
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of low-cost stainless-steel electrodes for efficient and stable anion-exchange membrane water electrolysis†
Tao
Jiang
 *a,
Leila
Zouridi
bc,
Nannan
Li
a,
Vassilios
Binas
cd,
Marc. C. A.
Stuart
e,
P. V.
Aravind
f,
Bayu
Jayawardhana
a,
Paolo P.
Pescarmona
a and
Vasileios
Kyriakou
*a
*a,
Leila
Zouridi
bc,
Nannan
Li
a,
Vassilios
Binas
cd,
Marc. C. A.
Stuart
e,
P. V.
Aravind
f,
Bayu
Jayawardhana
a,
Paolo P.
Pescarmona
a and
Vasileios
Kyriakou
*a
aEngineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: taojiang0510@gmail.com; v.kyriakou@rug.nl
bDepartment of Materials Science and Technology, University of Crete, Heraklion, Greece
cInstitute of Electronic Structure and Laser, Foundation for Research and Technology-Hellas, Greece
dDepartment of Chemistry, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece
eGroningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
fEnergy and Sustainability Research Institute Groningen (ESRIG), University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 24th September 2024
Abstract
Commercial stainless steel is gaining interest as a promising, low-cost electrode material for green hydrogen production through water electrolysis in an alkaline environment. Herein, the electrocatalytic performance of 304-type stainless steel mesh in anion-exchange membrane (AEM) cells was enhanced through a simple two-step activation process, i.e., chemical etching followed by electrochemical activation. The modified electrodes exhibited comparable hydrogen and oxygen evolution reaction properties to noble metal-based electrodes, requiring >300 mV lower cell voltage than the unmodified stainless steel-based cells to sustain a constant current of 5.0 A (1.0 A cm−2). Electrochemical impedance spectroscopy studies and investigation of wettability and bubble dynamics demonstrated a significant decrease in interfacial contact, charge transfer, and mass transport resistances. Characterization by X-ray photoelectron spectroscopy and transmission electron microscopy of the modified stainless-steel surface revealed the presence of nanocrystalline Fe–NiCr LDH and Ni(OH)2/Fe(OH)2 species when used in hydrogen and oxygen evolution sides, respectively, which may explain the significantly higher performance. Moreover, the long-term durability of the modified electrodes was assessed in a continuous flow electrolyzer where exceptional stability was observed at a constant current of 5.0 A (1.0 A cm−2) for 250 h. Due to its simplicity and cost-effectiveness, the proposed modification of stainless-steel electrodes has the potential for upscaling and deployment in the next-generation, low-cost AEM systems.
Introduction
The increasing awareness of the need to mitigate the impact of global warming and climate change is spurring the adoption of clean and renewable energy sources.1 Among the promising technologies for harnessing renewable energies (such as solar, wind, and hydro) to foster a sustainable future, water electrolysis stands out. This process can produce hydrogen gas, an efficient energy carrier and an alternative to fossil fuels for power generation.2,3 However, hydrogen production through electrolysis remains limited to less than 4% due to its high cost.4,5 Currently, three types of low-temperature water electrolyzers have been developed: conventional alkaline water electrolyzers (AWEs), proton exchange membrane (PEM) water electrolyzers, and anion exchange membrane (AEM) water electrolyzers.6 The AEM water electrolyzer can represent a significant advancement as it combines the benefits of both AWE and PEM electrolyzers. It enables the use of non-noble-metal electrocatalysts, similar to AWE, and incorporates a high-performance membrane separator, akin to PEM electrolyzers.7,8 Despite these advantages, the AEM electrolyzer is still in the early stages of development, and there is ample room for reducing the total device capital costs, as well as improving its efficiency, particularly when employing non-noble-metal catalysts.9–13 Extensive research on alkaline electrolysis has focused on non-noble metal-based electrocatalysts, including transition metals such as Ni, Fe, Co, and Cu.14–23 However, in recent years, the growing demand for these metals in energy storage and conversion applications has caused their prices to rise, eroding their initial cost advantages. This trend is expected to continue, posing challenges for future implementations. Moreover, most of them require complex synthesis processes to obtain small amounts of catalysts, which are not suitable for large-scale industrial applications. Consequently, there is a significant endeavor to explore alternative materials that should offer the combined benefit of modest price and easy fabrication on a large scale. This pursuit aims to reduce the overall cost of the AEM water electrolyzer and optimize its efficiency, thereby promoting its widespread adoption. By achieving these goals, the obstacles posed by cost and scalability can be overcome, enabling the AEM water electrolyzers to emerge as a viable and sustainable solution for hydrogen production.Commercial stainless steel, such as the widely used 316-/304-type stainless steel, is known for its large-scale availability and versatility across various industries. In the context of alkaline water electrolysis, stainless steel exhibits several advantages that make it an ideal choice as a current collector or conductive substrate.5 One key advantage of stainless steel is its mechanical and chemical stability, which allows it to withstand the harsh operating conditions of electrolysis. Additionally, stainless steel possesses good electrical conductivity, ensuring efficient electron transfer during the electrolysis process.24 One of the most appealing aspects of stainless steel for water electrolysis applications is its affordability. Compared to other specialized materials such as nickel mesh (80 mesh, which costs around 100 USD per m2) and carbon cloth (which costs approximately 875 USD per m2), the price of 304-type stainless steel mesh (80 mesh) is considerably lower, at around 40 USD per m2.25 Moreover, stainless steel, the composition of which includes elements that are active in catalyzing water electrolysis such as nickel (Ni), chromium (Cr), and iron (Fe), provides a foundation for potential functionalization and enhancement as high-performance electrocatalysts specifically tailored for alkaline water electrolysis applications.24,26 Significant endeavors have been undertaken to tailor the surface properties and composition of stainless steel, harnessing its inherent elements to enhance its electrocatalytic activity in water electrolysis.5,24–32 However, its catalytic performance still falls short of meeting industrial requirements of bearing large current densities (>500 mA cm−2) at competitive potentials and displaying long-term durability.24,27–29,31,32 Moreover, there is a scarcity of reports on the hydrogen evolution reaction (HER) performance of stainless steel that satisfies industrial application standards.5,25,26 Addressing these limitations and improving the electrocatalytic performance of stainless steel in both the oxygen evolution reaction (OER) and the HER are essential for advancing the industrial application of stainless steel in alkaline water electrolysis. It is important to highlight that, up to now, no research has been conducted on: (1) modifying stainless steel as bifunctional electrodes using simple methods; (2) evaluating the modified electrodes in advanced AEM water electrolysis at industrial-scale current densities; (3) revealing the electrolysis mechanism through in situ EIS analysis and a thorough investigation of wettability and bubble dynamics.
