
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving long-term capacity retention of NMC811 via lithium aluminate coatings using mixed-metal alkoxides†
Víctor
Riesgo-González‡
 ab,
Christopher A.
O'Keefe
a,
Clare P.
Grey
*a and
Dominic S.
Wright
*a
ab,
Christopher A.
O'Keefe
a,
Clare P.
Grey
*a and
Dominic S.
Wright
*a
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. E-mail: cpg27@cam.ac.uk; dsw1000@cam.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot OX1 10RA, UK
First published on 22nd July 2024
Abstract
Ni-rich LiNixMnyCozO2 (NMC) materials such as NMC811 (80% Ni) are promising Li-ion battery cathodes due to their high energy density and low cost. However, their reactive surfaces pose a practical challenge for their handling and storage, due to their fast degradation under operation or storage in ambient air. To tackle these problems, lithium aluminum alkoxides are used here for the first time to coat NMC811 with lithium aluminate (Li1−xAl1+x/3O2). This method allows the controlled deposition of lithium aluminates on the surface of NMC811 by hydrolysis of the alkoxide precursor with transition metal TM(O)–OH surface groups. Of the series of alkoxides investigated, Li[Al(OtBu)4] was found to have the best coating/deposition properties in terms of solubility, thermal decomposition, phases formed and reactivity with NMC811. The effects of different NMC811 particle morphologies and surface treatments on the solution deposition of lithium aluminate were carefully evaluated by testing two different substrates: pristine polycrystalline NMC811 (PC-NMC811) and Al2O3-coated single-crystal NMC811 (SC-NMC811). A 17% increase in capacity retention at C/2 was seen for Li1−xAl1+x/3O2/Al2O3 coated SC-NMC811 compared to Al2O3 coated SC-NMC811 after 100 charge–discharge cycles. Furthermore, coating PC-NMC811 that was degraded by soaking in water led to an impressive recovery of 50% of its C/20 capacity retention after 300 charge–discharge cycles while also showing higher specific capacities (25 mA h g−1 more on the 2nd cycle) due to surface Li enrichment and regeneration of degraded surfaces. This coating method is a significant step forward in the scalable processing of new-generation battery cathodes with higher energy density and longer useful life.
Introduction
In order for lithium-ion batteries to successfully replace fossil fuels in a wider range of transportation, their energy density and cycle life need to increase and their cost should be reduced without compromising on safety.1 To this end, Ni-rich LiNixMnyCozO2 (NMC) cathode active materials (CAM) were developed from the parent LiCoO2 (LCO) by partial substitution of Co by Ni and Mn. High-Ni content NMCs such as LiNi0.8Mn0.1Co0.1O2 (NMC811) have high energy densities, thermal stability and low cobalt content, which should reduce costs, toxicity and ethical mining concerns making them promising candidates for applications in electric vehicles.2One obstacle for the use of Ni-rich NMCs such as NMC811 is their poor stability in air, exposure of the powder and the preparation of electrode coatings outside a dry room leading to poor capacity retention and thus difficulties with practical handling, storage, transportation and electrode fabrication. The underlying reasons for this have been investigated in detail. A host of chemical and electrochemical reactions occur at the surface of NMC811 such as electrolyte oxidation during cycling,3,4 reaction with CO2 and moisture from air,5 transition metal dissolution,6,7 and reduction of the surface with the formation of an insulating rock-salt layer (NiO) accompanied by oxygen evolution.8–10 In addition to the surface reactivity, Ni-rich NMC materials have undesirable chemomechanical properties. In particular, they display large anisotropic volume changes in the unit cell at high states of charge (SoC), which causes mechanical strain and leads to secondary particle cracking, exposing fresh surfaces to electrolyte, which aggravates the surface degradation.11–14
As noted above, when NMC811 is exposed to air it reacts with CO2 and moisture forming surface impurities such as Li2CO3 and LiOH.5 This leads to a decrease in specific capacity and capacity retention by depletion of surface Li, which in turn encourages reduction of Ni3+ to Ni2+, the formation of rock-salt layers,5,15 and increased gassing during cycling.5 Furthermore, these surface impurities can increase the pH of the slurry during electrode preparation, which prevents gelation and leads to poor casting properties.16 To fully prevent the formation of these impurities, exposure to air should be minimized during storage, handling and electrode manufacturing. However, this is not always feasible in industrial-scale battery manufacture where large amounts of material are used.
To mitigate surface degradation, synthetic modifications of the cathode surface, via coating and surface doping, have been developed.17–19 However, it is still unclear whether coating with monometallic oxides or surface doping can prevent interfacial processes that lead to a loss of capacity in Ni-rich NMC materials effectively.20–22 Washing and annealing steps have also been explored as strategies to eliminate the surface impurities and regenerate the surfaces of air-exposed CAMs.16,23–31 The most common approach to remove surface impurities involves washing the CAM with water, which was shown to be effective in improving capacity retention in LCO.25 However, it is unlikely that this strategy alone will work for NMC811 due to its sensitivity to water,16 and the increased Li leaching observed when soaking NMCs with higher Ni content in water, which further deteriorated the capacity retention.27,32 To address this issue, washing methods involving simultaneous coating deposition from aqueous solutions have been recently applied to high Ni-content NMCs with some success.33,34 Treating Ni-rich NMC with water leads to a decrease in capacity retention due to lithium extraction by Li+/H+ exchange, forming LiOH and TM(O)–OH species on or close to the surface,16 and the removal of surface impurities by washing, which creates more exposed surfaces.30 Since delithiated surfaces are thought to be more reactive with the electrolyte,35 this may lead to more electrolyte degradation and rock-salt formation on cycling.30 Adding an annealing step after soaking NMC in water only leads to the formation of a NiO rock-salt layer which further reduces the long-term capacity retention.16 However, if excess lithium is added during this annealing step (for example in the form of Li2CO3), a more favorable surface which is less reactive with the electrolyte can be obtained.35 Washing NMC811 with water also affects its mechanical properties. In one study of polycrystalline NMC, stirring in water led to significant particle cracking.29 Furthermore, immersion without stirring did not cause cracking but led to a decrease of the compressive load for failure to half of the value of pristine NMC811.29
From these studies on NMC811 degradation and washing it becomes clear that coating Ni-rich NMC with a lithium-rich layer should improve its capacity retention and recover surfaces that were degraded due to exposure to air, which is unavoidable when processing NMC811 industrially. One way of achieving this is by coating NMC811 with lithium rich mixed-metal oxides,36–41 fluorides,42 phosphates,43 or silicates.44,45 In one study, ZrxPOy and LixZryPOz were deposited on NMC811 by atomic layer deposition (ALD).43 While both increased capacity retention at low cycling rates, the lithium-rich coating allowed higher initial capacities and improved rate capabilities.43 Li2ZrO3 coating has also been tested on a NMC with 70% Ni.40 Despite the fact that using two separate lithium and zirconium sources led to inhomogeneous distribution of the elements on the surface, both by sol–gel and co-precipitation methods, improved capacity retention and rate capability were observed.40 LiAlF4 was also deposited by ALD onto NMC811 and showed good stability and improved capacity retention over 300 cycles in half cells without compromising rate capability.42 Ternary oxides were coated onto Ni-rich NMCs too using mechanical mixing of nano-powders of the coating material with the CAM, generally obtaining positive results in capacity retention and rate capability.39,41 In a recent report, improvements in cycle life of an NMC with 85% Ni content and LNO were seen by solution deposition of an aluminosilicate using trimethyl aluminum (TMA) and tetraethylorthosilicate (TEOS) as precursors.45 However, it was necessary for the CAM to be wet in order for the coating reaction to take place, limiting the use of this method.45 Furthermore, these studies focused on coating pristine NMC cathodes, so the effect of coating Ni-rich NMC that has been degraded by moisture exposure using a Li-rich coating remains unknown.
Clearly the deposition of ternary coatings poses a synthetic challenge for conventional methods such as ALD, sol–gel, mechanical mixing, and co-precipitation. Most importantly, the need to use two precursors as the source of each of the metals leads to an inhomogeneous composition of the coating. A suitable single-source precursor (SSP)46 for the solution deposition of mixed-metal oxides onto NMC811 would overcome this problem since it would already contain the two metals in the desired stoichiometry and mixed at the atomic level.46 The SSP must display a series of properties: it should decompose at a low temperature to avoid its diffusion into NMC811 (T < 500–600 °C) and should be reactive with functional groups and/or adsorbed species present on the surface of NMC811 to allow selective surface deposition. Finally, the precursor should be soluble in innocent solvents (which do not react with the cathode).
In our previous work,20 we showed that an amorphous Al2O3 coating deposited by a solution-based method was not very effective at improving the capacity retention in half cells due to the insulating nature of Al2O3, which led to kinetic limitations, the extraction of Li from the NMC811 during annealing and the formation of a secondary phase containing electrochemically inactive Al3+ ions. This challenge prompted the search for other coating materials with more favourable properties. In this context, LiAlO2 emerges as a promising option. Like Al2O3, it contains cheap, non-toxic, and abundant elements. However, it is, in principle, a much better Li-ion conductor than Al2O3, specially in its amorphous form.47 More importantly, since it already contains Li, it is unlikely to extract Li from the surface of the NMC811 during annealing. Furthermore, it may be possible to reverse rock-salt formation by a surface Li treatment followed by annealing-induced Li diffusion and oxidation of Ni2+ to Ni3+.48 Therefore, by annealing NMC811 coated with a Li-rich layer, it may be possible to recover some of the capacity that has been lost due to exposure to air or water while also benefiting from having a surface coating.
Heterobimetallic alkoxides have been used extensively in other areas for the synthesis of ternary oxide coatings.46 This class of molecule is attractive due to their often simple synthesis, and favourable properties as SSPs such as the tendency to hydrolyse in the presence of water or thermolyze at relatively low temperatures. In this paper, a new method for coating NMC811 using these heterobimetallic alkoxides is developed as an effective strategy to improve the capacity retention of NMC811. Specifically, a lithium aluminate coating is prepared by developing appropriate mixed-metal alkoxides for use as SSPs for coating deposition. The coating of single-crystal NMC811 (SC-NMC811) and polycrystalline NMC811 (PC-NMC811) particles were then studied. SC-NMCs tend to show slower degradation compared to polycrystalline NMC,49 due to their reduced surface area compared to PC-NMC811.50,51 They also show less tendency to crack upon cycling – cracking in PCs primarily occurring between the fused primary particles that make up the larger secondary agglomerate. This cracking is proposed to be one of the degradation mechanisms of high Ni content NMC, exposing more NMC surfaces to the electrolyte.52 Of relevance here, these freshly exposed surfaces will not be coated by the aluminate coating.
Importantly, this coating method is then applied to SC- and PC-NMC811 samples that had been soaked in water and then dried. This was performed in order to explore whether coating a sample that had been exposed to extreme moisture conditions – and thus showed poor electrochemical performance – might help to recover its electrochemical activity and to see if soaking in water might affect the coating deposition.
