
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of hydrothermal etching and conversion on photocatalytic hydrogen evolution and overall water splitting with nanoparticulate and mesoporous TiO2 and SrTiO3/TiO2 composites†
Lion
Schumacher
 ,
Jana
Timm
and
Roland
Marschall
*
,
Jana
Timm
and
Roland
Marschall
*
Department of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: roland.marschall@uni-bayreuth.de
First published on 25th September 2024
Abstract
TiO2 and SrTiO3 are well-studied semiconductors for photocatalytic H2 evolution and overall water splitting, respectively. Hydrothermal conversion of different TiO2 starting materials enables the synthesis of SrTiO3/TiO2 composites with various morphological designs, which have so far shown increases in photocatalytic H2 evolution activity. Herein, we address this phenomenon and the underlying influences of alkaline and pH-neutral media during hydrothermal conversion reactions by detailed material characterization of SrTiO3/TiO2 composites and etched TiO2. Trends in photocatalytic H2 evolution activities can be related directly to morphological factors, which in turn determine the influence of hydrothermal treatment. TiO2 nanoparticles are mainly subjected to agglomeration, while mesoporous TiO2 benefits from increasing hydrophilicity and a broadening of pore size distributions. Taken together, both influences lead to “pore activation” that strongly enhances photocatalytic H2 evolution activities. Simple hydrothermal treatment in diluted NaOH solution or even plain H2O might be a promising strategy to enhance photocatalytic activities of mesoporous TiO2 and potentially other mesoporous semiconductors, but more importantly, it also needs to be taken into account in the case of hydrothermal conversion reactions, since the subtle impact of the reaction medium may be overlooked or misinterpreted as a composite effect.
Introduction
Affordable energy is socially and economically indispensable, but over the past few decades, our overall demands for energy sources and energy carriers have evolved beyond this basic requirement. Today and in the future, aspects of sustainability, reliability, cost effectiveness, and versatility must be considered to address one of the most pressing challenges of the 21st century: a complete transformation of the global economy away from fossil fuels.1,2Hydrogen (H2) is a promising energy carrier due to its high gravimetric energy density and foreseeable applications in fuel cells and various industrial processes.3–6 In this regard, abundant solar energy allows for scalable and potentially emission-free green H2 production.7 The production of green H2 could be mainly realized by two methods, namely electrocatalysis and photocatalysis. Both methods exhibit benefits and hindrances. In the case of electrocatalysis, electrocatalysts require an external potential to drive the electrolysis of water, which can be provided, for example, using photovoltaic cells. In this case, electrolyzers have a high efficiency for emission-free green H2 production or even the water splitting reaction, but the use of platinum group metals and the resulting high costs of this technology limit the competitiveness compared to conventional steam reforming.8,9 Furthermore, the replacement of noble metals is challenging due to the high demands on electrocatalysts such as high current densities, a large number of active sites, high conductivity, pH stability, and effective mass transfer properties.8,10–14 In contrast, semiconductor photocatalysts can be made from earth-abundant elements. In order to enhance the photocatalytic performance, only small amounts of noble metals or even earth-abundant elements as co-catalysts are combined with the semiconductor. In the long run, the competitiveness of photocatalysis compared to steam reforming and electrocatalysis will depend on the price and the availability of the used materials. Semiconductors are already largely made from comparably inexpensive alkaline earth metals, transition elements, and p-block elements.15–17 Examples are TiO2 and SrTiO3, which are model systems for photocatalytic H2 evolution and OWS, respectively.18–20 Kinetic limitations, electronic structures, and influences of morphologies, facets or surface treatments have been investigated for both semiconductors, and knowledge gained over decades is used to optimize photocatalysts.21–32
Despite various performance-limiting characteristics, photocatalytic activity can be exclusively limited by the surface area.33 Therefore, increasing the surface area by nanostructuring is a common strategy to enhance photocatalytic activity.33 In comparison to nanoparticles, porous photocatalysts benefit from enhanced light harvesting due to reflection and scattering by pores, diffusion of reactants and products through pores, efficient charge carrier transport, and easy reuse of the material.34,35 Antonelli et al. used a soft-templating approach and were the first to synthesize mesoporous TiO2 in 1995.36,37 Since then, TiO2 has also become a model system for porous photocatalysts. Over the last few decades, ordered mesoporous structures, larger pore sizes, and highly crystalline frameworks have been synthesized.35 Unfortunately, only some of this progress could be transferred to other semiconductors. More demanding synthesis conditions and higher calcination temperatures mainly limit the synthesis of mesoporous ternary oxides such as SrTiO3 or even quaternary semiconductors.38,39
Apart from intrinsic and morphological properties, TiO2 and SrTiO3 are frequently processed further for the deposition of co-catalysts or to form composite materials with other semiconductors.29,40–43 The latter allows for improved absorption of solar light if the oxides are combined with semiconductors with smaller band gaps. Enhancements in photocatalytic activities in composite systems are mostly attributed to heterojunctions or Z-scheme charge carrier separation mechanisms.44,45 Improvements have even been observed for composites such as SrTiO3/TiO2, ZnO/TiO2, or BaTiO3/SrTiO3, which consist of semiconductors with similar band gap values and similar potentials of valence and conduction band edges.46–52
Titanate perovskites (ATiO3, A = Ca, Sr, Ba) rank among the most active semiconductors in the field of photocatalytic water splitting.53 Limiting factors such as wide band gaps, insufficient charge carrier extraction, and inherent Ti3+ recombination centers have been tackled by different doping strategies, advanced combinations of co-catalysts, and formation of heterostructures.32,54–59 Commercial applications lie further in the future, but a first attempt at scaling solar H2 production has been established for SrTiO3 already.7 Aside from yet insufficient solar-to-hydrogen (STH) energy conversion efficiencies, basic research on morphological diversity and heterostructures is indispensable for the subsequent combination of existing research results. Lately, different SrTiO3/TiO2 composites synthesized by hydrothermal conversion of TiO2 materials have shown strongly enhanced performance in photocatalytic H2 evolution.46,47,60–67 The structural diversity includes nanospheres, MOF- and MXene-derived structures, nanotubes, and nanofibers/nanowires. However, simpler morphologies such as nanoparticles are noticeably underrepresented.63
In this work, we highlight the reason for the dependence of photocatalytic activities on morphological differences and we emphasize that influences of composite syntheses on individual semiconductors must be considered carefully for the correct attribution of observed activity enhancements. We used a simple hydrothermal method to convert mesoporous and nanoparticulate TiO2 into SrTiO3/TiO2 composites. In the case of the mesoporous starting material, considerable increases in photocatalytic H2 evolution rates have been detected, while partial conversion of nanoparticles only led to a decrease in photocatalytic activity. To address this phenomenon, we performed hydrothermal etching of the starting materials by omitting the Sr precursor. N2 and H2O physisorption measurements of mesoporous samples reveal that hydrothermal etching in diluted NaOH solution and even in H2O leads to a broadening of pore size distributions and more hydrophilic TiO2 surfaces. The latter effect already occurs in the first 30 minutes of the synthesis of mesoporous SrTiO3/TiO2 composites, and thus can be misinterpreted as a composite effect. On the other hand, hydrothermal treatment of mesoporous photocatalysts in diluted NaOH solution or H2O might be a simple and promising strategy to improve photocatalytic activities in general.
Experimental
Materials
Ti(OCH(CH3)2)4 (TTIP) (Sigma-Aldrich, 97%), PEG–PPG–PEG (Pluronic® P123) (Sigma-Aldrich), acetone (Fisher Chemical, ≥99.8%), ethanol absolute (VWR), 2-propanol (VWR, p.a.), TiO2 nanoparticles (Hombikat N100, Sachtleben), SrCl2·6H2O (abcr, 99%), NaOH (Grüssing, 99%), Na2SO4 (Roth, ≥99%), HCl (1 M) (Grüssing), methanol (Fisher Chemical, 99.99%), H2PtCl6·6H2O (Roth, 99.9995%), Co(NO3)2·6H2O (abcr, 99%), K2CrO4 (Alfa Aesar, 99.9%), and Na3RhCl6 (Sigma-Aldrich) were used without further purification. Demineralized H2O and ultrapure H2O were used in the synthesis and for photocatalytic measurements, respectively. Fluorine-doped SnO2 (FTO) samples were purchased from XOP glass with the specification TEC T10 (8–10 Ω sq−1) and were cleaned with ethanol, acetone, and isopropanol followed by ultrapure H2O for 30 min each in an ultrasonic bath. The FTO samples were dried with N2 for the subsequent spray-coating.Mesoporous TiO2
Mesoporous TiO2 (denoted as M0h) was synthesized via an adapted soft-templating approach.68Triblock copolymer Pluronic® P123 (3.00 g) was dissolved in absolute ethanol (20 g, 25.3 mL) and stirred vigorously for 30 minutes to obtain a clear solution. Titanium tetraisopropoxide (TTIP) (0.02 mol, 5.7 g, 5.9 mL) was added, and the solution was stirred for another 30 minutes. Afterwards, the solution was transferred to a 100 mL Teflon-cup, which was stored for approximately four days at 40 °C under a glass dome without stirring. Dried-up samples were calcined in air in a muffle furnace (Nabertherm). The temperature was kept at 350 °C for 10 h and 525 °C for 3 h. Heating rates were set to 3 K min−1.