Aiming at developing cost-competitive, high-performance, and stable bifunctional electrodes that will be attractive for industrial use, herein we attempted to address the challenges mentioned above by functionalizing stainless steel (SS) mesh with a simple and cost-effective strategy and validating its performance in an AEM electrolyzer cell. Specifically, we focused on engineering low-cost 304-type stainless steel mesh (SM) through a 2-step activation process. The first step involved chemical activation (CA), which created a suitable surface morphology and removed any impurities that could hinder the electrocatalytic activity (SM-CA). The second step of electrochemical activation involved an ion exchange strategy, which introduced catalytically active species on the surface of the SM-CA. As reported in most of the previous publications, we benchmarked the developed electrodes with noble metal-based electrodes to evaluate their performance.11,25,26,28,29,32,33 The modified electrode (SM-CA-H, at the cathodic side in electrochemical activation) exhibited comparable HER performance with that of Pt/C, as well as SM-CA-O (at the anodic side in electrochemical activation) showed higher OER performance than the commercial IrO2. More importantly, an AEM electrolyzer cell constructed with SM-CA-H(−)‖SM-CA-O(+) displayed more than 300 mV-voltage drop compared to a similar cell with bare SM electrodes, which was attributed to the great improvement of interfacial contact, charge transfer, and mass transport of the AEM electrolyzer. Additionally, the obtained AEM electrolyzer possessed outstanding durability for 250 h at 5.0 A (1.0 A cm−2). The results are attributed to the generation of nanocrystalline Fe–NiCr LDH and nanocrystalline Ni(OH)2/Fe(OH)2 active species in the SM-CA-H and SM-CA-O electrodes. Such cost-effective and high-performance electrodes compete with state-of-the-art noble-based electrodes and display great potential to be deployed in the next-generation AEM electrolyzer.
Results and discussion
Modification and characterization of the SS electrodes
The electrodes were prepared by functionalizing commercial 304-type stainless steel mesh (SM) through a 2-step activation process (described in detail in the Experimental section of the ESI†). In brief, the first step of chemical etching helped create a rough surface morphology on SMs and remove any impurities that could hinder the electrocatalytic activity.25,26,34 The samples subjected to this chemical activation were labeled SM-CA (Steel Mesh-Chemical Activation). The second step of ion exchange strategy was realized by a simple electrochemical treatment, which allowed the introduction of the desired catalytic species onto the surface of the target substrate.5,29 The resulting cathodic SM-CA sample and anodic SM-CA sample through electrochemical activation were identified as SM-CA-H and SM-CA-O, respectively.The X-ray diffraction (XRD) technique was employed to characterize the crystalline phase of SM-CA-H and SM-CA-O. Fig. 1a shows the XRD patterns, revealing diffraction peaks corresponding to austenite (Cr0.19Fe0.7Ni0.11, JCPDS no. 33-0379), which were also present in the parent SM material.28,30 Moreover, the SM-CA-H sample displayed a weak diffraction peak corresponding to (110) of Ni–Cr–Fe (JCPDS no. 35-1375), which was not observed in the XRD patterns of SM, SM-CA and SM-CA-O. This observation suggests that the reductive conditions experienced by the cathodic SM-CA-H electrode during electrochemical activation might play a role in the formation of Ni–Cr–Fe species.30 In addition, scanning electron microscopy (SEM) was used to reveal the surface morphology of the prepared samples. A distinctively cracked morphology was observed for SM-CA, compared to the relatively smooth surface of SM, as depicted in Fig. 1b and c. This change in morphology could be attributed to the etching process during the initial activation, which resulted in the significant removal of iron species from the exposed SM, as evidenced by elemental mapping and EDS (Energy Dispersive Spectroscopy) analysis (see Fig. S1, S2 and Table S1 in the ESI†). Fig. 1d and g illustrate the surface morphology of SM-CA-H and SM-CA-O, respectively. These images show the in situ generation of nanoparticles on the pre-catalyst of SM-CA during the second activation process, which involved ion exchange and electrochemical reconstruction strategies.29,35–37 To gain further insight into the surface layer of SM-CA-H and SM-CA-O electrodes, the exfoliated flakes obtained from the surface of SM-CA-H (inset of Fig. 1e) and SM-CA-O (inset of Fig. 1h) were analyzed through transmission electron microscopy (TEM). As a comparison, the high-resolution TEM (HR-TEM) image of SM-CA, displaying a thick amorphous layer, is shown in Fig. S3 (ESI†). HAADF-STEM-EDS elemental contents of SM-CA, SM-CA-H, and SM-CA samples are exhibited in Table S2.† The high-angle annular dark-field (HAADF)-TEM elemental mapping of SM-CA (Fig. S4, ESI†) indicates a uniform distribution of Fe, Ni, Cr, O, and S elements in this material. It is worth noting that the amorphous phases exhibit a large electrochemical surface area but possess poor electrical conductivity, thereby impeding the electrochemical kinetics associated with the HER and OER processes.38,39 To overcome this limitation, electrochemical activation was utilized to further refine the surface and structure of the SM-CA pre-catalyst. As a result, the high-angle annular dark-field (HAADF)-TEM elemental mapping (Fig. S5, ESI†) revealed a uniform distribution of Fe, Ni, Cr, O, and S elements within the SM-CA-H sample. The high-resolution TEM (HR-TEM) image of SM-CA-H (Fig. 1f) revealed lattice fringes with distances of approximately 2.03 Å, corresponding to the (110) plane of Ni–Cr–Fe (JCPDS no. 35-1375), in agreement with the XRD results. Furthermore, a layer of approximately 10 nm-thick nanocrystalline NiCr LDH (which was not observed by XRD, possibly due to each limited thickness), surrounded by amorphous phases was observed to cover the metallic Ni–Cr–Fe. The NiCr LDH phase was identified based on the observed lattice fringes with distances of approximately 4.37 Å, corresponding to the (003) plane of NiCr LDH (JCPDS no. 52-1626). The specific structure of SM-CA-H offers significant advantages, including excellent electrical conductivity and sufficient electrochemical surface area, which collectively contribute to its remarkable catalytic activity towards the hydrogen evolution reaction (HER).40,41 NiCr LDH was probably doped by iron (Fe–NiCr LDH), due to the 6.4 atom% of iron element uniformly distributed in the exfoliated SM-CA-H flake. The fast Fourier transform (FFT) pattern (inset of Fig. 1f) further confirmed the formation of Ni–Cr–Fe and Fe–NiCr LDH. In parallel, the HAADF-TEM elemental mapping (Fig. S6, ESI†) of SM-CA-O exhibited a uniform distribution of Fe, Ni, and O elements. Additionally, the lattice image (Fig. 1i) of SM-CA-O revealed the presence of nanocrystalline Ni(OH)2 and Fe(OH)2 in the amorphous phases, with lattice distances of approximately 7.50 Å and 4.45 Å, respectively, matching well with the (001) plane of Ni(OH)2 (JCPDS no. 22-0444) and the (010) plane of Fe(OH)2 (JCPDS no. 13-0089). These phases were not observed by XRD, most likely due to their limited size and defective nature. The large amount of amorphous phase provides a huge electrochemical active surface area. It is important to note that there is no evidence of NiFe hydroxide generation observed in the examined samples. This can be attributed to the tendency of atoms of the same element to aggregate and form crystal planes rather than forming mixed hydroxide phases during the electrochemical activation process. Additionally, the FFT pattern (inset of Fig. 1i) further confirmed the generation of Ni(OH)2 and Fe(OH)2, which is considered catalytically active for the OER.42,43
 | ||
| Fig. 1 Morphological and structural characterization of the SM-CA-H and SM-CA-O samples. (a) XRD patterns of the SM, SM-CA, SM-CA-H, and SM-CA-O samples; (b) typical SEM image of SM, inset: SEM image of SM at low magnification; (c) typical SEM image of SM-CA, inset: low magnification SEM image of SM-CA; (d) SEM image of SM-CA-H, inset: low-magnification SEM image of SM-CA-H; (e) TEM image of the SM-CA-H catalyst, inset: low-resolution TEM image of the flake exfoliated from the surface of SM-CA-H sample; (f) TEM lattice image of SM-CA-H in the yellow square area in (e), inset: FFT pattern of the interface between Ni–Cr–Fe (110) and NiCr LDH (003); (g) SEM image of SM-CA-O, inset: low-magnification SEM image of SM-CA-O; (h) TEM image of the SM-CA-O catalyst, inset: low-resolution TEM image of the flake exfoliated from the surface of the SM-CA-O sample; (i) TEM lattice image of SM-CA-O in the yellow square area in (h), inset: FFT pattern of the interface between Ni(OH)2 (001) and Fe(OH)2 (010). | ||
