
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature promotes selectivity during electrochemical CO2 reduction on NiO:SnO2 nanofibers†
M. A.
Rodriguez-Olguin‡
ab,
R.
Lipin‡
c,
M.
Suominen
d,
F.
Ruiz-Zepeda
 ef,
E.
Castañeda-Morales
g,
A.
Manzo-Robledo
g,
J. G. E.
Gardeniers
ab,
C.
Flox
*dh,
T.
Kallio
*d,
M.
Vandichel
*c and
A.
Susarrey-Arce
*ab
ef,
E.
Castañeda-Morales
g,
A.
Manzo-Robledo
g,
J. G. E.
Gardeniers
ab,
C.
Flox
*dh,
T.
Kallio
*d,
M.
Vandichel
*c and
A.
Susarrey-Arce
*ab
aMesoscale Chemical Systems, MESA+ Institute, University of Twente, P. O. Box 217, Enschede 7500AE, The Netherlands
bDepartment of Chemical Engineering, MESA+ Institute, University of Twente, P. O. Box 217, Enschede 7500AE, The Netherlands. E-mail: a.susarreyarce@utwente.nl
cSchool of Chemical Sciences and Chemical Engineering, Bernal Institute, University of Limerick, Limerick V94 T9PX, Republic of Ireland. E-mail: matthias.vandichel@ul.ie
dDepartment of Chemistry and Materials Science, Aalto University School of Chemical Engineering, Kemistintie 1, 02015 Espoo, Finland. E-mail: tanja.kallio@aalto.fi
eDepartment of Materials Chemistry, National Institute of Chemistry, Hajdrihova 19, 1000, Ljubljana, Slovenia
fDepartment of Physics and Chemistry of Materials, Institute of Metals and Technology, Lepi pot 11, Ljubljana, Slovenia
gInstituto Politécnico Nacional, Laboratorio de Electroquímica y Corrosión, Escuela Superior de Ingeniería Química e Industrias Extractivas, Av. Instituto Politécnico Nacional S/N, Unidad Profesional Adolfo López Mateos, CP 07708 CDMX, Mexico
hDepartment of Electrical Energy Storage, Iberian Centre for Research in Energy Storage, Campus University of Extremadura, Avda. de las Letras, s/n, 10004 Cáceres, Spain. E-mail: cristina.flox@ciiae.org
First published on 8th August 2024
Abstract
Electrolyzers operate over a range of temperatures; hence, it is crucial to design electrocatalysts that do not compromise the product distribution unless temperature can promote selectivity. This work reports a synthetic approach based on electrospinning to produce NiO:SnO2 nanofibers (NFs) for selectively reducing CO2 to formate above room temperature. The NFs comprise compact but disjoined NiO and SnO2 nanocrystals identified with STEM. The results are attributed to the segregation of NiO and SnO2 confirmed with XRD. The NFs are evaluated for the CO2 reduction reaction (CO2RR) over various temperatures (25, 30, 35, and 40 °C). The highest faradaic efficiencies to formate (FEHCOO−) are reached by NiO:SnO2 NFs containing 50% of NiO and 50% SnO2 (NiOSnO50NF), and 25% of NiO and 75% SnO2 (NiOSnO75NF), at an electroreduction temperature of 40 °C. At 40 °C, product distribution is assessed with in situ differential electrochemical mass spectrometry (DEMS), recognizing methane and other species, like formate, hydrogen, and carbon monoxide, identified in an electrochemical flow cell. XPS and EELS unveiled the FEHCOO− variations due to a synergistic effect between Ni and Sn. DFT-based calculations reveal the superior thermodynamic stability of Ni-containing SnO2 systems towards CO2RR over the pure oxide systems. Furthermore, computational surface Pourbaix diagrams showed that the presence of Ni as a surface dopant increases the reduction of the SnO2 surface and enables the production of formate. Our results highlight the synergy between NiO and SnO2, which can promote the electroreduction of CO2 at temperatures above room temperature.
1. Introduction
Anthropogenic CO2 emissions are a problem that must be addressed to decelerate greenhouse effects like global warming. According to the Intergovernmental Panel on Climate Change (IPCC) published in 2021, the planet's temperature may increase by 1.5 °C in the following decades due to the accelerated release of CO2 and other greenhouse gases, posing a threat to the global ecosystems and humankind.1 A call-to-action part of the energy transition goals is CO2 recycling and utilization. One way to recycle and utilize CO2 is by producing CO2-based value-added products electrochemically. For example, electrochemical CO2RR can enable the production of C1 (e.g., CO, HCOO−, CH4) and C2 (e.g., C2H4, alcohol) products. The selectivity of C1 or C2 products is often attributed to the catalyst type or the operation conditions during CO2 electrolysis. As for the catalysts, three different groups of metals have been recognized based on the product generated: formate (e.g., Sn,2 Pb,3 Bi,4,5 In,6 Hg7), CO (Au,8 Ag,9 Pd,10 Zn,11 Ni12), hydrocarbons and alcohols (Cu13,14) and other multi-carbon products like amino acids,15 which in combination with CO2 and nitrogenated sources (NH4HCO3) has been demonstrated to lead to the formation of serine.16,17 Among the mentioned metals, Cu is a catalyst capable of producing C1, C2, and higher multi-carbon products.18 However, copper exhibits low selectivity, resulting in a mixture of gaseous and liquid reaction compounds. Therefore, the selective production of CO2-reduction products is a key challenge that needs attention. One way to overcome this challenge is by synthetically designing a catalyst with earth-abundant elements that can cope with CO2RR on an industrial level aimed at 90% conversion to a single product and elevated electrolyzer temperatures for practical reasons (e.g., increased reaction rates or reduced overpotentials).The reaction temperature is a crucial yet sometimes overlooked parameter in electrochemistry that can compensate for the thermal losses during CO2 electrolysis. Mizuno et al. have delved into the electrochemical reduction of CO2 with various electrode compositions.19 In a different study, Kim et al. analyzed the effect of operating conditions, including temperature, on the electrochemical conversion of CO2 to formic acid.20 Like Mizuno et al.,19 the results underscore the intricate relationship between temperature and selectivity for CO2RR. Recently, Löwe et al. addressed in depth how temperature variations can influence transport and, thus, formate production.21 According to the authors, the temperature notably impacts the reaction selectivity at an industrially accepted current density of 200 mA cm−2. Specifically, the FEHCOO− decreases from 89% at 20 °C to 85% at 70 °C, while the production of CO increases from around 7% to 11%. Interestingly, variations of about ±10% during FEHCOO− have been observed at the assessed temperatures with current densities of up to 1000 mA cm−2. The results from Löwe et al. indicate that with an increase in temperature, the selectivity towards formate decreases while it increases for other products like CO or H2. Other studies reported similar trends, ascribing the decreased formate production and selectivity with increasing temperature to the kinetic effects and CO2 solubility.22 Under other conditions, e.g., by varying the electrolyte solution, a fundamental understanding of the relationships between the surface coverage, pH, and kinetics has been proposed for Cu.18,23 The previously cited seminal works are therefore used as a stepping stone to understand the temperature effects over NiO:SnO2 catalyst, whose synergistic combinations can lead to tuning the selectivity of CO2RR products. It should be noted that other studies for NiO, SnO2, and their combinations do not deal with temperature variation conditions during CO2 electrolysis (Tables S1–S3†). NiO typically leads to CO, CH4, and, in some cases, to HCOO− (Table S1†), while SnO2 leads to HCOO− as the main product (Table S2†). However, their synergistic effect has only been shown HCOO− products near room temperature (Table S3†).
An additional expected challenge for a single oxide composition like SnO2 is its instability under cathodic electrochemical potentials.24 The instability of SnO2 during CO2RR can be attributed to the thermodynamic formation of various oxidized tin species.25 Mu et al. highlighted the dominance of hydroxyl radicals in the reoxidation of oxide-derived metals like Cu. Despite being thermodynamically unstable under cathodic conditions, the authors propose that the presence of Cuδ+ species enables the CO2RR.26 The results from Mu et al. emphasize the importance of the stability of metal oxide catalysts.26 Lately, Jiang et al. have discussed the importance of stabilizing the oxidation state of SnO2 to achieve highly selective CO2 electroreduction to formate.27 Their study emphasizes the necessity of maintaining the oxidation state of tin during the CO2RR, achieved by incorporating Cu single atoms into the SnO2 lattice. Like single-atom catalysis, other strategies such as engineering the defects in oxides, have been proposed to tune the selectivity of CO2RR28 and NOx reduction reactions.28,29 From the later examples, catalyst discovery should favor thermodynamically stable catalytic species that appropriately control product selectivity over various reaction conditions. This could be the case for catalysts that incorporate suitable supports or modifiers optimized for various temperature conditions to minimize the degradation of the active sites.
Aside from single Cu atoms and others like Pt and Bi,27 NiO is an exciting option to stabilize SnO2 because it can be incorporated in much higher proportions using electrospinning.30,31 The incorporation of NiO and SnO2 goes hand in hand with the possibility of achieving 1D nanofibrous structures to produce non-woven architectures ideal for transport control during electrochemical reactions.32,33 Herein, our working hypothesis is that NiO embedding will stabilize the SnO2 and increase the hydrogenation activity of SnO2, further enabling the formate (HCOO−) production. However, it is not ‘a priori’ clear how NiO and SnO2 combined in an NF can synergistically favor formate production and how temperature affects selectivity. Thus, it is essential to explore.
In this work, we employ electrospinning to synthesize different NiOSnO2 NFs by varying Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn precursor molar ratios between 75:25, 50:50, and 25:75. Among the applied Ni:Sn molar ratios, 50:50 and 75:25 resulted in an NF-like morphology after annealing at 550 °C while other molar ratios or single compositions did not lead to NFs. Therefore, a detailed structural and morphological characterization (XRD, HR-SEM, and STEM-EDX) is carried out for the 50:50 (NiOSnO50NF) and 25:75 (NiOSnO75NF) NFs. Additionally, XPS and EELS are used to understand the chemical environment before and after the CO2RR. The results show the presence of Ni3+ species and partially reduced SnO2 after CO2 electrolysis at 40 °C. The selectivity of the NFs is evaluated by using an electrochemical flow cell with a maximum heating capacity of 40 °C. The electrochemical experiments for NiOSnO50NF and NiOSnO75NF show HCOO− selectivities close to ∼25% at 25 °C, while at temperatures of 30 °C and 35 °C the HCOO− selectivity increased to ∼30% and ∼50%. An increased HCOO− selectivity is observed at an electroreduction temperature of 40 °C, ca. ∼85%, and ∼70% for NiOSnO50NF and NiOSnO75NF, respectively. The product distribution assessed with in situ DEMS aligns with the products observed in the flow cell experiments carried out at 40 °C, where, besides methane (CH4), HCOO−, hydrogen (H2), and carbon monoxide (CO) products have also been found. DFT modeling provides insights into the reaction mechanism and the effect of temperature during the CO2RR process. Furthermore, the computational surface Pourbaix diagram indicates that combining NiO and SnO2 increases the hydrogenation level in the catalyst model, enabling HCOO− production. Our results highlight the importance of catalyst discovery, as demonstrated by the synergistic effects between NiO and SnO2, which can boost the electroreduction of CO2 at temperatures higher than room temperature.
:Sn precursor molar ratios between 75:25, 50:50, and 25:75. Among the applied Ni:Sn molar ratios, 50:50 and 75:25 resulted in an NF-like morphology after annealing at 550 °C while other molar ratios or single compositions did not lead to NFs. Therefore, a detailed structural and morphological characterization (XRD, HR-SEM, and STEM-EDX) is carried out for the 50:50 (NiOSnO50NF) and 25:75 (NiOSnO75NF) NFs. Additionally, XPS and EELS are used to understand the chemical environment before and after the CO2RR. The results show the presence of Ni3+ species and partially reduced SnO2 after CO2 electrolysis at 40 °C. The selectivity of the NFs is evaluated by using an electrochemical flow cell with a maximum heating capacity of 40 °C. The electrochemical experiments for NiOSnO50NF and NiOSnO75NF show HCOO− selectivities close to ∼25% at 25 °C, while at temperatures of 30 °C and 35 °C the HCOO− selectivity increased to ∼30% and ∼50%. An increased HCOO− selectivity is observed at an electroreduction temperature of 40 °C, ca. ∼85%, and ∼70% for NiOSnO50NF and NiOSnO75NF, respectively. The product distribution assessed with in situ DEMS aligns with the products observed in the flow cell experiments carried out at 40 °C, where, besides methane (CH4), HCOO−, hydrogen (H2), and carbon monoxide (CO) products have also been found. DFT modeling provides insights into the reaction mechanism and the effect of temperature during the CO2RR process. Furthermore, the computational surface Pourbaix diagram indicates that combining NiO and SnO2 increases the hydrogenation level in the catalyst model, enabling HCOO− production. Our results highlight the importance of catalyst discovery, as demonstrated by the synergistic effects between NiO and SnO2, which can boost the electroreduction of CO2 at temperatures higher than room temperature.
2. Methods
2.1 Synthesis of NiO:SnO2 nanofibers
The electrospinning technique was used to synthesize metal oxide NFs. For the NiO:SnO2 NF, NiCl2·6H2O (ACS grade, Sigma Aldrich) and SnCl2·XH2O (ACS grade, Sigma Aldrich) were used as precursors. The stock solutions containing Ni, Sn, or their combinations were prepared by dissolving the metal salts in ethanol (100% Tech. grade, BOOM B.V., The Netherlands). Subsequently, polyvinylpyrrolidone (PVP, MW ∼1300000 by LS, Sigma Aldrich) is added to the solution and stirred magnetically overnight. The precursor solutions were spun using a commercial electrospinning system from IME Technologies (The Netherlands) at 2.0 mL h−1. NFs were obtained at 10.25 kV using a stainless-steel needle of 0.4 mm inner diameter. The collector was maintained at a separation distance of 12 cm from the needle to the aluminum collector plate. The NFs were collected at 22 °C and a relative humidity of 30%. After deposition, NFs were dried in a furnace at 80 °C for 12 h to remove the excess solvent. In a subsequent step, the NFs were annealed in two steps in air. First, the NFs were annealed at 350 °C (ramp-up rate of 1 °C min−1) for 3 h to remove the organic components. Second, the NFs were annealed at 550 °C (1 °C min−1) for 1 h to produce the NiO:SnO2 mixed oxides. Controls were produced using the electrospinning precursor mentioned above. The controls lead to nanoparticles (NP), hereafter labeled as NiOSnONP. In short, the control samples were prepared by directly pouring the electrospinning precursor into porcelain crucibles and calcined at 550 °C (1 °C min−1) for 1 h in air.
2.2 Morphological, structural and chemical characterization
2.3 Electrochemical characterization
:1 ratio by weight) and 600 μL of 2-propanol. Then, MWCNTs are added to the ink to increase the conductivity of the samples. In all samples, the final loading of the catalysts was 1.25 mg cm−2, covering a geometrical area of 1.8 cm2 of the cathode. The working electrode consists of carbon gas diffusion electrodes (GDE) (SIGRACET 25BC) sprayed with the catalyst ink and dried at 80 °C. Commercial Ir-MMO and leak-free Ag/AgCl electrodes were used as anode and reference electrodes, respectively. The Nafion 117 membrane separates the cathode and anode chambers. The potential was referred to against the reversible hydrogen electrode (RHE).| URHE = U(Ag/AgCl) + U0(Ag/AgCl) + 0.059pH | (1) |
The electroreduction was performed by applying a potential of −0.85 V (vs. RHE) for 2 h, using 0.5 M KHCO3 as the electrolyte in both chambers, circulated at 23 mL min−1. CO2 gas was fed at 11 mL min−1. The pH value of the CO2-saturated electrolyte at 40 °C was 7.9. The different temperatures for electroreduction (25, 30, 35, and 40 °C) were controlled by placing the electrolyte reservoirs in a hot water bath and keeping the cell insulated. The maximum temperature capacity of our electrochemical cell was 40 °C.
An online gas chromatograph (Agilent micro-GC) was connected to the electrochemical cell to analyze the gas products (H2 and CO). No CH4 was observed, possibly due to the low concentration, and thus, below the detection limit of the micro-GC. After electrolysis, liquid products were analyzed with HPLC (AMinex column HPX-87X from Bio-Rad). The eluent used was 5 mM of H2SO4 with a 0.6 mL min−1 flow rate at 65 °C. Typically, 10 mL of collected catholyte was mixed with 4 M of H2SO4 to decrease from pH 7.9 to pH 1–3, corresponding to formic acid formation. It should be noted that all current densities are expressed as cathodic currents. Thus, a negative value was used in the manuscript.
The faradaic efficiency (FE) for the products was calculated according to the following equation:
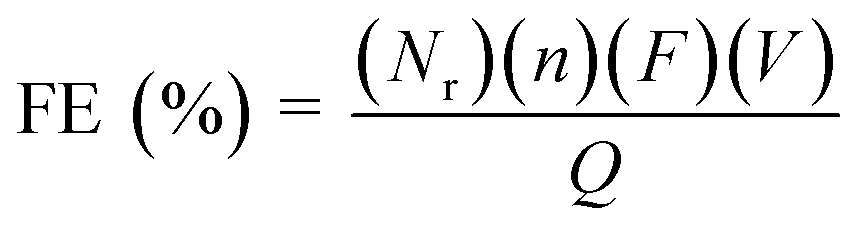 | (2) |
485 C mol−1), V is the molar flow rate of CO2 and Q is the total charge passed during electrolysis.
2.4 Theoretical methods
The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was employed to compute the total energies.36 The projector-augmented wave (PAW) pseudopotentials were used for the calculations with an energy cutoff of 400 eV for the plane waves.37,38 More in particular, 4, 6, 10, and 1 valence electrons were considered for Sn (5s25p2), O (2s22p4), Ni (3d84s2), and H (1s1) atoms, respectively. The Grimme-D3(BJ) method was implemented in VASP 5.4.4 to account for the van der Waals interactions.39,40 The atomic positions were optimized using the conjugate gradient algorithm with force and electronic convergence criteria of 0.01 eV Å−1 and 10−5 eV, a Gaussian smearing of 0.05 eV, and a 4 × 3 × 1 Monkhorst–Pack k-point grid.41 The Partial Hessian Vibrational Analysis (PHVA) was carried out only for the surface species and selected atoms at the active sites on the catalyst (involving three Sn and three bridging O atoms) surface while keeping the other atoms fixed (Fig. S1†). The numerical partial Hessian was calculated by displacing the unfixed atoms in x, y, and z-directions with ±0.01 Å, and the corresponding vibrational modes were obtained by a singular value diagonalization procedure as implemented in the post-processing toolkit TAMKIN.42 The zero-point corrections and free energy contributions to the reaction energies were determined from the PHVA-based partition functions to determine the pressure and temperature dependence of the Gibbs free energies.
2.4.1.1 Models. The xNi@SnO2(110) surface slabs (where x = 0, 1, 2) were constructed with 4 SnO2 layers, the same number of layers used in previous studies.43 We opted for the SnO2(110) surface since it was known for its thermodynamic stability and has garnered significant interest in experimental and theoretical investigations.44–48 The periodic slab models considered in the study were referred to as SnO2, Ni@SnO2, and 2Ni@SnO2 (Fig. 1). A vacuum of 15 Å was employed in the z-direction to avoid interactions between periodic images. The Ni@SnO2 systems were constructed by replacing surface Sn atoms with Ni. To understand the effect of H adsorption on the studied systems during the CO2 reduction at the cathode, the 2-fold coordinated O atoms (denoted as Sn–O–Sn) on the surface were hydroxylated, forming Sn–(OH)–Sn. A subsequent H addition to Sn–(OH)–Sn results in adsorbed water bonded to two Sn-sites, i.e., Sn–(OH2)–Sn. Additionally, larger supercell models were constructed to shed light onto the interface between NiO and SnO2 (see section 15 in ESI for details†).
 | ||
| Fig. 1 Top and side view of chosen SnO2(110) model systems for (a) bare SnO2, (b) single Ni-doped SnO2 (Ni@SnO2), and (c) double Ni-doped SnO2 (2Ni@SnO2) systems. | ||
The adsorption of n hydrogens on the SnO2 surfaces can be given by:
| n(H+ + e−) + * → nH* | (3) |
The change in Gibbs free energy upon n hydrogenation reactions with respect to the pristine SnO2(110) surface models (Fig. 1) is given by;
 | (4) |
However, to calculate and construct the Pourbaix diagrams, the change in Gibbs free energy corresponding to a sequential hydrogenation reaction is calculated as a function of electrode potential and pH as follows;
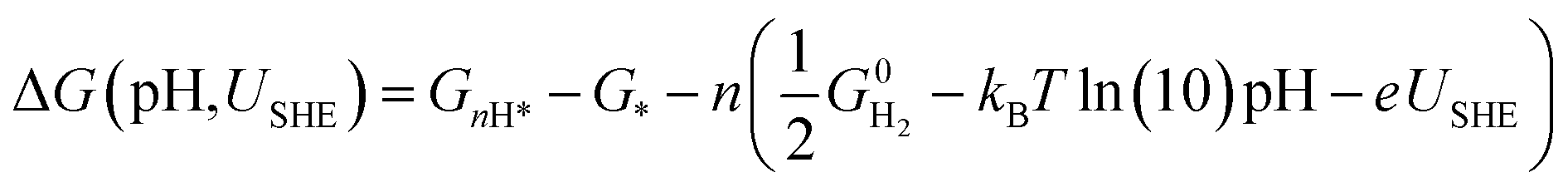 | (5) |
| GnH* = EDFT + EZPVE + ΔEvib(T) − TSvib ≈ EDFT + Fvib(T) | (6) |
 , TS, were obtained from the ideal gas approximations from a previous report (Table S4†). The Gibbs free energy for the gas molecules at 25 °C (i.e., 298 K) and 1 atm is given as:
, TS, were obtained from the ideal gas approximations from a previous report (Table S4†). The Gibbs free energy for the gas molecules at 25 °C (i.e., 298 K) and 1 atm is given as: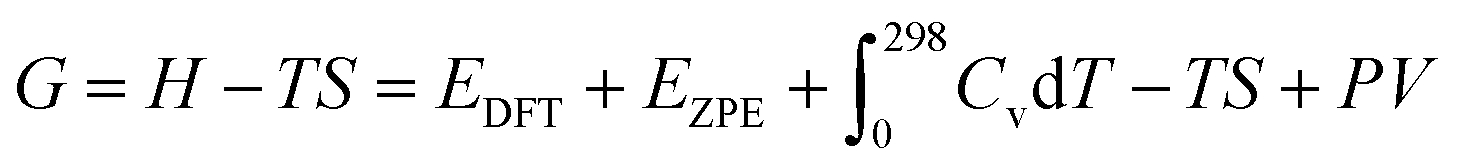 | (7) |
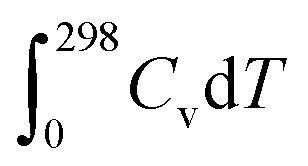 and S denote the electronic energy, zero-point vibrational energy, heat capacity, and entropy, respectively.
and S denote the electronic energy, zero-point vibrational energy, heat capacity, and entropy, respectively.
3. Results and discussions
3.1 Synthesis of NiO:SnO2 nanofibers
Electrospinning is used to produce NiO:SnO2 NFs. NFs are synthesized in various compositions, changing the molar ratios between Ni and Sn, i.e., 75%:25% (NiOSnO25NF), 50%:50% (NiOSnO50NF), and 25%:75% (NiOSnO75NF). Similarly, NiO and SnO2 single compositions have been electrospun. However, NiO and SnO2 do not lead to NFs under similar electrospinning conditions. The latter includes NiO and SnO2 formulations, which lose the NF shape after annealing. For instance, a single electrospun composition of NiO and SnO2 NF can lead to the formation of nanoparticles after annealing (Fig. S2†). Like NiO and SnO2, NiOSnO25NF led to nanoparticle formation after annealing (Fig. S3†). The previous results indicate that the spun NFs could not retain the fiber shape during annealing due to the unstable formation of metal oxide, leading to NF-shape coalescence. Similar results have been observed for NiO systems, indicating that our NiCl2-PVP formulation leads to nanoparticle (NP) upon annealing.12 This can also be the case for SnO2, which could occupy additional metal oxide agents to maintain the NF shape.50 Due to the lack of NF shape, NiOSnO25NF has not been analyzed further. However, NiO and SnO2 are still used as controls, along with NiOSnO50NP and NiOSnO75NP, which both lack NF shape.
NiOSnO50NF and NiOSnO75NF are inspected with STEM-ADF and STEM-EDX, as shown in Fig. 2. For NiOSnO50NF, Fig. 2a–d displays an NF-like shape with an NF diameter of 209 ± 45 nm. The NiOSnO50NF comprises ∼50 nm nanocrystals and multiple ∼10 nm nanocrystals or smaller in diameter. The inner structure corresponds to a polycrystalline arrangement of nanoparticles with some gaps between them. Interestingly, in some cases, a row of nanocrystals forms a string along the NF with higher contrast (e.g., Fig. 2d and e, yellow arrows). From STEM-EDX mapping Ni, Sn, and O (Fig. 2e′ and e′′′), we have identified that these nanoparticle rows primarily comprise oxidized Sn. The contrast over the rows is attributed to densified oxidized Sn. In addition, within the NiOSnO50NF, oxidized Ni has been found to a lesser extent over the NF body; however, larger NiO particles, typically ∼200 nm in diameter, decorate the NF morphology. A closer look at the interface between the NiO:SnO2 nanocrystallites is discussed in Fig. S4.† For NiOSnO75NF in Fig. 2f–i, the NF is more compact than in NiOSnO50NF, where nanocrystals are relatively smaller with a 35 nm diameter or less (e.g., Fig. 2i, yellow arrow). The presence of nanocrystals along the NF shape forming bright strings is less evident than in NiOSnO50NF. Moreover, NiO forms patches over the NF morphology (e.g., Fig. 2j, yellow arrow). The STEM-EDX mapping for Ni, Sn, and O in Fig. 2j′ and j′′′ supports these results (see yellow arrows). The presence of Ni within the NF morphology is also verified by the STEM-EDX line scan (Fig. 2i and k, see dashed yellow line). The STEM-EDX line scan demonstrates that the Ni remains within the NF body but to a lower extent when compared to Sn (Fig. 2k). The NiOSnO75NF chemical species have also been investigated with EELS after CO2 electroreduction (Fig. 2l and m). In Fig. 2l, the EELS signals for NiOSnO75NF after 22 h of CO2 electroreduction (NiOSnO75NF22h, black line) show that the oxidized tin resembles SnO2 (blue line). However, we should not disregard the possibility of partially reduced SnO2 since the NiOSnO75NF22h has some similarities to SnO (red line). It is then suggested that NiOSnO75NF22h contains multiple oxidized Sn species (SnOx). In Fig. 2m, the Ni L23 edge is used to obtain the L3/L2 ratio and determine the chemical environment of Ni-species in NiOSnO75NF after 2 h of CO2 electroreduction (NiOSnO75NF2h) and NiOSnO75NF22h. The EELS L3/L2 ratio shows that for NiOSnO75NF2h, more points have a wider L3/L2 ratio,12 indicating a higher fraction of Ni2+ and Ni3+ species than NiOSnO75NF22h, which reveals a narrower L3/L2 ratio, close to Ni3+ species. The results denote that uncoordinated Ni3+ species, e.g., defects, are formed more due to longer electrolysis time attributed to the loss of NF shape after CO2 electrolysis.12
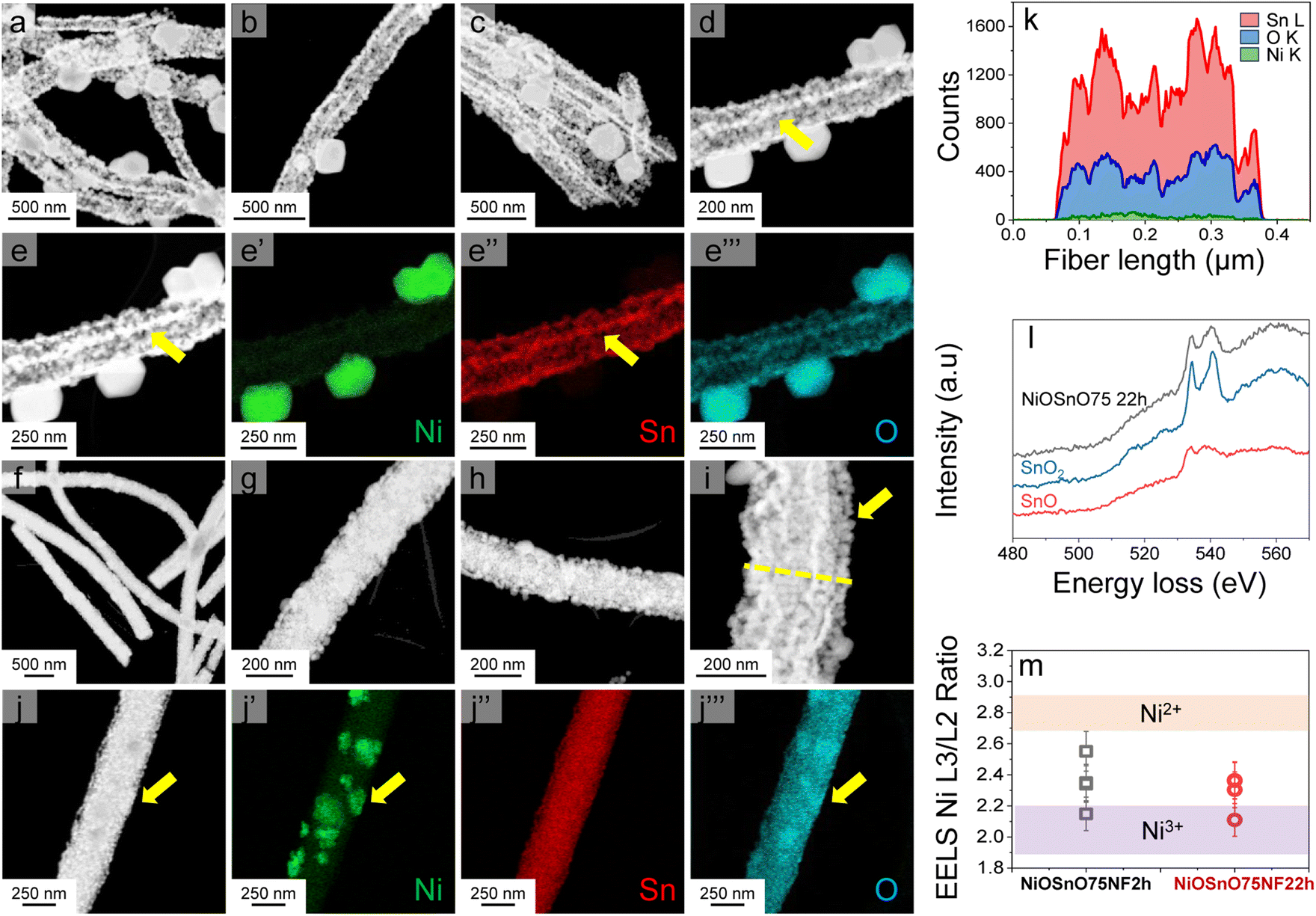 | ||
| Fig. 2 STEM-ADF images (a–e) and STEM-EDX maps (e′ and e′′′) for NiOSnO50NF. STEM-ADF images (f–j) and STEM-EDX maps (j′ and j′′′) for NiOSnO75NF. The STEM-EDX line scan for NiOSnO75NF22h is shown in (k). EELS measurements NiOSnO75NF22h are shown in (l) and (m) for Sn and Ni, respectively. SnO2 (blue line) and SnO (red line) controls are presented in (l). In (m), the L3/L2 ratio of Ni L23 edge EELS for NiOSnO75NF2h and NiOSnO75NF22h are carried out over different surface areas of the crystallites. | ||
From the STEM-EDX results in Fig. 2, we can verify the presence of metal oxides. To confirm the oxide type, we look at the structural characteristics of NiOSnO50NF and NiOSnO75NF using XRD (Fig. 3), with NiO and SnO2 as controls. First, we describe the diffractograms of NiO and SnO2. NiO has several diffraction peaks at 2θ = 37.2°, 43.3°, 62.9°, 75.4°, and 79.3°, which correspond to (111), (200), (220), (311), (222) crystallographic planes from NiO (JCPDS 65-6920), respectively.51 SnO2 also shows diffraction peaks at 2θ = 26.5°, 33.9°, 37.9°, 51.8°, 54.6°, 57.8°, 61.8°, 64.7°, 66.0°, 71.3°, and 78.7°, corresponding to (110), (101), (200), (211), (220), (002), (310), (112), (301), (220), and (321) crystallographic planes from SnO2 respectively (JCPDS No. 41-1445).52–54 Comparing NiO and SnO2 with NiOSnO50NF and NiOSnO75NF, we observed that the crystallographic phases correspond to NiO and SnO2. No changes in the diffraction peak positions for NiOSnO75NP used as control are observed. No evidence of a difference in crystallographic phase has been found, indicating that NiO and SnO2 prevail in separate phases within the NF (Fig. 2 and 3).
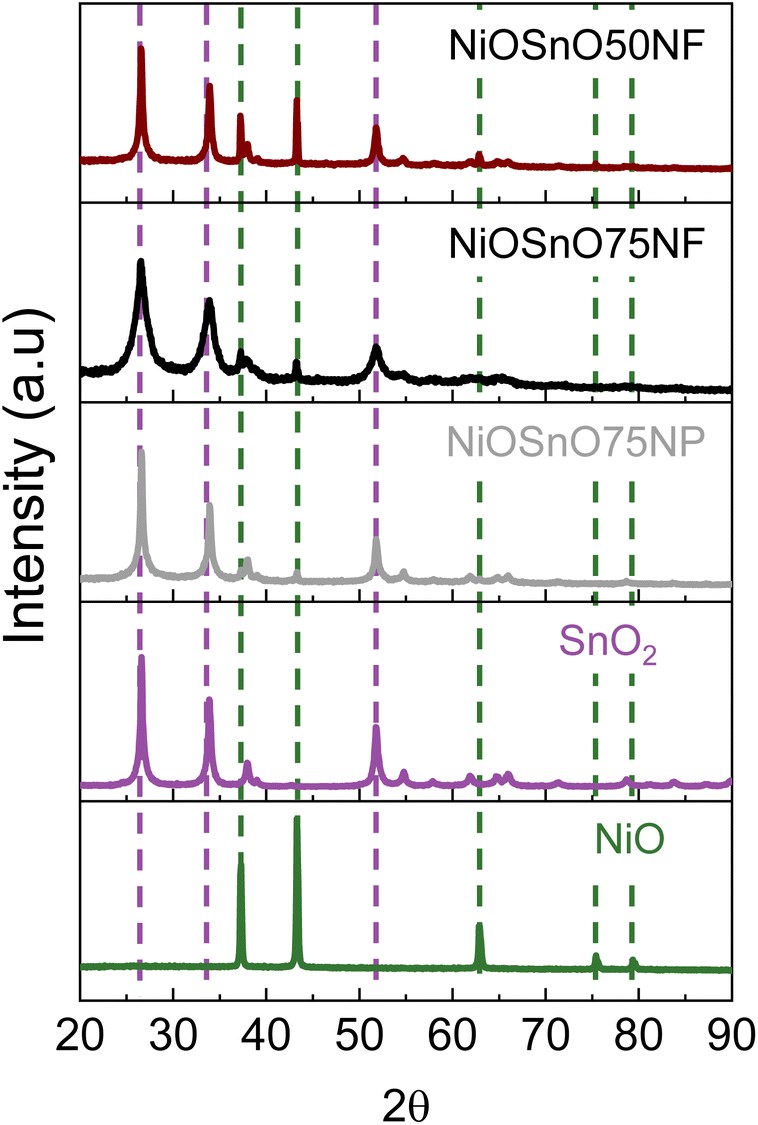 | ||
| Fig. 3 XRD diffraction patterns for NiOSnO50NF and NiOSnO75NF. XRD diffraction patterns of NiOSnO75NP, SnO2, and NiO are included for controls. | ||
The chemical environment is investigated with XPS to determine the type of species present over NiOSnO50NF and NiOSnO75NF. The XPS core spectra of Ni 2p, Sn 3d, O 1s, and Cl 2p for NiOSnO50NF and NiOSnO75NF are presented in Fig. 4. NiO and SnO2 controls are used for comparison. In Fig. 4a, Ni 2p comprises Ni 2p3/2 and Ni 2p1/2. The Ni 2p3/2 peak can be fitted into two components corresponding to Ni2+ and Ni3+ species, labeled in red. Ni2+ and Ni3+ peaks are located at 853.8 eV and 855.6 eV.55–57 The presence of Ni3+ can be ascribed to the uncoordinated species, like defects.12 The Ni3+/Ni2+ ratio for NiOSnO50NF is estimated to be close to 2.1, while NiOSnO75NF is around 8.5, indicating that Ni3+ is more prominent in NiOSnO75NF. Similar Ni3+/Ni2+ has been observed for NiOSnO50NP and NiOSnO75NP compared to the NF counterpart. Furthermore, a behavior opposite to NiOSnO50NF and NiOSnO75NF has been observed for NiO, in which the Ni3+/Ni2+ ratio is lower, ca. 1.3. Ni2+ and Ni3+ species have also been identified with EELS (Fig. 2), supporting our finding. The results demonstrate that the presence of Sn increases the amount of Ni3+ species. Next, we analyze the results for Sn 3d. In Fig. 4b, NiOSnO50NF and NiOSnO75NF show binding energy (BE) for Sn 3d5/2 around 486.3–486.5 eV, assigned to Sn4+ in SnO2.58–60 Compared to SnO2 control with a BE around 486.9 eV associated with Sn4+,61 a shift to lower BE has been found for NiOSnO50NF and NiOSnO75NF. This shift can be related to reduced Sn species (e.g., SnOx), similar to EELS, as shown in Fig. 2l.
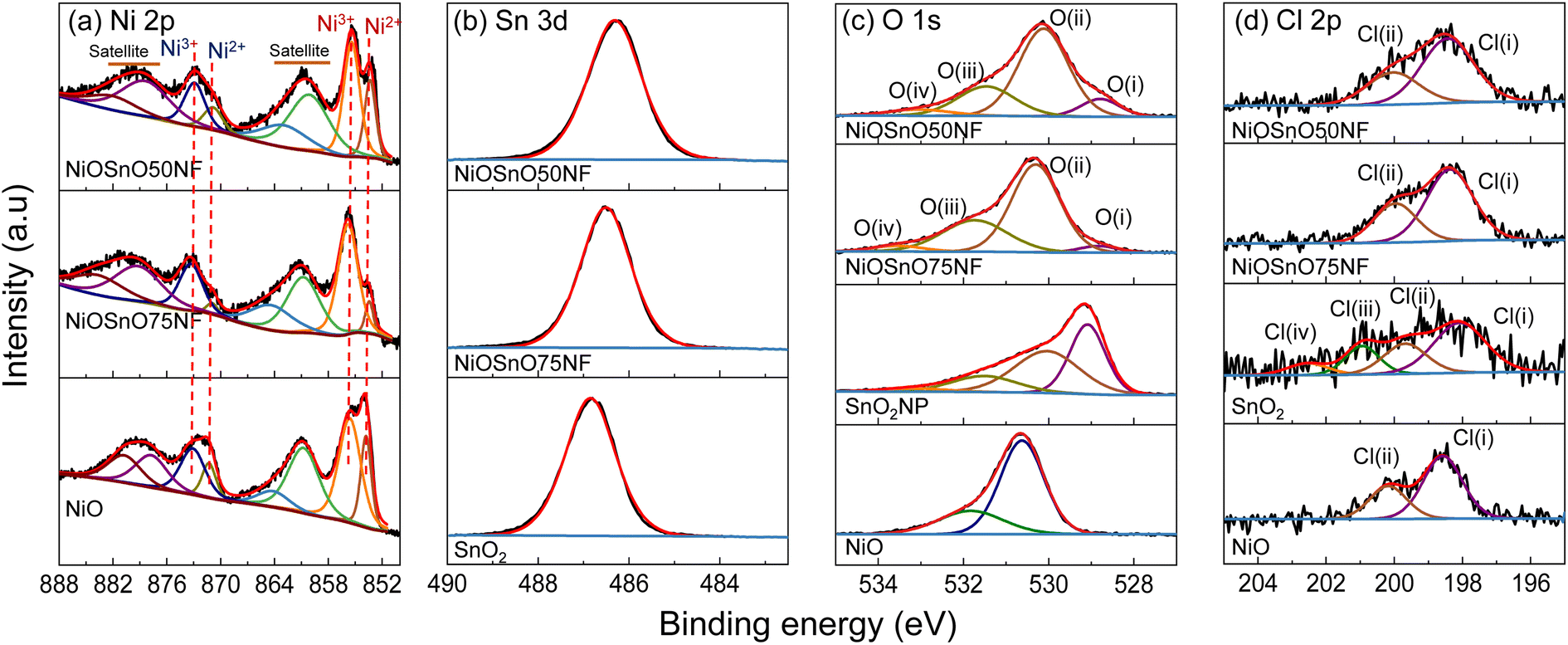 | ||
| Fig. 4 (a) Ni 2p, (b) Sn 3d, (c) O 1s, and (d) Cl 2p XPS core spectra for NiOSnO50NF, NiOSnO75NF, NiO, and SnO2. | ||
The XPS core spectra of O 1s and Cl 2p and the fitting curves have also been analyzed (Fig. 4c and d). O 1s core spectra for NiOSnO50NF and NiOSnO75NF show four different peaks resulting from NiO and SnO2 formation within the NF body. The O(I) peak (BE 528.8–529.1 eV) is attributed to oxygen in NiO.55,56 The O(II) peak (BE 530.0–530.3 eV) is attributed to mixed oxygen species from NiO and SnO2.57,62,63 The O(III) peak (BE 531.3–531.7 eV) is attributed to surface OH groups.56,63,64 The O(IV) peak (BE 532.9–533.5 eV) is attributed to chemisorbed water.63,64 Similar results have been obtained for NiO. The O 1s XPS spectrum of NiO shows BE at 529.1 eV, 530.0 eV, 531.5 eV, and 533.3 eV, attributed to oxygen in NiO,55,56 surface O2− species,65,66 surface OH groups, possibly from uncoordinated Ni3+ species present in NiO.12 As for SnO2, the O 1s XPS spectrum shows BE at 530.6 eV and 531.8 eV, corresponding to oxygen in SnO2 (ref. 67–69) and OH groups.68,69 Cl 2p core spectra for NiOSnO50NF, NiOSnO75NF, NiO, and SnO2 show several peaks labeled as Cl(I), Cl(II), Cl(III), and Cl(IV). Cl(I) between 198.6–198.1 eV and Cl(II) between 199.7–200.2 eV are assigned to inorganic chlorine species.70–73 NiO reveals two additional peaks at Cl(III) at 200.9 eV and Cl(IV) at 202.5 eV, both corresponding to Cl− from different decomposed chemical species of chlorine salt.12,74 The concentration of Cl− for all the samples remains similar, with an average atomic percentage of 1% ± 0.2.
3.2 CO2 electroreduction
The functionality of NiOSnO50NF and NiOSnO75NF for CO2RR is assessed in Fig. 5. To elucidate the effect of NF functionality, NiOSnO50NF and NiOSnO75NF are compared to NiOSnO50NP and NiOSnO75NP, which lack the NF shape. Additionally, the results are contrasted with NiO and SnO2. The NiOSnO50NF, NiOSnO75NF, NiOSnO50NP, NiOSnO75NP, NiO, and SnO2 comparatives are conducted in a flow cell to demonstrate the importance of the synthesized multimetal oxide NFs. Finally, in situ DEMS experiments are discussed to shed light on the product pathways.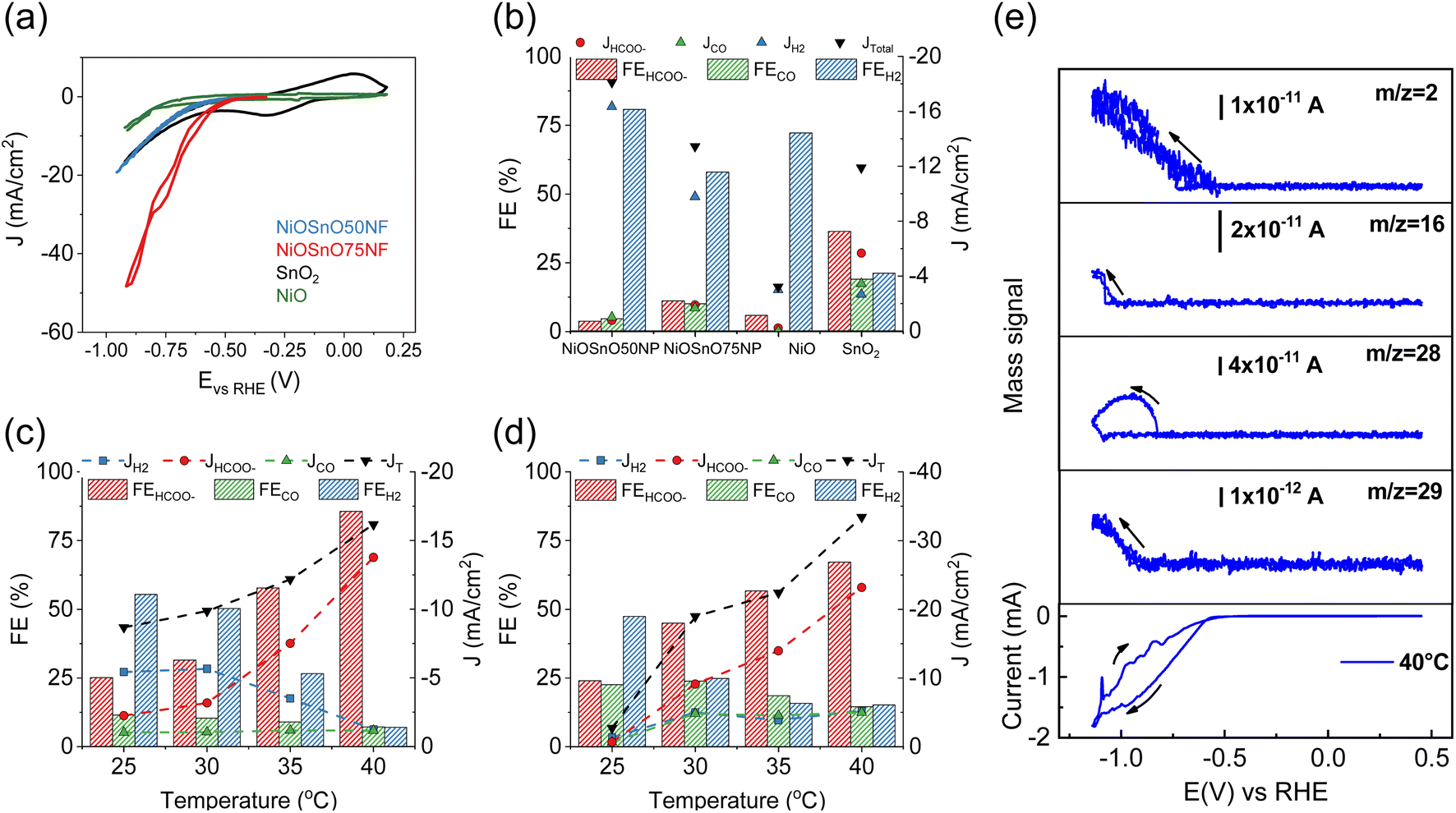 | ||
| Fig. 5 (a) CV characteristics of NiOSnO50NF, NiOSnO75NF, NiO, and SnO2 at 25 °C. FEs and partial current densities for (b) NiOSnO50NP, NiOSnO75NP, NiO, and SnO2 at 40 °C, along with (c) NiOSnO50NF and (d) NiOSnO75NF at −0.85 V vs. RHE for 2 h over various temperatures, i.e., 25, 30, 35, 40 °C. (e) CV characteristic for NiOSnO75NF at 40 °C. In situ DEMS mass signals recorded at 40 °C are shown as a function of the applied potential (1 mV s−1) for m/z = 2, m/z = 16, m/z = 28, and m/z = 29. In all cases, the pH of the bulk electrolyte remained at 7.9. | ||
The experiments start with preparing the CO2RR GDEs by spraying with ink containing MWCNTs, catalyst, and Nafion. The dried GDE is placed in the flow electrochemical cell using a three-electrode configuration containing a solution of 0.5 M KHCO3 as an electrolyte. The CV in the presence of CO2 shows the highest current density (J, mA cm−2) for the NiOSnO75NF, followed by NiOSnO50NF and SnO2, with NiO showing the lowest in J (Fig. 5a). At 40 °C and −0.85 V vs. RHE, the product distribution for NiOSnO50NP, NiOSnO75NP, NiO, and SnO2 are also evaluated (Fig. 5b). It should be noted that three different potentials have been used, i.e., −0.75, −0.85, and −0.95 V vs. RHE, and −0.85 V is selected since it produced the highest FEHCOO− and JHCOO−. Compared to Fig. 5c and d, the results highlight the advantage of the synthesized NFs.
In Fig. 5b, FE for NiOSnO50NP and NiOSnO75NP at 40 °C displays the product distribution, where HCOO−, H2, and CO are formed during CO2 electroreduction. The FE for HCOO−, H2, and CO for NiOSnO50NP are FEHCOO− = 3.8%, FEH2 = 80.7% and FECO = 4.7% with partial J values of JHCOO− = −0.7 mA cm−2, JH2 = −14.7 mA cm−2 and JCO of = −0.9 mA cm−2. Similar product distribution is observed for NiOSnO75NP with FEHCOO− = 11.2%, FEH2 = 58.0%, and FECO = 10.1% have been found with JHCOO− = −1.7 mA cm−2, JH2 = −8.8 mA cm−2 and JCO = −1.5 mA cm−2. For NiO, a prominent generation of H2 is observed with FEH2 = 72.2% and JH2 = −2.7 mA cm−2. For the same electrocatalyst, FEHCOO− = 6% and JHCOO− = −0.2 mA cm−2, no CO has been detected. It could be argued that to increase HCOO− formation, NiO should be selectively shaped as octahedra and not as mixed particle shapes (Fig. 2).12 SnO2 FE for HCOO−, H2, and CO are 37%, 21%, and 19% with JHCOO− = −5.1 mA cm−2, JH2 = −2.4 mA cm−2 and JCO of = −3.1 mA cm−2. The results demonstrate the importance of synergistic effects between NiO and SnO2, particularly for the NF morphology (Fig. 5c and d).
The effect of the temperature (25, 30, 35, and 40 °C) during CO2R for NiOSnO50NF and NiOSnO75NF is evaluated in Fig. 5c and d. For NiOSnO50NF (Fig. 5c), the HCOO− has a gradual increase in selectivity as the temperature increases, starting from FEHCOO− = 25.2% at 25 °C and reaching a maximum of FEHCOO− = 85.7% at 40 °C. At 40 °C, the highest JHCOO− is observed (−13.8 mA cm−2). For FEH2, we observe a gradual decrease in H2 generation as a function of temperature, starting from FEH2 = 55.4% at 25 °C and reaching a minimum of 7% at 40 °C. The JH2 value for NiOSnO50NF follows a similar trend, from JH2 = −5 mA cm−2 at 25 °C, reaching a minimum JH2 = −1.2 mA cm−2 at 40 °C. FECO does not drastically decrease with increasing temperature, maintaining JCO around −1 mA cm−2 across the various conditions, with a FECO = 7.2% at 40 °C. Overall, in NiOSnO50NF, FEHCOO− is favored as temperature increases while maintaining FECO constant and suppressing FEH2. NiOSnO50NF and NiOSnO75NF increase the total current densities (JT) with temperature.
Next, the electrochemical performance of NiOSnO75NF during CO2 electroreduction is discussed (Fig. 5d). For HCOO−, a gradual increase in selectivity, with FEHCOO− = 25% at 25 °C and FEHCOO− = 70% at 40 °C, is observed. At 40 °C, the highest JHCOO− is observed with −26.1 mA cm−2. The FEH2 in NiOSnO75NF also follows a gradual decrease with increasing temperature, with FEH2 = 47.4% at 25 °C and 15.2% at 40 °C. For H2, J remains at JH2 = −5 mA cm−2 from 30 °C to 40 °C. Lastly, FECO presents a gradual decrease with 22.6% at 25 °C and 14.5% at 40 °C with JCO = −5 mA cm−2 for temperatures similar to or higher than 30 °C. Although FEHCOO− for NiOSnO75NF remained 15% lower than for NiOSnO50NF at 40 °C, NiOSnO75NF has a 2-fold increase in JHCOO−. This 2-fold increase can be attributed to an increase in the electrochemical surface area (ECSA) as the obtained double-capacitance is higher for NiOSnO75NF (4.68 × 10−4 mF cm−2) than NiOSnO50NF (3.78 × 10−4 mF cm−2). Furthermore, the results at 40 °C for NiOSnO75NF are substantiated with EIS. EIS reveals less charge transfer resistance and an increased affinity in the presence of CO2 for NiOSnO75NF (Fig. S5 and Table S5†). Likewise, there is no significant effect when looking at the Tafel slopes in the presence of CO2 (Fig. S6 and Table S6†). The Tafel slopes are somehow similar. Hence, the results indicate the existence of similar rate-determining steps in the presence of CO2 for temperatures close to 40 °C. Slight variations in the Tafel slopes are observed for temperatures close to 45 °C, suggesting a different rate-determining step associated with other processes, e.g., H2 competition. The results of the chronoamperometry from Fig. 5c and d are shown in Fig. S7.†
In short, a trade-off between selectivity and product yield should be found when assessing catalyst performance. However, NiOSnO50NF and NiOSnO75NF resulted in similar trends, elucidating temperature effects, which could be reasonably associated with favored reaction kinetics at high temperatures.19–21 Such effects have not been observed during CO2 electroreduction using synergistic catalysts shaped as NFs. Hence, the synergistic effects require an understanding of the reaction product to the fullest. Therefore, an in situ DEMS is assessed to generate insight into the reaction product pathway by detecting the formic acid (HCOOH) mass fragments for NiOSnO75NF, as it yielded the highest HCOO− production at 40 °C (Fig. 5d). Mass spectrometric signals corresponding to H2 (m/z = 2), methane (CH4, m/z = 16), CO (m/z = 28), and HCO− (m/z = 29) from HCOOH,75 and CV are recorded simultaneously (Fig. 5e). It should be noted that mass m/z = 29 is selected as it is the most pronounced for HCOOH, and in the absence of CO2, no CO2 reaction products are observed (Fig. S8†). Additionally, to corroborate the detection of HCOOH, formic acid is added to the electrolyte, and the mass signals associated with this organic compound are shown in Fig. S9.† Overall, the distribution of DEMS products confirmed our flow cell observations in Fig. 5d, except for CH4, which could be expected to be below the detection limit of our gas chromatograph but captured by DEMS. It should be noted that other factors that might change reaction product selectivity to CH4 can be related to the DEMS cell configuration as it can impact pH, generating some gradients.75
Lastly, we discuss the effect of uncoordinated Ni species found in NiOSnO NFs, which could have enabled the formation of HCOO−.12 We could expect NiO species to enhance hydrogenation over SnO2, which is more likely to be as partially reduced SnO2, i.e., SnOx, after 2 or 22 h CO2 electrolysis. Although at 2 h, the NF shape drastically changed its morphology (Fig. S10†), Ni and Sn species remained present even after 22 h of CO2 electrolysis (Fig. 2l and m). Furthermore, the Ni3+/Ni2+ ratios for NFs connect with the improved FEHCOO− at 40 °C. However, we should not disregard the Ni3+/Ni2+ ratio in NPs. For example, the Ni3+/Ni2+ ratio for NiOSnO75NP is 7.5, close to NiOSnO75NF (i.e., 8.5). The benefit of structuring becomes evident when comparing the Raman spectra in Fig. S11† for NiOSnO75NP and NiOSnO75NF. NiOSnO75NP contains more organic species than NiOSnO75NF, compromising the CO2 reduction reaction activity (Fig. 2b and d).
The results highlight the advantage of the NF morphology as carbon is removed from the NiOSnO precursor due to the open fibrous structure. Similar effects have been observed for polymer-derived metal oxides, such as 3D-printed structures where carbon remnants are found.76 Hence, the carbon remnant could act as a blocking layer during CO2RR, affecting NiOSnO75NP selectivity. This hypothesis is well aligned with NiOSnO75NF loaded with t-octylphenoxypolyethoxyethanol (Triton ×100) used as a surfactant, acting as a carbon-blocking agent without compromising the NF morphology after annealing (Fig. S12†). The electrochemical results of NiOSnO75NF loaded with surfactant demonstrate a change in the product distribution with low HCOO− selectivity over the explored temperature ranges (Fig. S13†). The results are substantiated further by ECSA. ECSA result for NiOSnO75NF is 4.68 × 10−4 mF cm−2, while in the presence of a surfactant or NiOSnO75NP, it decreases to 8.39 × 10−5 mF cm−2. Now that we have identified the importance of blocking agent-free catalysts, we propose a mechanism for the NiOSnONF using the most significant products (H2 and HCOO−), as shown in Fig. 5c and d.
3.3 CO2RR mechanism
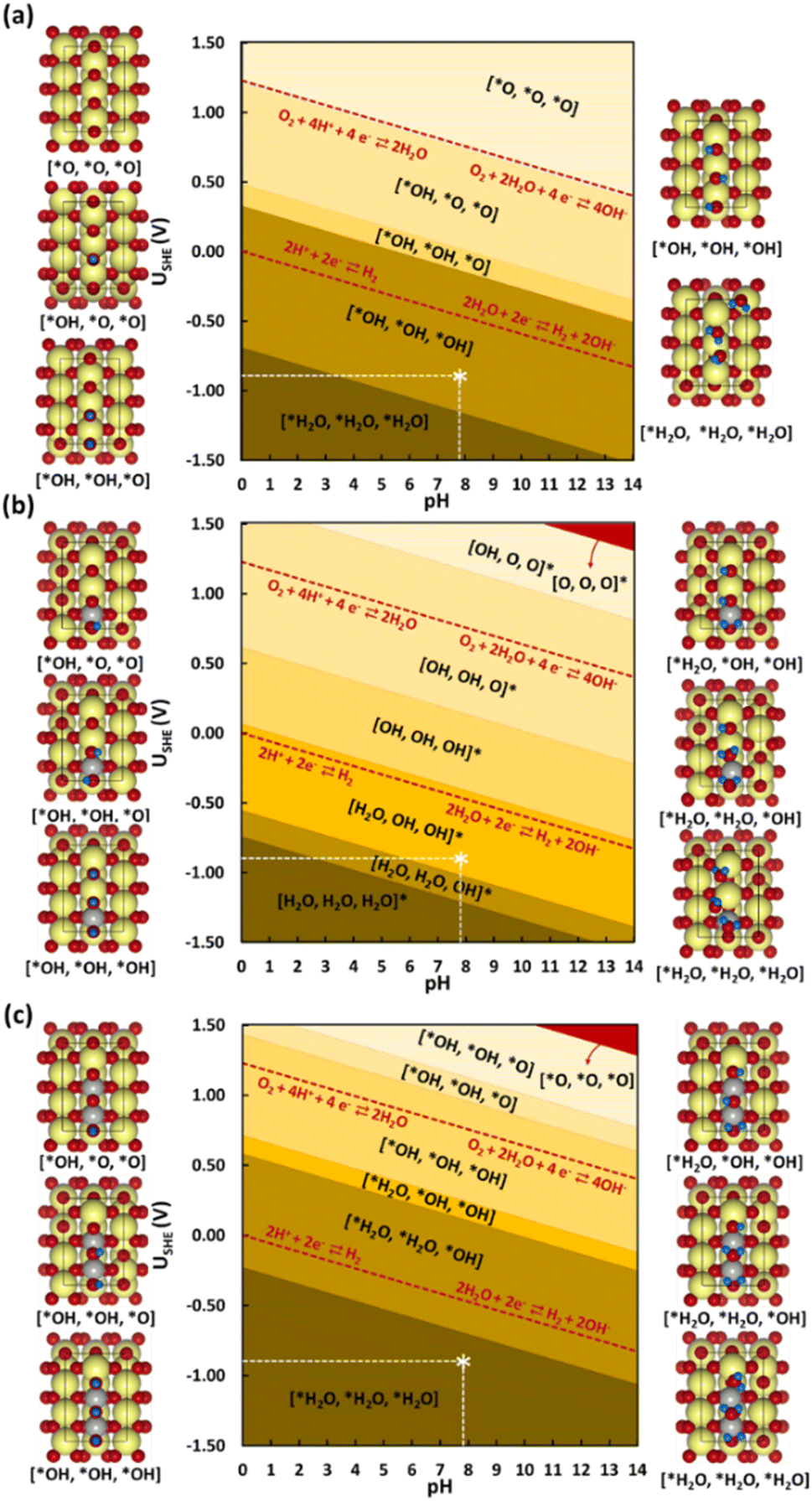 | ||
| Fig. 6 Surface Pourbaix diagrams for the studied (110) surface models (a) SnO2, (b) Ni@SnO2, and (c) 2Ni@SnO2. Surfaces with no H adsorbed are colored in red. Color codes: Sn (yellow), O (red), H (blue), and Ni (grey). The white star * in the diagram corresponds to typical experimental conditions used (USHE = 0.85 V, pH = 7.9). | ||
Next, the influence of Ni-doping of the SnO2(110) model system on the Pourbaix diagram is depicted in Fig. 6b and c. Unlike for SnO2, all H-covered terminations from 0.33 to 2.00 ML H* are present in the Pourbaix diagram for Ni-doped SnO2 models at potentials between −1.5 and 1.5 V. In the case of a single Ni-doped SnO2(110) system, the surface with no H adsorbed is stable only at higher potentials (>2.14 V) and pH (>13). Between 1.5 and 0.06 V (at pH = 0), the two-fold bridging O* atoms (Sn–O–Sn) tend to get fully hydrogenated. At lower potentials (<0.06 V), the hydrogenated O atoms can adsorb H to form adsorbed water molecules. Below −0.74 V versus RHE, the surface is completely reduced with all two-fold bridging O atoms (Sn–O–Sn) forming water molecules. As a characteristic of H adsorption, on moving to a higher pH, the stable H terminations occur at lower potentials in the SPD due to the shift of −59 meV per pH unit. Finally, another Sn atom is replaced with Ni to understand the effect of Ni concentration on the SPDs. For the 2Ni@SnO2 surface model, all H terminations from 0.33 to 2.00 ML H* appeared in the Pourbaix diagram. The surface with no hydrogen appears only above 2.10 V and high pH. The termination with 1.00 ML coverage of H is favorable under OER conditions, while the surface with 1.66 ML H* coverage (2× H2O*) appears at the HER limit. Interestingly, at the experimental conditions of U = −0.85 V and pH = 7.9 (Fig. 5), the surface with 2.00 ML (3× H2O*) is likely to be thermodynamically preferred. This also highlights that in CO2RR conditions, the surface of the Ni-doped SnO2 is partially reduced (e.g., SnOx, Fig. 2 and 4), which could further tailor the electrocatalytic activity at the surface.80,81 Overall, the SPDs emphasize the reduction of the SnO2-based catalyst surface, specifically the co-adsorption of water molecules at potentials of experimental interest.
| CO2 + * → *CO2 | (8) |
| *CO2 + H+ + e− → *HCOO | (9) |
| *HCOO + H+ + e− → HCOOH + * | (10) |
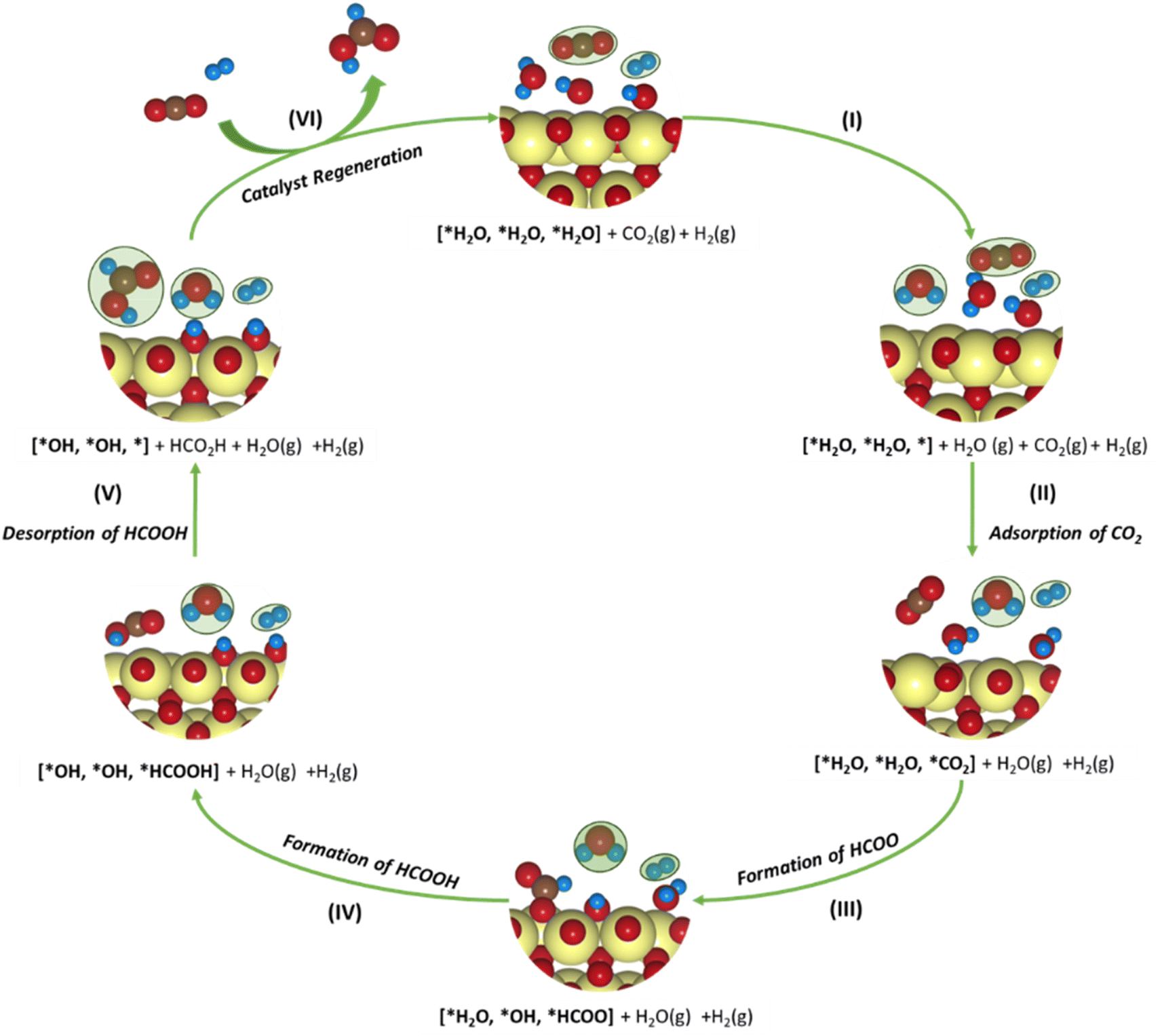 | ||
| Fig. 7 Schematic representation of the plausible CO2 reduction mechanism for the SnO2-based model systems. The adsorbates in gaseous states are marked in green. Color codes: Sn (yellow), C (brown), O (red), H (blue). | ||
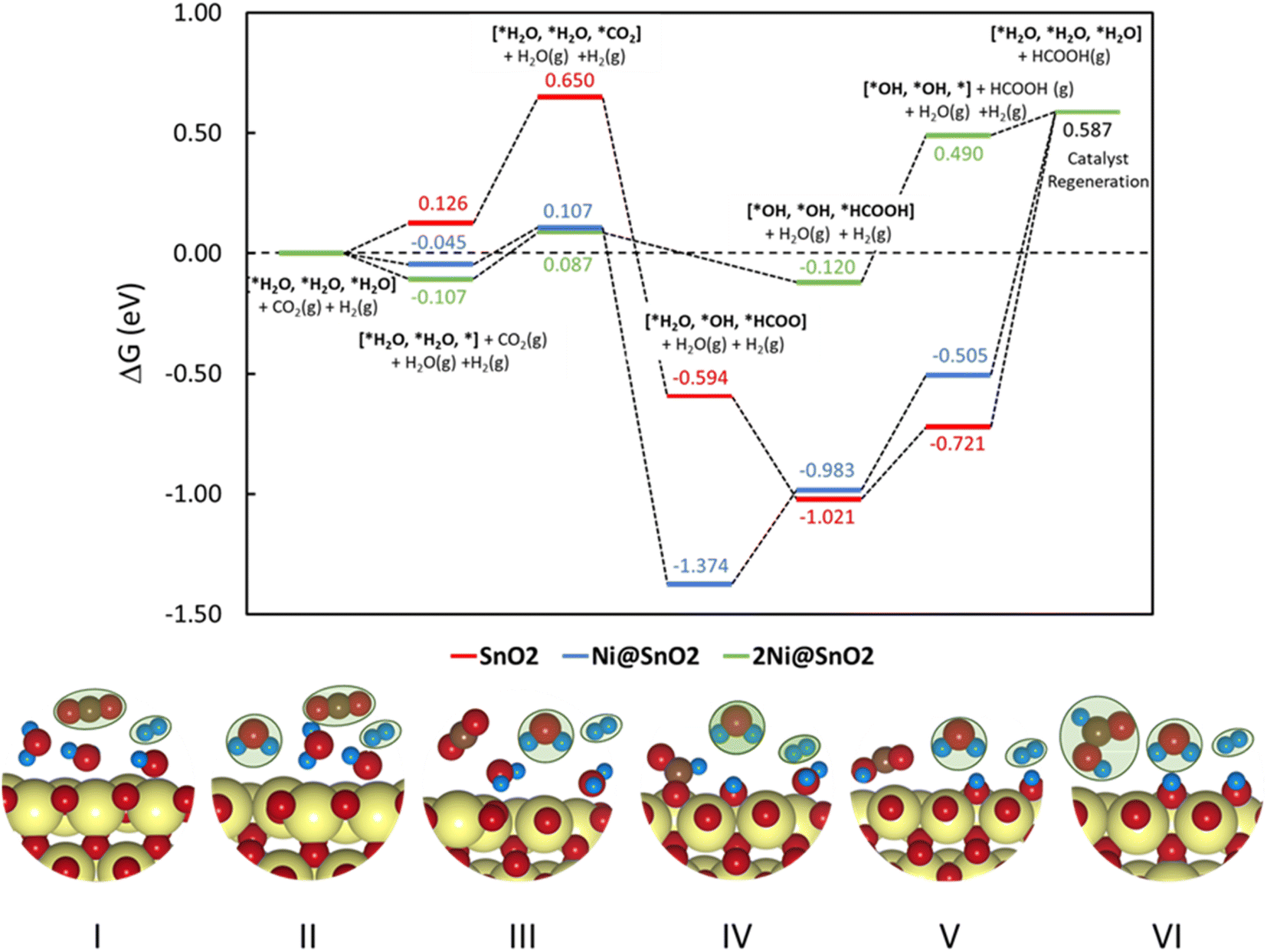 | ||
| Fig. 8 Free energy diagram for CO2 reduction to HCOOH over SnO2(110), Ni@SnO2(110), and 2Ni@SnO2(110) electrocatalyst models. (Bottom panel) The binding modes of the adsorbates on the catalyst at different reaction states and the adsorbates in the gaseous state are marked in green. Color codes: Sn (yellow), C (brown), O (red), H (blue). | ||
To understand the thermodynamic feasibility of the reaction pathway proposed, we calculated the Gibbs free energy profile for each model system. The mechanism starts with the desorption of one of the H2O molecules on 2Ni@SnO2, leaving an empty site for CO2 adsorption [*H2O, *H2O, *] (Fig. 8). From Fig. 8, the process of water desorption is exothermic and exergonic for the Ni-doped SnO2 (110) systems compared to pure SnO2. However, the subsequent CO2 adsorption on the empty site is endergonic (Fig. 8), with positive reaction-free energies for SnO2 (0.524 eV), Ni@SnO2 (0.152 eV), and 2Ni@SnO2 (0.194 eV) model systems which becomes more feasible if SnO2 is Ni doped. Further, upon abstracting an H from a co-adsorbed water molecule, *CO2 can form *HCOO. For Ni@SnO2, the *HCOO intermediate is thermodynamically more stable (−1.374 eV, Fig. 8) compared to pure SnO2 (−0.594 eV, Fig. 8). Interestingly, for 2Ni@SnO2, the CO2 molecule directly tends to form a stable HCOOH, surpassing the *HCOO intermediate state. The formation of *HCOOH from *HCOO and neighboring *OH2, is exergonic for SnO2 with a reaction-free energy of −1.021 eV, whereas for the Ni@SnO2 system, this process is endergonic (+0.391 eV) energy. For 2Ni@SnO2 the *HCOO intermediate is found to be protonated directly and form *HCOOH, an overall exergonic process with a reaction-free energy of −0.227 eV. From *HCOOH, the desorption of HCOOH is endergonic and requires 0.300 eV, 0.478 eV, and 0.610 eV for SnO2, Ni@SnO2, and 2Ni@SnO2, respectively (Fig. 8). Therefore, it is clear that under certain reaction conditions, the desorption of HCOOH can become the rate-limiting step in the reaction. Especially, for the 2Ni@SnO2 system, with the highest adsorption free energy barrier for *HCOOH desorption, temperature facilitates the desorption process given the decreasing free energy differences from 25 °C to 40 °C (Fig. 9). These findings agree with the temperature-dependent faradaic efficiencies and partial current densities of CO2 electroreduction (Fig. 5c and d). Finally, after the production of HCOOH, the catalyst needs to regenerate to continue with the catalytic cycle. The catalyst regeneration with two H+/e− pairs is endergonic with the ΔG increasing in the order: 2Ni@SnO2 (0.097 eV) < Ni@SnO2 (1.092 eV) < SnO2 (1.308 eV), however, this regeneration can happen electrochemically, and under cathodic potentials of −0.90 V vs. RHE this regeneration process will be spontaneous. The regeneration process is more favorable for the surfaces with more Ni species (e.g., Ni2+ and Ni3+), which indicates the influence of Ni doping on the catalytic activity of SnO2.
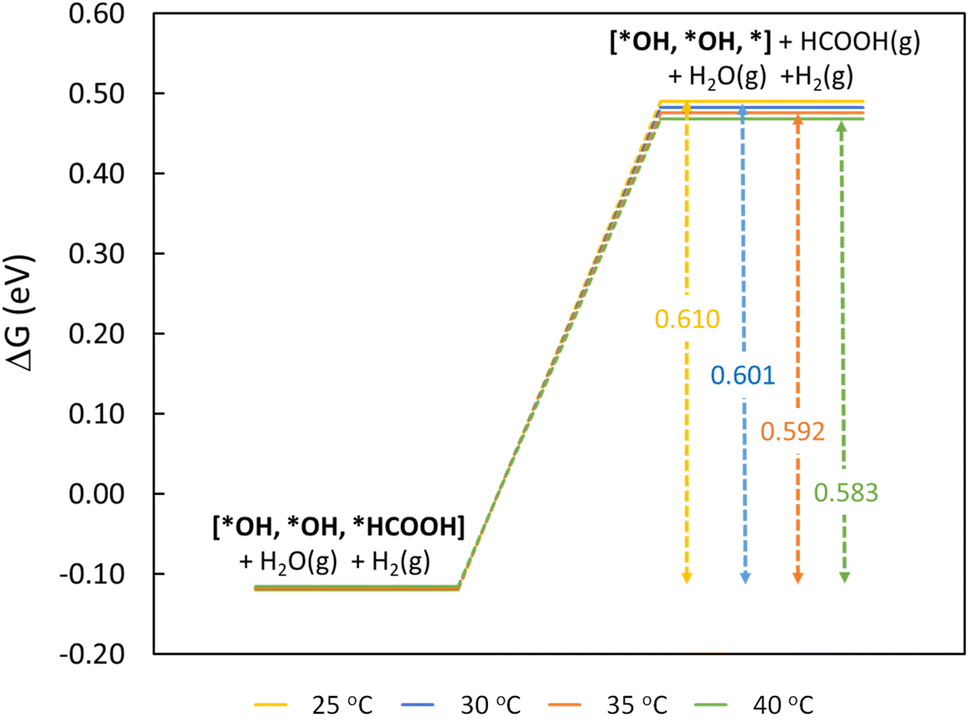 | ||
| Fig. 9 Free energy diagram for rate-limiting step of HCOOH desorption as a function of temperature on the 2Ni@SnO2(110) model. | ||
4. Conclusions
NiOSnO NFs have been synthesized by electrospinning. NiOSnO NFs effectively function as electrocatalysts for the electrochemical CO2RR, yielding HCOO− beyond the room temperature suitable to current electrolyzers. The highest faradaic efficiencies to formate are achieved with NiOSnO50NF and NiOSnO75NF at an electroreduction temperature of 40 °C. XPS and EELS analyses reveal a synergistic effect between the Ni and Sn species. Electrochemical measurements and in situ DEMS provide insights into product distribution during CO2RR. Computational Pourbaix diagrams show that this synergistic effect arises from the dissolution of NiO under reducing conditions. DFT calculations show that embedding Ni in SnO2 is energetically more favorable in addition to aiding the reduction of the SnO2 surface under relevant electroreduction conditions. The desorption of HCOOH is the rate-limiting step whose free energy decreases with increasing temperature from 25 °C to 40 °C, which agrees with the temperature-dependent faradaic efficiencies and partial current densities found during the experiments. Looking into the future, it is clear that catalysts like NiOSnO NFs can be further designed for other temperature conditions rather than room temperature and will, in the future, be used in CO2 electrolyzer technologies over various temperature ranges. These findings underscore the significance of catalyst discovery and explore the potential for temperature-driven synergistic effects in metal oxide catalysts for CO2 electroreduction.Data availability
Data are available upon request from the authors.Author contributions
M. A. R. O., R. L., M. V., and A. S. A. designed the experiments, analyzed the data, and wrote the first draft of the manuscript. M. A. R. O., M. S., C. F., and E. C. -M. synthesized the material and carried out electrochemical measurements. R. L. and M. V. performed the DFT calculations (1) to construct the computational Pourbaix diagrams, and (2) to unravel the relevant CO2 reduction pathways. F. R. -Z. performed STEM-related analysis. The initial idea was coined by A. M., C. F., T. K., J. G. E., M. V., and A. S. A. All authors contributed to the final draft.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors thank Mark Smithers and Gerard Kip (MESA+ Institute, University of Twente) for their support. The research leading to this report's results has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 742004). R. L. (GOIPG/2022/442) thanks the Irish Research Council (IRC) for an IRC postgraduate fellowship. R. L. and M. V. acknowledge the Irish Centre for High-End Computing (ICHEC) for the computational facilities and support. M. S. and T. K. acknowledge funding from the Jane and Aatos Erkko foundation (USVA project). In addition, F. R.-Z. acknowledges the European Research Council (ERC) Starting Grant 123STABLE (Grant agreement ID: 852208).References
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, IPCC, 2021: Climate Change 2021: the Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2021 Search PubMed.
- X. Bai, W. Chen, C. Zhao, S. Li, Y. Song, R. Ge, W. Wei and Y. Sun, Angew. Chem., 2017, 129, 12387–12391 CrossRef.
- F. Köleli and D. Balun, Appl. Catal., A, 2004, 274, 237–242 CrossRef.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2020, 10, 358–364 CrossRef CAS.
- B.-Q. Miao, W.-S. Fang, B. Sun, F.-M. Li, X.-C. Wang, B.-Y. Xia and Y. Chen, Chin. J. Struct. Chem., 2023, 42, 100095 CrossRef.
- R. Hegner, L. F. M. Rosa and F. Harnisch, Appl. Catal., B, 2018, 238, 546–556 CrossRef CAS.
- W. Yang, S. Chen, W. Ren, Y. Zhao, X. Chen, C. Jia, J. Liu and C. Zhao, J. Mater. Chem. A, 2019, 7, 15907–15912 RSC.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- M. A. Rodriguez-Olguin, C. Flox, R. Ponce-Pérez, R. Lipin, F. Ruiz-Zepeda, J. P. Winczewski, T. Kallio, M. Vandichel, J. Guerrero-Sánchez, J. G. E. Gardeniers, N. Takeuchi and A. Susarrey-Arce, Appl. Mater. Today, 2022, 28, 101528 CrossRef.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- C. Englezos, A. Raman, D. Jonker, N. A. Ramos-Delgado, M. Altomare, H. Gardeniers and A. Susarrey, Chempluschem, 2024, 89, e202300763 CrossRef CAS PubMed.
- Y. Fang, X. Liu, Z. Liu, L. Han, J. Ai, G. Zhao, O. Terasaki, C. Cui, J. Yang, C. Liu, Z. Zhou, L. Chen and S. Che, Chem, 2023, 9, 460–471 CAS.
- Y. Fang, L. Han and S. Che, Chin. J. Struct. Chem., 2023, 42, 100107 CrossRef.
- R. E. Vos, K. E. Kolmeijer, T. S. Jacobs, W. Van Der Stam, B. M. Weckhuysen and M. T. M. Koper, ACS Catal., 2023, 13, 8080–8091 CrossRef CAS PubMed.
- T. Mizuno, K. Ohta, A. Sasaki, T. Akai, M. Hirano and A. Kawabe, Energy Sources, 1995, 17, 503–508 CrossRef CAS.
- H. Y. Kim, I. Choi, S. H. Ahn, S. J. Hwang, S. J. Yoo, J. Han, J. Kim, H. Park, J. H. Jang and S. K. Kim, Int J Hydrogen Energy, 2014, 39, 16506–16512 CrossRef CAS.
- A. Löwe, C. Rieg, T. Hierlemann, N. Salas, D. Kopljar, N. Wagner and E. Klemm, ChemElectroChem, 2019, 6, 4497–4506 CrossRef.
- S. T. Ahn, S. Sen and G. T. R. Palmore, Nanoscale, 2022, 14, 13132–13140 RSC.
- R. E. Vos and M. T. M. Koper, ChemElectroChem, 2022, 9, e202200239 CrossRef CAS.
- K. Bejtka, J. Zeng, A. Sacco, M. Castellino, S. Hernández, M. A. Farkhondehfal, U. Savino, S. Ansaloni, C. F. Pirri and A. Chiodoni, ACS Appl. Energy Mater., 2019, 2, 3081–3091 CrossRef CAS.
- M. He, B. Xu and Q. Lu, Chin. J. Catal., 2022, 43, 1473–1477 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- Y. Jiang, J. Shan, P. Wang, L. Huang, Y. Zheng and S. Z. Qiao, ACS Catal., 2023, 13, 3101–3108 CrossRef CAS.
- T. S. Bui, E. C. Lovell, R. Daiyan and R. Amal, Adv. Mater., 2023, 35, 2205814 CrossRef CAS PubMed.
- E. Castañeda-Morales, J. O. Peralta-Cruz, F. Ruiz-Zepeda, A. Susarrey-Arce, M. L. Hernández-Pichardo and A. Manzo-Robledo, Mater. Today Energy, 2024, 41, 101525 CrossRef.
- M. A. Rodriguez-Olguin, R. N. Cruz-Herbert, H. Atia, M. Bosco, E. L. Fornero, R. Eckelt, D. A. De Haro Del Río, A. Aguirre, J. G. E. Gardeniers and A. Susarrey-Arce, Catal. Sci. Technol., 2022, 12, 4243–4254 RSC.
- M. A. Rodriguez-Olguin, H. Atia, M. Bosco, A. Aguirre, R. Eckelt, E. D. Asuquo, M. Vandichel, J. G. E. Gardeniers and A. Susarrey-Arce, J. Catal., 2022, 405, 520–533 CrossRef CAS.
- W. Ju, F. Jiang, H. Ma, Z. Pan, Y. B. Zhao, F. Pagani, D. Rentsch, J. Wang and C. Battaglia, Adv. Energy Mater., 2019, 9, 1901514 CrossRef.
- X. Zong, Y. Jin, C. Liu, Y. Yao, J. Zhang, W. Luo, A. Züttel and Y. Xiong, Electrochem. Commun., 2021, 124, 106968 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- D. G. A. Smith, L. A. Burns, K. Patkowski and C. D. Sherrill, J. Phys. Chem. Lett., 2016, 7, 2197–2203 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Ghysels, T. Verstraelen, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, J. Chem. Inf. Model., 2010, 50, 1736–1750 CrossRef CAS PubMed.
- S. Ning, J. Wang, D. Xiang, S. Huang, W. Chen, S. Chen and X. Kang, J. Catal., 2021, 399, 67–74 CrossRef CAS.
- D. Koziej, K. Thomas, N. Barsan, F. Thibault-Starzyk and U. Weimar, Catal. Today, 2007, 126, 211–218 CrossRef CAS.
- I. Manassidis, J. Goniakowski, L. N. Kantorovich and M. J. Gillan, Surf. Sci., 1995, 339, 258–271 CrossRef CAS.
- J. Oviedo and M. J. Gillan, Surf. Sci., 2000, 463, 93–101 CrossRef CAS.
- L. Braglia, M. Fracchia, P. Ghigna, A. Minguzzi, D. Meroni, R. Edla, M. Vandichel, E. Ahlberg, G. Cerrato and P. Torelli, J. Phys. Chem. C, 2020, 124, 14202–14212 CrossRef CAS PubMed.
- C. Salvini, M. Re Fiorentin, F. Risplendi, F. Raffone and G. Cicero, J. Phys. Chem. C, 2022, 2022, 126–14441 Search PubMed.
- H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Phys. Chem. Chem. Phys., 2008, 10, 3722–3730 RSC.
- M. Safari, J. Mazloom, K. Boustani and A. Monemdjou, Sci. Rep., 2022, 12, 1–15 CrossRef PubMed.
- Y. Luo, M. Weng, J. Zheng, Q. Zhang, B. Xu, S. Song, Y. Shen, Y. Lin, F. Pan and C. Nan, J. Alloys Compd., 2018, 750, 17–22 CrossRef CAS.
- X. Ye, W. Zhang, Q. Liu, S. Wang, Y. Yang and H. Wei, New J. Chem., 2015, 39, 130–135 RSC.
- P. P. Dorneanu, A. Airinei, M. Grigoras, N. Fifere, L. Sacarescu, N. Lupu and L. Stoleriu, J. Alloys Compd., 2016, 668, 65–72 CrossRef.
- N. Van Hieu, P. Thi Hong Van, L. Tien Nhan, N. Van Duy and N. Duc Hoa, Appl. Phys. Lett., 2012, 101, 1–5 CrossRef.
- A. Kotta, E.-B. Kim, S. Ameen, H.-S. Shin and H. K. Seo, J. Electrochem. Soc., 2020, 167, 167517 CrossRef CAS.
- X. Xu, L. Li, J. Huang, H. Jin, X. Fang, W. Liu, N. Zhang, H. Wang and X. Wang, ACS Catal., 2018, 8, 8033–8045 CrossRef CAS.
- X. Xu, H. Zhang, Y. Tong, Y. Sun, X. Fang, J. Xu and X. Wang, Appl. Surf. Sci., 2021, 550, 149316 CrossRef CAS.
- H. Liu, F. Wang, K. Hu, B. Zhang, L. He and Q. Zhou, Nanomaterials, 2019, 9, 1250 CrossRef CAS PubMed.
- M. Taeño, D. Maestre, J. Ramírez-Castellanos, S. Li, P. S. Lee and A. Cremades, Nanomaterials, 2021, 11, 1–13 CrossRef PubMed.
- Q. Ma, H. Li, J. Guo, S. Chu, Q. Zhang and Z. Lin, Mater. Sci. Semicond. Process., 2021, 128, 105762 CrossRef CAS.
- M. Kwoka, L. Ottaviano, M. Passacantando, S. Santucci, G. Czempik and J. Szuber, Thin Solid Films, 2005, 490, 36–42 CrossRef CAS.
- M. Kandasamy, A. Seetharaman, D. Sivasubramanian, A. Nithya, K. Jothivenkatachalam, N. Maheswari, M. Gopalan, S. Dillibabu and A. Eftekhari, ACS Appl. Nano Mater., 2018, 1, 5823–5836 CrossRef CAS.
- C. N. R. Rao, V. Vijayakrishnan, G. U. Kulkarni and M. K. Rajumon, Appl. Surf. Sci., 1995, 84, 285–289 CrossRef CAS.
- Z. Chen, T. Dedova, I. O. Acik, M. Danilson and M. Krunks, Appl. Surf. Sci., 2021, 548, 149118 CrossRef CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- B. Payne, M. Biesinger and N. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 159–166 CrossRef CAS.
- J. Jeong and B. J. Lee, J. Nanosci. Nanotechnol., 2013, 13, 711–713 CrossRef CAS PubMed.
- R. Zhang, Z. Xu, T. Zhou, T. Fei, R. Wang and T. Zhang, J. Colloid Interface Sci., 2019, 557, 673–682 CrossRef CAS PubMed.
- P. G. Choi, N. Izu, N. Shirahata and Y. Masuda, Sens. Actuators, B, 2019, 296, 126655 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
- C. D. Wagner, W. M. Briggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-Ray Photoelectron Spectroscopy, Perkin Elmer Corp, Physical Electronics Division, 1992 Search PubMed.
- C. A. Tolman, W. M. Riggs, W. J. Linn, C. M. King and R. C. Wendt, Inorg. Chem., 1973, 12, 2770–2778 CrossRef CAS.
- R. Félix, N. Llobera-Vila, C. Hartmann, C. Klimm, M. Hartig, R. G. Wilks and M. Bär, RSC Adv., 2018, 8, 67–73 RSC.
- Y. C. Lin, Y. Y. Chen, B. Y. Yu, W. C. Lin, C. H. Kuo and J. J. Shyue, Analyst, 2009, 134, 945–951 RSC.
- J. M. Mora-Hernandez, W. I. González-Suárez, A. Manzo-Robledo and M. Luna-Trujillo, J. CO2 Util., 2021, 47, 101504 CrossRef CAS.
- J. P. Winczewski, S. Zeiler, S. Gabel, A. Susarrey-Arce, J. G. E. Gardeniers and B. Merle, Mater. Des., 2023, 232, 112142 CrossRef CAS.
- A. Ngoipala, R. Lipin, R. L. Arevalo and M. Vandichel, Int. J. Hydrogen Energy, 2024, 53, 829–839 CrossRef CAS.
- R. Lipin, A. Ngoipala, R. L. Arevalo and M. Vandichel, Int. J. Hydrogen Energy, 2024, 61, 460–472 CrossRef CAS.
- J. W. Liao, X. Lu, B. Y. Huang, G. Q. Yu and X. B. Li, Int. J. Hydrogen Energy, 2021, 46, 9077–9086 CrossRef CAS.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- H. Hu, L. Gui, W. Zhou, J. Sun, J. Xu, Q. Wang, B. He and L. Zhao, Electrochim. Acta, 2018, 285, 70–77 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04116j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |