
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of water loading on the stability of pristine and defective UiO-66†
E.
Acuna-Yeomans
 ab,
P. J.
Goosen
c,
J. J.
Gutiérrez-Sevillano
*d,
D.
Dubbeldam
e and
S.
Calero
*ab
ab,
P. J.
Goosen
c,
J. J.
Gutiérrez-Sevillano
*d,
D.
Dubbeldam
e and
S.
Calero
*ab
aMaterials Simulation and Modelling, Department of Applied Physics and Science Education, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: s.calero@tue.nl
bEindhoven Institute for Renewable Energy Systems (EIRES), Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands
cEindhoven University of Technology, Eindhoven, Netherlands
dCenter for Nanoscience and Sustainable Technologies (CNATS), Universidad Pablo de Olavide, 41013 Seville, Spain. E-mail: jjgutierrez@upo.es
eVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Amsterdam, Netherlands
First published on 16th August 2024
Abstract
Materials used for water treatment purposes need to be stable for easy handling and cost-effectiveness. UiO-66 has been identified as a promising option. In this work, we investigate the impact of water loading on the structural and mechanical properties of pristine and defective UiO-66 using classical molecular simulations. We employ and compare two approaches for modeling non-bonded interactions between the framework and water molecules: direct Lorentz–Berthelot (L–B) mixing and hybrid mixing. We conducted molecular dynamics simulations to examine the spatial arrangement of water molecules within the framework, water affinity for specific framework interaction sites, and their impact on the framework's structural parameters under atmospheric conditions, high hydrostatic pressures, and increased water loading. Our results indicate that both methods predict water affinity near zirconium clusters, but differ in identifying principal interaction sites and interaction strength. L–B mixing predicts strong binding to linker oxygen atoms, restricting water movement, while hybrid mixing indicated dynamic water behavior, with site-to-site hopping and pore-to-pore movement observed at moderate and high loadings. Structural analysis at increased water loadings showed adsorption-induced expansion using L–B mixing due to linker–cluster bond stretching, contrasting with slight system contraction predicted by hybrid mixing. High-pressure NPT simulations evidence that water loading reduces amorphization pressure, although values obtained using both approaches differ significantly at moderate and high loadings.
Introduction
Water pollution is an escalating concern in the world, as diverse sources such as sewage, wastewater, agricultural pesticides, and industrial waste continually introduce harmful substances into natural water bodies.1–3 This contamination poses significant threats to ecosystems, the environment, and human health. Recent research has highlighted the potential of metal–organic frameworks (MOFs) in addressing wastewater treatment challenges.4–7 MOFs have demonstrated effectiveness in eliminating a variety of pollutants, including phosphates,8,9 fluorides,10,11 heavy metals,12,13 pharmaceuticals,14,15 and chemical warfare agents.16,17 The limitations of traditional water harvesting methods, such as their reliance on high humidity and energy inputs, further underscore the need for innovative and efficient approaches like using MOFs.18–20For industrial applications, it is crucial that materials used in water treatment exhibit structural stability under moist conditions to ensure ease of handling and cost-effectiveness. Despite the development of numerous MOFs, only a select few are chemically stable enough for such applications.21–24 Among these, UiO-66, a Zr-based MOF, is particularly noteworthy for its exceptional water stability, which is attributed to the robustness of the Zr–O bond and a unique geometry that minimizes water inclusion and reduces hydrolysis reactions.13,21,25–27 UiO-66 features zirconium-based Zr6O4(OH)4 clusters interconnected by 1,4-benzenedicarboxylic acid (BDC) ligands. In its pristine, defect-free state, each inorganic cluster is 12-fold coordinated, contributing to its remarkable structural stability under high pressure conditions.25,28–32 Functionalized derivatives of UiO-66, developed to tune hydrophobicity, also exhibit impressive stability, further underscoring the potential of Zr-based MOFs in advanced water treatment applications.7,17,33–36 Most synthesis procedures for UiO-66 result in a defective structure with missing linkers, typically reducing the coordination number of each zirconium cluster to 11, indicating one missing linker per unit cell on average.30,37–40 Defects increase the porosity and surface area, enhancing adsorption behavior,31,32,41,42 catalytic properties,37,43 and proton conductivity.44,45 However, defects also reduce structural stability, creating a trade-off between improved adsorption and mechanical integrity.38,41,46
Molecular simulations provide a reliable means to analyze the relationships between the material structure and specific properties, particularly when adsorbates or defects are present. Over the past decade, computational studies have extensively examined the adsorption properties of pristine and defective UiO-66, focusing primarily on water and CO2 adsorption.47–51 These simulations have offered valuable insights for industrial applications such as water harvesting and gas separation, often modeling UiO-66 as a rigid structure interacting with guest molecules solely via non-bonded potentials.
To accurately study the structural and mechanical properties of UiO-66, a flexible model of the framework is essential. Flexible models, such as the Rogge et al. force fields developed in 2016,46 have been extensively validated for predicting the properties of unloaded UiO-66. These models employ ab initio derived parameters tailored to each specific structure, constructed through the QuickFF procedure52,53 developed by the same group. However, these force fields were primarily validated by reproducing the structural and mechanical properties of evacuated systems, where interactions with adsorbate molecules were not considered. Recent studies54,55 have demonstrated that when adsorbates such as acetone and isopropyl alcohol are introduced, the Rogge potential inadequately captures adsorbate–framework interactions, particularly hydrogen bonding with the inorganic clusters. This limitation underscores the need for modifications to the force field to accurately model the interaction between Zr clusters and adsorbates in loaded UiO-66 systems.