To gain further insight into the surface chemical composition and oxidation states of SM-CA-H and SM-CA-O samples, X-ray photoelectron spectroscopy (XPS) analysis was carried out. Control samples comprising SM and SM-CA were also included in the study to assess the influence of each activation step. Concretely, Fig. 2a illustrates the deconvoluted Fe 2p spectrum of all the samples. Among them, only the deconvoluted Fe 2p spectrum of SM exhibited a peak at 706.3 eV, indicating the presence of metallic Fe.44 This result further demonstrates that a significant amount of metallic Fe was removed from the surface of SM through chemical etching. The binding energies of all samples at 711.7 eV and 725.1 eV correspond to Fe 2p3/2 and Fe 2p1/2 peaks, respectively, which are typical for Fe(II).45 The noticeable shakeup satellites (abbreviated as “sat.”) at 715.1 eV and 727.3 eV observed in the SM-CA-O sample reveal the emergence of Fe(OH)2,45 consistent with the above results of HR-TEM. In Fig. 2b, the deconvoluted Ni 2p spectrum of all the samples is presented. It can be concluded that there is no detectable Ni on the outermost surface of SM, as there is no Ni signal observed in the SM sample. However, for the remaining samples, the fitting peaks of Ni 2p3/2 and 2p1/2 at 855.6 and 873.3 eV, respectively, are characteristic of Ni(II). Additionally, peaks at 862.2 and 880.3 eV correspond to Ni(II) satellite peaks (abbreviated as “sat.”), manifesting the presence of Ni oxide/hydroxide. This finding provides further evidence for the formation of NiCr LDH in SM-CA-H and Ni(OH)2 in SM-CA-O, as supported by previous research on Ni compounds.3,46,47 The minor peaks at 857.5 and 874.7 eV in the SM-CA-O sample are associated with Ni(III),47 suggesting the presence of a small amount of Ni3+ during the electrochemical activation step. However, the observation of this phenomenon in TEM is challenging due to the limited amount of Ni(III) present and the tendency to be reduced to Ni(II). In the Cr 2p spectrum depicted in Fig. 2c, distinctive peaks of Cr(0) are observed at approximately 575.1 and 585.2 eV, while peaks corresponding to Cr(III) are observed at around 577.4 and 587.5 eV in the SM, SM-CA, and SM-CA-H samples.24 These findings align well with the formation of NiCr LDH in SM-CA-H, as corroborated by the HR-TEM analysis. Importantly, it is worth noting that the presence of Cr was found to leach during the electrochemical activation process, as evidenced by the absence of any Cr peak in the SM-CA-O sample. This finding is consistent with a previous report.34 In the high-resolution S 2p XPS spectrum (Fig. 2d) of SM-CA, the peaks at 162.3 eV and 163.3 eV confirm the bonding of sulfur and metal atoms, while the peak at 167.6 eV is assigned to oxidized sulfur species because of surface oxidation.34 However, following the second electrochemical activation process, the presence of sulfur in SM-CA-H is minimal, which might originate from the electrolyte during the electrochemical activation process. Additionally, no sulfur was detected in the SM-CA-O sample. This can be attributed to the ion exchange processes that take place during electrochemical activation, resulting in the depletion of sulfur species in both the SM-CA-H and SM-CA-O. These findings further support the results obtained from SEM-EDS and TEM-EDS analyses.
 | ||
| Fig. 2 The high-resolution XPS spectra of the SM, SM-CA, SM-CA-H, and SM-CA-O samples. (a) Fe 2p; (b) Ni 2p; (c) Cr 2p; (d) S 2p. | ||
Electrocatalytic evaluation of the SM-CA-H electrode for the HER
The HER electrocatalytic activity of the developed electrodes was evaluated using a three-electrode in-house H-cell with 1 M KOH as the electrolyte. Linear sweep voltammetry (LSV) curves were recorded with a scan rate of 3 mV s−1, as described in the Experimental section of the ESI.† LSV curves without iR correction are shown in Fig. S7.† The performance of the developed electrodes was benchmarked with the bare SM, pure Ni mesh (NM), and noble-metal-based Pt/C@SM electrodes (noble-metal-based Pt or Pt/C is usually used as the benchmark HER catalyst12,29,30,36). The current density at an overpotential of 400 mV was measured to assess the HER catalytic activity of the samples. As shown in Fig. 3a, the SM-CA-H electrode exhibited significantly improved HER activity compared to the bare SM electrode. Specifically, at an overpotential of 400 mV, the SM-CA-H sample displayed a much higher current density of −58.6 mA cm−2, whereas the bare SM electrode only displayed −3.49 mA cm−2. Since the electrodes were compared by using the same cell reactor, this 10-fold increase in current density can be only attributed to the activation of SM through the two-step activation process. Furthermore, the current density of SM-CA-H was even higher than that of the NM (−47.1 mA cm−2). The Pt/C@SM sample exhibited the highest current density of −69.5 mA cm−2. These findings clearly demonstrate that the HER activity of the modified steel mesh is greatly improved compared to bare steel and even exceeds that of the commercially available Ni mesh, which has typically 2.5 times higher cost.25 Additionally, the SM-CA-H catalyst outperforms the commercial Ni mesh at high current densities, underscoring its strong potential for industrial applications as a cathode material in alkaline water electrolysis. | ||
| Fig. 3 Electrocatalytic HER performance of the SM-CA-H catalyst. (a) LSV curves; (b) Tafel plots obtained from the LSV curves; (c) Nyquist plots at an overpotential of 400 mV; (d) chronopotentiometric analysis of the SM-CA-H and SM electrodes at different current densities. Test conditions: 1 M KOH at RT. | ||
The Tafel slope of SM-CA-H is illustrated in Fig. 3b and was found to be 111 mV dec−1, which is remarkably lower than the values obtained for SM (189 mV dec−1) and NM (141 mV dec−1) samples. This indicates that the SM-CA-H sample has the most favorable HER kinetics, as reflected by its lowest Tafel slope. In Fig. 3c, electrochemical impedance spectroscopy (EIS) measurements were conducted on the SM-CA-H sample along with the reference samples. The EIS data were fitted using an equivalent circuit with one time constant (inset of Fig. 3c), and the optimized values of solution resistance (Rs) and charge-transfer resistance (Rct) are summarized in Table S3.† As anticipated, the SM-CA-H sample exhibited a significantly smaller Rct value of 3.98 Ω cm2, which is considerably lower than those of SM (23.52 Ω cm2) and NM (6.75 Ω cm2). The Pt/C@SM sample displayed the lowest Rct value of 2.51 Ω cm2. To evaluate the operational stability of the SM-CA-H catalyst compared to bare SM, a chronopotentiometry test (Fig. 3d) was performed at various current densities of −10, −50, −100, −200, and −300 mA cm−2 in a 1 M KOH solution. It is evident that the overpotentials of SM-CA-H at each applied current density are lower than those of SM. Furthermore, the difference in the overpotential becomes increasingly pronounced as the current density increases. These results further confirm the excellent potential of the modified steel mesh as a cathode material for practical alkaline water electrolyzers. These impressive performances of SM-CA-H can be attributed to the presence of nanocrystalline NiCr LDH surrounded by amorphous phases through a two-step activation process. The specific structure of the modified steel mesh not only exhibits excellent electronic conductivity attributed to the presence of nanocrystalline NiCr LDH but also possesses a significantly large electrochemical surface area (Fig. S8†), thanks to the abundant lattice-scale defect-rich amorphous phases. This unique combination of characteristics results in finely tuned surface chemical and electronic properties, which greatly enhance the catalytic kinetics towards the HER.38,39 The HER activity of the prepared catalysts was compared with state-of-the-art HER catalysts reported earlier, as shown in Table S4.† Because of the use of an inactive nickel plate as the counter electrode, the resulting activity was not as high as some of the reported catalysts.