Conversion of TiO2
A single-step direct hydrothermal synthesis was employed to convert mesoporous TiO2 (M0h) or commercial Hombikat N100 nanoparticles (denoted as N0h) to SrTiO3 or SrTiO3/TiO2 composites. The procedure by Zhang et al. was adapted in terms of starting materials, temperature, holding time and concentration of the NaOH solution.69TiO2 (0.500 g, 6.260 mmol, 1 eq.) was mixed with a slight excess of SrCl2·6H2O (1.8361 g, 6.887 mmol, 1.1 eq.). In the case of M0h, the material was ground beforehand for 10 minutes to obtain a fine powder. Then, NaOH solution (0.2 M, 25 mL) was added to the mixture, and the dispersion was transferred to a PTFE-lined stainless-steel autoclave. The autoclave was heated to 150 °C for 0.5 h, 1 h, 2 h, 5 h, 10 h, 20 h, or 1 day, respectively.
In the following, samples are abbreviated related to the respective starting material and conversion time. Converted mesoporous TiO2 is denoted as MCxh, x showing the reaction time. The respective samples converted from nanoparticles are denoted as NCxh.
Higher concentrated NaOH solution (5 M) and a longer reaction time of 6 days (144 h) were applied for the synthesis of the samples MC144h(5M) and NC144h(5M).
The autoclave was quenched in ice water and the product was washed two times with 10 mL H2O to remove excess alkali. Subsequently, HCl (1 M, 10 mL) was added and the obtained dispersion was treated in an ultrasonic bath for 10 minutes at room temperature to eliminate unreacted hydroxides. The centrate was removed after centrifugation and the sediment was washed again two times with 10 mL H2O. The product was transferred with 4 mL H2O to a glass vial and dried in an oven at 80 °C.
Etching of TiO2
Etching of M0h and N0h was performed analogously to the conversion reactions but without the addition of SrCl2·6H2O. Etching was conducted for reaction times of 0.5 h, 1 h, 2 h, 5 h, 10 h, and 20 h. The work-up procedure was performed analogously to the above stated steps for converted samples. Samples are denoted as MExh and NExh, respectively.H2O treatment
H2O treatment of M0h and N0h was performed analogously to the conversion reactions but without the addition of SrCl2·6H2O and in 25 mL H2O instead of NaOH solution. The treatment was conducted for reaction times of 0.5 h and 20 h. The work-up procedure was performed analogously to the above stated steps for converted samples. Samples are denoted as M0.5hH2O, M20hH2O, N0.5hH2O, and N20hH2O, respectively.HCl work-up procedure and H2O work-up procedure
HCl work-up and H2O work-up were employed to investigate the influence of the above stated work-up procedure after hydrothermal conversion, etching or H2O treatment reactions.HCl work-up was conducted by performing the above stated washing steps and ultrasonic treatment directly on the TiO2 starting materials M0h and N0h without any previous hydrothermal treatment. The respective samples are denoted as M0hHCl and N0hHCl.
The H2O work-up procedure was performed analogously to the HCl work-up but H2O was added before ultrasonic treatment instead of 1 M HCl. The respective samples are denoted as M0hH2O and N0hH2O.
Material characterization
Samples were characterized with powder X-ray diffraction (XRD) using a Malvern PANalytical Empyrean XPert Pro device equipped with a PIXcel1D detector. Copper Kα irradiation (λ1 = 1.5406 Å; λ2 = 1.54443 Å) was generated using an emission current of 40 mA and an acceleration voltage of 40 kV. Measurements were carried out with a scattering slit of 1°, a step size of 0.02626° or 0.0393908° and a scan step time of 147.390 s or 296.31 s, respectively. Reference Bragg reflections were obtained from the Crystallography Open Database (COD) (anatase: 1526931, rutile: 9001681, SrTiO3: 1512124). Average crystallite sizes were estimated using the Scherrer equation:where D is the crystallite size, λ is the X-ray wavelength of the diffractometer, K is a dimensionless shape factor, B is the full width at half maximum (FWHM) in radians, and θ is the diffraction peak angle. Average crystallite sizes were calculated based on fits (Lorentz function) of the (101) anatase reflections and (110) reflections of SrTiO3.
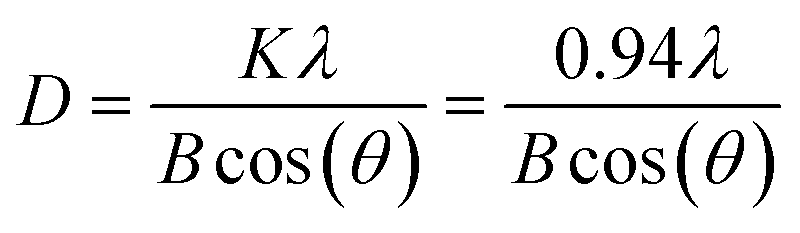
Transmission electron microscopy (TEM) was employed for the determination of sample size, crystallinity, and morphology using a 200 kV JEOL JEM-2200FS EFTEM, equipped with a Schottky FEG and an omega in-column energy filter. Lattice planes were evaluated with ImageJ 1.53e.
The nanoparticulate and porous morphologies were further examined by scanning electron microscopy (SEM). Imaging was performed on a Zeiss Leo 1530 device at an acceleration voltage of 3.0 kV after sputter coating with platinum (Cressington Sputter Coater 208 HR). The same instrument was used for energy-dispersive X-ray spectroscopy (EDX) using an acceleration voltage of 20.0 kV and a Thermo Fisher Scientific NS7 UltraDry-EDX detector.
X-ray photoelectron spectroscopy (XPS) was performed on a Physical Electronics (PHI) VersaProbe III scanning XPS microprobe instrument equipped with a monochromatized Al Kα source. The beam voltage was set to 15 kV, the X-ray power was set to 25 W and a beam diameter of 100 μm was used. Survey scans were recorded in the range of 1200 eV to 0 eV with a step size of 0.4 eV and step time of 50 ms at a pass energy of 224 eV. High-resolution spectra were recorded with a pass energy of 26 eV, a step size of 0.1 eV, and a time per step of 50 ms. To avoid charging effects, samples were continuously flooded with electrons and Ar+ ions at low energy. XP (X-ray photoelectron) spectra were evaluated with CASA XPS 2.3.25 using Shirley backgrounds.70 Gaussian–Lorentzian line shapes with a 30% Lorentz ratio (GL30) were assumed for peak fitting. The C–C fit of the carbon 1s signal was set to 284.8 eV for charge correction. Curve-fitting of adventitious carbon was performed based on parameters reported by Biesinger et al.71 Fitting of Sr 3d, Ti 2p, and O 1s signals is based on different parameters reported for TiO2 (anatase),72 SrTiO3,73–75 SrCO3,73,76 and SrCl2.73,77
Diffuse reflectance infrared Fourier transform (DRIFT) measurements were performed on a Bruker Alpha II spectrometer using the software OPUS. The resolution was set to 4 cm−1 and measurements were conducted from 4000 cm−1 to 350 cm−1 with 24 scans per sample.
Raman measurements were conducted using Labspec software and a LabRam Horiba Jobin Yvon spectrometer, that was equipped with an Olympus BX41 microscope. A He–Ne-laser (λ = 633 nm; 11.5 mW) was employed for excitation. Measurements were conducted from 65 cm−1 to 1450 cm−1 (Raman shift) with an exposure time of 0.5 to 2 s and an accumulation number of 20.