The primary objective of this work is to determine how water loading affects the structural and mechanical properties of flexible UiO-66. Given the established reliability of the Rogge force fields in accurately predicting these properties, we prioritized maintaining their original parameters for simulating water-loaded frameworks. Instead of altering these parameters, we assessed and compared two approaches for modeling the interaction between the framework and water molecules: direct Lorentz–Berthelot (L–B) mixing and hybrid mixing. The hybrid mixing method has been previously employed to study the pore filling process and water diffusion within UiO-66,56 using separate water force fields to model water–water interactions (MB-pol57) and water–framework interactions (TIP4P2005 (ref. 58)). In our study, the hybrid mixing method utilizes the Rogge force field for interactions between framework atoms and the UFF59 force field for interactions between the framework and water molecules. By systematically evaluating these approaches, we aim to provide a deeper understanding of how water impacts the stability and performance of UiO-66, with significant implications for its use in industrial water treatment applications.
Methodology
Along with the pristine structure, we considered the eight defective structures (and corresponding force fields) proposed by Rogge et al.,46 each containing linker defects: one of them (type 0) corresponds to a system with one linker vacancy per unit cell, while the rest (types 1–7) represent structures in which an additional, distinct linker defect is introduced to the type 0 parent structure (Fig. S1†). Further details on the force fields and the construction of the structures are provided in the first portion of the ESI.† Before the production simulations, these structures were loaded with different amounts of water molecules, from 10 molecules per unit cell of the framework (corresponding to a relative humidity of 5%) to 120 molecules per unit cell (80% RH), near the water saturation point at room temperature. As was done in recent published work,47,56,60 the quantity of water molecules within the MOF structure at various relative humidity levels was determined using the experimental water adsorption isotherm for UiO-66.51,61Force fields and models
We model the pristine and defective frameworks using the group of force fields proposed by Rogge et al.46 which have proven to provide an accurate prediction of the structural and mechanical properties of the unloaded systems. The functional form for the interactions between framework atoms, regardless of the inclusion of linker vacancies, is the following:| U = Ubonded + Unon-bonded | (1) |
 | (2) |
 | (3) |
 | (4) |
 | (5) |
Two terms contribute to the non-bonded portion of the potential energy function. The electrostatics are described by the Coulomb interaction between spherical Gaussian densities with distinct radii di and dj containing charges qi and qj, respectively. The potential energy term for particles separated by a distance of rij is:
 | (6) |
The second term models the van der Waals interactions between two atoms i and j separated by a distance rij using the two-parameter molecular mechanics (MM3)62,63 Buckingham potential:
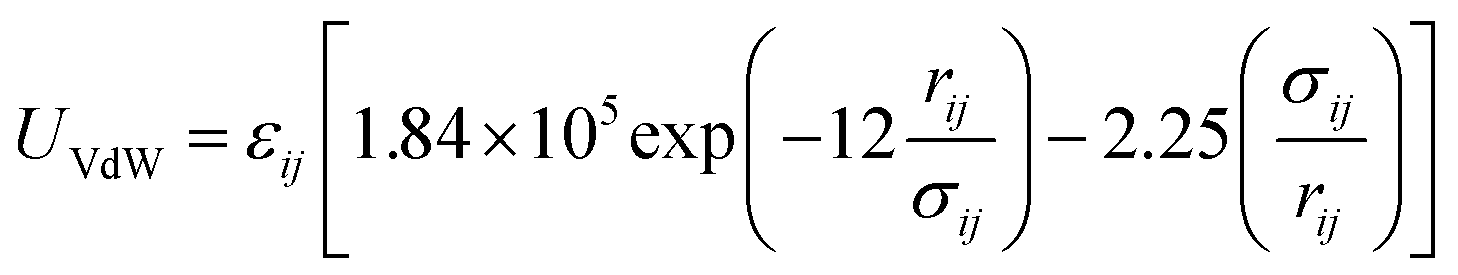 | (7) |
For bonded atoms, different scaling and exclusion rules are applied for each contribution to the non-bonded part of the potential energy. As defined by the MM3 force field, 1–2 and 1–3 exclusion rules for bonded pairs are applied to VdW interactions, with no scaling applied to atoms separated by two or more atoms (1–4). For framework atoms of different species, the equilibrium distance (σij) and well depth (εij) parameters are determined via the empirical Lorentz–Berthelot mixing rules:
 | (8) |
The interaction parameters of the energy equation depend on the particular structure to be modelled, that is, each of the 9 structures simulated (pristine, type 0 defective and type 1–7 defective) has distinct associated sets of parameters, and they are provided in the original publication of the force field.46
The water molecules were modeled using the extended SPC model,64,65 although the Tip4P2005 (ref. 58) force field was initially implemented and considered as well. The SPC/E model was selected primarily due to its lower computational cost, as it involves taking into account fewer interactions, and because early results showed that both models influenced the framework similarly. The non-bonded parameters for the description of water–water interactions were derived using Lorentz–Berthelot mixing rules as well.
Two distinct methods for modeling the interaction between water molecules and the framework atoms were employed; in both cases the VdW interactions are modelled using the Lennard-Jones potential and the electrostatics are modelled using the canonical Coulomb potential for point charges. In the first method, the Lennard-Jones parameters for the description of the water–framework interaction were derived by applying Lorentz–Berthelot (L–B) mixing rules directly. Here, it should be noted that although the empirical combination of L-J parameters from popular rigid water models and generic force fields have been used extensively in published work to study this and similar systems, this is not the case for the combination of SPC/E and MM3 parameters. This first mixing methodology is thus implemented as an intuitive point of reference, reflective of the usual default choice in the simulation community. In the second method, the Lennard-Jones contribution of the water–framework interaction was modified by employing universal force field (UFF)59ε and σ coefficients for the framework atoms instead of the MM3 force field (MM3)62,63 parameters used for intra-framework non-bonded interactions, therefore making use of two framework force fields in a hybrid manner.