Electrocatalytic performance of the SM-CA-O electrode for the OER
Intriguingly, the SM-CA-O demonstrates remarkable activity towards the OER. As depicted in Fig. 4, the SM-CA-O electrode exhibits significantly faster OER kinetics compared to all the control electrodes. Noble-metal-based IrO2 was also employed as a benchmark OER catalyst.29,30,32,33 To provide reliable electrochemical data and avoid the overlap between Ni2+/Ni3+ oxidation and the OER, polarization curves were recorded from high initial potentials to low final potentials. In particular, the SM-CA-O sample displays the lowest overpotential of 245.8 mV to achieve a current density of 10 mA cm−2. This value is 63.5, 103.1, and 122.9 mV lower than those of IrO2@SM (309.3 mV), NM (348.9 mV), and SM (368.7 mV), respectively. Moreover, at a higher current density of 50 mA cm−2, the overpotential differences are further amplified for all electrodes. Remarkably, the SM-CA-O electrode demonstrates the lowest overpotential of 276.5 mV to deliver a current density of 50 mA cm−2. This value is 55.4, 115.7, and 165.7 mV lower than those of IrO2@SM (331.9 mV), NM (392.2 mV), and SM (442.2 mV), respectively (Fig. 4a). LSV curves without iR correction are shown in Fig. S9.† These results highlight the superior OER activity of the SM-CA-O electrode, which maintains its excellent performance even at high current densities. Specifically, a peak marked is attributed to the reduction of Ni3+ to Ni2+ on the polarization curve of SM-CA-O that appeared at the potential 1.37 V vs. RHE.47 The Tafel slope of SM-CA-H is illustrated in Fig. 4b and is found to be only 27 mV dec−1, significantly lower than the values obtained for the IrO2@SM (39 mV dec−1), NM (73 mV dec−1), and SM (140 mV dec−1) samples. This indicates that the SM-CA-O sample exhibits the most favorable OER kinetics, primarily attributed to the strong cooperative effect between the nanocrystalline Ni(OH)2 and Fe(OH)2. The OER activity of the prepared catalysts was compared with that of state-of-the-art OER catalysts reported earlier, as shown in Table S6.† The resulting activity surpasses most of the reported catalysts. | ||
| Fig. 4 Electrocatalytic OER performance of the SM-CA-O catalyst. (a) LSV curves; (b) Tafel plots from LSV curves; (c) Nyquist plots at an overpotential of 300 mV; (d) chronopotentiometric analysis of the SM-CA-O and SM electrodes at different current densities. Test conditions: 1 M KOH at RT. | ||
As shown in Fig. 4c, EIS measurements were conducted on the SM, NM, IrO2@SM, and SM-CA-O samples. The EIS data were fitted using an equivalent circuit with a one-time constant (inset of Fig. 4c), and the optimized values of solution resistance (Rs) and charge-transfer resistance (Rct) are summarized in Table S4.† Consistent with expectations, the SM-CA-O sample showed the smallest Rct value of 0.91 Ω cm2, significantly lower than those of IrO2@SM (2.02 Ω cm2), NM (4.18 Ω cm2) and SM (8.25 Ω cm2). The highest Rct of 8.25 Ω cm2 obtained for SM indicates its poor OER activity. SM and NM show slightly smaller values of Rs (1.60 Ω cm2) compared to SM-CA-O (1.62 Ω cm2) and IrO2@SM (1.64 Ω cm2). A chronopotentiometry test (Fig. 4d) was carried out on the SM-CA-O catalyst in comparison to bare SM at various current densities of 10, 50, 100, 200, and 300 mA cm−2 in a 1 M KOH solution. The overpotentials of SM-CA-O at each applied current density are consistently lower than those of SM. Moreover, the difference in overpotential becomes more pronounced as the current density increases. The superior performance demonstrated by the SM-CA-O catalyst, particularly in terms of lower overpotentials at various current densities, highlights its excellent operational stability. These findings underscore the suitability of the modified steel mesh for real-world applications, further supporting its viability as a reliable and efficient component in alkaline water electrolysis systems. The remarkable performance of SM-CA-O can be due to its unique structure, characterized by the uniform distribution of nanocrystalline Ni(OH)2 and Fe(OH)2 within amorphous phases. This specific structure offers two key advantages: first, it exhibits excellent electronic conductivity, owing to the presence of nanocrystalline Ni(OH)2 and Fe(OH)2. Second, it provides a large electrochemical surface area (as shown in Fig. S10†) due to the abundance of amorphous phases. The combination of these attributes results in finely tuned surface chemical and electronic properties, ultimately leading to significantly enhanced catalytic kinetics for the OER.