Diffuse-reflectance UV/vis measurements were performed on a PerkinElmer Lambda 750 UV/vis/NIR spectrometer with a Praying Mantis geometry (Harrick). A step width of 1 nm and a range of 200 nm to 800 nm were used. A Spectralon tablet was used as the white standard. Diffuse reflection was converted to pseudo-absorption using the Kubelka–Munk equation. Band gaps were estimated from Tauc-plots assuming indirect semiconductors.
DLS measurements were performed on a Litesizer 500 by Anton Paar. Kalliope software was used and measurements were conducted in quartz cuvettes and aqueous methanol dispersions (10% methanol; 0.33 mg nanoparticulate photocatalyst per mL, analogous to photocatalytic H2 evolution experiments). The measurement angle was set to automatic. Titania was assumed to be the analyzed material, since its refractive index is expected to be similar to the refractive indices of the measured TiO2 and SrTiO3/TiO2 samples. Measurements of mesoporous samples could not be conducted due to the large secondary particle sizes and the resulting instability of the dispersions.
Electrochemical impedance spectroscopy (EIS) measurements were carried out with a Zahner Zennium CIMPS-PCS system and Thales software in a three-electrode photoelectrochemical cell filled with aqueous 0.1 M Na2SO4 electrolyte. The working electrodes were prepared via spray-coating of nanoparticulate TiO2 dispersions (6 mg in 6 mL ethanol) on FTOs and subsequent heating on a heating plate at 80 °C for 5 minutes. Higher temperatures were avoided to preserve the surface characteristics of the samples. A platinum wire was used as the counter electrode and an Ag/AgCl (3 M NaCl) electrode was used as the reference electrode. The working electrode was irradiated with simulated solar light using a 300 W Xe lamp (LOT-QuantumDesign) with an air mass 1.5G filter and the EIS measurements were performed from 0.1 Hz to 100 kHz.
For the characterization of specific surface area and gas adsorption properties, N2 physisorption isotherms were measured at 77 K with a Quadrasorb evo surface area & pore size analyzer (Anton Paar) or a Nova 800 surface area & pore analyzer (Anton Paar) or an Autosorb iQ-MP-MP-AG instrument (Anton Paar QuantaTec). Samples were degassed for 12 h at 120 °C prior to the measurements. For surface area calculations, the Brunauer–Emmet–Teller (BET) model was used, and the data evaluation was conducted with ASiQwin™ from Quantachrome Instruments and Kaomi for Nova from Anton Paar. Data fitting of full isotherms was performed with a QSDFT adsorption model and adsorption of N2 on carbon and slit/cylindrical shaped pores was assumed.
H2O vapor isotherms were recorded with an Autosorb iQ-MP-MP-AG (Anton Paar QuantaTec). Samples were degassed for 12 h at 30 °C before the measurement. The measurement temperature was 20 °C and the saturation vapor pressure (p0) was set to 17.38 torr.
Photocatalytic activity regarding H2 evolution from aqueous methanol solution was tested under simulated solar light irradiation. A dispersion of H2O (135 mL), methanol (15 mL), and the respective sample (50 mg) was added to a glass reactor with a quartz window on top. In advance, the sample was dispersed in a small amount of H2O and treated in an ultrasonic bath (Emmi® 20 HC, EMAG) for 10 minutes to support the dispersal. The reactor was placed under a solar simulator (Newport), equipped with a 150 W Xe lamp and an air mass 1.5G filter. The setup was cooled and kept at 20 °C with a thermostat (Eco RE 1050, LAUDA). The suspension was stirred with a magnetic stirrer. O2 was removed by flushing the suspension with Ar. H2 evolution was recorded via a gas chromatograph (Shimadzu GC-2014), which was equipped with a thermal conductivity detector (TCD) and a shin carbon ST column. Pt was used as the co-catalyst and added via an aqueous H2PtCl6·6H2O solution (0.1 wt% Pt; photodeposition) after two hours of measuring time through a rubber sealing. The measurement was then performed for another two hours. The lamp was switched off and the measurement continued until no more H2 was detected. The amount of H2 was converted to μmol h−1 using the ideal gas law. An additional long-term measurement was performed analogously but the irradiation time after addition of H2PtCl6·6H2O solution was extended to 20 h.
Photocatalytic overall water splitting (OWS) activity was measured under UV light irradiation in an immersion lamp setup. Ultrasonic treatment was conducted as described above. A dispersion of the sample (300 mg) in H2O (600 mL) was added to a glass reactor. The reactor was equipped with a double-walled quartz glass envelope into which an Hg vapor lamp (700 W, TQ 718 Peschl Ultraviolet) was inserted. The suspension was stirred with a magnetic stirrer and cooled to 10 °C with a thermostat (Proline RP845, LAUDA). O2 was removed by flushing the suspension with Ar overnight. Gas evolution was measured using a mass spectrometer (HIDEN Analytical HPR20). The lamp was switched on and set to a power of 71.4% (500 W). After 30 min, the lamp was switched off for 5 min to add Na3RhCl6 solution (1.0 wt% Rh). After 2 h of irradiation, the lamp was switched off again for 5 min to add K2CrO4 solution (0.5 wt% Cr2O3). The dispersion was irradiated for another 2 h until the lamp was switched off again for 5 min. Co(NO3)2 solution (0.5 wt% assuming CoOOH) was added and the lamp was switched on for at least 2 h or until steady H2 and O2 evolutions were measured. After the lamp was switched off, the measurement was continued for at least 45 min. Detected amounts of H2 and O2 were converted to μmol h−1 using the ideal gas law.
Results and discussion
To differentiate between composite effects and different morphological aspects, we conducted hydrothermal conversion reactions on mesoporous TiO2 (M0h) and on commercial anatase nanoparticles (Sachtleben Hombikat N100, N0h). Fig. S1† illustrates the schematic structure of all conducted reactions and treatments. The synthesis of mesoporous starting material M0h is based on an evaporation-induced self-assembly (EISA) approach but adapted in terms of synthesis parameters to obtain high yield and crystallite sizes comparable to those of the nanoparticulate starting material N0h.SrTiO3 reflexes become visible in XRD patterns of converted nanoparticles (NC series) and converted mesoporous TiO2 (MC series) after 2 h of reaction time (Fig. 1). A prolonged reaction time of 6 days (144 h) in higher concentrated NaOH solution (5 M) leads to a seemingly complete conversion of the nanoparticles (NC144h(5M)), whereas anatase with only a minor decrease in crystallite size (Tables S1 and S2†) remains present in the case of the mesoporous sample MC144h(5M). A phase transformation from anatase into rutile is not observed.
 | ||
| Fig. 1 XRD patterns (top) and Tauc plots (bottom) of converted mesoporous (left) and nanoparticulate (right) TiO2. | ||
All samples absorb UV light as visible from Kubelka–Munk plots (Fig. S2†). Band gap values derived from Tauc plots are 3.14 eV and 3.31 eV for the starting materials M0h and N0h, respectively. The divergence can be attributed to the presence of defect states and a more pronounced absorption tail (Urbach tail) in the case of M0h.78 Band gap values tend to approach 3.2 eV with increasing reaction time, which is the expected value for SrTiO3.53 Only the Tauc plot of NC144h(5M) shows another linear range in the absorption edge, which can be assigned to a small portion of amorphous TiO2. Experimental data reported for the optical band gap of amorphous TiO2 lie between 3.2 and 3.4 eV.79 Limited mass transport during the unstirred hydrothermal reaction could be the reason for incomplete conversion after 144 h in 5 M NaOH solution.
DRIFT spectra of the NC and MC series (Fig. 2) differ in the presence of a CO2 signal at 2350 cm−1.80 Calcination of the as-synthesized M0h most likely leads to CO2 residues trapped in pores without connection to the surface. Thus, hydrothermal conversion does not lead to the disappearance of the CO2 signal but only to a reduced intensity due to the growing SrTiO3 layer on the surface. SrTiO3 formation can be followed by the appearance of a band at around 500 cm−1.81 The less intense signal in the spectrum of MC144h(5M) in comparison to NC144h(5M) corresponds to the incomplete conversion of M0h, which is in accordance with the XRD results. Intense absorbance at around 900 cm−1 can be ascribed to Ti–O and Sr–O vibrations.82,83 Differences in the range of 1250 cm−1 to 1750 cm−1 within a series and between both series can be attributed to surface H2O (1620 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching of carbonate species (SrCO3, 1450 cm−1), respective protonated species, and signals of other organics.84–87 No clear trend is observed from the intensities of the broad OH vibrations (2600 cm−1 to 3800 cm−1).
O bond stretching of carbonate species (SrCO3, 1450 cm−1), respective protonated species, and signals of other organics.84–87 No clear trend is observed from the intensities of the broad OH vibrations (2600 cm−1 to 3800 cm−1).
 | ||
| Fig. 2 DRIFT (top) and Raman spectra (bottom) of converted mesoporous (left) and nanoparticulate (right) TiO2. | ||
Raman spectra (Fig. 2) are dominated by intense anatase signals.88 Only the highly converted sample NC144h(5M) shows a significantly different spectrum. The distinct peak at 149 cm−1 can be ascribed to either the anatase or rutile phase. A lack of long range order is indicated by peak broadening and the observed shift of 5 cm−1.89 SrTiO3 spectra generally show relatively broad signals, but the individual course of the spectra is quite diverse and allows for conclusions regarding strains or defects. Due to its theoretically ideal cubic perovskite structure, SrTiO3 exhibits no first-order Raman activity. Instead, spectra are dominated by second-order scattering.90 Aside from broad intensities, the spectrum of NC144h(5M) exhibits two further signals at 176 cm−1 and 542 cm−1. Both bands arise from first-order Raman scattering due to local loss of inversion symmetry. The peak at 176 cm−1 corresponds to the TO2 polar phonon and seems to exhibit Fano asymmetry, which hints at the presence of polar domains. The feature at 542 cm−1 can be assigned to the TO4 Raman band and is likewise a consequence of distortion from the long range cubic structure.90,91
SEM images of the mesoporous starting material M0h show large micrometer sized secondary particles as they are obtained from the EISA synthesis (Fig. 3). The mesoporous structure with slit-like pores can be surmised only at maximum magnification. Hydrothermal conversion leads to less pronounced edges and rougher surfaces at the micrometer scale. SrTiO3 at the surface can be observed as small crystallite structures (Fig. S3†). The generally smaller secondary particles are the result of grinding of M0h before hydrothermal conversion.
 | ||
| Fig. 3 SEM images (top) of the mesoporous TiO2 starting material (M0h) and a SrTiO3/TiO2 composite (MC144h(5M)). EDX mapping of MC144h(5M) is shown at the bottom. | ||
Fig. S3† shows the respective images for nanoparticulate samples. The EDX results (Table S3†) confirm the incomplete conversion of MC144h(5M) with a Sr/Ti ratio of 0.24. A noticeably higher value of 0.87 was determined for the NC144h(5M) sample. However, the deviation from the expected value of 1 hints at the presence of a small amorphous portion of TiO2, which is in accordance with the UV-vis and Raman results. Primary particles and lattice planes of SrTiO3 are visible in TEM images in Fig. S4.†
XPS survey data (Fig. 4a and S5†) show the presence of O, C, and Ti and the increase of the Sr 3d signal after 2 h of conversion time. Small amounts of Na and Cl can be detected in some samples due to the work-up procedure (Tables S4 and S5†). The Sr/Ti ratios of the MC and NC series show a completely different behavior with increasing conversion time (Fig. 4b). The micrometer sized TiO2 particles of M0h are quickly covered with Sr species. The Sr/Ti ratio for the MC series surpassed 1 after 5 h of reaction time, while that for the NC series surpassed 1 only in the case of the longest reaction time of 144 h in 5 M NaOH. The incomplete conversion of M0h can therefore be attributed to the large secondary particle sizes but also to the dissolution–precipitation mechanism of the hydrothermal conversion reaction. Zhang et al. described the process by progressive dissolution of TiO2 and subsequent precipitation of SrTiO3 due to the reaction of [Ti(OH)6]2− with Sr2+ in the reaction medium.69 Sr/Ti ratios surpassed 1 because of the presence of SrCO3 at the surface. Respective ratios obtained from the EDX results are generally lower due to a larger escape depth. The phenomenon of surface SrCO3 is known for SrTiO3 surfaces because Sr(OH)2 and other surface-bound Sr2+ species will form SrCO3 during drying or calcination processes. Low amounts of CO2 in the atmosphere and surface-bound organics are enough to promote the formation of SrCO3 on the surface.32,92
 | ||
| Fig. 4 XP survey spectra of the MC series (a); derived Sr/Ti ratios of the MC and NC series (b); and O 1s (c), Ti 2p (d), and Sr 3d spectrum (e) of MC144h(5M). | ||
High-resolution XP (X-ray photoelectron) spectra clearly show the presence of SrTiO3 after 2 h of conversion time (Fig. S6 and S7†), which is in accordance with XRD data. Shorter reaction times of 1 h and 0.5 h and subsequent drying, however, already lead to SrCO3 and possibly small amounts of SrCl2(H2O)x on the surface. In the case of longer reaction times, two Sr species had to be considered for a reasonable fit of the Sr 3d signals. Based on binding energies, these two species can be assigned to SrTiO3 and SrCO3 (Fig. 4e). SrTiO3 formation can also be followed by a shift of the O 1s and Ti 2p signals to lower binding energies. In the case of MC144h(5M), TiO2 still had to be assumed to obtain reasonable fits of the O 1s and Ti 2p spectra, whereas spectra of NC144h(5M) indicate no TiO2 in surface proximity (Fig. 4c, d, S6, and S7†).
Photocatalytic H2 evolution activities of the MC and NC series were tested for conversion times up to 20 h (Table 1). Surprisingly, different trends were observed for both series. In the case of the converted nanoparticles, increasing conversion time led to a decrease in photocatalytic activity. In the case of the converted mesoporous TiO2, the same treatment led to an increase of 35% and 30% for short conversion times of 0.5 h and 1 h, respectively. Photocatalytic activity of the mesoporous starting material M0h was notably lower compared to the nanoparticulate starting material N0h, but the difference in the overall trend seems inexplicable based on the given characterization results. Similar results of an enhanced photocatalytic activity after short hydrothermal conversion have been observed elsewhere but without detailed comparison of different morphologies.46,47 We therefore decided to expand our study by performing analogous hydrothermal treatments but without the addition of SrCl2·6H2O. Thus, M0h and N0h were treated only in 0.2 M NaOH solution to account for any influences of the reaction medium that were so far undetected in the case of the MC and NC series. These etched samples, denoted as the ME and NE series, were tested under the same conditions for their photocatalytic H2 evolution activity (Table 1).
| Synthesis reaction time | 0 h (starting material) | 0.5 h | 1 h | 2 h | 5 h | 10 h | 20 h |
|---|---|---|---|---|---|---|---|
| Converted N0h (NC series) | 118.1 | 111.9 | 103.9 | 90.8 | 59.9 | 66.1 | 74.3 |
| Converted M0h (MC series) | 52.8 | 71.5 | 68.5 | 45.4 | 21.1 | 21.0 | 22.3 |
| Etched N0h (NE series) | 118.1 | 110.1 | 101.1 | 98.6 | 84.5 | 80.8 | 76.1 |
| Etched M0h (ME series) | 52.8 | 70.0 | 67.1 | 65.8 | 86.5 | 91.3 | 106.7 |
Etched nanoparticles showed a decreasing activity with increasing synthesis reaction time, which is a similar trend compared to the NC series despite some considerable deviations for 5 h and 10 h reaction times. The photocatalytic activities of the ME series were unexpected and were comparable to those of the MC series for samples treated up to 1 h reaction time but increased even further with prolonged hydrothermal treatment. After 20 h, H2 evolution rates of up to 106.7 μmol h−1 were measured, which correspond to an increase of 102% compared to the starting material M0h. Furthermore, the sample ME20h almost reaches the activity of the nanoparticulate starting material N0h.
Standard characterization methods such as X-ray diffraction, UV-vis, DRIFT, and Raman spectroscopy, as well as scanning and transmission electron microscopy, were performed on samples of the ME and NE series. As expected, hydrothermal treatment in 0.2 M NaOH at 150 °C leads to no substantial changes. XRD patterns (Fig. S8†) continue to show anatase reflexes with similar crystallite sizes (Tables S6 and S7†). Only NE20h exhibits a slightly larger crystallite size of 23 nm instead of the 18 nm in the case of the starting material N0h. Again, a phase transformation from anatase into rutile is not observed. Kubelka–Munk plots (Fig. S9†) again show absorption in the UV range, and band gap values obtained from Tauc plots (Fig. S10†) are similar to those of the respective starting materials. DRIFT (Fig. S11†) and Raman spectra (Fig. S12†) show no significant differences. SEM images of ME20h reveal rounded edges of secondary particles and nanoparticle agglomerations in the case of NE20h (Fig. S13†), but no substantial differences are shown in their converted (MC or NC) counterparts on the micrometer scale. On the nanometer scale, ME20h appears rougher than the starting material M0h, which hampers display of the slit-like mesopores. XPS survey data (Fig. S14 and Table S8†) suggest no changes in surface compositions. High-resolution XP spectra of O 1s and Ti 2p show no noticeable shifts in binding energy (Fig. S15†). Overlaid O 1s spectra of the ME series (Fig. S16†) show exemplarily the comparability of the signals and indicate that fits of the signals are very similar. Thus, differences in the amount of hydroxides, hydrated oxygen, defective oxygen or hydroxyl groups on the outer surfaces are minor and cannot be detected systematically by XPS. Moreover, a more detailed evaluation would be complicated by the influences of organics on the O 1s signal. However, surface characteristics are known to be important for the photocatalytic activity of nanoparticulate and mesoporous materials. This includes influences of surface bound cations, which are known to modify the charge, surface potential, and photocatalytic proton reduction rate of suspended metal oxides.93 We therefore decided to expand our study by conducting hydrothermal treatment in pure H2O to eliminate the influence of Na+ and the influence of the alkaline medium (control samples M0.5hH2O, M20hH2O, N0.5hH2O, and N20hH2O). Furthermore, we wanted to assess the influence of hydrothermal treatment in general and the influence of the work-up procedure, which consists of multiple washing steps and ultrasonic treatment in hydrochloric acid. Control samples M0hHCl and N0hHCl were exposed to the standardized HCl work-up procedure but not to hydrothermal treatment. The study is completed using the control samples M0hH2O and N0hH2O, which were treated analogously but H2O instead of 1 M HCl was added before ultrasonic treatment.
N2 physisorption was used to study influences of hydrothermal treatment on BET surface areas and pore size distributions. BET surface areas (Fig. 5a and Tables S9–S11†) decreased for both the MC and NC series with increasing hydrothermal reaction time. However, conversion of N0h led to decreases in BET surface area even in the case of short hydrothermal reaction times, whereas samples of the MC series remained within the range of all M0h syntheses for conversion times up to 2 h before dropping below 10 m2 g−1. The ME series remained entirely within the range of M0h, which already indicates the general preservation of the mesoporous structure. In contrast, the NE series is subjected to a decrease in BET surface area but less pronounced compared to the NC series. Control samples show only minor deviations from their respective counterparts with the exception of N20hH2O, which exhibits a significantly higher surface area than NE20h or NC20h.
 | ||
| Fig. 5 BET surface areas (a), pore size distributions (each shifted by 0.03 cm3 nm−1 g−1) of the MC (b) and ME series (c), and maximum photocatalytic H2 evolution rates of all mesoporous (d) and nanoparticulate samples (e) up to synthesis reaction times of 20 h. | ||
Pore size distributions (PSDs) of the MC series show the influence of SrTiO3 crystallite growth on the mesoporous structure (Fig. 5b). The starting material M0h exhibits a maximum at around 6 nm, which is preserved for short conversion times of 0.5 h and 1 h. The beginning loss of the porous structure is clearly visible in the course of MC2h and goes along with the formation of SrTiO3 crystallites. After 5 h, the initial PSD has vanished due to a complete coverage of TiO2 with SrTiO3 and small amounts of SrCO3 as indicated by XPS analysis. The loss of the porous structure is consequently also visible by the disappearance of the hysteresis (Fig. S17†). The ME series is not subjected to a loss of porous structure as shown in Fig. 5c and S17.† The course of the PSDs, however, undergoes broadening, which seems to be a continuous process that starts after 2 h of reaction time. Isotherms of this series resemble type IV isotherms with type H2-like hystereses, which can result from cavitation or pore blocking effects.94,95 The presence of type H2-like hystereses confirms the mesoporous structure of the photocatalysts. Converted and etched N0h remain nanoparticles as visible from the course of the isotherms (Fig. S18†). The presence of hystereses, however, indicates considerable interparticle voids caused by nanoparticle agglomeration, which can also be seen in the resulting broad and varying PSDs of the NC and NE series (Fig. S18†). Control samples (Fig. S19†) behave similarly to the respective etched samples and show formation of hystereses in the case of nanoparticulate samples and retention of the initial hysteresis in the case of mesoporous samples. Interestingly, pore size broadening can also be observed for the mesoporous sample hydrothermally treated for 20 h in H2O (sample M20hH2O). Thus, hydrothermal treatment in H2O seems to have a similar but less pronounced effect than etching in 0.2 M NaOH solution. Furthermore, dynamic light scattering (DLS) analysis confirms the agglomeration behaviour of nanoparticulate samples (Fig. S20 and Table S12†). Measurements were performed in aqueous methanol solution, thus simulating the conditions during photocatalytic H2 evolution. A prolonged conversion time of 20 h leads to a maximum of the number weighted size distribution at approximately 2 μm, whereas the starting material N0h exhibits a maximum at only 44 nm. A similar but less pronounced trend can be observed for the NE series and nanoparticulate control samples with maxima at 77 nm and 66 nm for NE20h and N20hH2O, respectively.
All eight control samples show no significant changes in X-ray diffraction, UV-vis, DRIFT, or Raman spectroscopy compared to the starting samples M0h/N0h or the respective samples of the ME and NE series (Fig. S21 and Table S13†). Survey and high-resolution XP spectra are also unremarkable (Fig. S22, S23 and Table S14†).
Maximum photocatalytic H2 evolution of all tested samples is shown in Fig. 5d and e. The course of the individual measurements is shown in Fig. S24,† and post-photocatalytic characterization is depicted in Fig. S26–S30.† N2 physisorption is the first characterization method that allows for explanations of the observed changes in photocatalytic activity. Under conditions of optimal light absorption, long charge carrier lifetimes, and efficient charge carrier separation, surface area can be the only limiting factor in the performance of a photocatalyst.33 Correlations between BET surface area and photocatalytic activity can be seen for MC, NC, and NE series. In all three series, a general trend of decreasing photocatalytic activity with increasing hydrothermal reaction time can be observed. In the case of the NE series, this trend is consistent. Samples of the NC series decrease in photocatalytic activity up to conversion times of 5 h and slightly increase from 5 h to 20 h of conversion time. In the latter case, a beneficial effect of charge carrier separation by formation of heterojunctions cannot be entirely excluded. However, the effect is minor and the general divergence between samples of NC and NE series for samples after 5 h and 10 h of hydrothermal is rather attributed to the difference in BET surface area, which is largest between NC5h and NE5h as well as NC10h and NE10h. This explanation can also be applied for the observed higher activity of the control experiment N20hH2O compared to NC20h or NE20h. Furthermore, electrochemical impedance spectroscopy (EIS) of nanoparticulate samples indicates lower charge transfer resistance for the N0h starting material compared to converted or etched samples (Fig. S25†), which also corresponds to the photocatalytic results. EIS measurements of mesoporous samples could not be obtained due to insufficient contact of the large secondary particles with the FTO substrate.
The increase in photocatalytic activity observed for MC0.5h and MC1h compared to the starting material M0h cannot be explained using the N2 physisorption results. However, a correlation between BET surface area and photocatalytic activity is apparent as soon as SrTiO3 crystallization occurs. The increase in photocatalytic activity of the ME series is not correlated with BET surface area but can be attributed to pore size broadening at least for samples that were etched hydrothermally for 2 h or longer. Broadening of slit-like pores might benefit diffusion processes during photocatalysis, which are known to be restricted in narrow pores.96
Still, the N2 physisorption results are insufficient to explain the initial increase in photocatalytic activity of the MC and ME series and most control experiments. Yet, influences of hydrothermal treatments and work-up processes are not limited to BET surface areas and PSDs. In fact, changes in surface characteristics and hydrophilicity might influence photocatalytic performance directly and indirectly.
Direct influences of surface treatments are manifold and have been observed mainly in the context of photocatalytic degradation reactions.97–105 Most commonly observed is the formation of abundant surface hydroxyl groups, for example after treatments in NaOH solutions and alkaline H2O2 solution, or after a NaCl-assisted acidic EISA process and subsequent washing.97–100,105 During photocatalytic H2 evolution from methanol solution, methanol is photooxidized to carbon dioxide via the formation of the stable intermediates formaldehyde and formic acid. Photogenerated holes will either be trapped by reacting with surface Ti–OH groups or reacting with adsorbed H2O to form hydroxyl radicals. Alternatively, holes can be directly transferred to adsorbed methanol molecules.106 All three reaction pathways might be positively influenced by even very mild surface treatments. An enhanced water-dispersibility and formation of hydroxyl radicals have been observed by Wu et al.100 A noticeably higher mobility of hydroxyl radicals on anatase than on rutile has been detected by Kim et al.103,104 And furthermore, Nie et al. observed facilitated diffusion and transport of reactants and products in porous structures.99 Generally, direct and indirect influences are not universal and photocatalytic behavior needs to be understood with a broad view of the material and the conducted reaction.28,102
With regard to indirect effects, surface treatments can also influence morphological aspects.100,107 Nanoparticle agglomerations, for example, depend on surface charges and the density of hydroxyl groups on the catalyst surface.100 Thus, BET surface area and PSDs can be influenced by changes in hydrophilicity. Differences in PSDs are, for example, very apparent for the nanoparticulate control samples N0hHCl, N0hH2O, N0.5hH2O, and N20hH2O and might contribute to the varying photocatalytic performances (Fig. 5 and S19†). To further assess these influences, H2O physisorption measurements were conducted. Isotherms as well as tabular data are given in the ESI (Fig. S31–S33 and Tables S15–S17).† As an example, Fig. 6 shows H2O physisorption isotherms normalized to BET surface area of three samples of the MC and NC series, respectively. Normalization to N2 BET surface area allows for better comparability of hydrophilicity, due to the considerable variations in BET surface area. In the case of the MC series, H2O adsorption is significantly enhanced with longer conversion times, whereas changes are far less pronounced in the case of the NC series.
 | ||
| Fig. 6 Selected H2O physisorption isotherms normalized to N2 BET surface area. | ||
In general, etched and converted samples are difficult to compare because hydroxylation of SrTiO3 and TiO2 might differ despite the same treatment. Furthermore, hydroxylation of both semiconductor surfaces might influence the photocatalytic activity differently. SrTiO3 surfaces are also newly formed during the conversion, which is not directly the case for etched samples. Still, based on adsorbed volumes per H2O BET surface area in the monolayer range (approx. p/p0 = 0.3), all samples are more hydrophilic than the respective starting material M0h or N0h. Plain isotherms of the MC, NC, and NE series (Fig. S31 and S32†) show less differences because the surface gets more hydrophilic, but N2 BET surface areas decrease due to SrTiO3 crystallization or nanoparticle agglomeration.
In terms of photocatalytic activity, it is therefore essential to consider the samples that benefit from surface treatment without losing BET surface area. This only applies to the ME series, mesoporous control samples, and samples of the MC series before the start of SrTiO3 crystallization, namely MC0.5h and MC1h. Thus, these samples exhibit higher photocatalytic activities than M0h. As visible from the activities of the ME series, increased hydrophilicity and pore size broadening seem to be particularly advantageous. Both effects together could be described as “pore activation”. The long-lasting effect of this pore activation is visible in Fig. S34.† ME20h exhibits excellent stability under simulated solar light irradiation during a longtime measurement by maintaining 93.7%/87.4% of its initial activity 10 h/20 h after addition of H2PtCl6·6H2O solution.
Considering the enhanced hydrophilicity and the continuous pore size broadening, an increase in photocatalytically active surface area might occur if the smaller and less hydrophilic pores of M0h cannot fully contribute to the photocatalytic activity.96 An indication of the pore activation effect might also be visible in the course of the H2 evolution rate during the photocatalytic measurement of M0h: it is the only H2 evolution rate that increases noticeably over the course of the measurement (Fig. S24†), which could hint at an in situ increase in hydrophilicity.
In contrast to the existing literature on hydrothermally converted TiO2 materials (cf. comparison in Table S18†), little to no charge carrier separation effect was observed in this work for SrTiO3/TiO2 composites. The synthesized composites are heterostructures in the sense that SrTiO3 and TiO2 are in contact with each other. However, no substantial effects due to formation of a conceivable heterojunction could be observed. The reason might be a low thermodynamic driving force due to similar band gaps and similar band edge positions.44 According to literature data of undoped TiO2 (anatase) and SrTiO3, potential differences of valence band edges and conduction band edges amount to 0.55 V and 0.6 V, respectively. The difference of the Fermi level potentials amounts to 0.18 V and is therefore even smaller (Fig. S35†).51,108–111
Photocatalytic activities generally depend on band gap values, band edge positions, and other physicochemical properties. Therefore, the occurrence of composite effects also depends on the conducted photocatalytic redox reaction. Photocatalytic OWS is hence another possibility to check for beneficial composite effects. Gas evolution rates of OWS measurements under UV light irradiation are shown in Fig. 7 and S36.†
 | ||
| Fig. 7 (Near) steady-state gas evolution of M0h, N0h, MC24h, NC24h, MC144h(5M), and NC144h(5M) (top). The course of gas evolution during the experiment is shown exemplarily for NC144h(5M) (bottom). | ||
SrTiO3 is known to be an excellent photocatalyst for OWS, whereas TiO2 exhibits high photocatalytic H2 evolution activity but only minimal OWS activity, which is also known only for historical reasons.19,53,112 The absence of beneficial composite effects during photocatalytic OWS has been reported for SrTiO3/TiO2 materials earlier.47 (Near) steady-state gas evolution rates shown in Fig. 7 are in accordance with this report. NC144h(5M) as the highly converted sample exhibits by far the highest OWS activity and reaches 3.2 mmol h−1 H2 production after Rh/Cr2O3/CoOOH co-catalyst photodeposition. The samples MC24h and NC24h both consist of SrTiO3 and crystalline anatase (Fig. 1) and thus were chosen for OWS measurements as representatives for crystalline SrTiO3/TiO2 composites. Both composites exhibit much lower activity, and the starting materials M0h and N0h show no steady-state gas evolution. However, both starting materials exhibit O2 evolution after co-catalyst photodepositions (Fig. S36†). The O2 evolution is clearly photoresponsive but temporary and cannot be maintained. This might be attributed to the inability of TiO2 to perform the reduction reaction under the given conditions.
Post-photocatalytic characterization is shown in Fig. S37–S41.† Changes in UV-vis and Raman spectra can be related to photodeposited co-catalysts. XRD patterns show no differences.
In summary, similar to the H2 evolution measurements, photocatalytic OWS results show no indication of beneficial charge carrier separation by heterojunction or even Z-scheme formation. In contrast, H2 evolution activities are determined by the proportion of TiO2 and underlying etching effects, while OWS activities are determined by the proportion of SrTiO3. The pore activation effect, which is observed for photocatalytic H2 evolution of etched mesoporous TiO2 (ME series), likely enhances diffusion processes, as well as the photocatalytic reaction. Since hydroxyl groups play an important role in the oxidation of sacrificial agents, we assume that a higher density of surface hydroxyl groups within the pores increases the methanol oxidation rate.106 However, future studies will show whether this strategy can be transferred to other photocatalytic reactions. In particular, reactions that involve reduction and oxidation reactions on co-catalysts might benefit less from the pore activation effect. Furthermore, follow-up work could focus on transfer of the pore activation effect to nanoparticles. To obtain a similar effect, agglomeration must be prevented without reducing the effect of increasing hydrophilicity by the hydrothermal treatment.
Conclusions
Hydrothermal treatment of mesoporous TiO2 and other mesoporous photocatalysts might be a valuable strategy to enhance photocatalytic H2 evolution activity. We attribute improvements in photocatalytic activity to increased hydrophilicity of pore surfaces and broadening of pore size distributions. The latter was observed after longer hydrothermal treatments (≥2 h at 150 °C), while enhanced hydrophilicity was observed even after only 30 minutes. Advanced syntheses of mesoporous TiO2, which include, for example, carbonization steps of the surfactant using sulfuric acid or treatments with ammonia or ethylenediamine, might benefit from additional post-synthetic hydrothermal pore activation.113–117 Specifically, mesoporous materials with smaller pore sizes might benefit from this mild treatment in diluted NaOH solution or even just H2O due to pore size broadening and thus reduction of diffusion limitations.35 Potentially, this strategy can be transferred to other photocatalytic reactions, other semiconductor oxides, or even semiconductor non-oxides.Syntheses of composite materials, especially alleged heterojunctions and Z-schemes, need to be accompanied by control experiments to account for any changes of the individual components or starting materials. Standard characterization methods such as XRD, UV-vis, SEM/TEM, DRIFT, and Raman spectroscopy might not be sufficient to detect changes in surface characteristics or other properties. In the presented case, even XPS could not show the changes in hydrophilicity (OH groups) of converted or etched mesoporous TiO2 in comparison to the starting material. The potential for misinterpretation is high, since charge carrier separation effects are reported frequently and influences of diluted NaOH solution and even H2O during hydrothermal treatment are unexpected and subtle. Regarding hydrothermal conversions/treatments of mesoporous photocatalysts, we therefore emphasize the usage of N2 and H2O physisorption to investigate changes in pore size distribution and hydrophilicity, respectively.
Data availability
Data for this article are available at Open Science Framework, DOI: https://doi.org/10.17605/OSF.IO/NC835.Author contributions
Lion Schumacher: conceptualization, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing – original draft. Jana Timm: conceptualization, formal analysis, investigation, methodology, writing – review & editing. Roland Marschall: conceptualization, data curation, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. S. acknowledges financial support from the FCI (Fonds der Chemischen Industrie, Kekulé Scholarship). Furthermore, L. S. acknowledges support from the Elite Study Program Macromolecular Science within the Elite Network of Bavaria (ENB) and support from the BayNAT program within the UBTGS (University of Bayreuth Graduate School). The authors thank Dr Anja Hofmann and Mirco Ade for SEM/EDX analysis, Jonas Jungmann for N2 physisorption and TEM analysis, and Frederike von der Haar for advice on photoelectrode preparation and EIS measurements. Additionally, we thank the Bavarian Polymer Institute (BPI) for usage of the XPS, SEM, and TEM devices in the respective KeyLabs Device Engineering and Electron Microscopy.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strat. Rev., 2019, 24, 38–50 CrossRef.
- K. T. Møller, T. R. Jensen, E. Akiba and H. Li, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- P. P. Edwards, V. L. Kuznetsov, W. I. F. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- J. M. Thomas, P. P. Edwards, P. J. Dobson and G. P. Owen, J. Energy Chem., 2020, 51, 405–415 CrossRef PubMed.
- C. Yilmaz, J. Wendelstorf and T. Turek, J. Clean. Prod., 2017, 154, 488–501 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- W. J. Jiang, T. Tang, T. Tang, Y. Zhang and J. S. Hu, Acc. Chem. Res., 2020, 53, 1111–1123 CrossRef CAS PubMed.
- R. Marschall, Eur. J. Inorg. Chem., 2021, 2021, 2435–2441 CrossRef CAS.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61, e202103824 CrossRef CAS PubMed.
- M. Yu, E. Budiyanto and H. Tüysüz, Angew. Chem., 2022, 134, e202103824 CrossRef.
- Y. Chen, Y. Liu, L. Li, T. Sakthive, Z. Guo and Z. Dai, Adv. Funct. Mater., 2024, 2406587 CrossRef.
- Y. Chen, Y. Liu, L. Li, T. Sakthivel, Z. Guo and Z. Dai, Adv. Funct. Mater., 2024, 2401452 CrossRef CAS.
- Y. Liu, L. Li, L. Wang, N. Li, X. Zhao, Y. Chen, T. Sakthivel and Z. Dai, Nat. Commun., 2024, 15, 2851 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- S. Kahng, H. Yoo and J. H. Kim, Adv. Powder Technol., 2020, 31, 11–28 CrossRef CAS.
- J. Zander and R. Marschall, J. Mater. Chem. A, 2023, 11, 17066–17078 RSC.
- S. Fang and Y. H. Hu, Int. J. Energy Res., 2019, 43, 1082–1098 CrossRef.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc. Chem. Commun., 1980, 543–544 RSC.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1–26 Search PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- B. L. Phoon, C. W. Lai, J. C. Juan, P. L. Show and W. H. Chen, Int. J. Energy Res., 2019, 43, 5151–5174 CrossRef CAS.
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260–2288 CrossRef CAS.
- F. J. Knorr, C. C. Mercado and J. L. McHale, J. Phys. Chem. C, 2008, 112, 12786–12794 CrossRef CAS.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- R. Katal, S. Masudy-Panah, M. Tanhaei, M. H. D. A. Farahani and H. Jiangyong, Chem. Eng. J., 2020, 384, 123384 CrossRef CAS.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS.
- C. Avcıoǧlu, S. Avcıoǧlu, M. F. Bekheet and A. Gurlo, ACS Appl. Energy Mater., 2023, 6, 1134–1154 CrossRef.
- K. Kato, A. Yamakata and A. Yamakata, J. Phys. Chem. C, 2020, 124, 11057–11063 CrossRef CAS.
- S. Piskunov, E. Heifets, R. I. Eglitis and G. Borstel, Comput. Mater. Sci., 2004, 29, 165–178 CrossRef CAS.
- B. Moss, Q. Wang, K. T. Butler, R. Grau-Crespo, S. Selim, A. Regoutz, T. Hisatomi, R. Godin, D. J. Payne, A. Kafizas, K. Domen, L. Steier and J. R. Durrant, Nat. Mater., 2021, 20, 511–517 CrossRef CAS PubMed.
- F. E. Osterloh, ACS Energy Lett., 2017, 2, 445–453 CrossRef CAS.
- J. H. Pan, H. Dou, Z. Xiong, C. Xu, J. Ma and X. S. Zhao, J. Mater. Chem., 2010, 20, 4512–4528 RSC.
- W. Zhou and H. Fu, ChemCatChem, 2013, 5, 885–894 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed. Engl., 1995, 34, 2014–2017 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., 1995, 107, 2202–2206 CrossRef.
- K. Yu, C. Zhang, Y. Chang, Y. Feng, Z. Yang, T. Yang, L. L. Lou and S. Liu, Appl. Catal., B, 2017, 200, 514–520 CrossRef CAS.
- T. Weller, L. Deilmann, J. Timm, T. S. Dörr, P. A. Beaucage, A. S. Cherevan, U. B. Wiesner, D. Eder and R. Marschall, Nanoscale, 2018, 10, 3225–3234 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., 2006, 118, 7970–7973 CrossRef.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. He, L. Zhang, Y. Wei, X. Zhang, Z. Wang and R. Yu, Small Methods, 2022, 6, 2101567 CrossRef CAS PubMed.
- Z. Zhuo, X. Wang, C. Shen, M. Cai, Y. Jiang, Z. Xue, Z. Fu, Q. Wang, Y. Wei and S. Sun, Chem.–Eur. J., 2023, 29, e202203450 CrossRef CAS PubMed.
- T. Cao, Y. Li, C. Wang, C. Shao and Y. Liu, Langmuir, 2011, 27, 2946–2952 CrossRef CAS PubMed.
- D. Ramírez-Ortega, D. Guerrero-Araque, P. Acevedo-Peña, E. Reguera, H. A. Calderon and R. Zanella, Int. J. Hydrogen Energy, 2021, 46, 34333–34343 CrossRef.
- Q. Chen, R. Tong, X. Chen, Y. Xue, Z. Xie, Q. Kuang and L. Zheng, Catal. Sci. Technol., 2018, 8, 1296–1303 RSC.
- J. i. Fujisawa, T. Eda and M. Hanaya, Chem. Phys. Lett., 2017, 685, 23–26 CrossRef CAS.
- L. Li, X. Liu, Y. Zhang, P. A. Salvador and G. S. Rohrer, Int. J. Hydrogen Energy, 2013, 38, 6948–6959 CrossRef CAS.
- S. Patial, V. Hasija, P. Raizada, P. Singh, A. A. P. K. Singh and A. M. Asiri, J. Environ. Chem. Eng., 2020, 8, 103791 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- R. Li, T. Takata, B. Zhang, C. Feng, Q. Wu, C. Cui, Z. Zhang, K. Domen and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202313537 CrossRef CAS PubMed.
- R. Li, T. Takata, B. Zhang, C. Feng, Q. Wu, C. Cui, Z. Zhang, K. Domen and Y. Li, Angew. Chem., 2023, 135, e202313537 CrossRef.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, N. Saito, Y. Inoue and K. Domen, J. Catal., 2006, 243, 303–308 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- Y. Yang, Y. Zhang, T. Wang, Y. Ma, S. Xing, J. Yan, J. Ran and X. Li, Catal. Lett., 2024, 154, 2537–2550 CrossRef CAS.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 1422–1426 CrossRef CAS PubMed.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., 2019, 131, 1436–1440 CrossRef.
- M. N. Ha, F. Zhu, Z. Liu, L. Wang, L. Liu, G. Lu and Z. Zhao, RSC Adv., 2016, 6, 21111–21118 RSC.
- L. Yang, Z. Chen, J. Zhang and C. A. Wang, Front. Mater. Sci., 2019, 13, 342–351 CrossRef.
- C. Hu, C. Tai, W. Zhang, Q. Lu, M. Wei, C. Si, E. Guo and Y. Pang, J. Alloys Compd., 2023, 930, 167449 CrossRef CAS.
- H. Bai, J. Juay, Z. Liu, X. Song, S. S. Lee and D. D. Sun, Appl. Catal., B, 2012, 125, 367–374 CrossRef CAS.
- E. C. Su, B. S. Huang, J. T. Lee and M. Y. Wey, Sol. Energy, 2018, 159, 751–759 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- Y. Zhang, L. Zhong and D. Duan, J. Mater. Sci., 2016, 51, 1142–1152 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- R. P. Vasquez, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 217–240 CrossRef CAS.
- N. Aas, T. J. Pringle and M. Bowker, J. Chem. Soc., Faraday Trans., 1994, 90, 1015 RSC.
- M. E. Pilleux, C. R. Grahmann and V. M. Fuenzalida, J. Am. Ceram. Soc., 1994, 77, 1601–1604 CrossRef CAS.
- M. I. Sosulnikov and Y. A. Teterin, J. Electron Spectrosc. Relat. Phenom., 1992, 59, 111–126 CrossRef CAS.
- K.-I. Aika and K. Aono, J. Chem. Soc., Faraday Trans., 1991, 87, 1273 RSC.
- B. Choudhury and A. Choudhury, Phys. E, 2014, 56, 364–371 CrossRef CAS.
- H. H. Pham and L. W. Wang, Phys. Chem. Chem. Phys., 2015, 17, 541–550 RSC.
- S. Harbeck, A. Szatvanyi, N. Barsan, U. Weimar and V. Hoffmann, Thin Solid Films, 2003, 436, 76–83 CrossRef CAS.
- T. Xie, Y. Wang, C. Liu and L. Xu, Materials, 2018, 11, 1–17 Search PubMed.
- A. Litke, Y. Su, I. Tranca, T. Weber, E. J. M. Hensen and J. P. Hofmann, J. Phys. Chem. C, 2017, 121, 7514–7524 CrossRef CAS PubMed.
- H. J. Jang, S. J. Park, J. H. Yang, S. M. Hong, C. K. Rhee, D. Kim and Y. Sohn, Mater. Sci. Semicond. Process., 2021, 132, 105919 CrossRef CAS.
- G. Jeantelot, S. Ould-Chikh, J. Sofack-Kreutzer, E. Abou-Hamad, D. H. Anjum, S. Lopatin, M. Harb, L. Cavallo and J. M. Basset, Phys. Chem. Chem. Phys., 2018, 20, 14362–14373 RSC.
- M. S. Falcão, M. A. S. Garcia, C. V. R. De Moura, S. Nicolodi and E. M. De Moura, J. Braz. Chem. Soc., 2018, 29, 845–855 Search PubMed.
- P. Lu, X. Hu, Y. Li, M. Zhang, X. Liu, Y. He, F. Dong, M. Fu and Z. Zhang, RSC Adv., 2018, 8, 6315–6325 RSC.
- Q. Zhang, S. Mo, J. Li, Y. Sun, M. Zhang, P. Chen, M. Fu, J. Wu, L. Chen and D. Ye, Catal. Sci. Technol., 2019, 9, 4538–4551 RSC.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- K. A. Smith, A. I. Savva, C. Deng, J. P. Wharry, S. Hwang, D. Su, Y. Wang, J. Gong, T. Xu, D. P. Butt and H. Xiong, J. Mater. Chem. A, 2017, 5, 11815–11824 RSC.
- F. A. Rabuffetti, H. S. Kim, J. A. Enterkin, Y. Wang, C. H. Lanier, L. D. Marks, K. R. Poeppelmeier and P. C. Stair, Chem. Mater., 2008, 20, 5628–5635 CrossRef CAS.
- I. Atkinson, V. Parvulescu, J. P. Cusu, E. M. Anghel, M. Voicescu, D. Culita, S. Somacescu, C. Munteanu, M. Šćepanović, Z. V. Popovic and V. Fruth, J. Photochem. Photobiol., A, 2019, 368, 41–51 CrossRef CAS.
- M. A. Alavi and A. Morsali, Ultrason. Sonochem., 2010, 17, 132–138 CrossRef CAS PubMed.
- R. L. Chamousis and F. E. Osterloh, Energy Environ. Sci., 2014, 7, 736–743 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- C. Schlumberger and M. Thommes, Adv. Mater. Interfaces, 2021, 8, 2002181 CrossRef CAS.
- A. S. Cherevan, L. Deilmann, T. Weller, D. Eder and R. Marschall, ACS Appl. Energy Mater., 2018, 1, 5787–5799 CrossRef CAS.
- H. Eskandarloo, A. Badiei, M. A. Behnajady and G. M. Ziarani, Photochem. Photobiol., 2015, 91, 797–806 CrossRef CAS PubMed.
- C. Y. Wu, K. J. Tu, Y. S. Lo, Y. L. Pang and C. H. Wu, Mater. Chem. Phys., 2016, 181, 82–89 CrossRef CAS.
- L. Nie, J. Yu, X. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777–2783 CrossRef CAS PubMed.
- C.-Y. Wu, K.-J. Tu, J.-P. Deng, Y.-S. Lo and C.-H. Wu, Materials, 2017, 10, 566 CrossRef PubMed.
- M. Trochowski, M. Kobielusz, K. Mróz, M. Surówka, J. Hämäläinen, T. Iivonen, M. Leskelä and W. Macyk, J. Mater. Chem. A, 2019, 7, 25142–25154 RSC.
- J. Yu, G. Wang, B. Cheng and M. Zhou, Appl. Catal., B, 2007, 69, 171–180 CrossRef CAS.
- W. Kim, T. Tachikawa, G. H. Moon, T. Majima and W. Choi, Angew. Chem., Int. Ed., 2014, 53, 14036–14041 CrossRef CAS PubMed.
- W. Kim, T. Tachikawa, G. Moon, T. Majima and W. Choi, Angew. Chem., 2014, 126, 14260–14265 CrossRef.
- J. Wang, X. Liu, R. Li, P. Qiao, L. Xiao and J. Fan, Catal. Commun., 2012, 19, 96–99 CrossRef CAS.
- T. A. Kandiel, R. Dillert, L. Robben and D. W. Bahnemann, Catal. Today, 2011, 161, 196–201 CrossRef CAS.
- E. Tsuji, K. I. Fukui and A. Imanishi, J. Phys. Chem. C, 2014, 118, 5406–5413 CrossRef CAS.
- V. J. Babu, S. Vempati, T. Uyar and S. Ramakrishna, Phys. Chem. Chem. Phys., 2015, 17, 2960–2986 RSC.
- Y. W. Chung and W. B. Weissbard, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 3456–3461 CrossRef CAS.
- W. Sun, S. Meng, S. Zhang, X. Zheng, X. Ye, X. Fu and S. Chen, J. Phys. Chem. C, 2018, 122, 15409–15420 CrossRef CAS.
- V. Pfeifer, P. Erhart, S. Li, K. Rachut, J. Morasch, J. Brötz, P. Reckers, T. Mayer, S. Rühle, A. Zaban, I. M. Seró, J. Bisquert, W. Jaegermann and A. Klein, J. Phys. Chem. Lett., 2013, 4, 4182–4187 CrossRef CAS.
- M. Ge, J. Cai, J. Iocozzia, C. Cao, J. Huang, X. Zhang, J. Shen, S. Wang, S. Zhang, K. Q. Zhang, Y. Lai and Z. Lin, Int. J. Hydrogen Energy, 2017, 42, 8418–8449 CrossRef CAS.
- R. Zhang, B. Tu and D. Zhao, Chem.–Eur. J., 2010, 16, 9977–9981 CrossRef CAS PubMed.
- G. Tian, H. Fu, L. Jing and C. Tian, J. Hazard. Mater., 2009, 161, 1122–1130 CrossRef CAS PubMed.
- T. Guohui, F. Honggang, J. Liqiang, X. Baifu and P. Kai, J. Phys. Chem. C, 2008, 112, 3083–3089 CrossRef.
- W. Zhou, W. Li, J. Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS PubMed.
- W. Zhou, F. Sun, K. Pan, G. Tian, B. Jiang, Z. Ren, C. Tian and H. Fu, Adv. Funct. Mater., 2011, 21, 1922–1930 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03416c |
| This journal is © The Royal Society of Chemistry 2024 |