
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnvironmentally responsible synthesis of high-performance P2-Na2/3[Ni1/3Mn2/3]O2 sodium-ion battery cathodes†
Jintao
Fu
 ,
Mohamed H.
Hassan
,
Jiaxin
Liu
,
Hyeongjun
Koh
,
Alexander K.
Ng
,
Chiara
Bruzzi
,
John S.
Corsi
and
Eric
Detsi
*
,
Mohamed H.
Hassan
,
Jiaxin
Liu
,
Hyeongjun
Koh
,
Alexander K.
Ng
,
Chiara
Bruzzi
,
John S.
Corsi
and
Eric
Detsi
*
Department of Materials Science & Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: detsi@seas.upenn.edu
First published on 13th September 2024
Abstract
P2-type Na2/3[Ni1/3Mn2/3]O2 (NNMO) is a promising Na-ion battery cathode for its high operation voltage, high gravimetric capacity, and low critical element content. Current methods to make NNMO, such as sol–gel, solid-state reaction, and spray pyrolysis, are not environmentally friendly, as they often use acetate and nitrate metal salt precursors, which release undesirable gaseous byproducts such as CO2 and HNO3 when these metal salts are heat-treated at high temperatures. Decarbonizing our industries and the transportation sector requires not only using batteries to store intermittent renewable energy, but also decarbonizing the synthesis methods to make key components of these batteries, i.e., the electrode materials. Here, we demonstrate an environmentally responsible synthesis of NNMO by direct conversion of metallic Ni powder with various particle sizes (10 nm, 100 nm, 1 μm, 5 μm), a process that only releases O2 and H2O as byproducts, as opposed to the harmful gases released when nickel salts are used. The resulting NNMO made from ≈1 μm commercial Ni powder exhibits an excellent cycling performance as a Na-ion battery cathode. When cycled between 2-4 V vs. Na/Na+, the material exhibits a rate-capability of ≈83 mA h g−1 at 10C (i.e., > 96% theoretical capacity) and ≈ 78 mA h g−1 at 20C (i.e., >90% theoretical capacity), together with a remarkable stability of 93% capacity retention after 500 cycles at 10C. When cycled between 1.3–4 V vs. Na/Na+, the material shows a high initial capacity of ≈134 mA h g−1 at 2C, with a capacity retention of 57% after 500 cycles.
1. Introduction
Sodium-ion batteries (SIBs) have emerged as a promising alternative to Li-ion batteries (LIBs) due to the abundance of their constituent elements, such as Na and Mn, in contrast to LIBs, which heavily rely on critical elements such as Li and Co.1 Among the different SIB components, cathode materials with fast Na storage kinetics, high operating potentials, and relatively high specific capacity are desirable.2 To date, various types of cathode materials have been developed, including Prussian blue analogues,3 polyanion compounds,4 and layered transition metal oxides.5 P2-type Na2/3[Ni1/3Mn2/3]O2 (NNMO), where “P” stands for Na occupying prismatic sites and “2” represents the number of transition metal layers in a repeating unit, is one of the most promising layered transition metal oxides offering a high operation voltage, a high specific capacity (173 mA h g−1 when all the 2/3 Na is (de)intercalated), and high air stability.6 Furthermore, NNMO only contains 33 at% of Ni, its only critical element, as opposed to the state-of-the-art NMC 811 cathode which contains 80 at% Ni together with Li and Co.7 Despite such advantages, the main challenge currently impeding the widespread adoption of NNMO as one of the next-generation cathode materials for SIBs is the structural change during cycling. Indeed, when cycled between 2–4.5 V vs. Na/Na+, NNMO undergoes a P2–O2 phase transition, accompanied by >20% volume change, leading to capacity decay and cell failure.8 Extensive research efforts have been directed towards mitigating issues related to this P2–O2 phase transition, which include strategies such as cation/anion doping,9 protective layer coating,10 and nanoengineering.6On the other hand, less attention has been paid to issues related to the synthesis of NNMO. Traditional methods to make NNMO, such as sol–gel,11 solid-state reaction,10 and spray pyrolysis,12 usually involve the use of metal salt precursors including nitrates and acetates, which subsequently release undesirable gaseous byproducts of NOx (from nitrates) and COx (from acetates) when annealed at high temperature in air.13,14 These emissions pose significant environmental hazards, contradicting the vision of clean and sustainable energy production. In this work, we introduce a novel protocol to fabricate P2-NNMO directly from metallic Ni and sodium permanganate (NaMnO4). This protocol eliminates the need for nickel salts and the release of toxic gases, a common drawback in conventional synthesis methods. In addition to the sustainable synthesis route, the resulting NNMO exhibits a remarkable cycling performance comparable to that of NNMO synthesized from a traditional approach. For example, when cycled between 2–4 V vs. Na/Na+ (utilizing half of Na in the formula, and yielding ≈86 mA h g−1 theoretical capacity15), NNMO synthesized from commercial Ni powder with an average particle size of 1 μm delivers a specific capacity of ≈83 mA h g−1 at 10C (i.e., >96% theoretical capacity) and ≈78 mA h g−1 at 20C (i.e., > 90% theoretical capacity). Further, it exhibits outstanding cycling stability as well, retaining 93% of its original capacity after 500 cycles at 10C. When cycled in a broader voltage window of 1.3–4 V vs. Na/Na+, the material shows a high initial capacity of ≈134 mA h g−1 at 2C, with a capacity retention of ≈80% after 180 cycles and ≈57% after 500 cycles. This combination of a sustainable synthesis approach and excellent electrochemical performance holds significant promise for advancing the development of next-generation SIB cathodes.