Validation of the activated SS electrodes in a 5 cm2 AEM electrolyzer flow cell
Motivated by the outstanding HER and OER activities exhibited by the fabricated electrodes, we constructed square samples SM-CA-H and SM-CA-O (with an effective area of 5 cm2) as the cathode and anode, respectively, in a commercial 5 cm2 AEM electrolyzer cell. These samples were denoted as SM-CA-H(–)‖SM-CA-O(+). To facilitate comparison, we also constructed two additional cells: one with two SM samples serving as both the cathode and anode (SM(−)‖SM(+)), and another with two NM samples serving as both the cathode and anode (NM(–)‖NM(+)). In all configurations, an anion exchange membrane (Sustainion® X37-50 Grade 60) was used to separate the anode and cathode. Notably, no ionomer was used in this work for cell assembly, as the ionomer-free approach is an advanced strategy for AEM cell assembly, which not only increases the exposure of catalytically active sites and enhances H2/O2 gas bubble transport but also reduces the overall cost of the electrolyzer.48,49 Fig. S11† shows the background performance: an AEM electrolyzer cell without electrodes. Fig. 5a illustrates the polarization curves of AEM electrolyzer cells operating at RT and atmospheric pressure in 1 M KOH. Remarkably, the SM-CA-H(–)‖SM-CA-O(+) exhibited a cell voltage of 2.33 V at a current density of 1 A cm−2, which is 320 mV and 250 mV lower than those of SM(−)‖SM(+) (2.65 V) and NM (–)‖NM(+) (2.58 V) cells. Even, at a current density of 0.5 A cm−2, the SM-CA-H(–)‖SM-CA-O(+) cell still exhibits a great voltage drop of 240 mV, compared to the SM(−)‖SM(+) cell (Fig. 5b). These findings are due to the excellent HER activity displayed by the SM-CA-H cathode and the exceptional OER activity displayed by the SM-CA-O anode. To further confirm the promising performance of the developed SM-CA-H cathode and SM-CA-O anode, SM-CA-H(–)‖SM-CA-O(+) was also constructed in a similar alkaline electrolyzer cell but with Zirfon PERL (500 mm, Agfa) instead of the AEM as the electrode separator. SM-CA-H(–)‖SM-CA-O(+) and SM(–)‖SM(+) were constructed in the alkaline electrolyzer cells operated at RT and atmospheric pressure in 1 M KOH. As recorded in Fig. S12,† the cell voltage for the SM-CA-H(–)‖SM-CA-O(+) configuration was measured to be 2.98 V. Impressively, this cell voltage is 300 mV lower than that of the SM(–)‖SM(+) cell, which displayed a voltage of 3.28 V. The significantly reduced cell voltage of SM-CA-H(–)‖SM-CA-O(+) can be ascribed to the great improvement in interfacial contact, charge transfer kinetics, and mass transport resistance facilitated by modified materials. To further investigate the positive effect of the SM-CA-H(–)‖SM-CA-O(+) configuration in the AEM cell, in situ EIS analysis was implemented. The EIS plots were analyzed using an equivalent circuit model that considers the physical processes and interactions in the AEM electrolyzer cell system. The model consisted of different components, including the ohmic resistance, charge transfer resistance (arising from HER and OER kinetics), and mass transport resistance.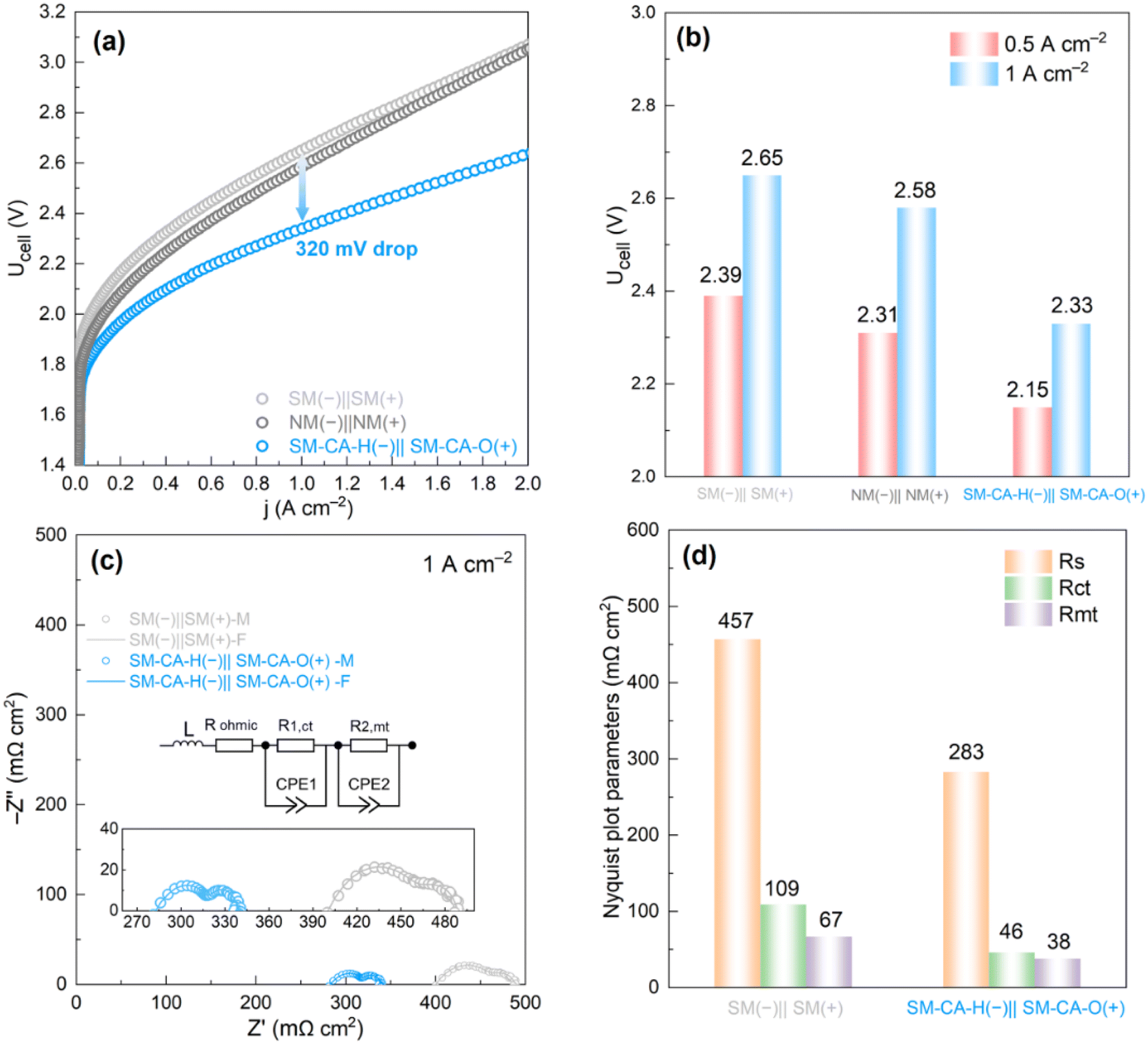 | ||
| Fig. 5 Impact of SM-CA-H(–)‖SM-CA-O(+) on the performance of the AEM electrolyzer cell (5 cm2). (a) Polarization curves for different cells: SM(−)‖SM(+), NM(−)‖NM(+), and SM-CA-H(–)‖SM-CA-O(+); (b) comparison of cell voltages under 0.5 and 1 A cm−2; (c) Nyquist plots from the in situ EIS analysis of different cells: SM(−)‖SM(+), and SM-CA-H(–)‖SM-CA-O(+) under 1 A cm−2 (from 100 kHz to 0.1 Hz), the magnified Nyquist plots and the equivalent circuit are shown in the inset; (d) comparison of Nyquist plot parameters obtained from (c). Test conditions: 1 M KOH at RT. | ||
In Fig. 5c, Nyquist plots are presented for two different cells: SM(−)‖SM(+) and SM-CA-H(–)‖SM-CA-O(+), at a current density of 1 A cm−2. Model fits are also shown for each cell, allowing for comparison between the experimental data and the theoretical predictions. The inserted figure presents an equivalent circuit model that highlights the individual contributions of each resistance loss. Two arc loops are visible across the range from the high frequency to low frequency, representing two resistor-constant phase element (CPE) pairs. The equivalent circuit consists of an ohmic resistance in series with two circuits, each composed of a resistance and a CPE (CPE1 and CPE2) in parallel to one another. Additionally, an inductor (L) in series with ohmic resistance (Rohmic) implies possible inductive contribution from cables and other equipment.3,50 The internal Rohmic appears as the x-axis intercept on the left side of the Nyquist plot, which encompasses all the ohmic resistance contributions, including those from the diaphragm, electrodes, bipolar plates (BPPs), and contact resistances.3 In the equivalent circuit model, the middle section represents the activation losses caused by the cathodic and anodic charge transfer kinetics (R1,ct). It is evident as an arc from the high frequency to middle frequency range. The CPE1 in this section is associated with the double-layer capacitances from both the HER and OER electrodes.51 The final arc in the equivalent circuit model corresponds to the mass-transport resistance (R2,mt), which is observable at the low frequencies of the EIS plot. Optimal bubble evolution and desorption during water electrolysis are crucial for efficient performance. As such, it is necessary to use electrodes with appropriate catalyst morphologies and nanostructures to achieve favorable outcomes. This will help to improve the overall efficiency of the electrolysis process.3,51Fig. 5d displays the results of the fitted equivalent circuit model for cells with the SM(−)‖SM(+) and SM-CA-H(–)‖SM-CA-O(+) configurations. Specifically, the incorporation of cathodic SM-CA-H and anodic SM-CA-O electrodes results in a dramatic decrease in Rohmic to 238 mΩ cm2 for the SM-CA-H(–)‖SM-CA-O(+) cell, compared to 457 mΩ cm2 for the SM(−)‖SM(+) cell. The lower Rohmic observed in the SM-CA-H(–)‖SM-CA-O(+) cell can be attributed to the increased surface area, which in turn improves the contact between the electrode, membrane, and electrolyte. The morphologies and nanostructures (Fig. 1(d–i)) of the SM-CA-H and SM-CA-O electrodes would minimize the size of the bubbles detaching from the electrode surface, resulting in lower contact resistance. At high to middle frequencies, there is a significant decrease in charge transfer resistances, from 109 mΩ cm2 in the SM(−)‖SM(+) cell to 46 mΩ cm2 in the SM-CA-H(–)‖SM-CA-O(+) cell, which was expected due to the high HER and OER activities of the cathodic SM-CA-H and anodic SM-CA-O electrodes, respectively. Remarkably, mass-transport limitation for the SM-CA-H(–)‖SM-CA-O(+) cell (38 mΩ cm2) considerably dropped compared to that of the SM(−)‖SM(+) cell (67 mΩ cm2). Notably, the use of cathodic SM-CA-H and anodic SM-CA-O electrodes with modified morphologies and generated nanostructures results in detectable changes in mass transport. Specifically, this can be caused by the favorable wettability and smaller bubble size on the cathodic SM-CA-H and anodic SM-CA-O electrodes.