Simulation details
The pressure-volume behavior of UiO-66 structures was determined using molecular dynamics (MD) simulations in the flexible NPT ensemble (N,P,σα = 0,T). This ensemble allows dynamic changes in both simulation cell volume and shape, enabling the estimation of the amorphization pressure (Pam) of the framework. For each system, flexible NPT simulations were conducted starting from a high pressure. The unloaded structures were first energy minimized and equilibrated under atmospheric conditions to record the reference volume (Vr). After loading with water molecules, the structures were equilibrated at high pressure, monitoring the instantaneous volume (Vi). Amorphization was deemed to occur if Vi dropped below a predefined threshold (Vt), proportional to Vr. The pressure was adjusted in subsequent simulations based on whether amorphization occurred, using a step size (ΔP) of 0.005 GPa. The final Pam was determined as the highest pressure at which the structure remained stable. Large simulation cells were used to mitigate fluctuations in instantaneous pressure, which can induce phase transitions at artificially low pressures, particularly in flexible MOFs.66,67All simulations were performed using the LAMMPS package.68 The Verlet integrator with a timestep of 1 fs ensured energy conservation, while independent thermostats optimized equilibration times by grouping atoms of the framework and water. A Nose–Hoover thermostat (300 K, 100 fs relaxation time) and an MTTK barostat (1000 fs relaxation time) controlled temperature and pressure, respectively. Radial distribution functions (RDFs) and 3D water density distribution maps69 were averaged from 500 ps NVT ensemble runs after equilibration. Simulations used a 2 × 2 × 2 supercell with non-bonded interactions truncated at 14.0 Å and long-range electrostatics calculated via Ewald summation. Periodic boundary conditions were applied in all dimensions, with specific scaling of 1–2, 1–3, and 1–4 non-bonded interactions as per the force field requirements.
Results and discussion
Effect of water loading on the pristine structures
In order to evaluate the performance of each parameter mixing method, we start by analyzing the spatial arrangement of water molecules inside the undefective framework at different water loadings. The Radial Distribution Function (RDF) describes how the average density varies as a function of distance from a chosen reference particle. By selecting framework atoms as references, they can be used to identify preferential adsorption sites within its surface and characterize the interaction environment in the vicinity of the metal clusters and organic ligands.Regarding the initial interaction sites, at low loadings both models predict that water molecules tend to agglomerate around the metal clusters while keeping away from the central portion of the organic ligands, in line with previously published research.47,56,70 However, each model suggests different species near the metal cluster as the primary interaction site for water. L–B mixing predicts that the oxygen atoms connecting the Zr atoms with the linkers are the main interaction site, whereas hybrid mixing points to the hydroxyl groups within the inorganic cluster.
For the L–B mixing simulations at low loadings, the RDF describes the spatial correlation between OH2O and OL peaks at a radial distance of 1.9 Å, contrasting with a clear separation from OOx and OOh sites (Fig. S2a†). This behavior is not consistent with previous computational studies indicating that water preferentially binds to the hydroxyl hydrogens in the zirconium clusters. Recently published computational studies of water diffusion and pore filling processes in UiO-66 predict an interaction site affinity order of OOh > OL > OOx.56 Furthermore, the peak distance indicates heavy perturbation of the Zr–OL bond electronic environment. While the interaction site water affinity order predicted by hybrid mixing (Fig. 1) is qualitatively congruent with said findings and the radial distance to OOh is the same (≈2.8 Å) the density of water molecules near the preferential site at low loadings is underestimated by 36%. The RDFs between water oxygen atoms (OH2O) and potential bonding sites were analyzed at higher loadings of 40 and 100 water molecules per unit cell using both mixing methods (Fig. S2†). Hybrid mixing predicts a broadening of RDF peaks with increasing water content, indicating a more dispersed distribution of water molecules. As loading increases, more water molecules shift away from the zirconium clusters toward the pore centers, forming one-dimensional water chains and clusters (Fig. S3†). In contrast, L–B mixing shows a slight decrease in water presence around zirconium clusters, with less pronounced occupancy within the pore volume.
 | ||
| Fig. 1 (a) RDF calculated using hybrid mixing between water molecules and framework interaction sites near the zirconium (Zr) cluster. Obtained at a temperature and pressure of 300 K and 1 atm, respectively, with a water loading of 10 molecules per unit cell. (b) The identified interaction sites in the inorganic cluster are the linker oxygen OL, the hydroxyl group OOh and the dehydroxylated group OOx within the cluster. | ||
A series of 3 ns simulations in the NVT ensemble were conducted at increasing loadings to monitor water molecule positions and analyze their trajectories. Fig. S4† compares the mean square displacement (MSD) of water molecules for the two interaction methods at a moderate loading of 40 water molecules per unit cell. As shown in Fig. 2, L–B mixing results in water molecules remaining highly localized near the linker oxygen, with minimal dimer formation. In contrast, hybrid mixing simulations exhibit diffusive behavior around the predicted adsorption sites, with occasional formation of water dimers.
