
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating performance degradation mechanisms in non-fullerene acceptor solar cells†
Vinod K.
Sangwan‡
 *a,
Zachary
Martin‡
a,
Guoping
Li
b,
Fei
Qin
b,
Shreyash
Hadke
a,
Robert M.
Pankow
b,
Woo Cheol
Jeon
b,
Ding
Zheng
b,
Yongjoon
Cho
b,
Ryan M.
Young
*b,
Kevin L.
Kohlstedt
*b,
Michael R.
Wasielewski
*b,
George C.
Schatz
*b,
Antonio
Facchetti
*bc,
Mark C.
Hersam
*abd and
Tobin J.
Marks
*b
*a,
Zachary
Martin‡
a,
Guoping
Li
b,
Fei
Qin
b,
Shreyash
Hadke
a,
Robert M.
Pankow
b,
Woo Cheol
Jeon
b,
Ding
Zheng
b,
Yongjoon
Cho
b,
Ryan M.
Young
*b,
Kevin L.
Kohlstedt
*b,
Michael R.
Wasielewski
*b,
George C.
Schatz
*b,
Antonio
Facchetti
*bc,
Mark C.
Hersam
*abd and
Tobin J.
Marks
*b
aDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA. E-mail: vinod.sangwan@northwestern.edu; m-hersam@northwestern.edu
bDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: ryan.young@northwestern.edu; kkohlstedt@northwestern.edu; m-wasielewski@northwestern.edu; g-schatz@northwestern.edu; a-facchetti@northwestern.edu; t-marks@northwestern.edu
cSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
dDepartment of Electrical and Computer Engineering, Northwestern University, Evanston, Illinois 60208, USA
First published on 9th July 2024
Abstract
Organic solar cells (OSCs) using non-fullerene acceptors (NFAs) afford exceptional photovoltaic performance metrics, however, their stability remains a significant challenge. Existing OSC stability studies focus on understanding degradation rate-performance relationships, improving interfacial layers, and suppressing degradative chemical reaction pathways. Nevertheless, there is a knowledge gap concerning how such degradation affects crystal structure, electronic states, and recombination dynamics that ultimately impact NFA performance. Here we seek a quantitative relationship between OSC metrics and blend morphology, trap density of states, charge carrier mobility, and recombination processes during the UV-light-induced degradation of PBDB-TF:Y6 inverted solar cells as the PCE (power conversion efficiency) falls from 17.3 to 5.0%. Temperature-dependent electrical and impedance measurements reveal deep traps at 0.48 eV below the conduction band that are unaffected by Y6 degradation, and shallow traps at 0.15 eV below the conduction band that undergo a three-fold density of states increase at the PCE degradation onset. Computational analysis correlates vinyl oxidation with a new trap state at 0.25 eV below the conduction band, likely involving charge transfer from the UV-absorbing ZnO electron transport layer. In situ integrated photocurrent analysis and transient absorption spectroscopy reveal that these traps lower electron mobility and increase recombination rates during degradation. Grazing-incidence wide-angle X-ray scattering and computational analysis reveal that the degraded Y6 crystallite morphology is largely preserved but that <1% of degraded Y6 molecules cause OSC PCE performance degradation by ≈50%. Together the detailed electrical, impedance, morphological, ultrafast spectroscopic, matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) spectroscopy, and computational data reveal that the trap state energies and densities accompanying Y6 vinyl oxidation are primarily responsible for the PCE degradation in these operating NFA-OSCs.
1 Introduction
Organic solar cells (OSCs) based on bulk heterojunctions (BHJs) continue to evolve as a next-generation photovoltaic technology due to unprecedented power conversion efficiencies (PCEs), low-cost fabrication, mechanical flexibility, and earth-abundant, non-toxic elemental composition.1–9 Among different molecular and polymeric systems, non-fullerene acceptors (NFAs) have emerged as one of the most promising electron acceptors that can be paired with electron donor polymers to achieve OSCs with PCEs in excess of 19%.10–19 NFAs such as ITIC, IT-4F, Y5, and Y6 not only possess favorable frontier energy levels that align well with donor polymers such as PBDB-TF (Fig. 1) and charge transport layers for efficient charge extraction, but they also possess band gaps that can harvest infrared photons to generate additional charge carriers.3,4,20–27 Furthermore, NFA molecules offer tunability of frontier orbital energies from modular design with a diverse range of end groups, strong π–π interactions, excellent mixing properties in the BHJ blend, and very low internal reorganization energies upon photon capture.24,27–31 This combination of properties has led to rapid progress in optimizing the device performance metrics by tailoring blend chemistry and morphology on demand. | ||
| Fig. 1 (a) Chemical structures of Y6 (top) and PBDB-TF (bottom) molecules. (b) Energy level alignment of the various layers in PBDB-TF:Y6 solar cells. (c) Current-density–voltage (J–V) characteristics of the solar cell at different PCE values during degradation. (d) External quantum efficiency (EQE) plot of the cell at different PCE values during degradation. (e) Dark J–V characteristics of a solar cell at varying temperatures from 140–295 K. Solid lines show fits to the Shockley diode eqn (1). (f) Plot of log(Rs) versus 1/T to extract Ers for pristine and degraded cells. | ||
Nevertheless, the practical deployment of OSCs will require improved stability to reach cost-effectiveness versus incumbent technologies.3,5,32–34 Thus far, NFA-based OSCs have shown limited PCE stability under continuous operation. Existing literature on stability either correlates performance metrics deterioration with various NFA chemical reaction pathways involving the final byproducts or reports empirical degradation rates of OSCs by varying constituent blends, interfacial layers, and device architecture.34–48 Thus, there is a knowledge gap in understanding the microscopic mechanism of how OSC degradation affects crystal structure, electronic structure, charge transport, and recombination dynamics in BHJ blends—properties that influence macroscopic performance metrics. Here, we study unencapsulated OSCs with an inverted geometry of ITO/ZnO/PBDB-TF:Y6/MoOx/Ag (Fig. 1) as a model system that are degraded by exposure to simulated solar irradiation, where UV exposure is expected to reduce PCE monotonically with increasing exposure duration.36 Here we systematically analyze the evolution of the trap density of states, charge carrier mobility, and recombination dynamics by in situ measurements under operating conditions with controlled degradation. Concomitantly, we describe the measured trends in terms of trap energy and recombination dynamics by proposing a putative degradation pathway and calculating the density of states for the degraded NFA.
It will be seen that degradation leads to three main results that we have modeled using quantum mechanical computational analysis. (i) Variable temperature current–voltage (IVT) characteristics reveal that forward bias charge transport is dominated by a series resistance originating from deep-lying trap states located at 0.48 eV below the lowest unoccupied Y6 molecular orbital (LUMO). Surprisingly, these trap states are not connected with the PCE degradation. (ii) Thermal admittance spectroscopy (TAS) of pristine PBDB-TF:Y6 cells identifies a trap energy level at 0.15 eV that moves deeper into the bandgap (0.21 eV) with at least a three-fold increase in the density of states upon initial PCE degradation from PCE = 17.3 to 11.1%. (iii) Integrated photocurrent device analysis (IPDA) reveals that electron mobility at the maximum power point falls roughly six-fold when PCE degrades by two-fold. As expected, increasing the number of trap states increases monomolecular recombination, whereas the bimolecular/geminate recombination coefficient under operating conditions decreases with degradation. (iv) Transient absorption spectroscopy of pure Y6 and the BHJ PBDB-TF:Y6 blend show increased kinetic rates of fast (ps) and slow (ns) processes upon degradation which is consistent with reduced mobility and increased geminate recombination. Thus, the trap energy levels from TAS correlate with the Y6 degradation such that a relatively low density of degraded molecules (<1%) can introduce recombination and charge trapping sites with major losses in the cell performance – PCE loss by more than 50%. These results are consistent with the role of the electron transport layer ZnO in generating radicals under UV exposure36,37 that can degrade Y6 with only minuscular changes in the blend morphology,36,37 as revealed here by grazing incidence wide-angle X-ray scattering (GIWAXS).
To complement the experimental input, parallel computational analysis provides a deeper understanding of the NFA degradation process, including the crystal structural changes and a molecular understanding of the trap states that are formed during degradation. In our model, we consider three likely degraded Y6 structures: (a) vinyl oxidation between the core and end group, (b) acceptor end group scission, and (c) dicyanomethylene scission on the end group (see computational methods for structural details). These candidate structures represent three likely degradation pathways induced by UV-generated free radicals that can occur even some distance from the ETL interface. Of the three proposed degradation pathways, it will be seen that the vinyl oxidation pathway is the most likely degradation product based on the calculated density of states reproducing the measured trap energetics. Using the crystal structure of Y6, we generate a relaxed crystal of degraded Y6 molecules and determine the frontier orbital energies of degraded Y6 molecules. Based on the degraded crystal structure and density of states, we characterize the vinyl oxidized crystal structure and describe the evolution of a LUMO trap state 0.25 eV into the bandgap. Therefore, while the chemical degradation pathways further corroborate existing molecular degradation hypotheses,35,36,39,49 this study provides new insight into the evolution of NFA electronic states under PCE degrading irradiation.