To investigate the behavior of the modified electrodes closer to industrial application conditions, the AEM cell was tested at 60 °C in 1 M KOH (Fig. S13†). The SM-CA-H(–)‖SM-CA-O(+) configuration exhibits cell voltages of 1.95 V and 2.08 V at current densities of 1 and 1.5 A cm−2, respectively. These voltages are 360 mV and 350 mV lower than the SM(−)‖SM(+) cell, which exhibited voltages of 2.31 V and 2.43 V at the same current densities. Furthermore, the SM-CA-H(–)‖SM-CA-O(+) configuration was tested under a constant current of 1.0 A cm−2 for 120 h where promising stability was observed with no signs of degradation. These encouraging results further demonstrate the advantages of the modified steel mesh electrodes in AEM electrolyzer cell application. In addition, the performance of the modified electrodes in the AEM cell was compared with state-of-the-art electrodes, as shown in Table S7.† Notably, the performance of the modified electrodes is comparable to that of most state-of-the-art electrodes reported. We also investigated the reproducibility of the electrode modification process. The procedure for fabricating the electrodes was repeated, and their performance was evaluated in a 5 cm2 AEM cell at 60 °C, as shown in Fig. S14.† The two cells with modified electrodes exhibited nearly identical performance, with slight variations likely attributable to differences in cell assembly.
Wettability and bubble dynamic analysis of the SM-CA-H and SM-CA-O electrodes
Typically, in practical water electrolysis applications, especially at high current densities, numerous gas bubbles are generated. These bubbles often do not detach promptly and can cover the electrode surface. This leads to two major issues: (1) the non-conductive bubble layer blocks the ion pathways necessary for current transport, increasing the ohmic resistance and (2) the attached bubbles hinder access to the catalytically active sites, thereby increasing the charge transfer resistance of the catalysts. Additionally, the accumulation of large gas bubbles obstructs the electrolyte flow, resulting in increased mass transfer resistance. This is particularly problematic in water-splitting processes, such as in commonly used water electrolyzer configurations with a zero-gap design, where efficient ion transport is highly dependent on the flow of the electrolyte.52–57 As demonstrated by the in situ EIS analysis, the significantly improved resistances of the SM-CA-H(–)‖SM-CA-O(+) cell would be primarily attributed to the more favorable surface wettability and more efficient gas bubble release mechanism displayed by the cathodic SM-CA-H and anodic SM-CA-O electrodes when compared to those of the SM electrodes. Therefore, it is imperative to conduct a thorough surface wettability and bubble behavior release analysis of these developed electrodes.To investigate the wettability of the different electrodes, contact angle measurements were performed. As depicted in Fig. 6(a–c), the contact angles of SM (131°), SM-H (0°), and SM-CA-O (0°) were compared, and it was concluded that the surface of SM-CA-H and SM-CA-O electrodes became more hydrophilic due to the nanostructures covering their surface during the modification processes. This finding was further demonstrated by the dynamic contact angles recorded by videos. The contact angle of the SM sample still remains 131° after 20 s, while the corresponding times of the SM-CA-H and SM-CA-O samples were less than 1 s, as shown in the ESI video (SV1–SV3†). The super surface wettability of the SM-CA-H and SM-CA-O samples demonstrates that the modified electrodes greatly accelerate electrolyte transport and bubble removal, thereby directly improving the interfacial contact of the AEM electrolyzer cell constructed with modified electrodes.
 | ||
| Fig. 6 Wettability analysis and bubble release behavior. (a–c) Contact angle measurements of (a) SM, (b) SM-CA-H, and (c) SM-CA-O; (d and e) the average diameter of the H2 gas bubble generated on (b) SM and (e) SM-CA-H electrodes, inset: camera image showing H2 bubble release from the fabricated mesh electrodes; (f and g) the average O2 gas bubble diameter generated on (c) SM and (g) SM-CA-O electrodes, inset: camera image showing O2 bubble release from the fabricated mesh electrodes. H2 and O2 bubble release images of the samples were recorded under 200 mA cm−2 in 1 M KOH at RT. | ||
Furthermore, a camera was utilized to capture the water-splitting process and analyze the size distribution and detachment behavior of the produced gas bubbles on various electrodes at a current density of 200 mA cm−2. The H2 bubble size distribution histogram (Fig. 6(d and e)) revealed that the H2 bubbles released from SM-CA-H (49.9 ± 17.6 μm) were significantly smaller than those from SM (86.9 ± 29.5 μm), and similarly the O2 bubbles (Fig. 6(f and g)) released from SM-CA-O (43.8 ± 13.4 μm) were also much smaller than those from SM (77.5 ± 23.6 μm), suggesting that both the SM-CA-H and SM-CA-O electrodes possess more aerophobic features and promote easy detachment of gas bubbles. The presence of larger gas bubbles can block the electrode surface area and require a greater detachment force from the surface, resulting in increased diffusion resistance and overpotential during water splitting.58 The modified surfaces on the SM-CA-H and SM-CA-O electrodes enhanced their hydrophilic and aerophobic properties, successfully impeding the buildup and firm attachment of gas bubbles on the electrode surface, which would significantly enhance the interfacial contact and mass transport, as well as increase the charge transfer because of the exposure of more active sites after prompt detachment of gas bubbles from the electrode surface. In conclusion, these results lead to a great improvement in the hydrogen production efficiency for the SM-CA-H(–)‖SM-CA-O(+) AEM electrolyzer cell.