 | ||
| Fig. 2 Trajectories described by water molecules inside the framework over a simulation period of 500 picoseconds at moderate loading (40 molecules per uc) using (a) L–B and (b) hybrid mixing under atmospheric conditions. On the left side, the cumulative trajectories of all O(H2O) atoms are pictured. On the right side a subset of trajectories is presented, representative of 3 types of observed behavior: (A) molecules bound to an interaction site, (B) molecules orbiting already occupied sites, and (C) molecules of the previous type hopping from one interaction site to another within the same pore. Trajectories are colored based on a gradient from red (start of the trajectory) to blue (end of the trajectory). | ||
To assess how differences in predicted adsorption sites affect the framework structure, NPT simulations were performed under atmospheric conditions with increased water loadings. System volume and bond distances between inner-cluster atoms were monitored and averaged. As shown in Fig. 3, L–B mixing predicts a significant increase in the unit cell lattice parameter with loading, reaching a 7% volume increase at 80 molecules per unit cell compared to the unloaded structure. In contrast, hybrid mixing simulations indicate a slight decrease in lattice parameters, suggesting water-induced framework contraction.
 | ||
| Fig. 3 Average bond lengths between (a) Zr and OL and (b) OOh and its bonded hydrogen. The unit cell lattice parameter is indicated above each bar. The data for (a) and (b) at a given loading are from the same four simulation sets. | ||
The expansion correlates with stretching of Zr–OL bonds. In L–B mixing, increased water presence near OL results in noticeable elongation of Zr–OL bonds, while hybrid simulations show a more pronounced effect on OOh–H bonds. Non-preferential site bonds, such as Zr–Ox and Zr–Oh, experience minimal changes in L–B mixing (Fig. S5†). The observed weakening of framework bonds, particularly Zr–OL, suggests a reduction in cluster–linker coordination and a decrease in mechanical stability upon water loading.
The UiO-66 crystal structure is known for its stability under high pressure, remaining intact up to approximately 1.4 GPa.33 To assess the impact of water loading on structural stability, NPT simulations were conducted under varying hydrostatic pressures and water loadings. Fig. 4 shows that the amorphization pressure (Pam) decreases with increasing water loading. At low loadings (10–20H2O molecules per unit cell), Pam values are similar for both models. However, at intermediate loadings (30–40 H2O molecules), significant differences emerge, due to the hybrid model predicting water dimer and cluster formation at lower water concentrations than the L–B model. The water groupings tend to occupy space closer to the middle of the pores and away from the metal clusters, thus contributing less to cluster–linker weakening. L–B mixing likely predicts unrealistically low Pam values, and the use of hybrid mixing is suggested at moderate and low loadings.
 | ||
| Fig. 4 Amorphization pressure (Pam) with increased water loading at 300 K. Both models show similar Pam values at low loadings but diverge at higher loadings. Hybrid mixing predicts a higher Pam at high loadings, reflecting methodological limitations. | ||
At high loadings (>80 molecules per uc), simulations using hybrid mixing exhibit a noticeable increase in Pam values, a behavior absent in L–B mixing simulations. This difference stems from the water distribution within the crystal and highlights a limitation in our framework collapse detection methodology, in which a framework collapse is defined by a system volume drop below a threshold during equilibration. At high particle loadings, the collapse may be impeded or masked, preventing detection using our criteria.
Our findings indicate that in a system as complex as UiO-66 and water, the selection of guest–host interaction parameters significantly influences the distribution of the former within the available pore volume. When comparing the employed methodologies, hybrid mixing predicts water molecule behavior more in line with previously published simulation studies on the diffusion and pore-filling process of different adsorbates in UiO-66 and analogous structures.47,54–56 The results obtained when employing direct Lorentz–Berthelot mixing rules predict an unusually strong coordination between the water molecules and the framework oxygen atoms connecting the organic linkers with the zirconium cluster, heavily restricting the movement of individual water molecules and inhibiting the formation of water clusters at moderate loadings. Moreover, in both cases, the results indicate that the particular spatial arrangement of the water molecules at different loadings influences both inner-cluster and framework structural parameters. This is more evident when employing L–B mixing, as water coordination causes a stretching of the linker–cluster bonds and a subsequent increase in system volume. This mechanical response to loading also impacts loss of crystallinity pressures and is uncharacteristic of a MOF as rigid as UiO-66, and it might be evidence of poor cross-interaction parameter selection.
Effect of water loading on defective structures
Previous simulation studies show that although the inclusion of defects corresponds to a negligible decrease in equilibrium volume at a particular temperature, they introduce substantial decreases in both amorphization pressure and bulk moduli.46,71 The effect of water loading at high pressure was investigated for structures containing one and two linker defects per unit cell. NPT simulations were conducted at 300 K, systematically varying the water loading from 10 molecules per unit cell to 80. The resulting Pam data obtained using both methods are visually presented in Fig. 5. | ||
| Fig. 5 Amorphization pressure predicted with increased water loading for structures containing linker defects using (a) L–B mixing and (b) hybrid mixing. The color of the curve indicates the average linker–cluster coordination per unit cell. The area in red indicates the Pam range of values obtained for structures with an average cluster coordination of 11.0, where the limiting upper and lower red curves indicate the most stable (type 3) and least stable (type 5) structures. The results for the pristine structures (blue) are included for comparison. | ||
A decrease in amorphization pressure is predicted upon increasing water inclusion for all defective structures, using both parameter mixing methods. In terms of discerning the relative stability of 11-fold coordination structures under increasing water loading, both methodologies predict that inclusion of type 3 vacancies yields the most stable configuration, while type 5 defects exhibit the least stability to high hydrostatic pressure. A close inspection of the results obtained for the structure with 1 missing linker per unit cell (Fig. S6a†) reveals a similar comparative behavior to the pristine structures, where the amorphization values predicted at low loadings using both models are almost identical, followed by deviations at middle and high loadings. Interestingly, a different trend is observed for the 11-fold coordinated structures (Fig. S6b–d†), where the hybrid mixing simulations predict a sharper decrease in Pam values at low loadings when compared to the L–B mixing results. Furthermore, unlike with the less-defective structures, both methods predict comparable high-loading values for all structures with two linker vacancies per unit cell.