From the combined experimental and theoretical work reported here, the shallow Y6 trap states are identified and shown to become deeper even with minute amounts of degraded NFAs by well-structured in situ measurements. Moreover, the electronic structure analysis of the NFA crystal structures and matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) spectroscopy clarifies that these trap states arise from vinyl oxidation, among the three aforementioned plausible reaction pathways that can be initiated by free radicals under UV exposure. These findings should provide insight into the design of future more stable NFA molecules and suggest replacing ZnO with other electron transport layers, passivating the ZnO, and/or designing NFA molecules with functionalized end groups to suppress vinyl linkage oxidation.
2 Results and discussion
In view of the variety of photophysical and structural characterizations described in this section, we have organized the main results into six subsections. The organization of this section is as follows: First discussed (Section 2.1) are procedures for creating and monitoring PBDB-TF:Y6 degradation by variable temperature current-density–voltage measurements over a broad voltage range. The data are fit to a Shockley diode and a thermal activation model to extract key parameters including the location of deep traps in the energy band structure. Next, Section 2.2 focuses on quantifying the number, energy, and attempt frequency of shallow trap states in the pristine blend and then tracking their evolution with degradation by TAS, and optical absorption spectroscopic measurements. Section 2.3 employs in situ IPDA to study the evolution of mobility and recombination coefficient as the OSCs are systematically degraded. In Section 2.4, we then use transient absorption spectroscopy to probe the effects of OSC degradation on the excited-state dynamics, to assess the origins of the degraded OSC spectral signatures, hole transfer rates, and recombination kinetics. Section 2.5 employs grazing-incidence wide-angle X-ray scattering (GIWAXS) to assess the structural integrity of the OSC bulk-heterojunction blend during the degradation process and Section 2.6 uses electronic structure computation and the experimental photophysical, diffraction, and MALDI-ToF data to understand the energies/chemistry of the trap states and how the Y6 molecule degrades. Here, we propose a putative degradation pathway by using computational modeling of the NFA band structure before and after degradation through three possible degradation pathways and by considering the experimental characterizations discussed above.2.1 Photophysical measurements
Inverted BDB-TF:Y6 OSCs were fabricated in an inert argon glove box by following the procedure outlined in a previous study (Fig. 1a and b), see ESI Sections 1 and 2† for details.24 An active layer thickness of 120 nm was used to optimize the performance of the pristine cells that were measured in the same glove box. Typical OSCs (area = 0.0625 cm2) show PCEs of greater than 16% (Fig. 1c) with a champion device delivering a PCE of 17.3%, an open circuit voltage (VOC) of 0.81 V, a short-circuit current density (JSC) of 18.39 mA cm−2, and a fill factor (FF) of 73.4%. Pristine OSCs were then removed to ambient conditions and exposed to AM 1.5G, 100 mW cm−2, and the current-density–voltage (J–V) characteristics were again measured in the glove box by increasing the exposure durations in incremental steps of 5 minutes (Fig. 1c). Most of the loss in PCE comes from decreased JSC and FF while VOC is relatively unchanged. The total integrated photocurrent from external quantum efficiency (EQE) measured in a glove box matches the JSC of OSCs from J–V characteristics within 95% (Fig. 1d) while the residual mismatch is likely attributable to the continued slow degradation during the interval between the two measurements.To understand the effect of contacts on the degradation process, we measured J–V characteristics of pristine and degraded OSCs in the dark. The evolution of device performance metrics with exposure time is shown in ESI Fig. S1.†Fig. 1e shows representative curves of a pristine device (PCE = 17.3%) at temperatures ranging from 295 to 140 K for IVT analysis (ESI Section 3†). All optoelectronic measurements in this and the next section were conducted on three different devices showing qualitatively similar behavior. To determine the physical processes involved in dark characteristics we fitted the J–V curves to the general Shockley diode eqn (1),
 | (1) |
Since the present model does not make any assumptions about the physical processes in charge transport, we first analyze the temperature dependence of the fit parameters to gain further insight. We find that the ideality factor n0 for PBDB-TF:Y6 BHJ diodes to be less than 2 at room temperature for all the devices and only becomes greater than 2 at lower temperatures following a well-established behavior in semiconductor diodes (ESI Fig. S5†). The temperature dependence of n0 in OSCs has been reported and can be attributed to three factors: (i) band gap variation at low temperatures, (ii) interfacial recombination temperature dependence, and (iii) increased tunneling processes at low temperatures that do not pose an upper bound of 2 for n.52–55 Also, note that an ideal Shockley equation for the diode does not include series or shunt resistance. The generalized Shockley eqn (1) has been extensively used to fit the temperature dependence of OPV IV curves in the dark and under illumination despite being judged “very non-ideal”.51,53
Here, we note that the degradation process does not significantly affect the temperature dependence of n0 (<10%), thus, ruling out large changes in the effective bandgap. In contrast, the fitted value of J0 falls by four orders of magnitude from 1.37 × 10−11 to 2.23 × 10−15 A on lowering the temperature from 295 to 150 K, which might be ascribed to thermal activation across the mobility gap. However, estimation of the mobility gap from an Arrhenius-type plot of J0T−2vs. T gives unreasonable values due to the rather small Rp varying from 4.47 to 17.62 MΩ over the same temperature range and makes the cell ‘leaky’ even at low temperatures.51 The model fits better at relatively large voltages (V > 0.5 V) where Rs dominates and thus, the extracted temperature dependence is analyzed using the eqn (2):
 | (2) |
 , where e is the fundamental electronic charge and εs is the dielectric constant of the active layer. We obtain εs = 2.5 for the PBDB-TF:Y6 blend from capacitance–voltage measurements at room temperature on fully depleted OSCs (i.e., geometrical capacitance), and the extracted value of εs is comparable to that of blends from other NFAs (≈3) and Y6 (≈3.5–3.74) reported in the literature.56–58 This underestimation may reflect uncertainty in thickness, roughness, and dielectric properties of PBDB-TF, and interfaces.
, where e is the fundamental electronic charge and εs is the dielectric constant of the active layer. We obtain εs = 2.5 for the PBDB-TF:Y6 blend from capacitance–voltage measurements at room temperature on fully depleted OSCs (i.e., geometrical capacitance), and the extracted value of εs is comparable to that of blends from other NFAs (≈3) and Y6 (≈3.5–3.74) reported in the literature.56–58 This underestimation may reflect uncertainty in thickness, roughness, and dielectric properties of PBDB-TF, and interfaces.
The temperature dependence of Rs is critical for fitting at large voltages and assuming a constant Rs results in poor agreement with the fits. The flattened J–V curves suggest a barrier lowering either from the Schottky effect at the contacts or the Poole–Frenkel effect in the bulk of the active layer. First, we extract Ers = 0.48 eV (taking εs = 3.74) for the undegraded cell and Ers value changes by only 3% upon PCE degradation from 17.3 to 5% (Fig. 1f). Second, we obtain the lowest possible current density of 4.34 mA cm−2 at 295 K from Schottky barrier lowering using A0T2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ers/kBT), where A0 is the Richardson constant (120 A cm−2 K−2). Since this value is less than the measured current density of 6.04 mA cm−2 at 1 V, either the Schottky or Poole–Frenkel effect can explain the diode behavior.51 Therefore, the series resistance arises either from the contact resistance between the blend and charge transport layer or charge trapping within the bulk of the solar cell. In either case, the activation energy does not change with PCE degradation. This suggests these deep traps at the contact regions or in the BHJ only begin to emerge in the last stages of degradation of these NFA-based OSCs and most of the initial PCE degradation comes from other factors. To conclude, dc-based IVT measurements reveal deep traps in the charge transport layer that do not change significantly upon rapid PCE degradation from fresh cells. Therefore, in the following, we employ impedance-based methods to probe shallow trap states within the BHJ to understand the mechanism of initial PCE degradation of OSCs.
exp(−Ers/kBT), where A0 is the Richardson constant (120 A cm−2 K−2). Since this value is less than the measured current density of 6.04 mA cm−2 at 1 V, either the Schottky or Poole–Frenkel effect can explain the diode behavior.51 Therefore, the series resistance arises either from the contact resistance between the blend and charge transport layer or charge trapping within the bulk of the solar cell. In either case, the activation energy does not change with PCE degradation. This suggests these deep traps at the contact regions or in the BHJ only begin to emerge in the last stages of degradation of these NFA-based OSCs and most of the initial PCE degradation comes from other factors. To conclude, dc-based IVT measurements reveal deep traps in the charge transport layer that do not change significantly upon rapid PCE degradation from fresh cells. Therefore, in the following, we employ impedance-based methods to probe shallow trap states within the BHJ to understand the mechanism of initial PCE degradation of OSCs.