Finally, the durability of the SM-CA-H(–)‖SM-CA-O(+)and SM(−)‖SM(+) cells was examined by subjecting them to a current density of 1 A cm−2 at room temperature in 1 M KOH for 250 hours. The cell voltage was monitored during the test, and the results are presented in Fig. 7a. Remarkably, after 250 hours of operation, there was no performance degradation for the SM-CA-H(–)‖SM-CA-O(+) cell. The result displays excellent durability of the SM-CA-H(–)‖SM-CA-O(+) cell. Furthermore, Nyquist plot results of the post electrode (post-SM-CA-H(–)‖SM-CA-O(+))-based cell showed improved interfacial resistance and mass transfer resistance, which explicate its high efficiency and outstanding stability, as shown in Fig. 7b. Additionally, after 250 hours of operation, the SEM and optical images of the SM-CA-H and SM-CA-O electrodes demonstrate remarkable durability in the AEM cell, even at high current densities, as shown in Fig. 7c, d, and S15.† The SM-CA-H(–)‖SM-CA-O(+) configuration was also assembled into an alkaline electrolyzer cell with a Zirfon PERL (500 mm, Agfa) diaphragm to further evaluate its stability. Notably, the SM-CA-H(–)‖SM-CA-O(+) cell exhibited exceptional stability, as demonstrated in Fig. S16.† These findings indicate that incorporating cathodic SM-CA-H and anodic SM-CA-O electrodes in the AEM electrolyzer cell yields significant performance enhancements. As discussed above, the high catalytic activity for OER and HER is enabled by the successful functionalization of the SM-based electrodes.
 | ||
| Fig. 7 Long-term stability tests of the 5 cm2 AEM electrolyzer cell constructed with SM-CA-H(–)‖SM-CA-O(+) and SM(–)‖SM(+). (a) U–t curve with 250 h under 5.0 A (1 A cm−2); (b) Nyquist plots of cells before and after the 250 h test; (c) SEM image of the post-SM-CA-H cathode; (d) SEM image of the post-SM-CA-O anode. | ||
Conclusions
In this work, we developed a simple and effective way to functionalize stainless-steel mesh for its use as a bifunctional electrocatalyst for alkaline water electrolysis. The obtained SM-CA-H exhibited comparable HER properties with those of Pt/C, as well as SM-CA-O, showing higher OER properties than the commercial IrO2. More importantly, our results exemplify that with simple activation processes, SS can be used both as an OER and a HER electrocatalyst for AEM electrolyzers and with significantly lower electrode costs. Specifically, the AEM electrolyzer cell constructed with the developed SM-CA-H(–)‖SM-CA-O(+) displayed more than 300 mV-voltage drop compared to a similar cell with bare SM electrodes, which is attributed to the great improvement of interfacial contact, charge transfer, and mass transfer processes, as revealed by the in situ electrochemical impedance spectroscopic analysis. Characterization of the used samples revealed the generation of nanocrystalline Fe–NiCr LDH and nanocrystalline Ni(OH)2/Fe(OH)2 species in the SM-CA-H and SM-CA-O electrodes, respectively, which could explain the enhanced performance. In addition, the obtained AEM electrolyzer cell can run for 250 h at a very high current density of 1.0 A cm−2 (5 A in total) in which no sign of degradation was observed. The method employed in this work has the potential for scalability due to its cost-effectiveness and simplicity.Abbreviations
| SS | Stainless steel |
| SM | Steel mesh |
| CA | Chemical activation |
| SM-CA | SM sample after the chemical activation process |
| SM-CA-H | SM-CA sample as the cathodic side in the electrochemical activation process |
| SM-CA-O | SM-CA sample as the anodic side in the electrochemical activation process |
| AWE | Alkaline water electrolyzer |
| PEM | Proton-exchange membrane |
| AEM | Anion-exchange membrane |
| HER | Hydrogen evolution reaction |
| OER | Oxygen evolution reaction |
| HAADF | High-angle annular dark field |
| FFT | Fast Fourier transform |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the financial support from the Samenwerkingsverband Noord-Nederland (SNN) and the EU in the context of the Waviater project.References
- W. Jiang, A. Y. Faid, B. F. Gomes, I. Galkina, L. Xia, C. M. S. Lobo, M. Desmau, P. Borowski, H. Hartmann, A. Maljusch, A. Besmehn, C. Roth, S. Sunde, W. Lehnert and M. Shviro, Adv. Funct. Mater., 2022, 32, 2203520 CrossRef.
- A. Hodges, A. L. Hoang, G. Tsekouras, K. Wagner, C. Y. Lee, G. F. Swiegers and G. G. Wallace, Nat. Commun., 2022, 13, 1304 CrossRef.
- T. Jiang, X. Jiang, J. Hnát, A. Michalcova, I. Biswas, R. Reissner, V. Kyriakou, F. Razmjooei, H. Liao, K. Bouzek and S.-a. Ansar, J. Mater. Chem. A, 2022, 10, 23863–23873 RSC.
- A. Khataee, A. Shirole, P. Jannasch, A. Krüger and A. Cornell, J. Mater. Chem. A, 2022, 10, 16061–16070 RSC.
- P. Zhou, P. Niu, J. Liu, N. Zhang, H. Bai, M. Chen and H. Pan, Adv. Funct. Mater., 2022, 32, 2202068 CrossRef.
- G. Ding, H. Lee, Z. Li, J. Du, L. Wang, D. Chen and L. Sun, Adv. Energy Sustainability Res., 2022, 4, 2200130 CrossRef.
- M. J. Jang, S. H. Yang, M. G. Park, J. Jeong, M. S. Cha, S.-H. Shin, K. H. Lee, Z. Bai, Z. Chen, J. Y. Lee and S. M. Choi, ACS Energy Lett., 2022, 7, 2576–2583 CrossRef.
- F.-L. Wang, Y.-W. Dong, C.-J. Yu, B. Dong, X.-Y. Zhang, R.-Y. Fan, J.-Y. Xie, Y.-N. Zhou and Y.-M. Chai, Appl. Catal., B, 2023, 331, 122660 CrossRef CAS.
- P. Chen and X. Hu, Adv. Energy Mater., 2020, 10, 2002285 CrossRef.
- L. Wan, Z. Xu, P. Wang, P.-F. Liu, Q. Xu and B. Wang, Chem. Eng. J., 2022, 431, 133942 CrossRef.
- B. Chen, A. L. G. Biancolli, C. L. Radford and S. Holdcroft, ACS Energy Lett., 2023, 8, 2661–2667 CrossRef.
- F.-L. Wang, N. Xu, C.-J. Yu, J.-Y. Xie, B. Dong, X.-Y. Zhang, Y.-W. Dong, Y.-L. Zhou and Y.-M. Chai, Appl. Catal., B, 2023, 330, 122633 CrossRef.
- M. Ďurovič, J. Hnát, M. Strečková and K. Bouzek, J. Power Sources, 2023, 556, 232506 CrossRef.
- H. Chu, P. Feng, B. Jin, G. Ye, S. Cui, M. Zheng, G.-X. Zhang and M. Yang, Chem. Eng. J., 2022, 433, 133523 CrossRef.
- J. Jiang, Y.-J. Zhang, X.-J. Zhu, S. Lu, L.-L. Long and J.-J. Chen, Nano Energy, 2021, 81, 105619 CrossRef CAS.
- W. H. Lee, M. H. Han, Y. J. Ko, B. K. Min, K. H. Chae and H. S. Oh, Nat. Commun., 2022, 13, 605 CrossRef.
- X. Liu, K. Ni, B. Wen, R. Guo, C. Niu, J. Meng, Q. Li, P. Wu, Y. Zhu, X. Wu and L. Mai, ACS Energy Lett., 2019, 4, 2585–2592 CrossRef.
- G. Solomon, A. Landström, R. Mazzaro, M. Jugovac, P. Moras, E. Cattaruzza, V. Morandi, I. Concina and A. Vomiero, Adv. Energy Mater., 2021, 11, 2101324 CrossRef.
- Y. Wu, Y. Li, M. Yuan, H. Hao, X. San, Z. Lv, L. Xu and B. Wei, Chem. Eng. J., 2022, 427, 131944 CrossRef.
- C. Yang, W. Zhong, K. Shen, Q. Zhang, R. Zhao, H. Xiang, J. Wu, X. Li and N. Yang, Adv. Energy Mater., 2022, 12, 2200077 CrossRef.
- K. Beliaeva, N. Grimaldos-Osorio, E. Ruiz-López, L. Burel, P. Vernoux and A. Caravaca, Int. J. Hydrogen Energy, 2021, 46, 35752–35764 CrossRef.
- I. A. Poimenidis, M. Lykaki, S. Moustaizis, P. Loukakos and M. Konsolakis, Mater. Chem. Phys., 2023, 305, 128007 CrossRef CAS.
- I. A. Poimenidis, N. Papakosta, A. Klini, M. Farsari, M. Konsolakis, P. A. Loukakos and S. D. Moustaizis, Fuel, 2023, 342, 127798 CrossRef.
- A. K. Gomaa, B. S. Shaheen, G. E. Khedr, A. M. Mokhtar and N. K. Allam, Energy Fuels, 2022, 36, 7025–7034 CrossRef.
- M. S. Balogun, W. Qiu, Y. Huang, H. Yang, R. Xu, W. Zhao, G. R. Li, H. Ji and Y. Tong, Adv. Mater., 2017, 29, 1702095 CrossRef PubMed.
- V. R. Jothi, K. Karuppasamy, T. Maiyalagan, H. Rajan, C. Y. Jung and S. C. Yi, Adv. Energy Mater., 2020, 10, 1904020 CrossRef.
- C. Zhou, T. Gao, J. Tan, Z. Luo, L. Mutallip and D. Xiao, Sustainable Energy Fuels, 2021, 5, 2649–2659 RSC.
- L. Zhong, L. He, N. Wang, Y. Chen, X. Xie, B. Sun, J. Qian, S. Komarneni and W. Hu, Appl. Catal., B, 2023, 325, 122343 CrossRef.
- H. Zhong, J. Wang, F. Meng and X. Zhang, Angew. Chem., 2016, 128, 10091–10095 CrossRef.
- T. Zhou, H. Gao, Y. Hu, W. Huang, F. Yang, W. Sun and X. Yi, J. Power Sources, 2023, 577, 233241 CrossRef CAS.
- Y. Lyu, R. Wang, L. Tao, Y. Zou, H. Zhou, T. Liu, Y. Zhou, J. Huo, S. P. Jiang, J. Zheng and S. Wang, Appl. Catal., B, 2019, 248, 277–285 CrossRef.
- M. Kim, J. Ha, Y.-T. Kim and J. Choi, Chem. Eng. J., 2022, 440, 135459 CrossRef.
- P. Liu, Y. Shi, X. Zhang, J. Yin, D. Zhang, T. Wang, J. Fei, T. Zhan, G. Li, J. Lai and L. Wang, Appl. Catal., B, 2024, 341, 123332 CrossRef.
- H. Chen, J. Li, Y. Shen, W. Jiao, J. Wang, Y. Zou and X. Zou, Appl. Catal., B, 2022, 316, 121605 CrossRef.
- Y. Wang, S. Liu, X. Hao, S. Luan, H. You, J. Zhou, D. Song, D. Wang, H. Li and F. Gao, J. Mater. Chem. A, 2019, 7, 10572–10580 RSC.
- P. W. Menezes, C. Walter, J. N. Hausmann, R. Beltran-Suito, C. Schlesiger, S. Praetz, V. Yu Verchenko, A. V. Shevelkov and M. Driess, Angew Chem. Int. Ed. Engl., 2019, 58, 16569–16574 CrossRef.
- J. Li, H. Chen, Y. Liu, R. Gao and X. Zou, J. Mater. Chem. A, 2019, 7, 5288–5294 RSC.
- Z. Gong, R. Liu, H. Gong, G. Ye, J. Liu, J. Dong and H. Fei, ACS Catal., 2021, 11, 12284–12292 CrossRef.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef.
- W. Ye, X. Fang, X. Chen and D. Yan, Nanoscale, 2018, 10, 19484–19491 RSC.
- A. A. Lobinsky, D. S. Dmitriev and V. I. Popkov, Int. J. Hydrogen Energy, 2023, 48, 22495–22501 CrossRef.
- X. Cheng, J. Yuan, J. Cao, C. Lei, B. Yang, Z. Li, X. Zhang, C. Yuan, L. Lei and Y. Hou, J. Colloid Interface Sci., 2020, 579, 340–346 CrossRef PubMed.
- B. Wu, Z. Yang, X. Dai, X. Yin, Y. Gan, F. Nie, Z. Ren, Y. Cao, Z. Li and X. Zhang, Dalton Trans., 2021, 50, 12547–12554 RSC.
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- Q. Liang, L. Zhong, C. Du, Y. Luo, Y. Zheng, S. Li and Q. Yan, Nano Energy, 2018, 47, 257–265 CrossRef.
- T. Jiang, S. A. Ansar, X. Yan, C. Chen, X. Fan, F. Razmjooei and H. Liao, ACS Appl. Energy Mater., 2019, 2, 8809–8817 CrossRef.
- T. Jiang, T. Liu, S. Liu, Y. Wang, S. Gao, G. Montavon, S. Ansar and H. Liao, Appl. Mater. Today, 2022, 28, 101526 CrossRef.
- T.-H. Kong, P. Thangavel, S. Shin, S. Kwon, H. Choi, H. Lee, N. Park, J.-J. Woo and Y. Kwon, ACS Energy Lett., 2023, 8, 4666–4673 CrossRef.
- E. López-Fernández, C. Gómez-Sacedón, J. Gil-Rostra, J. P. Espinós, A. R. González-Elipe, F. Yubero and A. de Lucas-Consuegra, Chem. Eng. J., 2022, 433, 133774 CrossRef.
- S. H. Frensch, A. C. Olesen, S. S. Araya and S. K. Kær, Electrochim. Acta, 2018, 263, 228–236 CrossRef.
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle and K. A. Friedrich, Joule, 2021, 5, 1776–1799 CrossRef.
- T. Jiang, X. Jiang, V. Kyriakou, K. Bouzek and H. Liao, J. Mater. Chem. A, 2023, 11, 26011 RSC.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Rivas, Joule, 2020, 4, 555–579 CrossRef.
- S. Park, L. Liu, Ç. Demirkır, O. van der Heijden, D. Lohse, D. Krug and M. T. Koper, Nat. Chem., 2023, 15, 1532–1540 CrossRef.
- T. Kou, S. Wang and Y. Li, ACS Mater. Lett., 2021, 3, 224–234 CrossRef.
- X. Jiang, V. Kyriakou, C. Song, X. Wang, S. Costil, C. Deng, T. Liu, T. Jiang and H. Liao, J. Energy Chem., 2024, 93, 511–518 CrossRef.
- X. Jiang, V. Kyriakou, B. Wang, S. Deng, S. Costil, C. Chen, T. Liu, C. Deng, H. Liao and T. Jiang, Chem. Eng. J., 2024, 486, 150180 CrossRef.
- X. Qin, B. Yan, D. Kim, Z. Teng, T. Chen, J. Choi, L. Xu and Y. Piao, Appl. Catal., B, 2022, 304, 120923 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04762a |
| This journal is © The Royal Society of Chemistry 2024 |