2. Experimental
2.1 Materials preparation
Four types of metallic Ni were utilized as precursors for the synthesis of P2-Na2/3[Ni1/3Mn2/3]O2. Three commercial Ni powders purchased from Sigma Aldrich and Thermo Scientific, comprised of (i) Ni nanopowder with 80–150 nm average particle size (99.8%, Thermo Scientific Chemicals), denoted as 100 nm Ni in this article; (ii) 1 μm Ni powder (99.8%, Sigma Aldrich); and (iii) Ni powder with 3–7 μm mean particle size (99.8%, Sigma Aldrich), denoted as 5 μm Ni in this article. The fourth type of Ni was nanoporous Ni (NP-Ni) fabricated by dealloying.16 In doing so, we first prepared a Ni30Mn70 at% alloy ingot by mixing Ni powder (99.8%, Sigma Aldrich) and Mn powder (99.99%, Fisher Scientific) with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 and melting this mixture under an argon atmosphere using an electric arc-melter (EQ-SP-MSM207 from MTI). Afterward, the ingot was cold rolled into thin foils and immersed in 1 M ammonium sulfate solution for 6 days to fully dissolve the sacrificial Mn atoms by free corrosion dealloying. After dealloying, the product was thoroughly rinsed 5 times with Deionized (DI) H2O, dried under vacuum overnight, and stored in air for NNMO synthesis.
:2.2 and melting this mixture under an argon atmosphere using an electric arc-melter (EQ-SP-MSM207 from MTI). Afterward, the ingot was cold rolled into thin foils and immersed in 1 M ammonium sulfate solution for 6 days to fully dissolve the sacrificial Mn atoms by free corrosion dealloying. After dealloying, the product was thoroughly rinsed 5 times with Deionized (DI) H2O, dried under vacuum overnight, and stored in air for NNMO synthesis.
In a typical NNMO synthesis process, 100 mg of metallic Ni powder was mixed with 560 mg of sodium permanganate monohydrate (NaMnO4·H2O) (97%, Sigma Aldrich) and 1 mL 1 M NaOH solution. The NaOH was used to provide an extra Na source to compensate for Na loss during high-temperature solid-state conversion.10,12 The mixture was heated on a hot plate at 150 °C in air to evaporate water, followed by ball-milling in an agate jar (Across International) for one hour. An exception was made for the mixture containing 100 nm Ni as the sample was put into an agate jar without milling balls to avoid particle coarsening.17 Finally, solid-state conversion was performed between 800 and 900 °C in a muffle furnace (Thermo Scientific) in air, followed by rapid cooling to room temperature.18
For comparison, NNMO was also synthesized using the traditional approach, by mixing metal salts of sodium, nickel, and manganese acetates. Specifically, sodium acetate trihydrate (2.1 mmol, Sigma Aldrich), nickel acetate tetrahydrate (1 mmol, Sigma Aldrich), and manganese acetate tetrahydrate (2 mmol, Sigma Aldrich) were dissolved in 3 mL DI H2O. Oxalic acid (5.1 mmol, Sigma Aldrich) dissolved in 4 mL DI H2O was added to the previous solution under stirring, forming a white precipitate. This mixture was heated to 90 °C until it became dry, and the collected powder was annealed at 500 °C for 5 h and then at 950 °C for 15 h in air with a heating rate of 2 °C min−1.
In the following sections, the NNMO converted from NP-Ni, 100 nm Ni, 1 μm Ni, 5 μm Ni, and Ni acetate (NiAc) are denoted as NP-Ni-NNMO, 100 nm Ni-NNMO, 1 μm Ni-NNMO, 5 μm Ni-NNMO, and NiAc-NNMO, respectively.
2.2 Materials characterization
X-ray diffraction (XRD) was performed using a Rigaku Miniflex powder diffractometer (Cu X-ray source, operated at 40 kV/15 mA) with a 0.02° step size and a 4° min−1 scanning speed. The morphology of the starting metallic Ni and converted NNMO was studied by scanning electron microscopy (SEM) using a JEOL 7500F equipped with energy-dispersive X-ray spectroscopy (EDS). The chemical compositions of 1 μm Ni-NNMO and NiAc-NNMO were determined by inductively coupled plasma optical emission spectroscopy (Spectro Genesis ICP-OES device). Small-angle X-ray scattering (SAXS) was performed using a XENOCS Xeuss 2.0 Environmental X-ray Scattering instrument with dual copper and molybdenum X-ray sources at Penn, which has a detection limit between 1 Å and 570 nm.19–23 Transmission electron microscopy (TEM) characterization was performed with JEOL F200 operated at 200 kV, with NNMO samples mixed with 3 mL of DI H2O and drop-casted on lacey carbon TEM grids (Ted Pella). Zone axes such as [100] and [001] were carefully selected based on simulated Kikuchi patterns.24,25 The selected area electron diffraction (SAED) patterns were captured at a dose rate of 5 e−/Å2 s for 1.2 s with a OneView camera (Gatan). High-resolution images were acquired at 400 e−/Å2 s for 0.6 s.2.3 Electrochemical measurements
2032 type coin cells were assembled using NNMO slurry as working electrodes, Na metal as counter and reference electrodes, and glass microfiber (Whatman, Grade GF/C) as the separator in an argon-filled glove box (MBraun) with H2O/O2 content below 0.1 ppm. The NNMO slurry was prepared by homogeneously mixing 70 wt% active material (NNMO powder), 20 wt% carbon additives (equal mass of carbon black, carbon nanofibers, and graphene nanosheets), and 10 wt% binder (40 mg mL−1 of polyvinylidene fluoride dissolved in N-methyl-2-pyrrolidone), and was cast on Al foil current collector (18 μm thick, Fisher Scientific). The slurry coating was then dried under vacuum at room temperature overnight, followed by heating at 150 °C under argon atmosphere for 2 h to remove residual water. After drying, circular disks of the working electrode with active material loading 1.6 ± 0.4 mg cm−2 were cut with a disc cutter (MSK-T-09, MTI Corp). The electrolyte used in coin cells was 1 M sodium perchlorate in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 by volume) with 5 vol% addition of fluoroethylene carbonate (FEC). All electrochemical tests were performed using a Biologic VMP-300 multichannel potentiostat. Electrochemical impedance spectroscopy (EIS) data were taken using AC voltage with an amplitude of 10 mV in a frequency range between 1 MHz and 10 mHz.
3. Results and discussion
3.1 Impact of the size of Ni metal precursor on the formation of NNMO
To make NNMO starting with metallic Ni, one needs to identify an appropriate source of Na, Mn, and O. Here, we use NaMnO4 as the source of Na, Mn, and O, since NaMnO4 can react with Ni at a Ni-to-NaMnO4 molar ratio of 1:2 to form NNMO with the overall reaction as shown in eqn (1):| Ni (s) + 2NaMnO4·H2O (s) → 3Na2/3[Ni1/3Mn2/3]O2 (s) + 2H2O (g) + O2 (g) | (1) |
The initial stage of the reaction in eqn (1) involves the thermal decomposition of NaMnO4 to form MnO2 between 120-180 °C and Na2MnO3 between 540–580 °C, both of which are stable in air.26 The major challenge of using metallic Ni as a precursor arises from the limited diffusion of Na and Mn species into the core of the Ni particles. To address this, we investigated the effect of metallic Ni precursors with various sizes, namely 10 nm Ni (NP-Ni), 100 nm Ni, 1 μm Ni, and 5 μm Ni, on the synthesis of NNMO. Fig. 1 shows the XRD profiles of converted NNMO. The relatively small sizes of NP-Ni and 1 μm Ni facilitated their complete conversion to NNMO after 24 h at 800 °C, as shown by the magenta and orange XRD curves, respectively. Notably, the present annealing condition (i.e., 800 °C and 24 h) was identified as the minimal time and temperature to achieve the full conversion of NNMO, as lower temperature or shorter annealing time did not result in a full conversion (Fig. S1a†). While the 100 nm Ni nanopowder was converted to NNMO within only 1 h at 850 °C (Fig. S1b†) owing to its short diffusion length, persistent impurity peaks emerged even after extending the annealing time to 24 h at 800 °C (Fig. 1, green pattern). The impurity peaks match with NaMnO2 (PDF#25-0845), as marked with triangle labels, and the origin of such impurities will be discussed in the next section. The 5 μm Ni was not completely converted to NNMO, even after 24 h at 800 °C, due to the increased diffusion length in bigger Ni particles. However, a higher temperature (900 °C) and a longer annealing time (72 h) led to enhanced (while not yet full) conversion, as shown by the dashed and solid blue curves, respectively. For comparison, the traditional synthesis route utilizing Ni acetate salt achieved complete conversion after 5 h at 500 °C followed by 15 h at 950 °C, as indicated by the purple pattern. Rietveld refinement is further performed on two samples, namely the 1 μm Ni-NNMO and NiAc-NNMO, Fig. S2.† The d(002) layer spacing is then extracted from the refined spectrum, which shows 5.57 Å for the 1 μm Ni-NNMO and 5.59 Å for the NiAc-NNMO (Fig. S2c†). These results are in agreement with the d(002) distance derived from Si powder calibrated XRD spectrum, which shows 5.59 Å for both the 1 μm Ni-NNMO and the NiAc-NNMO (Fig. S2d†). The values are smaller compared to the nominal (002) distance of 5.60 Å for NNMO, suggesting that our NNMO contains excess Na (enhanced electrostatic cohesions of O–Na–O leading to decreased layer spacing), as will be confirmed with the EDS/ICP composition analysis in a latter section.
 | ||
| Fig. 1 XRD patterns of NNMO converted from different metallic Ni precursors. Under the annealing condition of 800 °C for 24 h, the 100 nm Ni (green) and 5 μm Ni (dashed blue) showed impurity peaks corresponding to NaMnO2 (triangle labels), while the NP-Ni (magenta) and 1 μm Ni (orange) yielded full conversion. An enhanced (but not yet full) conversion was obtained for 5 μm Ni under the annealing condition of 900 °C for 72 h (solid blue). The NNMO prepared from Ni acetate salt using the traditional synthesis method yielded full conversion (purple). | ||
The size of converted NNMO particles is critical for its cycling performance, as it directly impacts the Na diffusion length, which in turn impacts the rate capability. Therefore, SEM was used to investigate the morphology of the starting metallic Ni and the converted NNMO, as shown in Fig. 2. Although the initial size of the 100 nm Ni particles was in the nanoscale, the converted NNMO particles were much larger, with a characteristic “diameter” of approximately 1 μm (Fig. 2a). Further details on the variation of NNMO particle size obtained from 100 nm Ni under different heat-treatment conditions (1 h or 3 h at 850 °C) are provided in Fig. S3,† where it is seen that the ≈1 μm NNMO particles formed after 1 h of annealing remained relatively unchanged thereafter. Based on the reports on coarsening and densification of nanopowders during sintering,27 we speculate that the 100 nm Ni particles coalesced into larger particles at the early stage of solid-state conversion, hindering the formation of nanoscale NNMO. In addition, this densification might impede Na and Mn diffusion, leading to incomplete conversion and the persistence of impurity phases even after extended annealing. The morphology of NP-Ni is characterized by nanoscale ligaments and pores with an approximate size of 10 nm (Fig. 2b). The presence of these nanoscale features was further confirmed with SAXS analysis, as shown in Fig. 2c. The green curve representing the SAXS pattern of NP-Ni displays a characteristic peak centered around q = 0.045 Å−1, which is absent in the dense Ni-Mn parent alloy before dealloying (black curve). This peak corresponds to a ligament–ligament distance (or pore–pore distance) of approximately 17 nm, as calculated using eqn (2):23,28
| d (ligament–ligament distance) = 1.23 × 2π/q | (2) |
 | ||
| Fig. 2 SEM images comparing the morphology and size of starting Ni precursor and converted NNMO particles: (a) 100 nm Ni (left) and converted NNMO (right). (b) NP-Ni (left) and converted NNMO (right). (c) SAXS pattern confirming the presence of nanoscale features in NP-Ni. Compared to the absence of a scattering peak in the dense Ni-Mn parent alloy (black), NP-Ni (green) showed a characteristic peak centered around q = 0.045 Å−1, which corresponds to a ligament/pore size of ≈9 nm. (d) 1 μm Ni (left) and converted NNMO (right). (e) 5 μm Ni (left) and converted NNMO (right). (f) Ni acetate salt converted NNMO. Despite various types of starting Ni, the minimal “diameter” of converted NNMO particles scales around 1 μm. | ||
Thus, the ligament/pore size is approximately 9 nm (corresponding to half of d in eqn (2)), which agrees with the SEM images. Following solid-state conversion, the previously observed nanoscale structures vanished, and were replaced by NNMO with plate-like morphology having a characteristic “diameter” of approximately 1 μm (Fig. 2b). The annealing temperature influenced the synthesized NNMO particle size, with higher temperatures (850 °C and 900 °C) leading to significantly larger NNMO particles (Fig. S4†). NNMO derived from 1 μm Ni (Figure 2d) and 5 μm Ni (Fig. 2e) precursors closely resembled the size of these starting materials. Regardless of the initial Ni precursor size (nano or micro), the minimum achievable characteristic “diameter” of the converted NNMO particles remained around 1 μm, which is among the smallest reported.12,29,30 NNMO synthesized from Ni acetate salt also gave an average particle size of 1 μm (Fig. 2f). Dynamic light scattering (DLS) analysis is further performed on two samples, namely the 1 μm Ni converted NNMO and NiAc salt converted NNMO, Fig. S5.† The particle sizes derived from DLS analysis are in agreement with the SEM results. EDS and ICP were subsequently used to analyze the elemental composition of the synthesized NNMOs, Table 1. Compared to the stoichiometric ratio of Na:Ni:Mn = 40:20:40, the NP-Ni-NNMO and 1 μm Ni-NNMO have an average ratio of 42:18:40 and 48:15:37 from EDS, respectively. This indicates that while the NP-Ni-NNMO was close to the stoichiometric ratio, the 1 μm Ni-NNMO was rich in Na. The Na-rich composition in 1 μm Ni-NNMO is further confirmed with ICP, showing an average ratio of 62:13:25. A similar Na-rich composition is also observed for the NiAc-NNMO control sample, with an average ratio of 61:13:26. Such excess Na is responsible for their excellent electrochemical performances, as will be discussed in the next section.
| Sample | Method | Na (at%) | Ni (at%) | Mn (at%) |
|---|---|---|---|---|
| Nominal | — | 40 | 20 | 40 |
| NP-Ni-NNMO | EDS | 42 | 18 | 40 |
| 1 μm Ni-NNMO | EDS | 48 | 15 | 37 |
| ICP | 62 | 13 | 25 | |
| NiAc-NNMO | ICP | 61 | 13 | 26 |
Considering the limited conversion of 100 nm Ni-NNMO and the increased particle size of 5 μm Ni-NNMO, NNMOs derived from NP-Ni and 1 μm Ni appear particularly promising for usage as SIB cathodes. Thus, the nanostructures of NP-Ni-NNMO and 1 μm Ni-NNMO were further investigated using TEM. Both materials showed well-defined lattice fringes (Fig. 3a and d), indicative of a layered oxide structure. Additionally, EDS mapping revealed the homogeneous distribution of Na, Ni, and Mn elements across the structure, Fig. S6.† The SAED pattern acquired along the [001] axis (Fig. 3b and e) confirms the formation of a hexagonal structure, with the presence of satellite spots that are associated with Na+-vacancy ordering.31,32 The SAED pattern collected along the [100] axis (Fig. 3c and f) reveals the interlayer spacing available for Na intercalation. The (002) spacing is calculated to be 5.76 Å and 5.98 Å for NP-Ni-NNMO and 1 μm Ni-NNMO, respectively. This value is slightly larger than the nominal value of 5.60 Å,12 likely due to variation in Na content within each cathode particle, resulting in small change in interlayer spacing. However, our XRD result in Fig. S2† suggests that the average interspacing is generally smaller among the cathode materials synthesized from different starting precursors.
 | ||
| Fig. 3 TEM images of NNMO converted from NP-Ni (1st row) and 1 μm Ni (2nd row). Lattice fringes observed under high-resolution TEM (a and d), and corresponding SAED pattern along the [001] axis (b and e) showing hexagonal structure with Na+-vacancy ordering. SAED patterns along the [100] axis reveal (002) layer spacings of 5.76 Å (c) and 5.98 Å (f), respectively. | ||
3.2 Electrochemical performance of NNMO as SIB cathode material
 | ||
| Fig. 4 (a) Cyclic voltammetry of NNMO converted from NP-Ni (magenta), 1 μm Ni (orange), 5 μm Ni (blue), and Ni acetate salt (purple) cycled between 2–4 V vs. Na/Na+ under 1 mV s−1. (b) Rate-capability of NNMO converted from NP-Ni (magenta), 1 μm Ni (orange), 5 μm Ni (blue), and Ni acetate salt (purple) cycled between 2-4 V vs. Na/Na+, with the corresponding galvanostatic curves of 1 μm Ni-NNMO in (c). (d) Long-term cycling of NNMO at 10C between 2–4 V vs. Na/Na+. NiAc-NNMO (purple) exhibited 88% capacity retention, and 1 μm Ni-NNMO (orange) achieved 93% capacity retention after 500 cycles. | ||
The rate-capability of 1 μm Ni-NNMO showed the best performance, followed by NNMO converted from Ni acetate salt, NP-Ni, and 5 μm Ni (Fig. 4b). The 5 μm Ni-NNMO showed a capacity decay at high rates (20C and 40C), which arise from the sluggish kinetics caused by the large particle size. The 1 μm Ni-NNMO achieved ≈83, 78, 65, and 90 mA h g−1 at 10C, 20C, 40C, and 2C, corresponding to ≈96, 90, 76, and 104% of the theoretical capacity, respectively. The excess capacity at 2C might be attributed to a minor capacitive contribution.33 When the C-rate returned from 2C to 10C, the original capacity was fully recovered. The corresponding galvanostatic (dis)charge curves are shown in Fig. 4c for the 1 μm Ni-NNMO, with further data for NP-Ni-NNMO and NiAc-NNMO provided in Fig. S7b and S8,† respectively. The clear plateaus observed under all C-rates suggest that the charge storage process remains mostly faradaic, even during high-rate cycling.34 Since the 1 μm Ni-NNMO and NiAc-NNMO showed the best (and comparable) performance, galvanostatic charge–discharge profiles of these NNMOs under slower C-rates (i.e., 0.1C, 0.2C, 0.5C, and 1C) are also evaluated (Fig. S9†), and a normalized energy efficiency (taking the energy density of C/10 as 100% efficiency) is calculated based on the integration of the E–Q profile (Fig. S10†). The results show that both 1 μm Ni-NNMO and NiAc-NNMO can maintain >95% energy efficiency up to 1C, and >60% energy efficiency even at 40C, confirming their excellent rate-performance. Kinetic analysis performed on the NP-Ni-NNMO further reveals that the characteristic “b-value” is higher compared to literature reports, confirming the excellent rate performance (details on kinetics analysis shown in the ESI†). Fig. 5 represents a comparative analysis of the obtained rate-capability with previously reported data for P2-NNMO cycled within the same voltage window.8,11,12,15,30,31,35–37 Our 1 μm Ni-NNMO achieved the highest capacity at C-rates exceeding 2C. This superior performance is attributed to the presence of excess Na in the NNMO structure,38,39 as confirmed by EDS, ICP, XRD, and Rietveld refinement. Specifically, while a Na-rich composition in NNMO does not favor the layer spacing for Na+ migration, it can contribute to the rate performance from two aspects: (i) Promote Ni2+ oxidation, such that more Ni2+ can participate in the charge compensation upon desodiation, thus providing more capacity within a given C-rate.38 (ii) Increased Nae/Naf ratio, where Nae and Naf stand for Na+ occupying the edge-sharing and face-sharing prismatic sites, respectively. Since Nae sites have a higher Na+ mobility, this increase in Nae/Naf ratio leads to improved rate performance.39 Finally, the cycling stability was tested at 10C, evaluated for 1 μm Ni-NNMO and NiAc-NNMO, as shown in Fig. 4d, as well as in Fig. S5c† for NP-Ni-NNMO. Both exhibited similar initial capacity and stability, with 1 μm Ni-NNMO retaining ≈93% of its capacity after 500 cycles, corresponding to a minimal capacity decay of only ≈0.014% per cycle.
 | ||
| Fig. 5 Rate capability reported for P2-NNMO. All electrodes are cycled between 2–4 V vs. Na/Na+. | ||
Electrochemical impedance spectroscopy (EIS) is further performed on the 1 μm Ni-NNMO and NiAc-NNMO, to evaluate the charge transfer resistance associated with the (de)sodiation at 2C. Fig. 6 shows the Nyquist plots consisting of depressed semicircles in the high and medium frequency regions, and a linear curve in the low frequency region. The former corresponds to the charge transfer resistance, while the latter corresponds to the Warburg diffusion.40 The first cycle (pristine NNMO) shows a high charge transfer resistance, likely due to the poor electrolyte wetting in the as-assembled cell. After 30 cycles of charge–discharge at 2C, both NNMOs showed a similar resistance value of ≈350 Ω for discharge (i.e. sodiation, Fig. 6a and c) and ≈75 Ω for charge (i.e., desodiation, Fig. 6b and d). The similar resistance seen in both NNMOs agrees with their comparable electrochemical performance. Notably, in both samples the impedance during discharge (Fig. 6a and c) was much larger than during charge (Fig. 6b and d). Similar observations have been reported previously in the case of LiCoO2 tested as a LIB cathode.41 This phenomenon can be attributed to the concentration gradient of Na+ formed at the electrode–electrolyte interface, leading to an increased Na+ intercalation resistance seen during discharge of NNMO.
 | ||
| Fig. 6 Nyquist plots of NNMO electrodes after various (de)sodiation cycles. (a) 1 μm Ni-NNMO, discharge (sodiation), (b) 1 μm Ni-NNMO, charge (desodiation), (c) NiAc-NNMO, discharge (sodiation), (d) NiAc-NNMO, charge (desodiation). After stabilization, both samples showed similar charge transfer resistance of ≈350 Ω for discharge and ≈75 Ω for charge. | ||
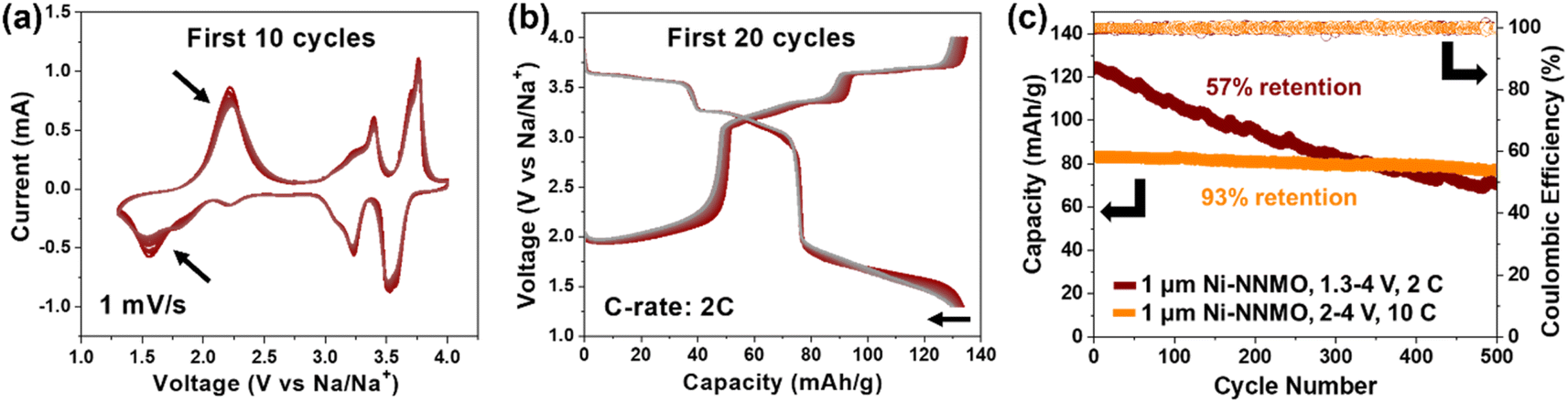 | ||
| Fig. 7 (a) Cyclic voltammetry of 1 μm Ni-NNMO cycled between 1.3–4 V vs. Na/Na+ under 1 mV s−1. The peak intensity decay over 10 cycles (black arrows) indicates structural distortion caused by Jahn–Teller effect of Mn3+. (b) Galvanostatic curves of 1 μm Ni-NNMO cycled between 1.3–4 V vs. Na/Na+ under a C-rate of 2C. Gradual capacity decay is observed over 20 cycles. (c) Long-term cycling of 1 μm Ni-NNMO between 1.3–4 V vs. Na/Na+ under a C-rate of 2C (brown), achieving 57% capacity retention after 500 cycles. In contrast, 1 μm Ni-NNMO cycled between 2–4 V vs. Na/Na+ under a C-rate of 10C (orange) achieved 93% capacity retention after 500 cycles. | ||
4. Conclusion
This work presents an environmentally responsible synthesis route to P2-type Na2/3[Ni1/3Mn2/3]O2 by direct conversion of metallic Ni powder in the presence of sodium permanganate, a process that only releases O2 and H2O gases. Thus, the proposed method prevents the formation of undesirable byproducts such as HNO3 and CO2, commonly released when nickel salts (nitrates and acetates) are used as precursors. We have systematically studied the formation of P2-Na2/3[Ni1/3Mn2/3]O2 using metallic Ni powders with various average particle sizes, including 10 nm, 100 nm, 1 μm, and 5 μm, and found that P2-Na2/3[Ni1/3Mn2/3]O2 synthesized from Ni powder with an average particle size of 1 μm showed an excellent performance as a Na-ion battery cathode. When cycled between 2–4 V vs. Na/Na+, the material exhibited a rate-capability of ≈83 mA h g−1 at 10C (i.e., >96% theoretical capacity) and ≈78 mA h g−1 at 20C (i.e., >90% theoretical capacity), together with a remarkable cycling stability of ≈93% capacity retention after 500 cycles at 10C. When cycled in a broader voltage window of 1.3–4 V vs. Na/Na+, the material showed a high initial capacity of ≈134 mA h g−1 at 2C, with a capacity retention of ≈80% after 180 cycles and ≈57% after 500 cycles. These findings pave the way for the design and development of next-generation Na-ion battery cathodes.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the National Science Foundation (NSF) for their financial support through NSF-CAREER CMMI-2047851 and NSF FM-2134715. We gratefully acknowledge the National Science Foundation (NSFMRI-1725969), which funded the Dual Source (Cu and Mo) and Environmental X-ray Scattering (DEXS) facility used in this research. This work was carried out in part at the Singh Center for Nanotechnology, part of the National Nanotechnology Coordinated Infrastructure Program, which is supported by the NSF grant NNCI-2025608. Lastly, AI-assisted manuscript proofreading was performed using Grammarly.References
- C. Helbig, A. M. Bradshaw, L. Wietschel, A. Thorenz and A. Tuma, J. Cleaner Prod., 2018, 172, 274–286 CrossRef CAS.
- W.-J. Lv, Z. Huang, Y.-X. Yin, H.-R. Yao, H.-L. Zhu and Y.-G. Guo, ChemNanoMat, 2019, 5, 1253–1262 CrossRef CAS.
- B. Xie, B. Sun, T. Gao, Y. Ma, G. Yin and P. Zuo, Coord. Chem. Rev., 2022, 460, 214478 CrossRef CAS.
- Q. Ni, Y. Bai, F. Wu and C. Wu, Advanced Science, 2017, 4, 1600275 CrossRef PubMed.
- P. Gupta, S. Pushpakanth, M. A. Haider and S. Basu, ACS Omega, 2022, 7, 5605–5614 CrossRef CAS PubMed.
- Y. Liu, Q. Shen, X. Zhao, J. Zhang, X. Liu, T. Wang, N. Zhang, L. Jiao, J. Chen and L.-Z. Fan, Adv. Funct. Mater., 2020, 30, 1907837 CrossRef CAS.
- C. M. Julien and A. Mauger, Energies, 2020, 13, 6363 CrossRef CAS.
- D. H. Lee, J. Xu and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC.
- J. Zhang, W. Wang, W. Wang, S. Wang and B. Li, ACS Appl. Mater. Interfaces, 2019, 11, 22051–22066 CrossRef CAS PubMed.
- Y. Liu, X. Fang, A. Zhang, C. Shen, Q. Liu, H. A. Enaya and C. Zhou, Nano Energy, 2016, 27, 27–34 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, Q. Gu and S. Chou, J. Mater. Chem. A, 2019, 7, 9215–9221 RSC.
- S. Y. Lee, J. H. Kim and Y. C. Kang, Electrochim. Acta, 2017, 225, 86–92 CrossRef CAS.
- W. Brockner, C. Ehrhardt and M. Gjikaj, Thermochim. Acta, 2007, 456, 64–68 CrossRef CAS.
- G. A. M. Hussein, A. K. H. Nohman and K. M. A. Attyia, J. Therm. Anal., 1994, 42, 1155–1165 CrossRef CAS.
- D. Yang, X.-Z. Liao, J. Shen, Y.-S. He and Z.-F. Ma, J. Mater. Chem. A, 2014, 2, 6723–6726 RSC.
- E. Detsi, J. B. Cook, B. K. Lesel, C. L. Turner, Y.-L. Liang, S. Robbennolt and S. H. Tolbert, Energy Environ. Sci., 2016, 9, 540–549 RSC.
- B. Madavali, J.-H. Lee, J. K. Lee, K. Y. Cho, S. Challapalli and S.-J. Hong, Powder Technol., 2014, 256, 251–256 CrossRef CAS.
- O. Ruiz, M. Cochrane, M. Li, Y. Yan, K. Ma, J. Fu, Z. Wang, S. H. Tolbert, V. B. Shenoy and E. Detsi, Adv. Energy Mater., 2018, 8, 1801781 CrossRef.
- S. S. Welborn and E. Detsi, Nanoscale Horiz., 2020, 5, 12–24 RSC.
- J. Fu, J. S. Corsi, S. S. Welborn, V. Basile, L. Wang, A. K. Ng and E. Detsi, ACS Sustain. Chem. Eng., 2021, 9, 2762–2769 CrossRef CAS.
- T. Lee, H. Koh, A. K. Ng, J. Liu, E. A. Stach and E. Detsi, Scr. Mater., 2022, 221, 114959 CrossRef CAS.
- S. S. Welborn, J. S. Corsi, L. Wang, A. Lee, J. Fu and E. Detsi, J. Mater. Chem. A, 2021, 9, 19994–20005 RSC.
- A. K. Ng, S. S. Welborn and E. Detsi, Scr. Mater., 2022, 206, 114215 CrossRef CAS.
- A. D. Herron, S. P. Coleman, K. Q. Dang, D. E. Spearot and E. R. Homer, MethodsX, 2018, 5, 1187–1203 CrossRef PubMed.
- D. B. Williams and C. B. Carter, in Transmission Electron Microscopy: A Textbook for Materials Science, ed. D. B. Williams and C. B. Carter, Springer US, Boston, MA, 1996, pp. 3–17 Search PubMed.
- Z. Gontarz, R. Grzybowska and R. Sabaliński, J. Therm. Anal., 1995, 45, 1125–1133 CrossRef CAS.
- Z. Z. Fang, H. Wang and V. Kumar, Int. J. Refract. Met. Hard Mater., 2017, 62, 110–117 CrossRef CAS.
- J. Fu, S. Mooraj, A. K. Ng, C. Zhu, W. Chen and E. Detsi, ACS Appl. Mater. Interfaces, 2023, 15, 27905–27914 CrossRef CAS PubMed.
- J. Mao, X. Liu, J. Liu, H. Jiang, T. Zhang, G. Shao, G. Ai, W. Mao, Y. Feng, W. Yang, G. Liu and K. Dai, J. Electrochem. Soc., 2019, 166, A3980 CrossRef CAS.
- G. Liu, L. Wen, Y. Li and Y. Kou, Ionics, 2015, 21, 1011–1016 CrossRef CAS.
- Y. Shi, P. Jiang, S. Wang, W. Chen, B. Wei, X. Lu, G. Qian, W. H. Kan, H. Chen, W. Yin, Y. Sun and X. Lu, Nat. Commun., 2022, 13, 7888 CrossRef CAS PubMed.
- K. Kubota, T. Asari and S. Komaba, Adv. Mater., 2023, 35, 2300714 CrossRef CAS PubMed.
- H. Chu, Y. Pei, Z. Cui, C. Steven, P. Dong, P. M. Ajayan, M. Ye and J. Shen, Nanoscale, 2018, 10, 14697–14704 RSC.
- M. Li, T. Qiu, A. C. Foucher, J. Fu, Z. Wang, D. Zhang, A. M. Rappe, E. A. Stach and E. Detsi, ACS Appl. Energy Mater., 2020, 3, 11231–11241 CrossRef CAS.
- S. Wu, B. Su, K. Ni, F. Pan, C. Wang, K. Zhang, D. Y. W. Yu, Y. Zhu and W. Zhang, Adv. Energy Mater., 2021, 11, 2002737 CrossRef CAS.
- X. Zheng, P. Li, H. Zhu, K. Rui, G. Zhao, J. Shu, X. Xu, W. Sun and S. X. Dou, Energy Storage Mater., 2018, 15, 257–265 CrossRef.
- M. R. D. Bomio, P. Lavela, A. A. G. Santiago, F. V Motta and J. L. Tirado, Ceram. Int., 2023, 49, 12452–12461 CrossRef CAS.
- C. Zhao, Z. Yao, Q. Wang, H. Li, J. Wang, M. Liu, S. Ganapathy, Y. Lu, J. Cabana, B. Li, X. Bai, A. Aspuru-Guzik, M. Wagemaker, L. Chen and Y.-S. Hu, J. Am. Chem. Soc., 2020, 142, 5742–5750 CrossRef CAS PubMed.
- S. Liu, J. Wan, M. Ou, W. Zhang, M. Chang, F. Cheng, Y. Xu, S. Sun, C. Luo, K. Yang, C. Fang and J. Han, Adv. Energy Mater., 2023, 13, 2203521 CrossRef CAS.
- M. Li, T. Qiu, S. S. Welborn, A. C. Foucher, J. Fu, B. K. Lesel, Z. Wang, L. Wang, E. A. Stach, A. M. Rappe and E. Detsi, J. Mater. Chem. A, 2024, 12, 3671–3681 RSC.
- H. Watanabe, S. Omoto, Y. Hoshi, I. Shitanda and M. Itagaki, J. Power Sources, 2021, 507, 230258 CrossRef CAS.
- H. Wang, B. Yang, X.-Z. Liao, J. Xu, D. Yang, Y.-S. He and Z.-F. Ma, Electrochim. Acta, 2013, 113, 200–204 CrossRef CAS.
- J. Feng, D. Fang, J. Li and J. Li, Batteries Supercaps, 2024, 7, e202300476 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03708a |
| This journal is © The Royal Society of Chemistry 2024 |