2.2 Shallow charge trap energetics
We next employed TAS analysis of PBDB-TF:Y6 cells to further investigate degradation effects within the BHJ blend.59–65 Thus, we quantify the number, energy, and attempt frequency of the trap states in the pristine blend and then track their evolution with degradation. OSC frequency- and temperature-dependent capacitance (CfT) was measured at an applied DC voltage of 0.0 V using AC modulation of 50 mV over temperatures ranging from 295 to 120 K (Fig. 2a). First, the series resistance Rs, extracted from the impedance at 25 °C, shows a significant increase from 25.3 to 56.8 Ω (1.58 to 3.55 Ω cm2) when the PCE falls from 14.1 to 8.0% and increases more dramatically to 257 Ω (16.06 Ω cm2) at the lowest measured PCE value of 5.9% (ESI Fig. S6†). The capacitance step in CfT plots at low frequencies (40 Hz to 1 kHz, region A in Fig. 2a) is indicative of trap states in the bandgap. Thus, the variation in the inflection point of the capacitance step with temperature can be utilized to reconstruct the density of states and energy levels of the trap states.61 Note that the capacitance further decreases at higher frequencies (10–100 kHz, region B in Fig. 2a) which is not related to the trap states but is an artifact of the RC model, as explained in ESI Fig. S7.†66 We take the numerical derivative of the data in region A after smoothing the dataset at low frequencies (<100 Hz). The angular resonance frequency from the peak in
in region A after smoothing the dataset at low frequencies (<100 Hz). The angular resonance frequency from the peak in  vs. log(f) plots is given by w = 2ν0exp(−ΔET/kBT), where ν0 is the angular attempt frequency and ΔET is the trap activation energy. We include temperature dependence of the attempt frequency, ν0 = ν00T2 where ν00 is reduced attempt frequency.60 The peaks in the data in Fig. 2b are fit to Gaussian functions and the mean values are used to obtain an Arrhenius-type plot in Fig. 2c which yields ν00 ≈ 4.92 × 103 s−1 K−2. For comparison, the room temperature attempt frequency of the pristine PBDB-TF:Y6 cell ν0 ≈ 4.28 × 108 Hz is smaller than that in inorganic thin film solar cells (6.2 × 1011–1012 Hz),61 inorganic nanocrystal solar cells (≈1010 Hz),60 and organic–inorganic perovskite solar cells (1.35 × 109 Hz).59 The smaller PBDB-TF:Y6 attempt frequency likely reflects the relatively modest delocalization in π-stacked nanocrystals of Y6 molecules, evident in the single-crystal X-ray diffraction. The energy and the density of the trap state are calculated from eqn (3) and (4) where A is the area of cells,
vs. log(f) plots is given by w = 2ν0exp(−ΔET/kBT), where ν0 is the angular attempt frequency and ΔET is the trap activation energy. We include temperature dependence of the attempt frequency, ν0 = ν00T2 where ν00 is reduced attempt frequency.60 The peaks in the data in Fig. 2b are fit to Gaussian functions and the mean values are used to obtain an Arrhenius-type plot in Fig. 2c which yields ν00 ≈ 4.92 × 103 s−1 K−2. For comparison, the room temperature attempt frequency of the pristine PBDB-TF:Y6 cell ν0 ≈ 4.28 × 108 Hz is smaller than that in inorganic thin film solar cells (6.2 × 1011–1012 Hz),61 inorganic nanocrystal solar cells (≈1010 Hz),60 and organic–inorganic perovskite solar cells (1.35 × 109 Hz).59 The smaller PBDB-TF:Y6 attempt frequency likely reflects the relatively modest delocalization in π-stacked nanocrystals of Y6 molecules, evident in the single-crystal X-ray diffraction. The energy and the density of the trap state are calculated from eqn (3) and (4) where A is the area of cells,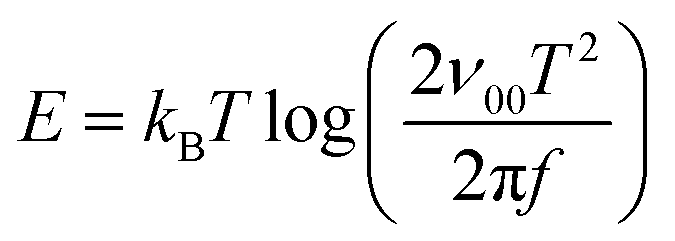 | (3) |
 | (4) |
 | ||
| Fig. 2 (a) Capacitance–frequency (C–f) characteristics of a pristine PBDB-TF:Y6 solar cell (PCE = 17.3%) at varying temperatures from 120 to 290 K. The A and B regions show two ‘knees’ in C–f curves. Solid lines show the smoothened curve in region A. (b) Plot of −fdC/df versus log(f) for different temperatures corresponding to region A of C–f plots for the pristine cell in (a). Solid lines show fits to Gaussian functions. (c) Plot of log(T2/fmax) vs. 1/T to extract the attempt frequency v00 and the energy ET for the dominant trap state using the thermal admittance spectroscopy (TAS) method. (d) Plot of the ‘collapsed’ density of trap states (NT) as a function of energy from LUMO of the Y6 molecule. (e) Comparison of energy dispersion of NT for the pristine and the degraded cell. (f) Schematic of the energy gap (Eg = 1.55 eV), activation energy in the charge transport layer (Ers = 0.48 eV), and trap energy level (ET = 0.15 eV) that changes to ∼0.21 eV after degradation. | ||
To investigate the optical signatures of the degraded molecules, we performed optical absorption spectroscopy of neat Y6, neat PBDB-TF, and PBDB-TF:Y6 blend films cast on ZnO/ITO substrates to simulate the photo-induced degradation environment of the OSCs. The spectra were collected before (pristine) and after (degraded) exposing the films to solar illumination for 30 min in ambient conditions (Fig. 3a and ESI Section 4, Fig. S9†). Overall, all films show minor decreases in characteristic peaks after degradation, however, neat Y6 and PBDB-TF:Y6 blend films also exhibit the emergence of a new absorbance feature at 952 nm (≈1.3 eV) after degradation. Surprisingly, this peak lies ≈0.25 eV below the Y6 LUMO, matching the peak location of degraded Y6 extracted from TAS analysis. However, the peak intensity is small which, compared with minor changes in other characteristic peaks after degradation, suggests that the molar fraction of degraded Y6 molecules is very small (<1%). Nevertheless, this optical observation provides a direct spectral signature of degraded Y6 molecules that matches the TAS trap state energy. Note that neat PBDB-TF and Y6 films show an 8% and 2.5% decline in the peak absorptions at 571 and 840 nm, respectively, during degradation. Note also that the PBDB-TF peak at 1150 nm does not significantly contribute to the OSC performance since the EQE is less than 1% in that regime (Fig. 1d). Thus, the neat PBDB-TF film shows a somewhat greater decline in absorption than Y6, and these changes are well-represented in the blend optical absorption spectra. While the reduction of net photon absorption in degraded films might decrease the photocurrent in proportion, it is not the main contributor to PCE reduction in the degraded devices since the majority of the photocurrent loss in OSCs comes from increased photocarrier recombination and decreased charge generation processes that are affected by the electronic structure and blend morphology.3–7 Thus, we have limited our analysis of the UV-Vis spectra to highlight the emergence of the new feature in the Y6 spectra. To conclude, the TAS and UV-vis analyses provide new insight beyond IVT into the evolution of charge traps in the BHJ during initial OSC degradation. However, these do not provide information on the charge transport and recombination dynamics in functional OSCs under illumination. In the following, we present an in situ measurement method to directly correlate PCE degradation with mobility and recombination coefficient while the cells are being operated at maximum power point.
 | ||
| Fig. 3 (a) Optical absorbance of a neat Y6 film spin-coated on ZnO/ITO before (pristine) and after exposure (degraded) to solar illumination for 30 min in ambient. The inset shows zoomed-in data from the blue rectangle showing the emergence of a new feature at ≈952 nm on degradation. (b) Plot of open circuit voltage (VOC) as a function of illumination intensity at different degrees of PBDB-TF:Y6 solar cell degradation. The legend corresponds to plots b–d. (c) A plot of effective carrier mobility (μeff) versus charge carrier density at maximum power point (nmp) extracted from IPDA data at different stages of degradation. (d) Plot of fill factor versus competition factor (θ) at different stages of degradation. The solid line is a guide to the eye. | ||
2.3 Integrated photocurrent analysis
Here, in situ IPDA measurements of PBDB-TF:Y6 cells at different stages of degradation (ESI Section 5†) were next conducted. The OSCs were degraded in a controlled manner by exposing the cells to AM-1.5G (100 mW cm−2) illumination for varying durations of 2 to 30 min and then, J–V and IPDA measurements were performed by bringing the cells into the glove box after each degradation step as PCE fell from 16.5 to 7.9%. First, VOC was measured as a function of illumination intensity (I) and the result is shown in Fig. 3b. The slope of VOCversus log(I) is ideally expected to be kBT/e (25 mV at room temperature) and 2kBT/e for bimolecular recombination and monomolecular recombination, respectively.31,68 However, space charge effects and other OSC non-idealities can yield larger values.69 In pristine PBDB-TF:Y6 cells (PCE = 16.5%) we obtain a slope of 57 mV per decade that gradually increases to 87 mV per decade in degraded cells with PCE = 7.9% (Fig. 3b and ESI Fig. S10a†). Although both values are larger than 2kBT/e, an increase in the slope by 30 mV per decade suggests significantly increased monomolecular recombination from the increased density of trap states in the degraded cells.From the IPDA data, the bimolecular recombination coefficient (kbr) is seen to fluctuate within instrumental noise for PCE changes from 16.5 to 12.6% and only then falls with the PCE degradation from 10.9 to 7.9%. This can be explained by dominant monomolecular recombination scavengers that reduce the fraction of photocarriers participating in the bimolecular processes (ESI Fig. S10b†). The reverse bias photocurrent shows only a slight reduction in the saturation behavior at higher voltages (>0.5 V) upon PCE degradation from 14.1 to 7.9%, again confirming minor changes in bimolecular recombination (ESI Fig. S11†). Note that this is in stark contrast to NFA-based OSCs with acceptors having end groups of varying degrees of fluorination where differences in PCE are inversely correlated with the IPDA-derived bimolecular recombination coefficient.24,28 Therefore, the PCE reduction by Y6 degradation does not follow the same morphology-dominated PCE loss mechanisms as NFAs having different end groups that strongly affect blend morphology.24,27 Instead, here the mean electron mobility falls more rapidly with degradation, by up to six-fold in PCE = 7.9% cells (Fig. 3c). Creation of new charge traps from degraded Y6 molecules is expected to strongly affect charge transport in Y6 nanocrystals even if the overall BHJ blend morphology is largely unaffected. Interestingly, the free charge carrier density and chemical capacitance at the maximum power point (nmp) do not increase significantly with degradation (Fig. 3c and ESI Fig. S12†). Thus, acceptor degradation affects the OSC performance through two major physical processes: increased monomolecular recombination and decreased electron mobility. The competition factor (θ) is defined as the ratio of the recombination rate to the charge extraction rate and captures the relative contributions from the two competing effects. In OSCs, FF scales inversely with θ and such a scaling relation has been observed in blends based on a wide range of functionalized NFAs.24,27–29,70 Here, the PBDB-TF:Y6 cell degradation scales with FF, almost linearly with log(θ) (Fig. 3d), suggesting poor charge extraction and increased recombination in degraded cells. Overall, the electrical measurements have quantified trap densities and correlated OSC degradation with steady-state competing processes of charge extraction and recombination. However, electrical measurements do not give insight into non-steady state photoexcited carrier dynamics that can be correlated with degradation at the molecular level. Ultrafast pump-probe spectroscopy has proven to be a powerful technique to understand excite state dynamics in OSCs and thus, we next employ this method to understand the effects of degradation.
