crosslinked polymer (copolymerization of DOL and GTE) and high voltage resistance fluorinated solvents was proposed. The totally amorphous crosslinked network structure endows CPGPE with high ionic conductivity and low activation energy. This electrolyte also exhibits high electrochemical stability due to the stable structure and fewer terminal hydroxyl groups of the crosslinked network. Fluorinated solvents (HFE and FEC) with low HOMO energy further enhance the oxide stability. In addition, it also exhibits favourable Li+ transfer number and thermal stability. The lithium symmetric battery assembled with CPGPE stably operates for 2700 h with no significant polarization growth at 0.2 mA cm−2. The LFP‖Li battery exhibits satisfactory capacity retentions of 88% after 1000 cycles at 1C and 91.7% after 500 cycles at 2C. Further, the NCM811‖Li battery assembled with CPGPE also exhibits stable cycling, and the XPS results reveal the mechanism of stabilizing the NCM811 cathode and lithium anode by the CPDOL crosslinked polymer. A 50 mA h semi-solid NCM811‖Li pouch cell with a NCM811 loading of 8 mg cm−2 can also work stably, demonstrating its potential for practical applications. It is believed that this semi-solid electrolyte supported by all the results above will provide a prospective reference for development of ether-based electrolytes in high safety and high energy density lithium metal batteries in the future.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial supports from the National Natural Science Foundation of China (No. 52072390), the National High-Level Talents Special Support Program (Leading Talent of Technological Innovation), the China Postdoctoral Science Foundation (No. 2023M743648), the Young Scientists Fund of the National Natural Science Foundation of China (No. 52302330). We also gratefully appreciate the support from the Shanghai Emperor of Cleaning Hi-Tech Co., LTD.References
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS.
- Y.-S. Hu, Nat. Energy, 2016, 1, 16042 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- F. Wu, K. Zhang, Y. Liu, H. Gao, Y. Bai, X. Wang and C. Wu, Energy Storage Mater., 2020, 33, 26–54 CrossRef.
- G. Xi, M. Xiao, S. Wang, D. Han, Y. Li and Y. Meng, Adv. Funct. Mater., 2020, 31, 2007598 CrossRef.
- D. M. Reinoso and M. A. Frechero, Energy Storage Mater., 2022, 52, 430–464 CrossRef.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- Y. Wu, Y. Li, Y. Wang, Q. Liu, Q. Chen and M. Chen, J. Energy Chem., 2022, 64, 62–84 CrossRef CAS.
- X. Fan, C. Zhong, J. Liu, J. Ding, Y. Deng, X. Han, L. Zhang, W. Hu, D. P. Wilkinson and J. Zhang, Chem. Rev., 2022, 122, 17155–17239 CrossRef CAS PubMed.
- Q. Liu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2300798 CrossRef CAS.
- M. Sun, Z. Zeng, W. Zhong, Z. Han, L. Peng, S. Cheng and J. Xie, Batteries Supercaps, 2022, 5, e202200338 CrossRef CAS.
- V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter and J. R. Nair, Energy Environ. Sci., 2021, 14, 2708–2788 RSC.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- F.-Q. Liu, W.-P. Wang, Y.-X. Yin, S.-F. Zhang, J.-L. Shi, L. Wang, X.-D. Zhang, Y. Zheng, J.-J. Zhou, L. Li and Y.-G. Guo, Sci. Adv., 2018, 4, eaat5383 CrossRef CAS PubMed.
- H. Yang, M. Jing, L. Wang, H. Xu, X. Yan and X. He, Nano-Micro Lett., 2024, 16, 127 CrossRef.
- Z. Ren, J. Li, M. Cai, R. Yin, J. Liang, Q. Zhang, C. He, X. Jiang and X. Ren, J. Mater. Chem. A, 2023, 11, 1966–1977 RSC.
- H. Yang, B. Zhang, M. Jing, X. Shen, L. Wang, H. Xu, X. Yan and X. He, Adv. Energy Mater., 2022, 12, 2201762 CrossRef CAS.
- W. Li, J. Gao, H. Tian, X. Li, S. He, J. Li, W. Wang, L. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2021, 61, e202114805 CrossRef.
- J. Cui, Y. Du, L. Zhao, X. Li, Z. Sun, D. Li and H. Li, Chem. Eng. J., 2023, 461, 141973 CrossRef CAS.
- S. Wen, C. Luo, Q. Wang, Z. Wei, Y. Zeng, Y. Jiang, G. Zhang, H. Xu, J. Wang, C. Wang, J. Chang and Y. Deng, Energy Storage Mater., 2022, 47, 453–461 CrossRef.
- Y. Du, L. Zhao, C. Xiong, Z. Sun, S. Liu, C. Li, S. Hao, W. Zhou and H. Li, Energy Storage Mater., 2023, 56, 310–318 CrossRef.
- J. Zhu, J. Zhang, R. Zhao, Y. Zhao, J. Liu, N. Xu, X. Wan, C. Li, Y. Ma, H. Zhang and Y. Chen, Energy Storage Mater., 2023, 57, 92–101 CrossRef.
- J. Zhu, R. Zhao, J. Zhang, X. Song, J. Liu, N. Xu, H. Zhang, X. Wan, X. Ji, Y. Ma, C. Li and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202400303 CrossRef CAS.
- C. Zhang, Z. Lu, M. Song, Y. Zhang, C. Jing, L. Chen, X. Ji and W. Wei, Adv. Energy Mater., 2023, 13, 2203870 CrossRef CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- K. Mu, D. Wang, W. Dong, Q. Liu, Z. Song, W. Xu, P. Yao, Y. a. Chen, B. Yang, C. Li, L. Tian, C. Zhu and J. Xu, Adv. Mater., 2023, 35, 2304686 CrossRef CAS.
- Q. Ma, J. Yue, M. Fan, S. J. Tan, J. Zhang, W. P. Wang, Y. Liu, Y. F. Tian, Q. Xu, Y. X. Yin, Y. You, A. Luo, S. Xin, X. W. Wu and Y. G. Guo, Angew. Chem., Int. Ed., 2021, 60, 16554–16560 CrossRef CAS.
- J.-Y. Liang, X.-D. Zhang, Y. Zhang, L.-B. Huang, M. Yan, Z.-Z. Shen, R. Wen, J. Tang, F. Wang, J.-L. Shi, L.-J. Wan and Y.-G. Guo, J. Am. Chem. Soc., 2021, 143, 16768–16776 CrossRef CAS.
- Y. Wang, T. Li, X. Yang, Q. Yin, S. Wang, H. Zhang and X. Li, Adv. Energy Mater., 2023, 14, 2303189 CrossRef.
- W. Ren, Y. Zhang, R. Lv, S. Guo, W. Wu, Y. Liu and J. Wang, J. Power Sources, 2022, 542, 231773 CrossRef CAS.
- X. Yang, M. Jiang, X. Gao, D. Bao, Q. Sun, N. Holmes, H. Duan, S. Mukherjee, K. Adair, C. Zhao, J. Liang, W. Li, J. Li, Y. Liu, H. Huang, L. Zhang, S. Lu, Q. Lu, R. Li, C. V. Singh and X. Sun, Energy Environ. Sci., 2020, 13, 1318–1325 RSC.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- Q. Hao, J. Yan, Y. Gao, F. Chen, X. Chen, Y. Qi and N. Li, ACS Appl. Mater. Interfaces, 2024, 16, 44689–44696 CrossRef CAS.
- W. Lee, C. K. Lyon, J. H. Seo, R. Lopez‐Hallman, Y. Leng, C. Y. Wang, M. A. Hickner, C. A. Randall and E. D. Gomez, Adv. Funct. Mater., 2019, 29, 1807872 CrossRef.
- Y.-N. Yang, C.-H. Cui, Z.-Q. Hou, Y.-Q. Li and T. Zhang, Energy Storage Mater., 2022, 52, 1–9 CrossRef.
- J. Zhou, H. Ji, Y. Qian, J. Liu, T. Yan, C. Yan and T. Qian, ACS Appl. Mater. Interfaces, 2021, 13, 48810–48817 CrossRef CAS PubMed.
- D. Chen, T. Zhu, M. Zhu, P. Kang, S. Yuan, Y. Li, J. Lan, X. Yang and G. Sui, Small Methods, 2022, 6, 2201114 CrossRef CAS.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2021, 12, 2102932 CrossRef.
- J. Y. Liang, X. D. Zhang, X. X. Zeng, M. Yan, Y. X. Yin, S. Xin, W. P. Wang, X. W. Wu, J. L. Shi, L. J. Wan and Y. G. Guo, Angew. Chem., Int. Ed., 2020, 59, 6585–6589 CrossRef CAS PubMed.
- X. Chen, W. Xu, M. H. Engelhard, J. Zheng, Y. Zhang, F. Ding, J. Qian and J.-G. Zhang, J. Mater. Chem. A, 2014, 2, 2346–2352 RSC.
- Q. Wang, Z. Yao, C. Zhao, T. Verhallen, D. P. Tabor, M. Liu, F. Ooms, F. Kang, A. Aspuru-Guzik, Y.-S. Hu, M. Wagemaker and B. Li, Nat. Commun., 2020, 11, 4188 CrossRef PubMed.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef.
- Y. Liu, H. Zou, Z. Huang, Q. Wen, J. Lai, Y. Zhang, J. Li, K. Ding, J. Wang, Y.-Q. Lan and Q. Zheng, Energy Environ. Sci., 2023, 16, 6110–6119 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04857a |
| This journal is © The Royal Society of Chemistry 2024 |