Results and discussion
Precursor synthesis
Lithium aluminum alkoxides LiAl(OR)4 were prepared by reaction of LiAlH4 with different alcohols in dry THF. This was done to test the effect of different alkoxy groups (–OCH2Ph, –OiPr, –OtBu) on the properties of the resulting molecules as precursors to LiAlO2 coatings. By changing these groups, it is possible to change critical properties of the precursor such as solubility, decomposition temperatures and coating properties. The synthetic route employed in the current work was first used by Pauls et al. to synthesize Li[Al(OCH2Ph)4] (1),53 and in this work is extended to two other heterobimetallic alkoxides: Li[Al(OiPr)4] (2) and Li[Al(OtBu)4] (3). The products are obtained by exothermic reactions (Scheme 1) of LiAlH4 (1 M solution in THF) with the appropriate alcohol, dissolved in THF at room temperature giving H2 and the mixed-metal alkoxide as the product. This simple, one-step reaction is appealing from a practical perspective; it generates useful heat, requires simple and widely available starting materials and yields H2(g) as the only side-product, which is essential for many industrial processes such as the synthesis ammonia and methanol.54 It was found that the choice of ligand affects the solubility, thermolysis and reactivity with the surface of NMC811 with compound 3 displaying the best properties as a SSP for LiAlO2 deposition onto NMC811. | ||
| Scheme 1 Synthesis of the three lithium aluminate precursors used in this work. A 1 M solution of LiAlH4 in THF is reacted with 4 equivalents of the alcohol to give hydrogen gas and the heterobimetallic alkoxide containing Li and Al. The yields of each of the products are stated in parenthesis. | ||
Compounds 1–3 were characterized by elemental analysis and nuclear magnetic resonance (NMR) spectroscopy (in solution, when possible, Fig. S3(1.1)–S3(1.9),† and in the solid state, Fig. S3(2.1)–S3(2.4) and Table S3(2.1)†). Elemental analysis (Synthetic procedures, Section S2(1)†) indicates that the formulae of complexes 1–3 is LiAl(OR)4 (with OR![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH2Ph, OiPr or OtBu) without any THF from the synthesis remaining after drying at 40 °C under vacuum. The use of bulkier groups such as tBuOH or PhCH2OH led to products that were soluble in THF, but not fully soluble in any other solvents tested, suggesting that THF coordination to Li+ is playing an important role in solubilizing these compounds (most likely by forming an ion-separated Li(THF)4+ Al(OR)4− pair). However, using a smaller alkoxy ligand (iPrOH) led to a product that was insoluble in THF. The 1H and 13C solution NMR spectra of the two soluble precursors (1 and 3, Fig. S3(1.2)–S3(1.7)†) show the expected presence of the alkoxy groups in the products (–OCH2Ph and –OtBu groups, respectively), with THF being absent in their 13C DEPT 135 NMR (Fig. S3(1.4) and S3(1.7)†) and solid-state NMR (SS-NMR) spectra (see Section S3(1)†) (consistent with elemental analysis).
OCH2Ph, OiPr or OtBu) without any THF from the synthesis remaining after drying at 40 °C under vacuum. The use of bulkier groups such as tBuOH or PhCH2OH led to products that were soluble in THF, but not fully soluble in any other solvents tested, suggesting that THF coordination to Li+ is playing an important role in solubilizing these compounds (most likely by forming an ion-separated Li(THF)4+ Al(OR)4− pair). However, using a smaller alkoxy ligand (iPrOH) led to a product that was insoluble in THF. The 1H and 13C solution NMR spectra of the two soluble precursors (1 and 3, Fig. S3(1.2)–S3(1.7)†) show the expected presence of the alkoxy groups in the products (–OCH2Ph and –OtBu groups, respectively), with THF being absent in their 13C DEPT 135 NMR (Fig. S3(1.4) and S3(1.7)†) and solid-state NMR (SS-NMR) spectra (see Section S3(1)†) (consistent with elemental analysis).
7Li (Fig. S3(1.1) and S3(1.8)†) and 27Al solution NMR measurements (Fig. S3(1.9)†) in d8-THF confirmed the presence of both elements in 1 and 3. Aluminium is typically found in 4-, 5- or 6- coordination environments (denoted here as [4]Al, [5]Al, and [6]Al). No peaks from LiAlH4 (sharp peak at 98 ppm)55 were observed in these samples indicating that the LiAlH4 is fully consumed during the synthesis. For precursor 1, 27Al solution NMR shows a single resonance centred at 69.76 ppm indicating the presence of a single [4]Al environment in solution.56 The 7Li solution NMR spectrum of 1 (Fig. S3(1.1)†) shows the presence of a main peak at −0.23 ppm and a broad peak at 0.35 ppm possibly indicating fast exchange between Li(THF)n+ cations of varying coordination number.55 In contrast with 1, compound 3 shows a single sharp resonance in the solution 7Li NMR spectrum at −0.26 ppm (Fig. S3(1.8)†) but three aluminium environments (77.68, 60.26 and 53.1 ppm, Fig. S3(1.9)†) indicating the presence of two [4]Al and potentially one [5]Al environment in solution.56 The presence of more than one aluminium or lithium environment in 1 and 3 suggests complicated equilibria in d8-THF solutions, which are not uncommon for aluminates of this type (e.g., involving ion-pairing of Li(THF)n+ with Al(OR)4−, and potentially coordination of THF to Al3+).55
Due to the insolubility of compound 2 (which prevented in-depth solution NMR studies), and in order to further characterize 1 and 3, SSNMR measurements were carried out on the three compounds (Fig. S3(2.1)–S3(2.4) and Table S3(2.1)†). SSNMR measurements confirmed that all of the precursor compounds have the formula LiAl(OR)4 in the solid state and also allowed an assessment of minor impurities and other phases present. The 7Li SSNMR spectra are shown in Fig. S3(2.1).† Compounds 1 and 2 show single resonances at −0.73 and −0.65 ppm, respectively. The main 7Li NMR resonance of 3 was shifted to higher frequency compared to the other two (0.23 ppm). The 27Al SSNMR spectra also show clear differences between the three compounds. The spectra were fitted as quadrupolar line shapes (Fig. S3(2.1)–S3(2.4) and Table S3(2.1)†) to obtain information about the number of environments present, their quadrupolar parameters (quadrupolar coupling constant, CQ and the asymmetry parameter, ηQ) as well as the isotropic shift (δiso). The 27Al SSNMR spectrum of 3 is dominated by one major [4]Al environment at 79 ppm (Fig. S3(2.4)†) with a high CQ of 6 MHz – higher than the [4]Al environments found in 1 and 2 (Table S3(2.1)†). Aluminium alkoxides can deviate significantly from ideal tetrahedral geometries,55 which would explain the large electric field gradient (EFG) and corresponding CQ values for the main peak at 79 ppm.57 Furthermore, deconvolution of the 7Li SSNMR spectrum of 3 (Fig. S3(2.4(b))†) shows that there is only one major Li environment at 0.23 ppm. Therefore, the SSNMR results are consistent with the presence of Li[Al(OtBu)4], in agreement with elemental analysis.
Precursor thermolysis
The NMC811 coating process involves the deposition of the precursors onto the surface of NMC811 particles from solution and the annealing of the coated materials under air to form LiAlO2 by thermolysis of the precursors. The thermolyses of 1–3 were studied using thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), SSNMR and inductively coupled plasma optical emission spectroscopy (ICP-OES). TGA performed under N2 (Fig. S4(1.1)†) shows that the % mass loss for the three compounds at 800 °C corresponds to the formation of LiAlO2 (Table S4(1.1)†), confirming that all three can successfully form the desired coating material. The thermolysis could proceed by elimination (LiAl(OR)4 → LiAlO2 + 2OR2) in dry atmosphere, although in ambient conditions this is likely to occur by reaction with ambient moisture (following the equation LiAl(OR)4 + 2H2O → LiAlO2 + 4ROH).Samples for PXRD, SSNMR and elemental analysis were prepared by annealing the precursors in an alumina crucible at 400–800 °C for 4 h under air (Fig. 1, Sections S4(2) and S4(3)†). The aluminium and lithium content in samples after thermolysis was determined by ICP-OES. The ICP-OES results clearly show that precursor 3 forms a solid with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Al to Li ratio, as expected for LiAlO2. Compounds 1 and 2, however, showed a Li:Al ratio of slightly less than 1 (0.876–0.976). Since lithium does not sublime under these conditions, the Li deficiency could result from a lower Li:Al ratio than 1:1 in 1 and 2 or the formation of volatile [Li(OR)]n oligomers.
:1 Al to Li ratio, as expected for LiAlO2. Compounds 1 and 2, however, showed a Li:Al ratio of slightly less than 1 (0.876–0.976). Since lithium does not sublime under these conditions, the Li deficiency could result from a lower Li:Al ratio than 1:1 in 1 and 2 or the formation of volatile [Li(OR)]n oligomers.
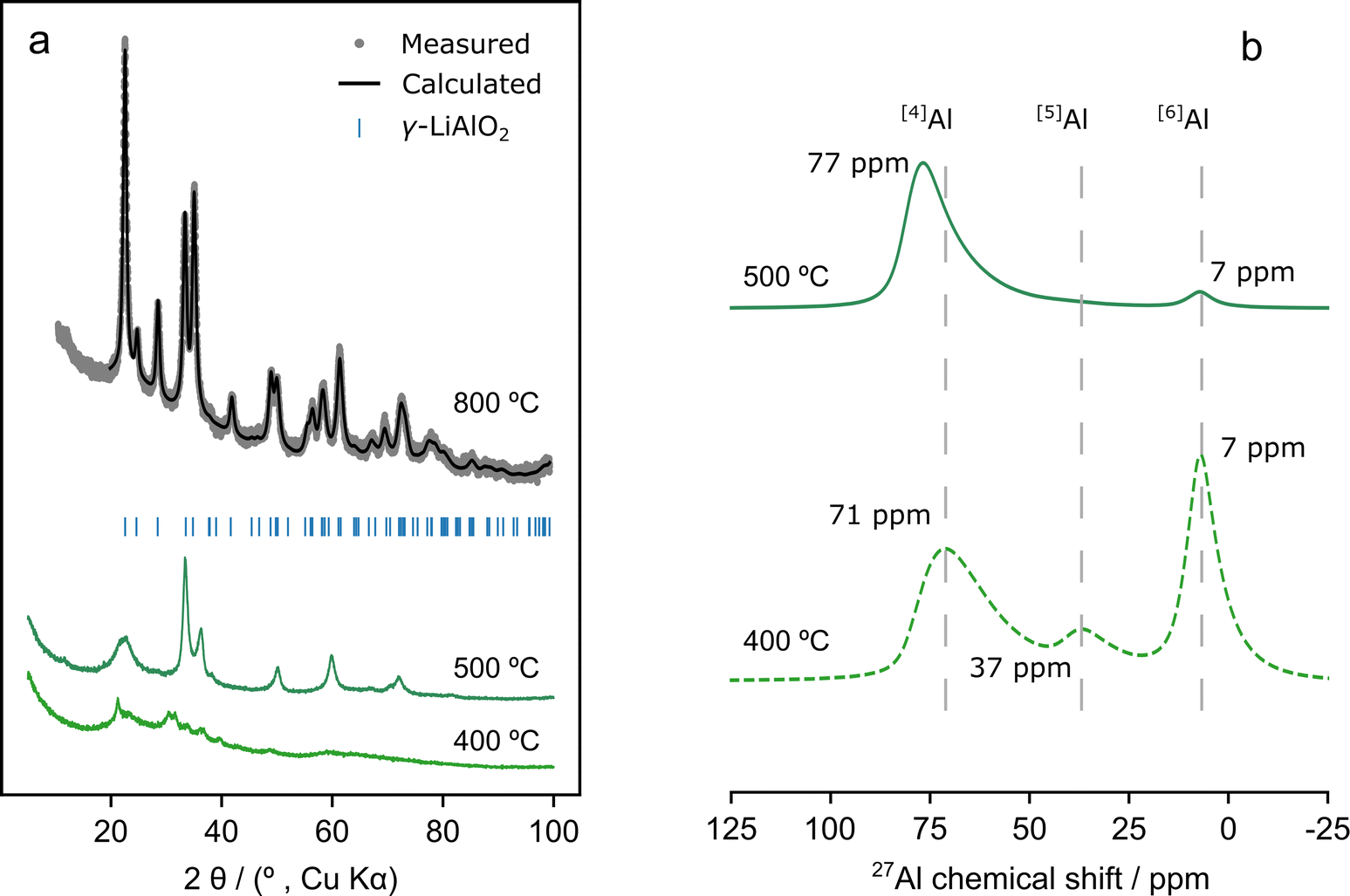 | ||
| Fig. 1 (a) PXRD patterns of compound 3 decomposed by annealing under air for 4 h at 400, 500 and 800 °C. Annealing at 800 °C produces a single phase γ-LiAlO2 material (rietveld refinement shown, see Section S4(2)† for refinement parameters). Annealing to 400 °C produces an amorphous product and annealing to 500 °C results in a disordered material. (b) Solid-state 27Al-MAS NMR spectra of compound 3 annealed under air at 400 and 500 °C. The spectra were recorded at a magnetic field strength of 16.4 T and a MAS frequency of 50 kHz. Only the central (isotropic) resonances are shown. | ||
Generally, the PXRD patterns show broad peaks below 800 °C, indicating significant disorder (Fig. 1 and Section S4(2)†). However, annealing 3 to 500 °C led to the formation of sharper peaks in the PXRD pattern compared to 1 and 2, suggesting that 3 starts to crystallise at this temperature (Fig. S4(2.1.1)†). There are three confirmed polymorphs of LiAlO2; α-, δ-, and γ-LiAlO2 which crystallize forming hexagonal (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), tetragonal (I41/amd), and tetragonal (P41212) structures, respectively.58,59 Of these only γ-LiAlO2 does not have [6]Al environments. 27Al-MAS SSNMR results showed almost exclusively [4]Al environments after annealing at 500 °C (Fig. 1b).
m), tetragonal (I41/amd), and tetragonal (P41212) structures, respectively.58,59 Of these only γ-LiAlO2 does not have [6]Al environments. 27Al-MAS SSNMR results showed almost exclusively [4]Al environments after annealing at 500 °C (Fig. 1b).
Thus, if the crystalline phase forming at this temperature retains the 1:1 Al:Li ratio of 3, then the NMR suggests that it is γ-LiAlO2. Furthermore, formation of crystalline Al2O3 polymorphs can be discarded since all of them contain [6]Al environments.60 Other stoichiometries were also considered: LiAl4O8 (cubic, P4332),61 metastable β-Li5AlO4 (orthorhombic, Pbca) and α-Li5AlO4 (orthorhombic, Pmmn).62 Of these, only Li5AlO4 is consistent with the 27Al-MAS SSNMR, but its formation is unlikely since its typically synthesized under lithium excess (Li:Al ratio of 9:1).62 PXRD peak positions (Fig. 1a) suggest the presence of γ-LiAlO2 after annealing at 500 °C but the disordered nature of the material (which led to peak broadening) prevented full Rietveld refinement of the PXRD pattern.
On the other hand, Rietveld refinements obtained from compounds 1–3 after annealing at 800 °C confirmed unambiguously the formation of γ-LiAlO2 at this temperature (Section S4(2.1)† and Fig. 1a). Materials 1 and 3 could be indexed to a single P41212 γ-LiAlO2 phase while for 2, a secondary α-LiAlO2 phase in the Rm space group (1.4% by weight) was detected (Tables S4(2.2.2), S4(2.2.3) and S4(2.2.5)†). Of these polymorphs, γ-LiAlO2 is particularly interesting as a lithium-ion conductor owing to the diffusion channels created by the 3D network of edge-sharing MO4 tetrahedra.59 Li+-ion diffusion experiments have shown that the presence of defects, or even a mixture of amorphous and crystalline LiAlO2, leads to an increase in lithium conductivity.58,59
TGA and PXRD results show that the three precursors can be used to synthesise γ-LiAlO2 by a simple thermolysis route at 800 °C (see Table S4(2.2.5)† for refined phases and lattice parameters). This is in contrast to more conventional synthetic methods using alkoxide precursors via sol–gel or using ceramic synthesis which generally proceeds at 900–1000 °C.63–66 This presents advantages in the synthesis of pure LiAlO2 samples, which is usually hampered by lithium sublimation at the temperatures at which the solid-state reaction takes place.66 The lower temperature of γ-LiAlO2 formation seen here can be attributed to the use of SSPs in which there is atomic mixing of Li and Al in the required 1:1 ratio before the start of the annealing process, which should eliminate diffusion limitations. Furthermore, precursor 3 showed greater crystallinity after thermolysis at 500 °C compared to 1 and 2 (Fig. S4(2.1.1)†), demonstrating that the nature of the precursor can have a noticeable impact on the temperature at which the desired oxide phase (γ-LiAlO2) starts to crystallise, which for 3 is ca. 500 °C.
The compositions of the products of 1–3 formed below 800 °C are of interest since these are the temperatures at which the annealing of the coated NMC811 particles will proceed. However, the fact that these materials are disordered and poorly crystalline/amorphous when annealing at these temperatures prevented the study of their structure by PXRD. For this reason, SSNMR spectroscopy was next used to determine their structures after annealing 1–3 at 500 °C (Fig. 1b and S4(3.1)†).
Compounds 2 and 3 form a material with very similar aluminium local environments (Fig. 1b and S4(3.1)†) at 500 °C. Both have mostly [4]Al and a small amount of [6]Al as expected for the formation of a largely γ-LiAlO2 type material. The tailing of [4]Al peaks to lower frequencies is a clear signature of disorder, in agreement with PXRD results, which showed broad peaks at annealing temperatures below 800 °C. Heating 3 to 400 °C clearly leads to an amorphous phase with a distribution of [4]Al, [5]Al and [6]Al environments similar to those found in amorphous Al2O3.20 Finally, the product formed from decomposing 1 at 500 °C has a very different structure from that of the other two (Fig. S4(3.1)†). [6]Al environments are present that only convert to the [4]Al characteristic of γ-LiAlO2 at temperatures higher than 500 °C (see Section S4, Fig. S4(2.1.1) and S4(3.1)†); the use of higher temperatures for coating annealing is avoided in this work, so as to avoid the coating (the Al3+ ions) diffusing into the NMC811, as observed previously for Al2O3 coatings.20,21,67
Coating deposition
A solution deposition method similar to that employed in our previous work was used here to prepare the LiAlO2-coated NMC811, with the whole process being carried out under inert atmosphere (Scheme 2).20 First, the SSP and NMC811 were loaded in a Schlenk flask (SSP being 1% of the total solid weight). Dry THF was added, and the reaction mixture stirred under N2 for 2 days at 60 °C. In this step, the alkoxy groups in the SSP react with metal-bonded hydroxyl groups or water molecules present on the surface of NMC811 by protonolysis, releasing the alcohol and covalently bonding to the surface of NMC811 (Scheme 2a).68 Then the coating layer grows by hydrolysis or slow thermolysis followed by condensation.68,69 For the coating layer to homogeneously cover the surface of the particles, the solvent needs to be dry to maximise surface deposition and limit hydrolysis which would promote the formation of aggregates and localised precipitates. To remove any unreacted precursor, the solvent was taken out by syringe, after which the sample was washed three times with dry THF (Scheme 2b). Finally, the coated NMC811 was dried under vacuum at 100 °C for 2 h and then calcined at 400 °C under air for 4 h to remove organic groups and form lithium aluminate. This annealing temperature (400 °C) was selected as it was previously found to lead to the best performing coatings,20 due to the formation of an amorphous oxide layer, which should help with Li-ion conductivity without increasing the cation mixing of the material or diffusing the coating in the near surface region.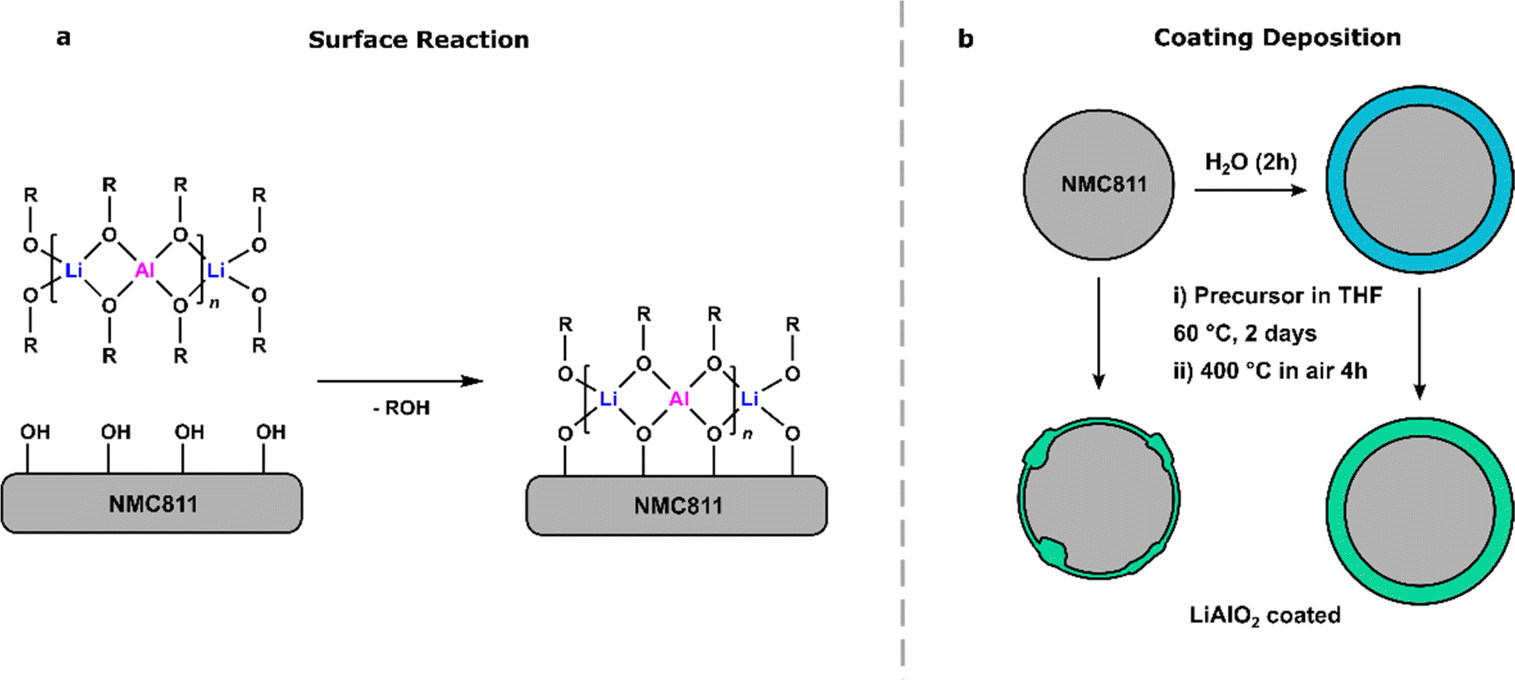 | ||
| Scheme 2 (a) Proposed surface reaction between NMC811 and heterobimetallic alkoxides. (b) Coating deposition: pristine and water-treated NMC811 materials are coated by reaction with precursors 1–3 in dry THF for 2 days followed by annealing at 400 °C under air for 4 h. | ||
Coating of both SC-NMC811 and PC-NMC811 particles were then studied using energy-dispersive X-ray spectroscopy (EDS, Fig. 2 and Section S5(1.2)†), and SS-NMR (Fig. 4 and Section S5(4)†). The pristine SC-NMC811 used in this work was already coated with a thin Al2O3 layer. Furthermore, SC- and PC-NMC811 samples were then soaked in deionized water for 2 h while stirring and then dried under vacuum for 2 h at 50 °C. These were also coated following the same procedure (Scheme 2b).
 | ||
| Fig. 2 Representative SEM images of (a) pristine PC-NMC811, (b) PC-NMC811 coated with 3, (c) PC-NMC811 soaked in water and coated with 3, (d) pristine SC-NMC811, (e) SC-NMC811 coated with 3, (f) SC-NMC811 soaked in water and coated with 3, (g) bar-chart showing the Al:Ni ratios determined for each of the samples using energy-dispersive X-ray spectroscopy (EDS) data. | ||
The SEM images in Fig. 2 show that the pristine PC-NMC811 is formed of 10–20 μm secondary particles that contain smaller 0.5–1 μm primary particles. Coating with 3 led to subtle differences in the morphology of the PC-NMC811 particles with some areas covered by “patches” of coating (Fig. S5(1.1.4)–S5(1.1.6)†). EDS showed that there is a consistent, although small, level of aluminium across different samples suggesting that the coating process was successful (Fig. S5(1.2.2)†). SEM and EDS provided no evidence of coating deposition taking place on PC-NMC811 when using 1, potentially due to the lower basicity of PhCH2O-compared to aliphatic groups (Fig. S5(1.1.2)†). Coating with 2 on the other hand, led to inhomogeneous coverage of some of the PC-NMC811 secondary particles with chunks of the insoluble precursor of 0.5–5 μm (Fig. S5(1.1.3)†). Based on this assessment and on the solubility of the SSPs, 3 was selected as the best candidate for the water soaking/coating experiments, coating of pristine SC-NMC811 and battery testing.
Fig. 2 shows SEM images of PC-NMC811 and SC-NMC811 coated with 3 with and without a pre-soaking step in water. From the SEM images it can be seen that SC-NMC811 is formed of monolithic 1–4 μm primary particles that do not form secondary aggregates. PC-NMC811, on the other hand, is formed of smaller (<1 μm) primary particles aggregated forming 10–20 μm secondary particles. In comparison to the PC-NMC811 sample, the SC-NMC811 material showed less obvious changes in morphology after coating with 3. The most striking change in morphology is seen for the PC-NMC811 sample soaked in water and coated using 3. The surface of the particles is clearly covered by a thick surface layer and the NMC811 primary particles can no longer be clearly distinguished. This suggests that the pre-soaking in water is promoting coating deposition either by increasing the moisture levels on the surface of the NMC811 or by forming surface TM(O)–OH species that are more reactive with the precursor. This change in morphology was not observed for the pre-soaked SC-NMC811 (Fig. 3f and S5(1.1.14)†).
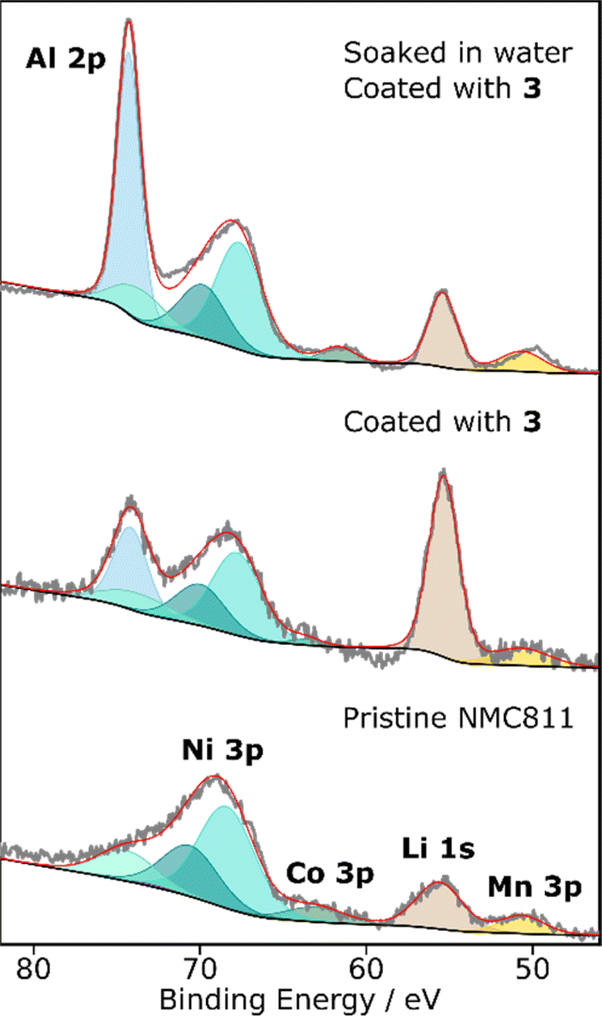 | ||
| Fig. 3 The Al 2p region of the XPS spectra of pristine PC-NMC811 and LiAlO2 coated PC-NMC811 (with and without a pre-soaking in water). The signals correspond to transition metals (nickel, cobalt, and manganese in turquoise, green and yellow, respectively), lithium (brown) and aluminium (blue). Nickel and cobalt show multiple peaks due to spin–orbit splitting (3p3/2 and 3p1/2) and the presence of a satellite peaks (see Table S5(3.2.3)†).20 The coating step results in the simultaneous deposition of aluminium and lithium on the surface of the particles as shown by the increased intensity of the Li 1s signal and the appearance of an Al 2p signal. The aluminium content of the coated NMC that was soaked in water is larger. | ||
EDS was used to determine the changes in aluminium content after coating using 3 for the samples shown in Fig. 2. First, the Al:Ni ratios in the pristine materials were measured to determine the baseline levels. No Al was detected for the pristine PC-NMC811. On the other hand, the commercially supplied sample of SC-NMC811 showed an aluminium signal (Al:Ni ratio of 0.012 ± 0.002) suggesting that it had already been coated or doped with aluminium. Coating PC-NMC811 using 3 led to a consistent, small increase in aluminium content (Al:Ni ratio of 0.009 ± 0.001). For the SC-NMC811, similar Al:Ni values were observed for the pristine and coated samples using 3. Control samples were prepared where both SC- and PC-NMC811 were soaked in THF and subjected to the same treatment as in the coating process but without addition of 3, i.e., they were stirred in THF at 60 °C for 2 days and then dried under vacuum at 100 °C and annealed under air at 400 °C for 4 h. This was done to determine what the effect of THF soaking and annealing under air on its own would be, this procedure itself leading to a decrease in the aluminium content in the case of SC-NMC811 (0.012 ± 0.002 vs. 0.008 ± 0.001). Since the Al:Ni ratio of the SC-NMC811 coated using 3 is greater than that of the SC-NMC811 soaked in THF (Fig. 2g), this suggests that coating material has been deposited. The increase in Al content after coating is greater for PC-NMC811 than for SC-NMC811, which could be because the pre-existing coating on the SC-NMC811 surface hinders further deposition but also because the PC materials have larger effective surface areas.50 EDS analysis of the coated samples pre-soaked in water showed that, compared to the coating of the pristine materials, a larger amount of aluminium is deposited for the soaked samples (both for the SC-NMC811 and the PC-NMC811) under the same conditions suggesting that soaking in water and drying at 50 °C under vacuum modifies the surface of the NMC811 and makes it more reactive with 3.
X-ray photoelectron spectroscopy (XPS) was next used to obtain a more detailed picture of the chemical species and elemental compositions of the surfaces (Fig. 3). The Al 2p region of the XPS spectra (85–45 eV) is of particular interest as it contains information about the transition metals (Ni, Co, and Mn) as well as the aluminium- and lithium-bearing species present on the surface. High-resolution spectra of the Al 2p region were recorded for pristine PC-NMC811, PC-NMC811 coated using 3 and PC-NMC811 soaked in water and coated using 3 (Fig. 3 and Tables S5(3.2.3) and S5(3.4.2)† for details on the fitting procedure). The transition metal peaks remain in the same positions and relative intensities across the different samples indicating that there has not been any significant change in transition metals content or oxidation state induced by the washing or coating procedures. The predominant oxidation states present on the surface of PC-NMC811 before electrochemical cycling are Ni2+, Ni3+, Co3+, and Mn4+.70 The Al 2p component is present in the sample coated with 3 further confirming the EDS results that the coating procedure results in the deposition of an aluminium-containing layer. The binding energy of the Al 2p peak is 74.18 eV, confirming the presence of Al3+ oxide species which typically appear at 73–74.5 eV.38,47,71,72 The literature values for the Al 2p binding energy of LiAlO2 coatings on NMC materials vary between 73.2 and 74.2 eV, whereas the values of Al2O3 coatings range between 73.0 and 73.5 eV.38,47,71,72 These small differences in binding energy are typical for non-ideal surfaces where imperfect charge correction can occur. Nevertheless, higher binding energies tend to be observed for LiAlO2 than for Al2O3,38,72 which is consistent with the assignment of the signal seen here (74.18 eV) to a lithiated alumina. More importantly, a sharp increase in the intensity of the Li 1s peak is observed for the sample coated using 3 compared to the pristine NMC811 material. This provides direct evidence that this precursor can successfully deposit Li and Al simultaneously, owing to its bimetallic nature.
After washing with water and annealing at 400 °C under air, a decrease in organic species is observed in the O 1s spectrum, which also shows the emergence of a lattice oxygen peak, suggesting that this treatment cleans the surface of impurities (Fig. S5(3.1.1)†). Furthermore, the C 1s spectrum shows that there is no Li2CO3 on the surface of the PC-NMC811 washed with water (Fig. S5(3.1.1)†). Coating PC-NMC811 that has been soaked in water leads to a much higher aluminium content compared to the unwashed, LiAlO2 coated PC-NMC811 (Fig. 3). This is in line with the SEM/EDS measurements which showed that the soaking step induces the deposition of a thicker, more homogeneous coating layer. The binding energy of the Al 2p is 74.28 eV in this sample, indicating that the same Al3+ bearing material is deposited regardless of water pre-treatment. Furthermore, the O 1s spectrum of the PC-NMC811 soaked in water and then coated using 3 is very different from that of the pristine PC-NMC811 or the pre-soaked NMC811 annealed without a coating step (Fig. S5(3.1.1)†). Since these differences cannot be attributed to the change in measurement parameters, they must be due to the coating deposition. In fact, the position of the main O 1s component is 531.4 eV, close to literature values for LiAlO2 O 1s peak position (530.6 eV).71
Finally, it is observed that the lithium content of the coated PC-NMC811 soaked in water is lower than that of the coated, untreated PC-NMC811 and similar to that of the pristine PC-NMC811. This may appear surprising considering that this precursor should lead to the simultaneous deposition of Li and Al. However, it should be remembered that the previous water treatment removed most of the Li-bearing impurities from the surface and may have delithiated some of the sub-surface bulk phase, so the Li signal observed in this case comes solely from the coating whereas in the case of the coated, untreated PC-NMC811 the lithium signal had contributions from both the coating, the impurities and bulk NMC811. Furthermore, surface water could induce the preferential deposition of Al, leading to an Al2O3/LiAlO2 coating in which the Al:Li atomic ratio is higher than one. Finally, if the annealing step is promoting re-insertion of lithium from the coating into the bulk of the de-lithiated PC-NMC811,48 this would decrease the surface lithium content detected by XPS.
A multinuclear SSNMR approach was then used to investigate the changes occurring in the surface and bulk of the material after soaking, annealing, and/or coating. 27Al SSNMR spectroscopy was first used to investigate the coatings produced with 1–3 (Fig. 4 and S5(4.3.1)–S5(4.3.3)†). Coating with any of the three precursors leads to the deposition of Al but the amount and structure of the coating changes significantly depending on the precursors used. Heating 3 at 400 °C led to a material with [4]Al, [5]Al and [6]Al environments in which the [4]Al signal appeared at 71.5 ppm (Fig. S4(3.1)†). Coating PC-NMC811 with 3 leads to the deposition of a coating layer with a single [4]Al environment at 68.2 ppm (Fig. 4). Although the presence of [4]Al is indicative of γ-LiAlO2, this compound gives rise to a [4]Al signal at 80–82 ppm,58,73 so the coating seen here is assigned to an amorphous “LiAlO2” phase bearing mostly [4]Al environments. The lower chemical shift suggest that this phase is likely lithium deficient in comparison to stoichiometric LiAlO2. Coating with 2 leads to two intense signals at 71 and 12.1 ppm, corresponding to [4]Al and [6]Al environments (Fig. S5(4.3.1)†) which result from the agglomeration of insoluble precursor particles on the NMC while coating with 1 result in two weak 27Al resonances at 67.1 and 10.3 ppm which are most likely due to a localized precipitate since no Al was detected by XPS on the NMC particles (Fig. 3).
 | ||
| Fig. 4 27Al MAS NMR of polycrystalline NMC811 materials coated using 3. The spectra were recorded at a magnetic field strength of 16.4 T and a MAS frequency of 50 kHz and normalized to the sample mass and the number of scans. The spinning sidebands are marked with asterisks. | ||
1H and 7Li SSNMR spectra of the pristine PC-NMC811 and PC-NMC811 soaked in water with and without an annealing step were then measured to study how the proton environments change after the water-soaking treatment and if Li is extracted during soaking (Fig. S5(4.1.1)†). After soaking in water and drying at 50 °C for 2 h a drastic increase in the intensity in 1H NMR environments corresponding to TM(O)–OH and carbonic acid (LiHCO3) is seen (Fig. S5(4.1.1)†). These changes indicate that water soaking promotes the formation of TM(O)–OH sites and that drying does not fully remove adsorbed water from the surface (Fig. S5(4.1.1)†). Lithium NMR is applied next to investigate the surface chemistry of these materials. This technique allows to distinguish between lithium in diamagnetic environments (surface) which resonates close to 0 ppm and lithium in paramagnetic environments (bulk), which are shifted by more than 100 ppm due to hyperfine interactions with the paramagnetic TM ions.74,75 Here, an increase in Li-containing surface species is seen in the 7Li NMR after soaking, suggesting that the soaking step extracts Li from the particles forming surface Li species (Fig. S5(4.1.1)†). After heating PC-NMC811 soaked in water at 400 °C, the 1H and 7Li (diamagnetic) resonances become less intense, as most of the surface water has been desorbed and surface lithium species have been re-incorporated in the lattice (Fig. S5(4.1.1)†).
Compared to the unsoaked PC-NMC811 coated with 3, coating the water-soaked PC-NMC811 with 3 results in a much more intense signal in the 27Al SSNMR spectrum (Fig. 4) in good agreement with the EDS and XPS results. The results do not support the presence of γ-Al2O3, which would have an [6]Al:[4]Al ratio higher than 1.60 Furthermore, our previous work showed that amorphous Al2O3 coatings deposited onto NMC811 by a solution method had an [6]Al:[4]Al ratio higher 1 but lithiation of those coatings by reaction with surface lithium impurities at 400 °C led to in an increase in [4]Al intensity so that [6]Al < [4]Al.20 The distribution of aluminium environments of the water-soaked PC-NMC811 coated with 3 is different from previous Al2O3 coatings formed by annealing at the same temperature (400 °C), with this coating showing a higher [4]Al content, as expected for LiAlO2.20 Two Al environments are seen at 72.6 and 14.1 ppm which are assigned to [4]Al and [6]Al, respectively (Fig. 4). Based on the peak positions ([4]Al signal expected at 80–82 ppm58,73 in γ-LiAlO2), the fact that annealing the precursor under air at this temperature led to an amorphous phase as determined by PXRD, and the higher Al content detected by XPS in the pre-soaked sample, these spectra are assigned to an amorphous LiAlO2/Al2O3 coating with the aluminium occupying [4]Al and [6]Al sites with [6]Al < [4]Al.
Pristine and coated SC-NMC811 materials were also studied by 27Al SSNMR (Fig. S5(4.3.3)†). In agreement with the EDS results, SSNMR shows that the pristine SC-NMC811 material contains aluminium. The line shape of the pristine SC-NMC811 spectrum is dominated by the [6]Al environments at 9.4 ppm, consistent with the presence of a (likely poorly) crystalline form of aluminium oxide, most likely boehmite (γ-AlOOH) based on the chemical shift.76 Furthermore, an extremely broad resonance centred at −800 ppm is present. The position and width of the peak is consistent with diffusion of Al3+ ions from the Al2O3 coating into the NMC811 induced by high-temperature annealing. This was further confirmed by PXRD which showed that there was splitting of all the reflections in the pristine SC-NMC811 material, which was particularly clear in reflections 108 and 110 (Fig. S5(2.3.4)†). This is indicative of the presence of two phases and becomes more pronounced after coating and annealing at 400 °C for 4 h under air (Fig. S5(2.3.6)†). The PXRD data was fitted to two phases by Rietveld refinement, and the two phases were assigned to NMC811 and an Al-doped NMC811.20,77 These results show that aluminium is present both in the surface of the pristine SC-NMC811 as a coating and in the bulk as dopant, suggesting that the as-received SC-NMC811 either have undergone a post-annealing treatment, or Al was introduced during mixing of the precursor starting materials.20 The 27Al SSNMR of the SC-NMC811 coated with 3 and dried at 100 °C under vacuum shows a new [4]Al environment at 67.4 ppm that was not observed in the pristine SC-NMC811 material. This, combined with the EDS results, which showed a uniform increase in surface aluminium content compared to the SC-NMC811 washed in THF, confirms that the coating deposition has taken place successfully. When the coated SC-NMC811 is heated under air at 400 °C for 4 h, the paramagnetic Al signal increases in intensity indicating growth of the Al-doped NMC811 phase.20
Electrochemical cycling
Long-term galvanostatic cycling of PC-NMC811 treated under the various conditions described in this study is shown in Fig. 5. First, the trends in capacity retention at slow (C/20) and fast (C/2) cycling rates will be discussed. Testing of the control sample prepared from PC-NMC811 soaked in THF but with no addition of 3 (Fig. 5a and four repeats, Fig. S5(5.1(b))†) consistently showed a reduction of capacity retention at both C/2 (13.7% less) and C/20 rates (10% less) as compared to the pristine NMC811 material. This is in contrast to the previous toluene washing experiments which showed that washing in toluene leads to a small improvement in capacity retention which was attributed to the removal of surface impurities.20 Unlike toluene, THF is a coordinating solvent with high affinity towards lithium so it could extract some of the lithium from the material. However, 7Li SSNMR does not support this hypothesis (Fig. S5(4.2.1)†) as the surface lithium peak does not change in intensity after soaking in THF. Alternatively, trace protic species present in the THF or the pristine material could be further reacting with NMC upon annealing reducing its capacity.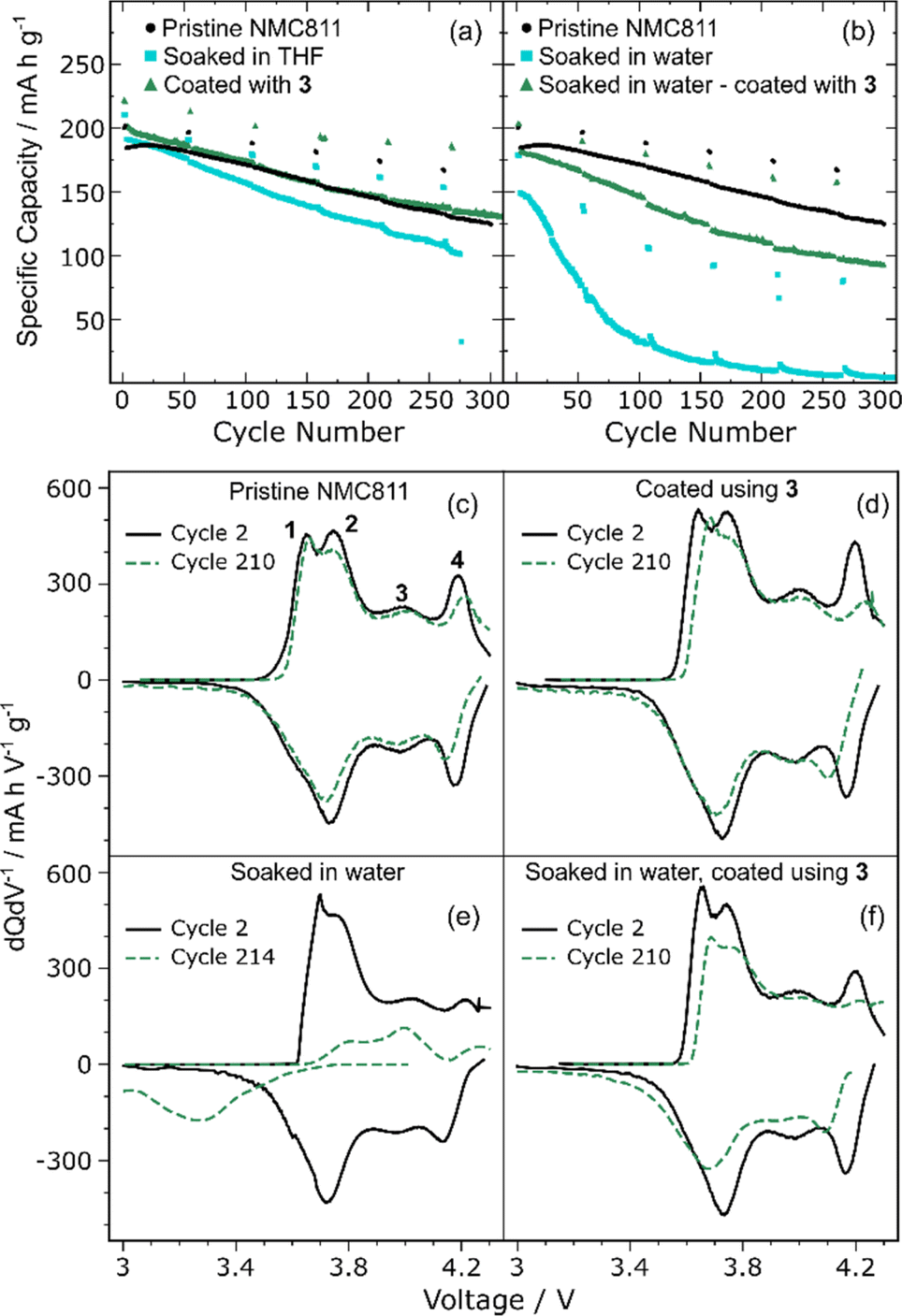 | ||
| Fig. 5 (a and b) Long-term electrochemical cycling data (specific discharge capacity/mA h g−1vs. cycle number). (a) Results for the pristine PC-NMC811, PC-NMC811 coated using 3 and control uncoated sample. (b) Results of PC-NMC811 soaked in water with and without a further coating step. These electrochemical measurements were carried out in half cell configuration (vs. Li) between 3 and 4.3 V. The cycling was performed at C/2 rate and slow C/20 cycles were included every 50 cycles. (c–f) dQ/dV profiles measured for the slow (C/20) cycles of PC-NMC811 samples. (c) Pristine PC-NMC811, (d) PC-NMC811 coated with 3, (e) PC-NMC811 soaked in water and then annealed at 400 °C under air. (f) PC-NMC811 soaked in water, coated with 3 and annealed under air at 400 °C. | ||
Coating PC-NMC811 with 3 led to discharge capacities and capacity retention values at C/2 and C/20 rates that were higher than or equal to that of pristine NMC811 (Fig. 5b, d and S5(5.1(d))†). For the data shown in Fig. 5, similar capacity retention is seen at C/20 for the coated and pristine sample (82.7 and 86.2% respectively) whereas a small improvement in capacity retention is seen at C/2. Compared to the control THF samples, there is a clear improvement in capacity retention for the coated materials which showed 6.5% more capacity retention at C/20 and 5.3% at C/2 (Fig. 5a). Furthermore, PC-NMC811 coated using 3 delivers higher capacity on charge and discharge on the second cycle without an increase in overpotential (Fig. S5(5.3(b))†). The coating process allows more Li to be extracted and re-inserted in the NMC811 without showing any sign of interfacial resistance in the C/20 cycling profile of cycle 2. These results provide evidence that coating polycrystalline NMC811 with LiAlO2 has a positive effect on the long-term capacity retention of the material during cycling in Li-ion half cells.
To investigate if long-term cycling had any effect on the coating structure, 27Al SSNMR was measured on cycled electrodes (Fig. 4 and S5(4.3.2)†). These were obtained after 300 charge–discharge cycles in half cells (more details on the electrochemical cycling in Section S2(2)†). The 27Al SSNMR spectra of the cycled samples is almost identical to that of the as-coated materials both in intensity and in chemical shift and distribution of aluminium environments, suggesting that the coating is quite stable under cycling conditions. The only difference was the appearance of a [6]Al environment in the cycled PC-NMC811 coated using 3 (not soaked) which could be the result of scraping some Al2O3 from the current collector or the deposition of some Al that was dissolved from the current collector into the electrolyte during cycling. In any case, the [4]Al peak remains unchanged.
Compared to the coating and THF soaking experiments, coating of the water-soaked NMC811 led to larger changes in the capacity retention of the materials. Soaking PC-NMC811 in water for 2 h at room temperature, followed by drying at 50 °C under vacuum led to a decrease in initial capacities (Fig. S5(5.1(c))†). Including an annealing step further decreases the capacity retention of the material with a 70.2% loss at C/2 and 47.3% at C/20 (Fig. 5b). These changes have been observed in the literature,16,29,30 and can be understood based on the lithium extraction that was seen by SSNMR. The long-term effect in the capacity retention indicates that the lithium extraction is accompanied by an irreversible structural change in the NMC811, presumably due to the evolution of Ni(O)–OH sites into NiO (giving H2O and O2 as side products).16 Furthermore, ion exchange of protons for lithium ions during soaking and subsequent drying may also contribute capacity fade.5,78 Although the rate of degradation is substantially increased both at C/2 and C/20 after soaking in water, it is much more marked at C/2 indicating that this loss of capacity is at least in part a kinetic effect, pointing to the formation of a resistive NiO (rock salt) layer on the surface.
If water-soaked PC-NMC811 is then coated, an impressive recovery of the capacity is observed (Fig. 5b, 37.8 vs. 78.8% capacity retention at C/20 and 22.8 vs. 58.2% at C/2). To ensure reproducibility, a duplicate of the measurement was performed on two different batches of PC-NMC811 materials subjected to the same soaking and coating treatment, and the same results were obtained (Fig. S5(5.1(e–h))†). To the best of our knowledge, this is the first example of regeneration of NMC degraded via exposure to water by using a surface coating. In order to see this recovery of capacity, our coating treatment must be reversing some of the structural changes happening during the water soaking and annealing. It is proposed that the improved capacity retention seen here proceeds by NiO oxidation (Ni2+ to Ni3+) and Li re-insertion,48 and possibly by removal of ion-exchanged protons and insertion of Li+ into the bulk of NMC811. Compared to previous work,48 which used LiOH to lithiate pristine NMC622 by annealing at 800 °C under oxygen, our method allows surface regeneration of degraded NMC811 at much lower temperature (400 °C) under air, and with the simultaneous formation of a beneficial amorphous “LiAlO2” coating. The formation of a lithium deficient lithium aluminate, which was promoted by water soaking (Fig. 4), was supported by 27Al SSNMR (and the observation of an [4]Al resonance at 58 ppm, i.e., at a chemical shift that is too low for stoichiometric LiAlO2, Fig. 4) and XPS (lower Li 1s:Al 2p ratio compared to unwashed PC-NMC811 coated with 3, Fig. 3). This is consistent with the idea that part of the lithium deposited from LiAl(OtBu)4 is inserted into PC-NMC811 beyond the depth of analysis of XPS, potentially explaining the recovery in electrochemical performance.
The pristine PC-NMC811 shows good reversibility in the dQ/dV profile (Fig. 5c) with highly symmetric charge–discharge curves. Clearly, most of the capacity fade comes from processes 2 and 4, which coincide with the start and end of charge states. The dQ/dV plot of cycle 2 of PC-NMC811 coated with 3 shows very similar features to the pristine PC-NMC811 suggesting that the coating treatment has not changed the electrochemistry of the material significantly on the first cycles. The dQ/dV peaks are more intense in cycle 2 (particularly processes 1 and 4, Fig. 5d) compared to the pristine NMC811 sample meaning that more capacity is obtained from these processes. The LiAlO2 coating may be enhancing this Li transport across the NMC/electrolyte interface thereby allowing higher specific capacities. Process 4 is suppressed after 210 cycles in both the pristine and the coated samples and the profile becomes less symmetric with the discharge peaks shifting to lower voltages. These results suggest that the coating is doing little to prevent the rock-salt formation which occurs spontaneously upon cycling, especially in PC-NMC811 where significant particle cracking is expected (exposing fresh surfaces to electrolyte). The improvements in capacity retention seen for PC-NMC811 coated with 3 (Fig. 5a) are likely to come from an initial rearrangement of the structure, with Li re-insertion and possibly less electrolyte oxidation, as the surface of the CAM is at least partially covered by LiAlO2.
The dQ/dV analysis of the PC-NMC811 soaked in water and annealed under air shows that the loss in capacity comes mainly from the suppression of the high voltage process while process 1 is shifted to higher voltages (Fig. 5e). Furthermore, significant polarization is observed even at cycle 2. This is all consistent with the hypothesis of the washing treatment creating a thick rock-salt/disordered and protonated NMC layer on the surface. At cycle 214, a completely different dQ/dV profile is observed, suggesting that the material is fully degraded. The peaks corresponding to processes 1 and 4 are almost fully suppressed on charge with the presence of a broad peak only at low voltages on discharge. Coating the PC-NMC811 soaked in water using 3 leads to a recovery of the capacity and an electrochemistry that is again much closer to that of the pristine PC-NMC811 (Fig. 5f). The recovery of capacity comes from processes, 2 and 4 which were suppressed after soaking in water but reappear after coating with 3. Furthermore, reversible electrochemical behaviour with the expected NMC811 peaks is observed at cycle 210, in contrast to the water-soaked, uncoated PC-NMC811.
Finally, the electrochemical cycling performance of pristine SC-NMC811, SC-NMC811 coated with 3 and the soaked in water/coated SC-NMC811 were also compared in half cells (Fig. 6, S5(5.2), S5(5.4) and S5(5.5)†). Specific discharge capacities as a function of cycle number are shown in Fig. 6. The y-axis has been changed to 100–250 mA h g−1 (as opposed to 0–300 mA h g−1 in the previous figure) to visualize the smaller changes in specific capacity and capacity retention better. Since the SC-NMC811 is already surface coated with Al2O3 and also because of the differences in morphology, the response to the soaking and coating treatments is quite different from that observed for PC-NMC811.
 | ||
| Fig. 6 Long-term cycling of SC-NMC811 materials over 100 cycles. Pristine, soaked in THF, soaked in water, coated with 3 and soaked in water/coated with 3 (all annealed at 400 °C) are shown in the figure. The y axis (specific capacity/mA h g−1) starts at 100 mA h g−1 in this figure to better visualize the differences in capacity retention between the samples. | ||
In contrast to PC-NMC811, soaking in THF does not harm the capacity retention of the SC-NMC811. The capacity retention was the same at C/20 (84.3–84.4%) for the pristine and THF-soaked SC-NMC811 and slightly higher for the THF soaked SC-NMC811 at C/2 (78.2 vs. 75.6%). Since SC-NMC811 was already coated and surface doped, it is possible that it has a surface that is less reactive towards THF or moisture that could have been introduced during the annealing step at 400 °C in air. The specific discharge capacities were ca. 10 mA h g−1 higher for the THF soaked SC-NMC811 over three repeats (Fig. S5(5.2(b))†). The EDS results (Fig. 3) showed lower Al content in SC-NMC811 after washing in THF suggesting partial removal of the insulating Al2O3 layer, which may be helping with the cycling performance.20–22
Soaking SC-NMC811 in water following the same procedure as for PC-NMC811 led to a larger gap in between the C/20 and C/2 first cycles indicating that the surface of SC-NMC811 has become more resistive after soaking in water, kinetically limiting the discharge capacity at C/2 (Fig. 6). Although this SC-NMC811 is less affected by water soaking than PC-NMC811, these results again suggest that the Al2O3 coating does not fully prevent the formation of rock-salt. Nevertheless, the capacity retention improved after soaking in water compared to pristine SC-NMC811, both at C/2 and C/20 C-rates (87.9 and 85.9% for the soaked SC-NMC811 at C/20 and C/2 vs. 84.4 and 75.6% for the pristine SC-NMC811). It is thought that the mechanism of degradation of PC-NMC811 by soaking in water proceeds by a combination of lithium leaching due to H+/Li+ exchange (with formation of LiOH as determined by HCl titrations),16 decreased mechanical strength as measured by load-compression testing,29 and cracking, since PC-NMC811 particle sizes obtained by SEM were observed to decrease after prolonged mixing in water.29 It is therefore possible that SC-NMC811 is less prone to degradation by soaking in water due to its better mechanical integrity. Furthermore, the Al2O3 coating and surface doping of the as-received SC-NMC811 may be preventing delithiation by H2O and formation of TM(O)–OH sites therefore reducing the degree of degradation caused in this soaking step. Finally, soaking in water may be removing some of the soluble surface lithium salts (Li2CO3, LiOH) which are known to cause capacity fade in NMC materials.
The SSNMR studies showed that coating SC-NMC811 using 3 leads to the deposition of an amorphous LiAlO2 layer on top of the Al2O3 coating (Fig. S5(4.3.3)†). This double coating is expected to cause a kinetic limitation during cycling owing to the two interphases present and the resulting increase in coating thickness. This is consistent with the cycling data, where a larger gap between the first C/2 and C/20 cycles is seen for the material coated using 3 compared to the pristine material (Fig. 6 and S5(5.2(d))†). Interestingly, the capacity retention improves both at C/2 and C/20 compared to both the pristine SC-NMC811 and the SC-NMC811 soaked in water (6.9% more capacity retention at C/2 and 6.1% more at C/20 compared to the SC-NMC811 soaked in water). It is possible that the coating deposition is covering regions of the particles that were not originally coated improving the surface coverage. Furthermore, the addition of surface Li+-ion containing species may stabilize the structure, improving the performance. SC-NMC811 coated with 3 showed 95% capacity retention after 100 cycles (17% improvement over the pristine material), placing it among one of the best performing LiAlO2 coated NMC materials with Ni >60% in terms of capacity retention, only matched by the work of Yu et al.,79 which reported a capacity retention of 97.4% for LiAlO2-coated LiNi0.9Co0.1O2 after 100 cycles in half cells although with a higher initial capacity of 199 mA h g−1 owing to the higher nickel content and lack of aluminium doping (see Tables S5(5.4.1) and S5(5.4.2)† for a full literature comparison). Finally, in the same way as with PC-NMC811, soaking SC-NMC811 in water induces more coating deposition (as shown by EDS, Fig. 2). This thick coating deposited on a coated NMC811 material leads to a very substantial kinetic limitation in the electrochemical cycling with a large gap in C/2–C/20 discharge capacity. This material still showed 3% more capacity retention than the pristine SC-NMC811 and the SC-NMC811 soaked in water at C/20, but similar capacity retention and lower specific capacities at C/2, owing to the thicker double coating present.
Conclusions
In this paper, the surface of NMC811 is coated with a LiAlO2-like amorphous coating to mitigate NMC811 degradation under electrochemical cycling conditions. LiAlO2 is an excellent coating material, as it creates a lithium-rich surface and is a Li-ion conductor composed of cheap, non-toxic elements. However, current methods to deposit ternary oxides present limitations in terms of scalability, and homogeneity of the deposited layer. To overcome these problems, a new solution deposition method using a heterobimetallic SSP is developed. First, the synthesis of the known alkoxide Li[Al(OCH2Ph)4] (1) was extended to two new compounds: Li[Al(OiPr)4] (2) and Li[Al(OtBu)4] (3). A study of the structure of the precursors as they thermolyze was then undertaken using solution and solid-state NMR, TGA, PXRD and elemental analysis. The –OR group (–OCH2Ph, –OiPr and –OtBu) was found to have a decisive influence on properties of the precursors such as solubility, thermal decomposition mechanism, crystallization temperature, phases formed upon decomposition and reactivity with the NMC surface. Although the three precursors formed γ-LiAlO2 at 800 °C and amorphous phases below this temperature, only Li[Al(OtBu)4] (3) had the required properties to act as a precursor to LiAlO2 coatings of NMC811, namely: it was soluble in an inert solvent, reacted with NMC811 forming a coating layer by a hydrolytic route, and decomposed at low temperatures (<400 °C) allowing the formation of an amorphous, LiAlO2-like coating without causing surface aluminium doping.The coating method was developed and tested on NMC811 materials with different morphologies (single-crystal vs. polycrystalline NMC) and surface chemistries (Al2O3 coated vs. pristine surface). By a combination of electron microscopy, XPS, PXRD and SSNMR, the changes in the surface and bulk of the material with soaking in THF and H2O, and after annealing at 400 °C under air were systematically investigated, in parallel to the surface changes due to the coating. 1H and 7Li SSNMR measurements showed that the surface of NCM811 is delithiated after soaking in water and that the number of TM(O)–OH sites increases. This surface change increased the amount of LiAlO2 deposited under the same conditions providing direct evidence that LiAlO2 deposition proceeds via hydrolysis of the alkoxides with surface TM(O)–OH groups and adsorbed water. Finally, the electrochemical properties of the materials were evaluated in Li-ion half-cells by long-term galvanostatic cycling. It was found that the coating improved the specific capacity and capacity retention of Al2O3 coated single-crystal NMC811 significantly (17% more capacity retained at C/2 after 100 cycles), while having only a small effect on the polycrystalline material. Interestingly, coating polycrystalline NMC811 that was soaked in water allowed an impressive recovery of 50% of the capacity retention while also showing higher specific capacities, possibly by regeneration of the surface layered structures that were reduced to rock-salt by the water treatment.
This work represents the first example of using heterobimetallic precursors to coat battery materials with a LiAlO2-like amorphous coatings. Our method is straightforward and uses environmentally benign and cheap precursors and should be easily scalable. This coating strategy improves the capacity retention of Al2O3 coated SC-NMC811 as well as regenerating fully degraded PC-NMC811. Furthermore, the work provides insight into the complex relationship between morphology, surface chemistry and electrochemical lifetime in Ni-rich cathode materials for Li-ion batteries. The most important implication of this work is that it provides a potential method for regenerating and recovering NMC811 that has been exposed to ambient moisture, potentially allowing its large-scale processing without the need for strict humidity control. The approach can readily be applied to other reactive Ni-rich layered cathode materials or more generally to lithium–metal oxides that suffer from similar degradation mechanisms.
Data availability
Data for this paper is available in the ESI† file.Author contributions
VRG conducted the research, analysed data, and wrote the original draft. CAO performed solid-state NMR experiments, analysed data, and reviewed/edited the draft. CPG and DSW acquired the funding, supervised VRG, analysed data and reviewed/edited the draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Faraday Institution Degradation Project (grant no. FIRG001 and FIRG024). VRG was supported by the EPSRC training grant EP/S514901/1. We wish to acknowledge the support of the Henry Royce Institute for advanced materials for VRG through the Student Equipment Access Scheme enabling access to the XPS facilities at the University of Oxford; EPSRC Grant Number EP/R00661X/1.References
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 Search PubMed.
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 Search PubMed.
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS PubMed.
- B. L. D. Rinkel, J. P. Vivek, N. Garcia-Araez and C. P. Grey, Energy Environ. Sci., 2022, 15, 3416–3438 RSC.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A378–A389 CrossRef CAS.
- Z. Ruff, C. Xu and C. P. Grey, J. Electrochem. Soc., 2021, 168, 060518 Search PubMed.
- C. Xu, K. Märker, J. Lee, A. Mahadevegowda, P. J. Reeves, S. J. Day, M. F. Groh, S. P. Emge, C. Ducati, B. Layla Mehdi, C. C. Tang and C. P. Grey, Nat. Mater., 2021, 20, 84–92 Search PubMed.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 Search PubMed.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 Search PubMed.
- H. Li, A. Liu, N. Zhang, Y. Wang, S. Yin, H. Wu and J. R. Dahn, Chem. Mater., 2019, 31, 7574–7583 Search PubMed.
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS.
- A. O. Kondrakov, A. Schmidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 3286–3294 CrossRef CAS.
- S. Schweidler, L. de Biasi, G. Garcia, A. Mazilkin, P. Hartmann, T. Brezesinski and J. Janek, ACS Appl. Energy Mater., 2019, 2, 7375–7384 CrossRef CAS.
- N. V. Faenza, L. Bruce, Z. W. Lebens-Higgins, I. Plitz, N. Pereira, L. F. J. Piper and G. G. Amatucci, J. Electrochem. Soc., 2017, 164, A3727–A3741 CrossRef CAS.
- D. Pritzl, T. Teufl, A. T. S. Freiberg, B. Strehle, J. Sicklinger, H. Sommer, P. Hartmann and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A4056–A4066 Search PubMed.
- C. Li, H. P. Zhang, L. J. Fu, H. Liu, Y. P. Wu, E. Rahm, R. Holze and H. Q. Wu, Electrochim. Acta, 2006, 51, 3872–3883 Search PubMed.
- Z. Chen, Y. Qin, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7606–7612 Search PubMed.
- S.-T. Myung, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7074–7095 RSC.
- V. Riesgo-González, D. S. Hall, K. Märker, J. Slaughter, D. S. Wright and C. P. Grey, Chem. Mater., 2022, 34, 9722–9735 Search PubMed.
- B. Han, B. Key, S. H. Lapidus, J. C. Garcia, H. Iddir, J. T. Vaughey and F. Dogan, ACS Appl. Mater. Interfaces, 2017, 9, 41291–41302 Search PubMed.
- N. Phattharasupakun, C. Geng, M. B. Johnson, R. Väli, A. Liu, Y. Liu, M. Sawangphruk and J. R. Dahn, J. Electrochem. Soc., 2020, 167, 120531 CrossRef CAS.
- X. Xiong, Z. Wang, P. Yue, H. Guo, F. Wu, J. Wang and X. Li, J. Power Sources, 2013, 222, 318–325 Search PubMed.
- J. Kim, Y. Hong, K. S. Ryu, M. G. Kim and J. Cho, Electrochem. Solid-State Lett., 2006, 9, A19–A23 Search PubMed.
- S. Jeong, J. Kim and J. Mun, J. Electrochem. Soc., 2019, 166, A5038–A5044 Search PubMed.
- X. Zheng, X. Li, Z. Wang, H. Guo, Z. Huang, G. Yan and D. Wang, Electrochim. Acta, 2016, 191, 832–840 Search PubMed.
- M. Hofmann, M. Kapuschinski, U. Guntow and G. A. Giffin, J. Electrochem. Soc., 2020, 167, 140535 Search PubMed.
- W. Lee, D. Lee, Y. Kim, W. Choi and W.-S. Yoon, J. Mater. Chem. A, 2020, 8, 10206–10216 Search PubMed.
- L. Azhari, X. Zhou, B. Sousa, Z. Yang, G. Gao and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 57963–57974 Search PubMed.
- W. Lee, S. Lee, E. Lee, M. Choi, R. Thangavel, Y. Lee and W.-S. Yoon, Energy Storage Mater., 2022, 44, 441–451 CrossRef.
- M. Wood, J. Li, R. E. Ruther, Z. Du, E. C. Self, H. M. Meyer, C. Daniel, I. Belharouak and D. L. Wood, Energy Storage Mater., 2020, 24, 188–197 Search PubMed.
- M. Hofmann, M. Kapuschinski, U. Guntow and G. A. Giffin, J. Electrochem. Soc., 2020, 167, 140512 Search PubMed.
- H.-H. Ryu, H.-W. Lim, S. G. Lee and Y.-K. Sun, Nat. Energy, 2023, 9, 47–56 Search PubMed.
- Y. Xie, J. Li, M. Li, J. Cai, X. Huang, H. Nguyen, L. Yu, D. Huo, Z. Yang, N. Karami, B. A. S. Sulaiman, N. A. Chernova, S. Upreti, B. Prevel, Y. Liu, F. Wang and Z. Chen, Adv. Funct. Mater., 2024, 34, 2311551 CrossRef CAS.
- S. E. Renfrew, L. A. Kaufman and B. D. McCloskey, ACS Appl. Mater. Interfaces, 2019, 11, 34913–34921 Search PubMed.
- S. Maiti, H. Sclar, R. Sharma, N. Vishkin, M. Fayena-Greenstein, J. Grinblat, M. Talianker, L. Burstein, N. Solomatin, O. Tiurin, Y. Ein-Eli, M. Noked, B. Markovsky and D. Aurbach, Adv. Funct. Mater., 2021, 31, 2008083 Search PubMed.
- C. Chen, W. Yao, Q. He, M. Ashuri, J. Kaduk, Y. Liu and L. Shaw, ACS Appl. Energy Mater., 2019, 2, 3098–3113 Search PubMed.
- W. Liu, X. Li, D. Xiong, Y. Hao, J. Li, H. Kou, B. Yan, D. Li, S. Lu, A. Koo, K. Adair and X. Sun, Nano Energy, 2018, 44, 111–120 Search PubMed.
- M. J. Herzog, N. Gauquelin, D. Esken, J. Verbeeck and J. Janek, ACS Appl. Energy Mater., 2021, 4, 8832–8848 Search PubMed.
- B. Song, W. Li, S.-M. Oh and A. Manthiram, ACS Appl. Mater. Interfaces, 2017, 9, 9718–9725 Search PubMed.
- S. Cangaz, F. Hippauf, R. Takata, F. Schmidt, S. Dörfler and S. Kaskel, Batteries Supercaps, 2022, 5, e202200100 Search PubMed.
- J. Xie, A. D. Sendek, E. D. Cubuk, X. Zhang, Z. Lu, Y. Gong, T. Wu, F. Shi, W. Liu, E. J. Reed and Y. Cui, ACS Nano, 2017, 11, 7019–7027 CrossRef CAS PubMed.
- S. H. Akella, S. Taragin, Y. Wang, H. Aviv, A. C. Kozen, M. Zysler, L. Wang, D. Sharon, S. B. Lee and M. Noked, ACS Appl. Mater. Interfaces, 2021, 13, 61733–61741 Search PubMed.
- M. Seenivasan, C. Yang, S. Wu, Y.-J. J. Li, W.-C. Chien, S. Piraman and S. J. Lue, Electrochim. Acta, 2021, 387, 138620 CrossRef CAS.
- S. L. Dreyer, K. R. Kretschmer, Đ. Tripković, A. Mazilkin, R. Chukwu, R. Azmi, P. Hartmann, M. Bianchini, T. Brezesinski and J. Janek, Adv. Mater. Interfaces, 2022, 9, 2101100 Search PubMed.
- H. Lu, D. S. Wright and S. D. Pike, Chem. Commun., 2020, 56, 854–871 RSC.
- J. S. Park, X. Meng, J. W. Elam, S. Hao, C. Wolverton, C. Kim and J. Cabana, Chem. Mater., 2014, 26, 3128–3134 Search PubMed.
- F. Zhang, S. Lou, S. Li, Z. Yu, Q. Liu, A. Dai, C. Cao, M. F. Toney, M. Ge, X. Xiao, W.-K. Lee, Y. Yao, J. Deng, T. Liu, Y. Tang, G. Yin, J. Lu, D. Su and J. Wang, Nat. Commun., 2020, 11, 3050 Search PubMed.
- E. R. Logan, H. Hebecker, X. Ma, J. Quinn, Y. HyeJeong, S. Kumakura, J. Paulsen and J. R. Dahn, J. Electrochem. Soc., 2020, 167, 060530 CrossRef CAS.
- H. Zhang, X. He, Z. Chen, Y. Yang, H. Xu, L. Wang and X. He, Adv. Energy Mater., 2022, 12, 2202022 Search PubMed.
- Q. Huang, L. Ma, A. Liu, X. Ma, J. Li, J. Wang and J. R. Dahn, J. Power Sources, 2018, 390, 78–86 CrossRef CAS.
- E. Trevisanello, R. Ruess, G. Conforto, F. H. Richter and J. Janek, Adv. Energy Mater., 2021, 11, 2003400 Search PubMed.
- J. Pauls and B. Neumüller, Z. Anorg. Allg. Chem., 2000, 626, 270–279 Search PubMed.
- The Future of Hydrogen, IEA, Paris, 2019, https://www.iea.org/reports/the-future-of-hydrogen Search PubMed.
- H. Nöth, A. Schlegel, J. Knizek, I. Krossing, W. Ponikwar and T. Seifert, Chem.–Eur. J., 1998, 4, 2191–2203 Search PubMed.
- M. Haouas, F. Taulelle and C. Martineau, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94–95, 11–36 Search PubMed.
- E. Lam, A. Comas-Vives and C. Copéret, J. Phys. Chem. C, 2017, 121, 19946–19957 Search PubMed.
- D. Wohlmuth, V. Epp, P. Bottke, I. Hanzu, B. Bitschnau, I. Letofsky-Papst, M. Kriechbaum, H. Amenitsch, F. Hofer and M. Wilkening, J. Mater. Chem. A, 2014, 2, 20295–20306 Search PubMed.
- D. Wiedemann, S. Nakhal, J. Rahn, E. Witt, M. M. Islam, S. Zander, P. Heitjans, H. Schmidt, T. Bredow, M. Wilkening and M. Lerch, Chem. Mater., 2016, 28, 915–924 Search PubMed.
- C. V. Chandran, C. E. A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Chem. Soc. Rev., 2019, 48, 134–156 Search PubMed.
- R. Famery, F. Queyroux, J.-C. Gilles and P. Herpin, J. Solid State Chem., 1979, 30, 257–263 Search PubMed.
- R. Hoppe and H. König, Z. Anorg. Allg. Chem., 1977, 430, 211–217 Search PubMed.
- L. M. Carrera, J. Jimenez-Becerril, P. Bosch and S. Bulbulian, J. Am. Ceram. Soc., 1995, 78, 933–938 Search PubMed.
- K. Kinoshita, J. W. Sim and J. P. Ackerman, Mater. Res. Bull., 1978, 13, 445–455 Search PubMed.
- M. A. Valenzuela, J. Jimenez-Becerril, P. Bosch, S. Bulbulian and V. H. Lara, J. Am. Ceram. Soc., 1996, 79, 455–460 Search PubMed.
- S.-I. Hirano, T. Hayashi and T. Kageyama, J. Am. Ceram. Soc., 1987, 70, 171–174 Search PubMed.
- B. Han, T. Paulauskas, B. Key, C. Peebles, J. S. Park, R. F. Klie, J. T. Vaughey and F. Dogan, ACS Appl. Mater. Interfaces, 2017, 9, 14769–14778 Search PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 Search PubMed.
- K. W. Terry, C. G. Lugmair and T. D. Tilley, J. Am. Chem. Soc., 1997, 119, 9745–9756 CrossRef CAS.
- E. Björklund, C. Xu, W. M. Dose, C. G. Sole, P. K. Thakur, T.-L. Lee, M. F. L. De Volder, C. P. Grey and R. S. Weatherup, Chem. Mater., 2022, 34, 2034–2048 CrossRef PubMed.
- A. T. Appapillai, A. N. Mansour, J. Cho and Y. Shao-Horn, Chem. Mater., 2007, 19, 5748–5757 CrossRef CAS.
- Y. Shu, Y. Xie, W. Yan, S. Meng, D. Sun, Y. Jin and L. Xiang, Ceram. Int., 2020, 46, 14840–14846 Search PubMed.
- D. Müller, W. Gessner and G. Scheler, Polyhedron, 1983, 2, 1195–1198 Search PubMed.
- C. P. Grey and N. Dupré, Chem. Rev., 2004, 104, 4493–4512 CrossRef CAS PubMed.
- K. Märker, P. J. Reeves, C. Xu, K. J. Griffith and C. P. Grey, Chem. Mater., 2019, 31, 2545–2554 CrossRef.
- K. J. D. MacKenzie, J. Temuujin, M. E. Smith, P. Angerer and Y. Kameshima, Thermochim. Acta, 2000, 359, 87–94 CrossRef CAS.
- F. Dogan, J. T. Vaughey, H. Iddir and B. Key, ACS Appl. Mater. Interfaces, 2016, 8, 16708–16717 Search PubMed.
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. T. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489–A1498 CrossRef CAS.
- H. Yu, Y. Cao, L. Chen, Y. Hu, X. Duan, S. Dai, C. Li and H. Jiang, Nat. Commun., 2021, 12, 4564 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03795b |
| ‡ VRG current address: Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA. |
| This journal is © The Royal Society of Chemistry 2024 |