2.4 Excited-state dynamics
The relationship between PCE degradation and excited-state dynamics for PBDB-TF:Y6 blends and neat Y6 films was investigated using transient absorption (TA) spectroscopy by exciting at 800 nm with a 20 μJ cm−2 pulse (ESI Section 6, Fig. 4, and S13–S15†). In the pristine blend, prominent excited-state absorption (ESA) bands appear at 690 and 760 nm (PBDB-TF) and 910 and 1500 nm (Y6) along with ground-state bleach (GSB) at 570 and 630 nm (PBDB-TF) and 730 and 800 nm (Y6). Following rapid (<1 ps) hole injection, geminate recombination occurs with a lifetime of 8 ps, followed by slower recombination between 300 ps and 150 ns (ESI Table S1†) with minimal spectral evolution. These longer-lived populations correspond to free carriers undergoing bimolecular recombination.71 | ||
| Fig. 4 (a) Transient absorption (TA) spectra of a pristine PBDB-TF:Y6 blend film taken at different delay times when excited at 800 nm (20 mJ cm−2). (b) Corresponding time-resolved differential absorption (ΔA) at indicated wavelengths (symbols). The solid lines show model fits from global analysis. (c) Model population dynamics of different species (A–F) in the pristine blend. (d) and (e) TA spectra and time-resolved ΔA of the same PBDB-TF:Y6 blend film after degradation. (f) A comparison of bleach kinetics shows the deepening of the bleach in a degraded blend over a few picoseconds. The instrument response function is 300 fs. | ||
For a direct comparison, the evolution-associated spectra of the pristine and degraded PBDB-TF:Y6 blends are shown in ESI Fig. S13.† The first spectral component (state A) shows the bleaching of Y6 (650–900 nm), but also some contribution from the bleaching of the PBDB-TF donor in the 500–650 nm region that indicates a contribution from ultrafast hole transfer. These features appear along with some excited-state absorption from the Y6 Frenkel exciton that diminishes with time in the 850–1000 nm and 1100–1400 nm regions.30 Similar excited-state absorptions are also observed in the neat pristine Y6 films (ESI Fig. S14†). The spectrum of state B resembles that of state A but with a lower amplitude of the Frenkel exciton features. These two states show distortion of the Y6 bleach below 800 nm due to contributions from the charge carrier absorptions and both show the hole injection process, with fast and slow contributions from optimal (A) and sub-optimal (B) donor–acceptor packing in the film, respectively. Neither of these states is dramatically affected by the degradation process, as their spectra and kinetics are very similar, although the sub-optimal charge injection is slightly faster following UV exposure of the film.
The spectra of the next two states (C and D) represent the charge carriers, with the polymer bleach in the 500–650 nm range, Y6 bleach in the 800–900 nm range, and the excited-state absorption of the carriers between 680–800 nm and 900–1000 nm.71 Additionally, at longer times there may also be some contribution from the Y6 triplet–triplet absorption beyond 1400 nm.72 These spectra decay with time constants ranging from ≈100 ps (state C) to ≈10 ns (state D) with minimal spectral evolution. From their short decay times, the populations associated with these spectra likely decay via geminate recombination, as the excitons do not have a sufficiently long lifetime to effectively dissociate and diffuse away. Both time constants are strongly affected by the degradation process, with each being accelerated by a factor of ≈4×. This likely reflects an increase in local trap states and changes in morphology that limit carrier lifetime.
The spectrum of state E is also similar to that of C and D and can be assigned to free carriers that decay via non-geminate recombination based on the minimal spectral evolution and much longer recombination time (≈100 ns). The decay of state E is moderately accelerated by the degradation process. The final state is unaffected by the UV irradiation; this may be a thermal artifact or photoproduct state, but it is very low in amplitude, and we, therefore, forego interpretation.
We emphasize here that the spectral signatures of the degraded blend films (Fig. 4d and e) and the hole transfer rate are changed, while the recombination kinetics are dramatically affected (Fig. 4f). Each recombination step captured by the sequential kinetic model is accelerated, with the fastest (5 ps) and slowest (153 ns) decays increasing by ≈50%, the intermediate processes (e.g., 8.9 ns) accelerate by as much as a factor of 4× upon degradation. This result is consistent with the loss of electron mobility and increased rate of geminate recombination discussed above, all of which will reduce the available charge collected at the electrodes thereby lowering device performance. In contrast, TA analysis of pristine Y6 films before and after degradation does not reveal conclusive trends as in the blend data (ESI Fig. S14 and S15†). Note, this sequential model is an oversimplification and does not account for many-body effects such as singlet–singlet- and triplet–triplet annihilation, both of which are bimolecular in nature.73,74 So far, we have focused on the evolution of electronic structure and charge dynamics upon OSC PCE degradation. A comprehensive picture of degradation can only emerge by also studying the structural and morphological evolution of the BHJ during degradation. X-ray measurements have been widely used in the structural analysis of NFA BHJs and we next leverage that to elucidate microstructural changes in crystalline OSC phases upon PCE degradation.
2.5 X-ray structural analysis
GIWAXS was employed to evaluate microstructural changes of neat Y6, neat PBDB-TF, and blend (PBDB-TF:Y6) films arising from photodegradation. Neat and blend films were prepared on Si substrates using identical fabrication procedures as described for OSC device fabrication (see ESI Section 7†). Note that GIWAXS measurements required the fabrication of films on Si substrates rather than on ZnO-coated ITO substrates to avoid background scattering that would complicate the NFA GIWAXS analysis. Note also that degradation has been shown to occur in the presence of O2 even without ZnO, e.g., in conventional OSCs with ITO substrates coated with PEDOT:PSS.46 Although the interface between the active layer and the charge transport layer plays a significant role in the degradation of OPV performance,75 photodegradation within the polymer/NFA blends can occur within 15 minutes in the absence of the ZnO ETL, and without significant disruption of the blend film morphology/microstructure, indicating that more nuanced and subtle structural changes lead to compromised device metrics.44–46,76 The primary reactive species in such instances are known to be superoxide and hydroxyl radicals generated from photolytic reactions with ambient moisture and O2.44–46,77,78Pristine films (neat and blend) were maintained under an inert environment before the GIWAXS measurements, while degraded films were prepared by exposing them to the same solar illumination under ambient conditions for 30 min. GIWAXS diffraction patterns and related line-cut profiles for the out-of-plane (OOP) and in-plane (IP) reflections are provided in Fig. 5, ESI Fig. S16, S17, and Table S2.† For all films, lamellar spacings (reflection = 100)/π–π distances (reflection = 010) and their corresponding crystal coherence lengths (CCLs) were determined from the IP/OOP reflections, respectively. For pristine (undegraded) neat Y6, neat PBDB-TF, and PBDB-TF:Y6 blend films, the diffraction patterns and trends for lamellar spacing (24.29/24.50/21.64 Å) and π–π distances (3.60/3.72/3.63 Å) and corresponding CCL values (38.67/33.92/59.32 Å and 25.66/18.18/20.74 Å) are similar to those of previous reports.25,79 Photodegradation of the neat PBDB-TF/Y6 films (ESI Fig. S16, S17, and Table S2†) induces slight changes in the lamellar spacings/CCL (23.36/41.21 Å and 23.42/33.49 Å) with no observable difference in the π–π distances/CCL (3.73/18.07 Å and 3.61/25.50 Å). The change in lamellar spacing is slightly greater for the degraded neat PBDB-TF films than for the degraded neat Y6 films. These changes agree with values reported in the literature,25,79 and they are not observed in the blend films. Morphological changes in the blends involve additional donor–acceptor interaction features that are not present in the neat films. For the degraded PBDB-TF:Y6 blend, the lamellar spacings/CCL (21.26/59.71 Å) and π–π distances/CCL (3.60/19.03 Å) are very similar to the pristine blend films indicating that the polymer/NFA structural changes due to photodegradation do not significantly contribute to observable changes in the morphology and microstructure, again consistent with previous reports.44–46 Rather, the photodegradation of PBDB-TF:Y6 blends have been attributed to a photo-oxidation process whereby oxidation of the exocyclic vinyl groups leads to epoxide formation and the subsequent degradation of the Y6 end-group, as reported for other NFAs.49 While complete scission of the Y6 end-group or scission of the dicyanomethylene group is expected to create catastrophic disruption of the 3D structure, these scenarios are inconsistent with GIWAXS analysis (Fig. 5).29 However, a reduction in conjugation length due to vinyl oxidation and the resulting changes in electronic structure could easily decrease electron mobility and increase recombination rates without disrupting the overall morphology of the PBDB-TF:Y6 BHJ. Thus, the X-ray measurements reveal that PCE degradation is not accompanied by significant structural changes in the BHJ morphology and that is consistent with the aforementioned observations that only a small mole fraction (<1%) of degraded NFA molecules is needed to reduce PCE by 50% upon degradation. Finally, we introduce a computational analysis to develop a complete picture of chemical degradation pathways that is consistent with the experimental analyses discussed so far.