Fig. 6 depicts the RDF of water molecules with respect to the metal sites in a structure containing type 3 defects at low loading. For this structure as well as all others containing two linker vacancies per unit cell, when hybrid mixing is employed, water molecules preferentially interact with open Zr atoms and thus tend to agglomerate around them, evidencing strong OH2O–Zr interactions between them. Furthermore, water molecules bonded to the open metal sites exhibit reduced translational and rotational movement, similarly to water molecules near the OL atoms when using L–B mixing in the pristine structures. Water molecules near defective inorganic clusters, but not directly bonded to Zr, frequently change hydrogen-bond partners and exhibit higher orientational mobility. As loading increases water begins to occupy the pore regions, with molecules near the center displaying less restricted movement compared to those near interaction sites.
 | ||
| Fig. 6 (a) RDF between water molecules and the zirconium atoms for the structure containing type 3 defects. The blue curve depicts the RDF between H2O and open Zr2 sites, where all others show the average density of water with respect to fully coordinated Zr sites. (b) Representative snapshot along the XY plane of the structure containing type 3 defects. Open metal sites are pictured in blue. 1D channels created by linker vacancies are highlighted in orange. Similar water molecule behavior observed in simulations with other defective structures at low loading, using hybrid mixing (Fig. S7–S12†). | ||
As was done for the pristine framework, the structural properties of the defective systems under atmospheric conditions, using both mixing methods, were also analyzed. Fig. 7a illustrates the change in unit cell volume for pristine, type 0 defective, and type 3 defective structures when using hybrid mixing. Although all three systems generally contract upon loading, the defective structures show a slight expansion at low loadings. This predicted volume expansion is more noticeable in structures with more linker vacancies, as seen when comparing the results for type 0 and type 3. The type 3 defective structure, representative of other structures with two linker defects per unit cell, exhibits a linear increase in system volume followed by contraction once a loading of 20 molecules per unit cell is reached. Comparing the trends for the type 0 and type 3 structures with that for the pristine one, the initial increase in system volume is likely related to the number of open metal sites available after linker vacancies are introduced. When employing hybrid mixing, water molecules bound to open metal sites contribute to the stretching of linker–cluster bonds, and once these sites are occupied, the general trend of volume contraction resumes.
 | ||
| Fig. 7 System volume change upon water loading under atmospheric conditions, using (a) hybrid mixing and (b) L–B mixing. The results for the pristine, type 0 (1 defect per uc) and type 3 (2 defects per uc) structures are presented. Unlike pristine behavior upon water loading, a slight increase in framework volume is observed at low loadings for defective structures when using hybrid mixing (highlighted in orange). | ||
For all three structures and over the loading range, the simulations employing hybrid mixing predict an overall gradual reduction in volume, eventually plateauing for loading values close to saturation. As was the case for the pristine structure, direct Lorentz–Berthelot mixing predicts a significant volume expansion (a 7% to 8% increase over the loading range) of framework volume upon water loading for defective systems (Fig. 7b). Although the results obtained using hybrid mixing are more in line with the expected behavior of UiO-66 and its employment over direct L–B mixing is recommended (comparisons in Fig. S13–S15†), further force field refinement might be necessary to more accurately capture the interactions between water molecules and open metal sites in the defective structures.
Conclusions
This work investigated the effect of water loading on the stability of pristine and defective UiO-66 via molecular simulations. We used two methodologies for modeling non-bonded interactions: direct Lorentz–Berthelot (L–B) mixing and hybrid mixing. L–B mixing combines Lennard-Jones parameters for framework–framework and water–water interactions, while hybrid mixing employs Universal Force Field (UFF) parameters for the framework. We examined water distribution within the framework under atmospheric conditions. Both methodologies predicted water clustering around zirconium clusters, but RDF analysis showed differences in interaction sites and strengths. L–B mixing suggests strong binding of water to linker oxygen atoms (OL), restricting movement and diffusion. Hybrid mixing, however, shows interactions with the hydroxyl groups of the inorganic cluster (OOh), resulting in dimer formation, site-to-site hopping at low loadings, and pore-to-pore diffusion at higher loadings, which aligns with recent studies on water diffusion in UiO-66.NPT simulations under atmospheric conditions demonstrated that interaction strength significantly influences structural parameters. At high water loadings, hybrid mixing predicted slight contraction of the system, whereas L–B mixing indicated over a 7 percent expansion due to stretching of linker–cluster bonds. This suggests that L–B mixing inadequately models the rigidity of UiO-66. High-pressure NPT simulations showed that water loading decreases amorphization pressure for both methodologies compared to the unloaded structure. L–B mixing consistently predicted lower amorphization pressures, with notable deviations at middle and high loadings, consistent with previous studies where water agglomeration reduces linker–cluster bond weakening. Similar trends were observed for structures with one missing linker per unit cell. For 11-fold coordinated structures, hybrid mixing predicted a sharper decrease in amorphization pressure at low loadings compared to L–B mixing, though both methods converged at high loadings for structures with two linker vacancies per unit cell.
For structures with two linker vacancies, hybrid mixing simulations showed water molecules preferentially interacting with open Zr atoms, leading to agglomeration and strong OH2O–Zr interactions. These water molecules exhibited reduced movement, similar to those near OL atoms in L–B mixing. Initial volume expansion was observed at low loadings due to interactions with open metal sites, which then resumed contraction once these sites were filled. This indicates a need for refining force field parameters to better model interactions with open Zr sites in defective UiO-66. Overall, our findings emphasize the significant impact of water loading on UiO-66's structural stability and the importance of selecting appropriate interaction parameters for accurate simulations. The differences between L–B and hybrid mixing highlight the need for careful parameter selection and further development of force fields for defective frameworks.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was funded by the Eindhoven Institute for Renewable Energy Systems (EIRES) and by the Spanish Ministerio de Ciencia e Innovacion (CNS2022-136163). We thank C3UPO for the HPC support.References
- W. M. Warren-Vega, A. Campos-Rodríguez, A. I. Zárate-Guzmán and L. A. Romero-Cano, A current review of water pollutants in American continent: trends and perspectives in detection, health risks, and treatment technologies, Int. J. Environ. Res. Public Health, 2023, 20, 4499 CrossRef CAS PubMed.
- X. Xu, H. Yang and C. Li, Theoretical model and actual characteristics of air pollution affecting health cost: a review, Int. J. Environ. Res. Public Health, 2022, 19, 3532 CrossRef CAS PubMed.
- L. Lin, H. Yang and X. Xu, Effects of water pollution on human health and disease heterogeneity: a review, Front. Environ. Sci., 2022, 10, 880246 CrossRef.
- J. Abdi, M. Vossoughi, N. M. Mahmoodi and I. Alemzadeh, Synthesis of metal-organic framework hybrid nanocomposites based on GO and CNT with high adsorption capacity for dye removal, Chem. Eng. J., 2017, 326, 1145–1158 CrossRef CAS.
- M. Raeiszadeh, A. Hakimian, A. Shojaei and H. Molavi, Nanodiamond-filled chitosan as an efficient adsorbent for anionic dye removal from aqueous solutions, J. Environ. Chem. Eng., 2018, 6, 3283–3294 CrossRef CAS.
- K. Yue, X. Zhang, S. Jiang, J. Chen, Y. Yang, F. Bi and Y. Wang, Recent advances in strategies to modify MIL-125 (Ti) and its environmental applications, J. Mol. Liq., 2021, 335, 116108 CrossRef CAS.
- C. Zhao, Y. Li, H. Chu, X. Pan, L. Ling, P. Wang, H. Fu, C.-C. Wang and Z. Wang, Construction of direct Z-scheme Bi5O7I/UiO-66-NH2 heterojunction photocatalysts for enhanced degradation of ciprofloxacin: mechanism insight, pathway analysis and toxicity evaluation, J. Hazard. Mater., 2021, 419, 126466 CrossRef CAS PubMed.
- X. Fang, D. Zhang, Z. Chang, R. Li and S. Meng, Phosphorus removal from water by the metal-organic frameworks (MOFs)-based adsorbents: a review for structure, mechanism, and current progress, Environ. Res., 2024, 243, 117816 CrossRef CAS PubMed.
- Q. Xie, Y. Li, Z. Lv, H. Zhou, X. Yang, J. Chen and H. Guo, Effective adsorption and removal of phosphate from aqueous solutions and eutrophic water by Fe-based MOFs of MIL-101, Sci. Rep., 2017, 7, 3316 CrossRef PubMed.
- L. Zhang, D. Mao, Y. Qu, X. Chen, J. Zhang, M. Huang and J. Wang, Facile synthesis of Ce-MOF for the removal of phosphate, fluoride, and arsenic, Nanomaterials, 2023, 13, 3048 CrossRef CAS PubMed.
- A. K. Tolkou and A. I. Zouboulis, Fluoride removal from water sources by adsorption on MOFs, Separations, 2023, 10, 467 CrossRef CAS.
- S. Essalmi, S. Lotfi, A. BaQais, M. Saadi, M. Arab and H. Ait Ahsaine, Design and application of metal organic frameworks for heavy metals adsorption in water: a review, RSC Adv., 2024, 14, 9365–9390 RSC.
- P. Chen, Y. Wang, X. Zhuang, H. Liu, G. Liu and W. Lv, Selective removal of heavy metals by Zr-based MOFs in wastewater: new acid and amino functionalization strategy, J. Environ. Sci., 2023, 124, 268–280 CrossRef CAS PubMed.
- E. Svensson Grape, et al., Removal of pharmaceutical pollutants from effluent by a plant-based metal–organic framework, Nat. Water, 2023, 1, 433–442 CrossRef.
- L. Huang, R. Shen and Q. Shuai, Adsorptive removal of pharmaceuticals from water using metal-organic frameworks: a review, J. Environ. Manage., 2021, 277, 111389 CrossRef CAS PubMed.
- Y. Kalinovskyy, N. J. Cooper, M. J. Main, S. J. Holder and B. A. Blight, Microwave-assisted activation and modulator removal in zirconium MOFs for buffer-free CWA hydrolysis, Dalton Trans., 2017, 46, 15704–15709 RSC.
- G. W. Peterson, A. X. Lu and T. H. Epps III, Tuning the morphology and activity of electrospun polystyrene/UiO-66-NH2 metal–organic framework composites to enhance chemical warfare agent removal, ACS Appl. Mater. Interfaces, 2017, 9, 32248–32254 CrossRef CAS PubMed.
- A. Lee, M.-W. Moon, H. Lim, W.-D. Kim and H.-Y. Kim, Water harvest via dewing, Langmuir, 2012, 28, 10183–10191 CrossRef CAS PubMed.
- M. Fessehaye, S. A. Abdul-Wahab, M. J. Savage, T. Kohler, T. Gherezghiher and H. Hurni, Fog-water collection for community use, Renewable Sustainable Energy Rev., 2014, 29, 52–62 CrossRef.
- M. Azeem, M. T. Noman, J. Wiener, M. Petru and P. Louda, Structural design of efficient fog collectors: a review, Environ. Technol. Innovation, 2020, 20, 101169 CrossRef.
- A. Jrad, P. Damacet, Z. Yaghi, M. Ahmad and M. Hmadeh, Zr-based metal–organic framework nanocrystals for water remediation, ACS Appl. Nano Mater., 2022, 5, 10795–10808 CrossRef CAS.
- X. Liu, Y. Shan, S. Zhang, Q. Kong and H. Pang, Application of metal organic framework in wastewater treatment, Green Energy Environ., 2023, 8, 698–721 CrossRef CAS.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, S. Tajahmadi, A. Bahi, F. Ko, T. M. Aminabhavi, J.-R. Li and M. Arjmand, UiO-66 metal–organic frameworks in water treatment: a critical review, Prog. Mater. Sci., 2022, 125, 100904 CrossRef CAS.
- N. C. Burtch, H. Jasuja and K. S. Walton, Water stability and adsorption in metal-organic frameworks, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y.-g. Huang and K. S. Walton, Stability and degradation mechanisms of metal–organic frameworks containing the Zr6O4(OH)4 secondary building unit, J. Mater. Chem. A, 2013, 1, 5642 RSC.
- D. Zou and D. Liu, Understanding the modifications and applications of highly stable porous frameworks via UiO-66, Mater. Today Chem., 2019, 12, 139–165 CrossRef CAS.
- D. Zou and D. Liu, Understanding the modifications and applications of highly stable porous frameworks via UiO-66, Mater. Today Chem., 2019, 12, 139–165 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- K. Leus, T. Bogaerts, J. De Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Systematic study of the chemical and hydrothermal stability of selected “stable” metal organic frameworks, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the complex structure of UiO-66 metal organic framework: a synergic combination of experiment and theory, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- H. Wu, T. Yildirim and W. Zhou, Exceptional mechanical stability of highly porous zirconium metal-organic framework UiO-66 and its important implications, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS PubMed.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F.-X. Coudert, Defects in metal-organic frameworks: a compromise between adsorption and stability?, Dalton Trans., 2016, 45, 4352–4359 RSC.
- P. G. Yot, K. Yang, F. Ragon, V. Dmitriev, T. Devic, P. Horcajada, C. Serre and G. Maurin, Exploration of the mechanical behavior of metal organic frameworks UiO-66(Zr) and MIL-125(Ti) and their NH2 functionalized versions, Dalton Trans., 2016, 45, 4283–4288 RSC.
- M. Sk, S. Kar, J. K. Dewangan and M. Chowdhury, Engineering linker defects in functionalized UiO-66 MOF nanoparticles for oil-in-water Pickering emulsion stabilization, Dalton Trans., 2023, 52, 11886–11896 RSC.
- Y. Huang, W. Qin, Z. Li and Y. Li, Enhanced stability and CO2 affinity of a UiO-66 type metal-organic framework decorated with dimethyl groups, Dalton Trans., 2012, 41, 9283–9285 RSC.
- H. G. T. Nguyen, N. M. Schweitzer, C.-Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, Vanadium-node-functionalized UiO-66: a thermally stable MOF-supported catalyst for the gas-phase oxidative dehydrogenation of cyclohexene, ACS Catal., 2014, 4, 2496–2500 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, Synthesis modulation as a tool to increase the catalytic activity of metal-organic frameworks: the unique case of UiO-66(Zr), J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Defect engineering: tuning the porosity and composition of the metal–organic framework UiO-66 via modulated synthesis, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- L. Liu, Z. Chen, J. Wang, D. Zhang, Y. Zhu, S. Ling, K.-W. Huang, Y. Belmabkhout, K. Adil, Y. Zhang, B. Slater, M. Eddaoudi and Y. Han, Imaging defects and their evolution in a metal-organic framework at sub-unit-cell resolution, Nat. Chem., 2019, 11, 622–628 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Tuned to perfection: ironing out the defects in metal–organic framework UiO-66, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- P. Ghosh, Y. J. Colón and R. Q. Snurr, Water adsorption in UiO-66: the importance of defects, Chem. Commun., 2014, 50, 11329–11331 RSC.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Defect engineering of UiO-66 for CO2 and H2O uptake – a combined experimental and simulation study, Dalton Trans., 2016, 45, 4496–4500 RSC.
- J. Canivet, M. Vandichel and D. Farrusseng, Origin of highly active metal-organic framework catalysts: defects? Defects!, Dalton Trans., 2016, 45, 4090–4099 RSC.
- J. M. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Defect control to enhance proton conductivity in a metal–organic framework, Chem. Mater., 2015, 27, 2286–2289 CrossRef CAS.
- J. M. Taylor, T. Komatsu, S. Dekura, K. Otsubo, M. Takata and H. Kitagawa, The role of a three dimensionally ordered defect sublattice on the acidity of a sulfonated metal-organic framework, J. Am. Chem. Soc., 2015, 137, 11498–11506 CrossRef CAS PubMed.
- S. M. J. Rogge, J. Wieme, L. Vanduyfhuys, S. Vandenbrande, G. Maurin, T. Verstraelen, M. Waroquier and V. Van Speybroeck, Thermodynamic Insight in the High-Pressure Behavior of UiO-66: Effect of Linker Defects and Linker Expansion, Chem. Mater., 2016, 28, 5721–5732 CrossRef CAS PubMed.
- S. Wang, G. Zhou, Y. Sun and L. Huang, A computational study of water in UiO-66 Zr-MOFs: diffusion, hydrogen bonding network, and confinement effect, AIChE J., 2021, 67, e17035 CrossRef CAS.
- Q. Yang, V. Guillerm, F. Ragon, A. D. Wiersum, P. L. Llewellyn, C. Zhong, T. Devic, C. Serre and G. Maurin, CH4 storage and CO2 capture in highly porous zirconium oxide based metal-organic frameworks, Chem. Commun., 2012, 48, 9831–9833 RSC.
- M. I. Hossain, J. D. Cunningham, T. M. Becker, B. E. Grabicka, K. S. Walton, B. D. Rabideau and T. G. Glover, Impact of MOF defects on the binary adsorption of CO2 and water in UiO-66, Chem. Eng. Sci., 2019, 203, 346–357 CrossRef CAS.
- A. Planchais, S. Devautour-Vinot, F. Salles, F. Ragon, T. Devic, C. Serre and G. Maurin, A joint experimental/computational exploration of the dynamics of confined water/Zr-based MOFs systems, J. Phys. Chem. C, 2014, 118, 14441–14448 CrossRef CAS.
- G. Jajko, J. J. Gutiérrez-Sevillano, A. Sławek, M. Szufla, P. Kozyra, D. Matoga, W. Makowski and S. Calero, Water adsorption in ideal and defective UiO-66 structures, Microporous Mesoporous Mater., 2022, 330, 111555 CrossRef CAS.
- L. Vanduyfhuys, S. Vandenbrande, T. Verstraelen, R. Schmid, M. Waroquier and V. Van Speybroeck, QuickFF: a program for a quick and easy derivation of force fields for metal-organic frameworks from ab initio input, J. Comput. Chem., 2015, 36, 1015–1027 CrossRef CAS PubMed.
- L. Vanduyfhuys, S. Vandenbrande, J. Wieme, M. Waroquier, T. Verstraelen and V. Van Speybroeck, Extension of the QuickFF force field protocol for an improved accuracy of structural, vibrational, mechanical and thermal properties of metal-organic frameworks, J. Comput. Chem., 2018, 39, 999–1011 CrossRef CAS PubMed.
- J. J. Wardzala, J. P. Ruffley, I. Goodenough, A. M. Schmidt, P. B. Shukla, X. Wei, A. Bagusetty, M. De Souza, P. Das, D. J. Thompson, C. J. Karwacki, C. E. Wilmer, E. Borguet, N. L. Rosi and J. K. Johnson, Modeling of diffusion of acetone in UiO-66, J. Phys. Chem. C, 2020, 124, 28469–28478 CrossRef CAS.
- C. V. Mhatre, J. J. Wardzala, P. B. Shukla, M. Agrawal and J. K. Johnson, Calculation of Self, Corrected, and Transport Diffusivities of Isopropyl Alcohol in UiO-66, Nanomaterials, 2023, 13, 1793 CrossRef CAS PubMed.
- J. Zhang, F. Paesani and M. Lessio, Computational insights into the interaction of water with the UiO-66 metal–organic framework and its functionalized derivatives, J. Mater. Chem. C, 2023, 11, 10247–10258 RSC.
- X. Zhu, M. Riera, E. F. Bull-Vulpe and F. Paesani, MB-pol(2023): sub-chemical accuracy for water simulations from the gas to the liquid phase, J. Chem. Theory Comput., 2023, 19, 3551–3566 CrossRef CAS PubMed.
- J. L. F. Abascal and C. Vega, A general purpose model for the condensed phases of water: TIP4P/2005, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- H. O. Frank and F. Paesani, Molecular driving forces for water adsorption in MOF-808: a comparative analysis with UiO-66, J. Chem. Phys., 2024, 160(9), 094703 CrossRef CAS PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, Water Adsorption in Porous Metal–Organic Frameworks and Related Materials, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, Molecular mechanics. The MM3 force field for hydrocarbons. 1, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- N. L. Allinger, X. Zhou and J. Bergsma, Molecular mechanics parameters, J. Mol. Struct.: THEOCHEM, 1994, 312, 69–83 CrossRef.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- S. Chatterjee, P. G. Debenedetti, F. H. Stillinger and R. M. Lynden-Bell, A computational investigation of thermodynamics, structure, dynamics and solvation behavior in modified water models, J. Chem. Phys., 2008, 128, 124511 CrossRef PubMed.
- S. M. J. Rogge, L. Vanduyfhuys, A. Ghysels, M. Waroquier, T. Verstraelen, G. Maurin and V. Van Speybroeck, A comparison of barostats for the mechanical characterization of metal-organic frameworks, J. Chem. Theory Comput., 2015, 11, 5583–5597 CrossRef CAS PubMed.
- S. M. J. Rogge, M. Waroquier and V. Van Speybroeck, Reliably modeling the mechanical stability of rigid and flexible metal–organic frameworks, Acc. Chem. Res., 2018, 51, 138–148 CrossRef CAS PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(33–8), 27–28 Search PubMed.
- M. I. Hossain and T. G. Glover, Kinetics of water adsorption in UiO-66 MOF, Ind. Eng. Chem. Res., 2019, 58, 10550–10558 CrossRef CAS.
- T. D. Bennett, T. K. Todorova, E. F. Baxter, D. G. Reid, C. Gervais, B. Bueken, B. Van de Voorde, D. De Vos, D. A. Keen and C. Mellot-Draznieks, Connecting defects and amorphization in UiO-66 and MIL-140 metal–organic frameworks: a combined experimental and computational study, Phys. Chem. Chem. Phys., 2016, 18, 2192–2201 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04252b |
| This journal is © The Royal Society of Chemistry 2024 |