 | ||
| Fig. 5 (a) and (b) GIWAXS diffraction patterns of pristine and degraded PBDB-TF:Y6 blends, respectively. (c) Line cuts along in-plane (IP) and out-of-plane (OOP) directions for pristine and degraded blend films. (d) π–π crystal coherence length (CCL) and π–π spacings for pure Y6, pure PBDB-TF, and PBDB-TF:Y6 blend films before and after degradation. | ||
2.6 Molecular and electronic structural analysis
To gain a molecular and electronic understanding of the PCE degradation process, we performed density functional-based tight binding (DFTB) calculations to generate possible Y6 degradation pathways (ESI Section 8†). We considered and optimized the Y6 and the structures (or fragments) of its three plausible UV-induced degradations in the calculations. The three pathways considered represent the three functional groups of Y6 most likely to be reactive to a free radical.36,38,39,46,49 The first is the end group-core linking the vinyl group oxidation (Fr1), as shown in Fig. 6a. The second is at that same position having a bond scission leading to end group scission from the core (Fr2) (Fig. 6a). The third degradation pathway considered is dicyanomethylene end group scission (Fr3), as shown in Fig. 6a. Further details of the three degraded structures are shown in ESI Fig. S18.† We additionally examine the doubly degraded fragments Fr1d, Fr2d, and Fr3d by considering degradation reactions on both acceptor end groups. Although two reactions on the same Y6 molecule would be statistically unlikely to happen under the solar fluxes considered here, we used the doubly reacted Y6 structures as a comparison tool. | ||
| Fig. 6 (a) Chemical structures of three types of possible single fragmentation products of a degraded Y6 molecule. Single (double) fragmentation via vinyl oxidation, acceptor end-group scission, and dicyanomethylene scission are labelled as Fr1, Fr2, and Fr3 (Fr1d, Fr2d, and Fr3d), respectively. The full chemical structures of all variations of the degraded Y6 molecules are shown in ESI Fig. S18.† (b) Energy levels of frontier molecular orbitals (HOMO−1, HOMO, LUMO, and LUMO+1) on pristine Y6 and degraded products after six kinds of degradation on Y6. | ||
From each degraded structure we selected frontier molecular orbitals (FMOs) of each molecule for comparison (Fig. 6b, 7a, b, ESI Table S3 and Fig. S19–S25†). Note that only the LUMO energy levels of Fr1 (−4.46 eV) have much lower energy levels versus the Y6 LUMO (−4.29 eV) (Fig. 6b). Furthermore, the HOMO energy levels shift upwards in all the fragments signifying the destabilization of the π orbitals (ESI Fig. S19–S25†). This lowered LUMO energy level and a relatively small change in HOMO corresponds to the experimental optical absorption spectrum where redshift is observed after the Y6 degradation and is due to the formation of hydrogen bonds around the hydroxyl group after vinyl oxidation, as discussed later.
 | ||
| Fig. 7 (a) Optimized molecular structures of the “Fr1 anion−” and “Fr1d anion2−” that are singly and doubly charged fragments, respectively. To create an anion from Fr1 and Fr1d, a hydrogen atom was removed from the nitrogen atom within the dicyanomethylene group. Charges have been stabilized by Zn2+ in “Fr1 anion− + Zn2+” and “Fr1d anion2− + 2Zn2+” fragments after additional removal of the hydrogen atom from the hydroxyl group. The atom color scheme is identical to that in Fig. 6a. Zn2+ is labeled as gray. (b) Frontier molecular orbital energy levels (HOMO−1, HOMO, LUMO, and LUMO+1) of pristine Y6 and degraded products Fr1 and Fr1d after each degradation on Y6, depending on the charge state and the existence of Zn2+. “Fr1 anion−” and “Fr1d anion2−” have −1 and −2 as total charges, respectively, and all other molecules are charge neutral. | ||
We also computed the FMOs of the singly and doubly charged anionic Fr1− and Fr2− and have stabilized the corresponding anions by incorporating Zn2+ cations in the vicinity, as shown in Fig. 7a. From our characterizations of the PBDB-TF:Y6 blend films (vide supra), we detail that minimal disruptions to the morphology and crystal structure can still contain trap states emerging due to photo-oxidation. The anions constructed here are entirely consistent with the ZnO acting as a photo-catalytic center for Y6 degradation, even away from the ZnO interface. The anionic fragments, stabilized with Zn2+, shown in Fig. 7a, offer a plausible Y6 degradation pathway within the BHJ active layer. These anions were constructed by removing each H atom connected to the N atom in the dicyanomethylene group, and neutral molecules with the Zn2+ cation were constructed after additional dehydrogenation of the hydroxyl group. The Fr1− anion with one additional electron shows unfavourable FMO energetics of about 2 eV compared to Fr1−. The Fr1− anion with two additional electrons has even more elevated FMO energies of about 4 eV versusFr1− because the electrons populate antibonding molecular orbitals. In the case of the neutral molecule paired with a Zn2+ cation, the molecule shows an overall tendency of much higher FMO energy levels versusFr1− (Fig. 7b). The molecule derived from Fr1d shows lower energy levels in the occupied molecular orbitals including the HOMO, with the LUMO and LUMO+1 energy levels even higher than those of original Fr1d. Therefore, anions or stabilized anions do not have lower LUMO levels compared to the charged neutral species. However, to go beyond gas-phase FMO calculations of the anion states is outside the scope of this manuscript. This is because the anion state in the solid crystal structure requires a polaronic treatment, and an additional term in the Hamiltonian to describe the charge aggregation must be considered to obtain an exact stabilized anion state.
We next investigated the crystalline packing of the three degraded Y6 structures to connect the molecular geometry to the generated trap states. The crystal structures were constructed by substituting the unit Y6 molecule of the original C12/c1 crystal structure24 with each Y6 and a degraded molecule, respectively, and they were subsequently optimized to match the stacking structure (Fig. 8, ESI Fig. S26–S30 and Table S4†). We note that the doubly oxidized crystal structure of Fr1d was not considered because of its unstable structure when it packs in the C12/c1 crystalline phase (not shown). The total density of states (TDOS) and partial density of states (PDOS) of each of the crystal structures are compared after optimization in Fig. 8b and ESI Fig. S31–S35.†
 | ||
| Fig. 8 (a) LUMO of Fr1 molecule after vinyl oxidation. The inset shows the formation of hydrogen bonds around the hydroxyl group. The HOMO of Fr1 is shown in ESI Fig. S20.† (b) TDOS and PDOS (p orbital of carbon, nitrogen, oxygen, and sulfur) of the Fr1 crystal structure. Energy levels of HOMO (EHOMO), LUMO (ELUMO), and trap energy levels are shown by blue, red, and grey solid vertical lines below the x-axis. Energy spectra are smeared with a value of 50 meV. (c) and (d) Optimized crystal structure of Fr1 in top and side views, respectively, based on the original pristine Y6 crystal structure. The color scheme on each element is identical to that in Fig. 6a. | ||
Note that the trap energy level of the Fr2d crystal occurs significantly far away from the Fermi energy level and is distinct from the other structures. This is because the π-structure in the Fr2d crystal is significantly truncated by the elimination of two end groups. Interestingly, a well-defined trap energy level of ET = 0.25 eV is observed in the Fr1 crystal (Fig. 8b) compared to a shallow trap level of ET = 0.12 eV in the pristine Y6 crystal (ESI Fig. S31†). This significant difference between these values agrees well with TAS observed ET measurement of 0.25 eV (vide infra; Fig. 2d–f). In contrast, the acceptor end-group and dicyanomethylene scissions either show minor evolution in the LUMO or a dramatically different TDOS profile that is inconsistent with experimental data. We find that the Fr1 mid-gap states can be explained by the PDOS, which shows the p orbitals of the oxygen causing the low-lying states in the Fr1 crystal. These states are red-shifted toward the Fermi level compared to that of the Y6 crystal while the LUMO energy level of Fr1 shows strongly localized molecular orbitals on the side where vinyl oxidation occurs (Fig. 8a). We note that these red-shifted mid-gap states are distinct from the shifted FMO energies between Y6 and the three proposed degraded fragments.
The Y6 degradation pathway of Fr1 is in agreement with a recent report detailing the chemical degradation pathway in Y6 involving breakage of conjugation with the vinyl group.80 The vinyl group cleavage was suppressed by fluorination of the end groups and incorporation of long side chains to achieve photostability in hot (300 K) outdoor climates. The present results also agree with previous reports on an ITIC-based OSC where Nuclear Magnetic Resonance and Fourier Transform Infrared spectroscopies implicated vinyl oxidation as the principal mechanism of ITIC molecule photo-induced degradation.36 As a proof-of-concept, we computed the UV/vis absorption spectra of the Y6 and ITIC molecules along with the three fragments using TDDFT (ESI Section 8†) and found transition state orbitals that are consistent with the destabilization of the vinyl group (Fig. S36–S41†).
An in situ Raman study on an ITIC molecule undergoing gradual degradation also suggested vinyl linkage breaking between the electron-donating core and the electron-deficient end groups.39 Computational and experimental studies of other fused and non-fused NFAs (such as IT-4F, PTIC, etc.) also showed photochemical degradation at the vinyl linkage.49 UV light can be absorbed in the ZnO bandgap of 3.37 eV, generating an electron–hole pair where the hole can initiate the desorption of neutral O2 and the electron then becomes available to break the double bond at the vinyl linkage.35 Indeed, X-ray Photoelectron Spectroscopy of degraded PBDB-TF:Y6:PC71BM OSCs confirms the role of ZnO in the degradation mechanism. We acknowledge that degradation of the donor polymer PBDB-TF is likely to have a minor effect on the overall performance degradation. In ref. 44, OSCs containing fullerene ([70]PCBM) and non-fullerene (ITIC) acceptors and the same donor polymer PBDB-T are compared to isolate the specific role of donor polymer degradation in cell performance. ITIC-containing blends show a 38% decrease in PCE upon degradation compared to 9% in those with [70]PCBM suggesting minor effects of the donor polymer degradation on overall cell performance loss.
The present study on high-PCE PBDB-TF:Y6 OSC degradation not only confirms earlier work on other NFA OSC compositions but goes beyond by comparing experimental and computational results, also quantifying the effects of degradation by correlating macroscopic parameters such as PCE with microscopic parameters such as trap density of states and recombination coefficient in operating cells. Importantly we find that the overall fraction of degraded molecules can be minute and still produce a significant PCE reduction. This is consistent with two observations here. First, the optical absorption spectroscopy of degraded neat Y6 and PBDB-TF:Y6 blend films show the emergence of a relatively minor peak corresponding to the degraded molecule. The absorption peak cross-section can be expected to approximately scale linearly with the fraction of degraded molecules and suggests that photo-degradation involves less than 1% mole fraction. Second, both GIWAXS measurements and computations show minor changes (4.3% change in volume and 2.5% change in lattice parameter) in morphology/metrical parameters between Y6 and Fr1 (ESI Tables S2 and S4†) and such volume-conserving degradation is consistent with vinyl oxidation but not with hypothetical routes of Y6 end-group and dicyanomethylene scissions.49 Such small mole fraction of degraded Y6 molecules reducing PCE by half also suggests the long-range interaction of trap states with charge carriers. Indeed, low-mobility semiconductors typically have highly localized interactions with trap states, and therefore, the present observations are consistent with high-mobility coherent channels that are predicted in NFA nanocrystals.81
Y6 degradation in the form of Fr1 does not lead to decomposition into two separated fragments but it weakens the bond which could then lead to complete fragmentation (Fr2) via subsequent activation. To identify the chemical structure of decomposed Y6 we performed matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) measurements of degraded Y6 films (ESI Section 9†). First, the MALDI-ToF spectra of pristine Y6 film directly deposited onto MALDI-ToF plates by drop casting reveals a strong peak at m/z = 1450.5 that agrees well with the calculated value for Y6 (ESI Table S5 and Fig. S42†). Next, Y6 films deposited onto ZnO/ITO substrates were subjected to the same solar illumination degradation procedure as used for OSCs, and the degraded Y6 film was deposited on a MALDI-ToF plate as a solution in 1,2-dichlorobenzene. The MALDI-ToF spectra of these films show a strong peak (m/z = 1225.4) assignable to Fr2 (ESI Table S5, Fig. S42 and S43†). This feature agrees with earlier MALDI-ToF measurements on degraded Y6-based solar cells, however, that study was limited to correlating device metrics and proposing a chemical degradation pathway.48 Here, in degraded cells, Y6 and Fr1 may decompose into Fr2 either during the film dissolution or during the MALDI-ToF measurement that exposes the Y6 film to UV light for molecular ionization. Therefore, subtle changes in Y6 during initial degradation via the formation of Fr1 should be sufficiently deleterious to decrease PCE by up to 50%. This does not eliminate the possibility of Fr2 formation in the partially degraded OSCs although energy level calculations suggest it is less likely to form than Fr1. We also note that double scissions on a single molecule are expected to be extremely rare since the overall volume fraction of vinyl oxidation is small and the computed crystal structures of Fr1d, Fr2d, and Fr3d are not consistent with the GIWAXS experimental data (ESI Tables S2 and S4†). Note that we do not observe any of the double scission fragments in MALDI-ToF spectra of the partially degraded Y6 (ESI Fig. S42†) and previous observations of Fr2d in degraded films are likely caused by a significantly greater degree of Y6 degradation.82 Here, we do not consider OSCs that are degraded beyond 50% of their original PCE values.
In principle, NFA degradation could be mitigated either by using an alternative electron transport layer83 or incorporating stabilizers such as dithiocarbamates or ET18,46,84 or by passivating the ZnO layer, e.g., by using polyethylenimine or self-assembled monolayers.35,46 Note that even suppressing the catalytic role of ZnO, NFA-based OSCs can degrade by other pathways due to inherent reactivity and aggregation of Y6 molecules, and such effects can be mitigated by designing stable end-groups (e.g., by fluorination) and/or by incorporating long side chains.33,35,44,80,85 While we considered a reactive NFA in the present study, practical OSCs will need to address the origins of other instabilities that may result from the donor polymer, such as the twisting of the polymer backbone.86
3 Conclusions
Here we have correlated the evolution of trap density of states, mobility, and recombination dynamics with the degradation processes of high-performance PBDB-TF:Y6 solar cells (PCE > 17%). Starting from the suggested and plausible role of the ZnO ETL layer in triggering OSC NFA degradation, we quantify the correlation between macroscopic device metrics such as PCE with microscopic processes such as trap energy and density of states, carrier mobility, and recombination rates. Two kinds of trap states are identified. The deep traps at 0.48 eV from the Y6 molecule LUMO are from interfacial states, contribute to series resistance, and are unaffected by the OSC degradation. The density of shallow trap states at 0.15 eV from the Y6 molecule LUMO in the BHJ rapidly increases (three-fold) with PCE degradation by 30% and shifts to a new energy level of 0.21 eV, thereby increasing monomolecular recombination and decreasing cell carrier mobility under operating conditions. The emergence of these trap states can be detected by absorption spectroscopy and a <1% fraction of degraded molecules is sufficient for severe OSC PCE performance degradation by 50%. Our experimental results and quantum calculations reveal vinyl oxidation as the putative chemical pathway of Y6 degradation and this volume-conserving process does not affect the crystal structure significantly while revealing the emergence of a new density of states peak at 0.25 eV below the LUMO of degraded Y6 that agrees with capacitance and optical measurements. Thus, chemical degradation of the Y6 molecule does not affect the performance through major changes in film morphology, which had been the traditional guiding principle to design NFA molecules and end-groups, but instead affects the charge transport and extraction processes through newly developed trap states in degraded Y6 molecules without full dissociation into separate fragments that determine the ultimate OSC performance. Such subtle changes were not evident in previous literature that mostly focused on dissociated fragments by mass spectroscopy in fully degraded cells. Thus, NFA-based stable OSC technologies would benefit from improved ETLs and mitigating vinyl oxidation by various strategies discussed earlier. Clearly, the first step in suppressing these processes is by understanding them.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
V. K. S. and Z. M. contributed equally. V. K. S., Z. M., A. F., T. J. M., and M. C. H. conceived the idea. G. L., F. Q., and Y. C. fabricated the solar cells. V. K. S. and Z. M. performed photophysical and trap energetics measurements and analysis. S. H. assisted in electrical data analysis. R. M. P. and D. Z. conducted GIWAXS measurements and data analysis. W. C. J. and K. L. K. performed computational analyses. R. M. Y. performed and analyzed transient absorption spectroscopy measurements. All authors discussed the results and assisted in writing the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by U.S. Office of Naval Research Contracts #No. N00014-20-1-2116 and N00014-24-1-2109 (G. L.: material synthesis and characterizations), by the U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design Award #70NANB19H005, and by the Northwestern University Materials Research Science and Engineering Center Award NSF DMR-2308691 (V. K. S., Z. M.: charge transport measurements). This work (IVT, TAS, and IPDA) made use of the MatCI Facility which receives support from the MRSEC Program (NSF DMR-2308691) of the Materials Research Center at Northwestern University. Transient optical spectroscopy was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-FG02-99ER14999 (MRW). Theory work (G. C. S.) was supported by NSF Grant CHE-2347622. This work made use of the GIANTFab core facility at Northwestern University. GIANTFab is supported by the Institute for Sustainability and Energy at Northwestern and the Office of the Vice President for Research at Northwestern. R. M. P. acknowledges support from the Intelligence Community Postdoctoral Research Fellowship Program at Northwestern University administered by Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy and the Office of the Director of National Intelligence (ODNI). This work made use of the IMSERC MS facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), the State of Illinois, and the International Institute for Nanotechnology (IIN). The computational resource was supported by Northwestern’s Quest High-Performance Computing Cluster. This work also acknowledges the U.S. Department of Energy under contract no. DE-AC02-05CH11231 at the beamline 8-ID-E of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.Notes and references
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 RSC.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew Chem. Int. Ed. Engl., 2019, 58, 4129–4142 CrossRef CAS.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS.
- D. Luo, W. Jang, D. D. Babu, M. S. Kim, D. H. Wang and A. K. K. Kyaw, J. Mater. Chem. A, 2022, 10, 3255–3295 RSC.
- H.-W. Cheng, Y. Zhao and Y. Yang, Adv. Eng. Mater., 2022, 12, 2102908 CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- A. S. Gertsen, M. F. Castro, R. R. Søndergaard and J. W. Andreasen, Flexible Printed Electron., 2020, 5, 014004 CrossRef CAS.
- T. Yan, W. Song, J. Huang, R. Peng, L. Huang and Z. Ge, Adv. Mater., 2019, 31, 1902210 CrossRef.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- L. Liu, Y. Kan, K. Gao, J. Wang, M. Zhao, H. Chen, C. Zhao, T. Jiu, A.-K.-Y. Jen and Y. Li, Adv. Mater., 2020, 32, 1907604 CrossRef CAS.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- H. Sun, T. Liu, J. Yu, T.-K. Lau, G. Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao and H. Yan, Mater. Chem. Front., 2019, 12, 3328–3337 CAS.
- D. Li, N. Deng, Y. Fu, C. Guo, B. Zhou, L. Wang, J. Zhou, D. Liu, W. Li, K. Wang, Y. Sun and T. Wang, Adv. Mater., 2023, 35, 2208211 CrossRef CAS.
- K. Liu, Y. Jiang, F. Liu, G. Ran, F. Huang, W. Wang, W. Zhang, C. Zhang, J. Hou and X. Zhu, Adv. Mater., 2023, 35, 2300363 CrossRef CAS PubMed.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K.-Y. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 3274–3287 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, C. Zhang, T.-K. Lau, G. Zhang, X. Lu, H.-L. Yip, S. K. So, S. Beaupré, M. Mainville, P. A. Johnson, M. Leclerc, H. Chen, H. Peng, Y. Li and Y. Zou, Adv. Mater., 2019, 31, 1807577 CrossRef.
- G. Li, X. Zhang, L. O. Jones, J. M. Alzola, S. Mukherjee, L.-w. Feng, W. Zhu, C. L. Stern, W. Huang, J. Yu, V. K. Sangwan, D. M. DeLongchamp, K. L. Kohlstedt, M. R. Wasielewski, M. C. Hersam, G. C. Schatz, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2021, 143, 6123–6139 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- F. Liu, Z. Zhou, C. Zhang, T. Vergote, H. Fan, F. Liu and X. Zhu, J. Am. Chem. Soc., 2016, 138, 15523–15526 CrossRef CAS PubMed.
- G. Li, L.-W. Feng, S. Mukherjee, L. O. Jones, R. M. Jacobberger, W. Huang, R. M. Young, R. M. Pankow, W. Zhu, N. Lu, K. L. Kohlstedt, V. K. Sangwan, M. R. Wasielewski, M. C. Hersam, G. C. Schatz, D. M. DeLongchamp, A. Facchetti and T. J. Marks, Mater. Chem. Front., 2022, 15, 645–659 CAS.
- X. Zhang, G. Li, S. Mukherjee, W. Huang, D. Zheng, L.-W. Feng, Y. Chen, J. Wu, V. K. Sangwan, M. C. Hersam, D. M. DeLongchamp, J. Yu, A. Facchetti and T. J. Marks, Adv. Eng. Mater., 2022, 12, 2102172 CAS.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- G. Li, F. Qin, R. M. Jacobberger, S. Mukherjee, L. O. Jones, R. M. Young, R. M. Pankow, B. P. Kerwin, L. Q. Flagg, D. Zheng, L.-W. Feng, K. L. Kohlstedt, V. K. Sangwan, M. C. Hersam, G. C. Schatz, D. M. DeLongchamp, M. R. Wasielewski, Y. Zhou, A. Facchetti and T. J. Marks, Joule, 2023, 7, 2152–2173 CrossRef CAS.
- S. M. Swick, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, W. Zhu, L. O. Jones, N. Powers-Riggs, A. Facchetti, K. L. Kohlstedt, G. C. Schatz, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Adv. Eng. Mater., 2020, 10, 2000635 CAS.
- Y. Li, T. Li and Y. Lin, Mater. Chem. Front., 2021, 5, 2907–2930 RSC.
- Y. Wang, J. Lee, X. Hou, C. Labanti, J. Yan, E. Mazzolini, A. Parhar, J. Nelson, J.-S. Kim and Z. Li, Adv. Eng. Mater., 2021, 11, 2003002 CAS.
- Y. Li, X. Huang, K. Ding, H. K. M. Sheriff, L. Ye, H. Liu, C.-Z. Li, H. Ade and S. R. Forrest, Nat. Commun., 2021, 12, 5419 CrossRef CAS.
- L. Hu, Y. Jiang, L. Sun, C. Xie, F. Qin, W. Wang and Y. Zhou, J. Phys. Chem. Lett., 2021, 12, 2607–2614 CrossRef CAS PubMed.
- S. Park and H. J. Son, J. Mater. Chem. A, 2019, 7, 25830–25837 RSC.
- Y. Zhao, Z. Wu, X. Liu, Z. Zhong, R. Zhu and J. Yu, J. Mater. Chem. C, 2021, 9, 13972–13980 RSC.
- M. A. Anderson, B. W. Larson and E. L. Ratcliff, ACS Appl. Mater. Interfaces, 2021, 13, 44641–44655 CrossRef CAS PubMed.
- A. J. Clarke, J. Luke, R. Meitzner, J. Wu, Y. Wang, H. K. H. Lee, E. M. Speller, H. Bristow, H. Cha, M. J. Newman, K. Hooper, A. Evans, F. Gao, H. Hoppe, I. McCulloch, U. S. Schubert, T. M. Watson, J. R. Durrant, W. C. Tsoi, J.-S. Kim and Z. Li, Cell Rep. Phys. Sci., 2021, 2, 100498 CrossRef CAS.
- A. Azeez and K. S. Narayan, J. Phys. Chem. C, 2021, 125, 12531–12540 CrossRef CAS.
- Y. Han, H. Dong, W. Pan, B. Liu, X. Chen, R. Huang, Z. Li, F. Li, Q. Luo, J. Zhang, Z. Wei and C.-Q. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 17869–17881 CrossRef CAS.
- X. Zhu, L. Hu, W. Wang, X. Jiang, L. Hu and Y. Zhou, ACS Appl. Energy Mater., 2019, 2, 7602–7608 CrossRef CAS.
- K. An, W. Zhong, F. Peng, W. Deng, Y. Shang, H. Quan, H. Qiu, C. Wang, F. Liu, H. Wu, N. Li, F. Huang and L. Ying, Nat. Commun., 2023, 14, 2688 CrossRef CAS.
- N. Y. Doumon, M. V. Dryzhov, F. V. Houard, V. M. Le Corre, A. Rahimi Chatri, P. Christodoulis and L. J. A. Koster, ACS Appl. Mater. Interfaces, 2019, 11, 8310–8318 CrossRef CAS PubMed.
- E. M. Speller, A. J. Clarke, N. Aristidou, M. F. Wyatt, L. Francàs, G. Fish, H. Cha, H. K. H. Lee, J. Luke, A. Wadsworth, A. D. Evans, I. McCulloch, J.-S. Kim, S. A. Haque, J. R. Durrant, S. D. Dimitrov, W. C. Tsoi and Z. Li, ACS Energy Lett., 2019, 4, 846–852 CrossRef CAS.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. Tang, C. Sun, Z. Luo, K. Chang, Z. Zhang, J. Yuan, T. Li, W. Tang, E. Zhou, Z. Xiao, L. Ding, Y. Zou, X. Zhan, C. Yang, Z. Li, C. J. Brabec, Y. Li and J. Min, J. Mater. Chem. A, 2019, 7, 25088–25101 RSC.
- W. Li, D. Liu and T. Wang, Adv. Funct. Mater., 2021, 31, 2104552 CrossRef CAS.
- T. Liu, Q. C. Burlingame, M. R. Ivancevic, X. Liu, J. Hu, B. P. Rand and Y.-L. Loo, Adv. Eng. Mater., 2023, 13, 2300046 CAS.
- Z.-X. Liu, Z.-P. Yu, Z. Shen, C. He, T.-K. Lau, Z. Chen, H. Zhu, X. Lu, Z. Xie, H. Chen and C.-Z. Li, Nat. Commun., 2021, 12, 3049 CrossRef CAS.
- U. Rau and H. W. Schock, Appl. Phys. A, 1999, 69, 131–147 CrossRef CAS.
- D. Bozyigit, W. M. M. Lin, N. Yazdani, O. Yarema and V. Wood, Nat. Commun., 2015, 6, 6180 CrossRef CAS PubMed.
- S. S. Hegedus and W. N. Shafarman, Progress in Photovoltaics: Research and Applications, 2004, vol. 12, pp. 155–176 Search PubMed.
- K. Tvingstedt and C. Deibel, Adv. Eng. Mater., 2016, 6, 1502230 Search PubMed.
- P. Singh and N. M. Ravindra, Sol. Energy Mater. Sol. Cells, 2012, 101, 36–45 CrossRef CAS.
- W. Dawidowski, B. Ściana, K. Bielak, M. Mikolášek, J. Drobný, J. Serafińczuk, I. Lombardero, D. Radziewicz, W. Kijaszek, A. Kósa, M. Florovič, J. Kováč, C. Algora and L. u. Stuchlíková, Energies, 2021, 14, 4651 CrossRef CAS.
- M. P. Hughes, K. D. Rosenthal, N. A. Ran, M. Seifrid, G. C. Bazan and T.-Q. Nguyen, Adv. Funct. Mater., 2018, 28, 1801542 CrossRef.
- X. Zhang, C. Li, J. Xu, R. Wang, J. Song, H. Zhang, Y. Li, Y.-N. Jing, S. Li, G. Wu, J. Zhou, X. Li, Y. Zhang, X. Li, J. Zhang, C. Zhang, H. Zhou, Y. Sun and Y. Zhang, Joule, 2022, 6, 444–457 CrossRef CAS.
- G. Zhang, X.-K. Chen, J. Xiao, P. C. Y. Chow, M. Ren, G. Kupgan, X. Jiao, C. C. S. Chan, X. Du, R. Xia, Z. Chen, J. Yuan, Y. Zhang, S. Zhang, Y. Liu, Y. Zou, H. Yan, K. S. Wong, V. Coropceanu, N. Li, C. J. Brabec, J.-L. Bredas, H.-L. Yip and Y. Cao, Nat. Commun., 2020, 11, 3943 CrossRef CAS.
- R. A. Awni, Z. Song, C. Chen, C. Li, C. Wang, M. A. Razooqi, L. Chen, X. Wang, R. J. Ellingson, J. V. Li and Y. Yan, Joule, 2020, 4, 644–657 CrossRef CAS.
- D. Bozyigit, S. Volk, O. Yarema and V. Wood, Nano Lett., 2013, 13, 5284–5288 CrossRef CAS PubMed.
- T. Walter, R. Herberholz, C. Müller and H. W. Schock, J. Appl. Phys., 1996, 80, 4411–4420 CrossRef CAS.
- S. Wang, P. Kaienburg, B. Klingebiel, D. Schillings and T. Kirchartz, J. Phys. Chem. C, 2018, 122, 9795–9803 CrossRef CAS.
- J. A. Carr, M. Elshobaki and S. Chaudhary, Appl. Phys. Lett., 2015, 107, 203302 CrossRef.
- G. Yang, R. A. C. M. M. v. Swaaij, S. Dobrovolskiy and M. Zeman, J. Appl. Phys., 2014, 115, 034512 CrossRef.
- H.-S. Duan, H. Zhou, Q. Chen, P. Sun, S. Luo, T.-B. Song, B. Bob and Y. Yang, Phys. Chem. Chem. Phys., 2015, 17, 112–116 RSC.
- V. K. Sangwan, M. Zhu, S. Clark, K. A. Luck, T. J. Marks, M. G. Kanatzidis and M. C. Hersam, ACS Appl. Mater. Interfaces, 2019, 11, 14166–14174 CrossRef CAS PubMed.
- D. Kroh, S. Athanasopoulos, V. Nádaždy, F.-J. Kahle, H. Bässler and A. Köhler, Adv. Funct. Mater., 2023, 2302520 CrossRef.
- J. Nelson, The Physics of Solar Cells, Imperial College Press, 2003 Search PubMed.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B, 2010, 82, 245207 CrossRef.
- L.-W. Feng, J. Chen, S. Mukherjee, V. K. Sangwan, W. Huang, Y. Chen, D. Zheng, J. W. Strzalka, G. Wang, M. C. Hersam, D. DeLongchamp, A. Facchetti and T. J. Marks, ACS Energy Lett., 2020, 5, 1780–1787 CrossRef CAS.
- N. D. Eastham, J. L. Logsdon, E. F. Manley, T. J. Aldrich, M. J. Leonardi, G. Wang, N. E. Powers-Riggs, R. M. Young, L. X. Chen, M. R. Wasielewski, F. S. Melkonyan, R. P. H. Chang and T. J. Marks, Adv. Mater., 2018, 30, 1704263 CrossRef PubMed.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS.
- G. Lo Gerfo M, L. Bolzonello, F. Bernal-Texca, J. Martorell and N. F. van Hulst, J. Phys. Chem. Lett., 2023, 14, 1999–2005 CrossRef CAS PubMed.
- A. J. Carrod, V. Gray and K. Börjesson, Mater. Chem. Front., 2022, 15, 4982–5016 CAS.
- P. Jiang, L. Hu, L. Sun, Z. a. Li, H. Han and Y. Zhou, Chem. Sci., 2022, 13, 4714–4739 RSC.
- F. C. Löhrer, C. Senfter, C. J. Schaffer, J. Schlipf, D. Moseguí González, P. Zhang, S. V. Roth and P. Müller-Buschbaum, Adv. Photonics Res., 2020, 1, 2000047 CrossRef.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- W. R. Mateker, T. Heumueller, R. Cheacharoen, I. T. Sachs-Quintana, M. D. McGehee, J. Warnan, P. M. Beaujuge, X. Liu and G. C. Bazan, Chem. Mater., 2015, 27, 6345–6353 CrossRef CAS.
- L. Zhu, M. Zhang, G. Q. Zhou, T. Y. Hao, J. Q. Xu, J. Wang, C. Q. Qiu, N. Prine, J. Ali, W. Feng, X. D. Gu, Z. F. Ma, Z. Tang, H. M. Zhu, L. Ying, Y. M. Zhang and F. Liu, Adv. Eng. Mater., 2020, 1904234 CAS.
- H. Xu, J. Han, S. Chen, Y. Liu, L. Huerta Hernandez, J. Bertrandie, M. Babics, S. Alam, D. R. Villalva, S. H. K. Paleti, J. Gorenflot, C. Herok, N. Ramos, J. Troughton, A. Sharma, T. B. Marder, B. Engels, J. Martin, S. De Wolf, F. Laquai and D. Baran, Joule, 2023, 7, 2135–2151 CrossRef CAS.
- B. Movaghar, L. O. Jones, M. A. Ratner, G. C. Schatz and K. L. Kohlstedt, J. Phys. Chem. C, 2019, 123, 29499–29512 CrossRef CAS.
- B. Zhou, L. Wang, Y. Liu, C. Guo, D. Li, J. Cai, Y. Fu, C. Chen, D. Liu, Y. Zhou, W. Li and T. Wang, Adv. Funct. Mater., 2022, 32, 2206042 CrossRef CAS.
- N. Zhou, M.-G. Kim, S. Loser, J. Smith, H. Yoshida, X. Guo, C. Song, H. Jin, Z. Chen, S. M. Yoon, A. J. Freeman, R. P. H. Chang, A. Facchetti and T. J. Marks, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 7897–7902 CrossRef CAS.
- E. Trippodo, V. Campisciano, L.-W. Feng, Y. Chen, W. Huang, J. M. Alzola, D. Zheng, V. K. Sangwan, M. C. Hersam, M. R. Wasielewski, B. Pignataro, F. Giacalone, T. J. Marks and A. Facchetti, J. Mater. Chem. C, 2023, 11, 8074–8083 RSC.
- N. Tanaka, M. Nakano, T. Kobayashi, A. Takahara, T. Fujinuki, M. Shahiduzzaman, M. Karakawa, T. Taima and K. Iiyama, Macromol. Rapid Commun., 2022, 43, 2100718 CrossRef CAS.
- Y. Wang, J. Luke, A. Privitera, N. Rolland, C. Labanti, G. Londi, V. Lemaur, D. T. W. Toolan, A. J. Sneyd, S. Jeong, D. Qian, Y. Olivier, L. Sorace, J.-S. Kim, D. Beljonne, Z. Li and A. J. Gillett, Joule, 2023, 7, 810–829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03501a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |