
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A well-defined supported Pt nanoparticle catalyst for heterogeneous catalytic surface science†
Taek-Seung
Kim
 a,
Christopher R.
O'Connor
a,
Samantha L.
Le
ab and
Christian
Reece
*a
a,
Christopher R.
O'Connor
a,
Samantha L.
Le
ab and
Christian
Reece
*a
aRowland Institute at Harvard, Harvard University, Cambridge, MA, USA. E-mail: christianreece@fas.harvard.edu
bDepartment of Chemistry, Tufts University, Medford, MA, USA
First published on 24th June 2024
Abstract
Our ability to rationalise the activity of heterogeneous catalytic materials relies on a precise understanding of both the structure and composition of the catalyst under reaction conditions to resolve active sites. Their highly dynamic nature means that catalysis suffers from an observer effect, where the act of measuring activity and/or structure will modify the structure itself. Herein, we present a well-defined catalyst consisting of 2 nm Pt nanoparticles supported on SiO2 that can be dynamically modified and regenerated without irreversibly changing the underlying structure or activity of the catalyst. Using in situ diffuse reflectance infrared Fourier transform spectroscopy and transmission electron microscopy we identify that the catalyst is resistant to oxidative and reductive treatments, but irreversibly restructures in reactive environments (CO + O2) at moderate pressures and temperatures. We show that by using transient pressure pulse experiments it is possible to minimise the restructuring in reactive environments, which when coupled with kinetic modelling can be used to identify site and state dependent activity while simultaneously quantifying the number of active sites. The well-defined nature of this 2 nm Pt/SiO2 catalyst means it can be utilised to gain molecular insight into catalytic processes and act as a new material for use in heterogeneous catalytic surface science. Further, the rapid restructuring of the catalyst during CO oxidation demonstrates that it is not a benign characterisation tool for comparison of catalysts.
 Christian Reece | Christian Reece is a Group Leader and Rowland Fellow at the Rowland Institute at Harvard University who specialises in heterogeneous catalytic surface science. After earning their PhD from Cardiff University in 2016, Christian joined Harvard University as a postdoctoral fellow under Prof. Robert J. Madix. Christian was awarded the Rowland Fellowship in 2019 where his group develops new instrumentation and methods for understanding catalytic processes in complex environments. Additionally, Christian is focused on enhancing accessibility in academia by developing low-cost open-source instrumentation. |
1. Introduction
The chemical industry, which relies extensively on heterogeneous catalytic processes, is vital to our modern infrastructure and economy. The massive scale in which the chemical industry operates means that it is a significant contributor to climate change, with most chemical processes being classified as unsustainable.1 One way to reduce the CO2 emissions of the chemical industry is by designing more efficient catalysts that can operate using greener fuels and feedstocks. This sustainable chemical production will require an approach where atomic and molecular scale understanding is combined with macroscopic insight to guide catalyst development.2 However, our ability to resolve an atomistic understanding of catalytic processes under reaction conditions is limited, making the design of catalysts particularly difficult.3One of the significant barriers to an atomistic understanding of catalysis is the highly dynamic nature of catalytic materials.4 Supported metal nanoparticle catalysts have long been known to sinter (where metal nanoparticles agglomerate) which decreases the surface area and number of active sites,5 while also changing the reaction kinetics due to different numbers of sites available at the surface.6 The reaction environment around the catalyst will also drastically effect its structure and composition.7 This means that catalysis suffers from an observer effect, where the act of measuring catalytic activity can irreversibly alter the state of the catalyst itself, making determining “active sites” particularly difficult.8 This dynamic nature is partially responsible for the well-known reproducibility issues in catalysis.9
To reliably identify active sites and reaction intermediates, an idealised catalyst would have three properties. First, the catalyst surface must be well-defined so that any heterogeneity in the system is minimised or precisely known. Second, it must be robust enough so that it does not irreversibly restructure in any significant fashion (e.g., via sintering) from the well-defined state. Finally, it should be regenerable such that any modification to the underlying catalyst state (e.g., oxidation, reduction, adsorbate coverage) can be reversed, and the initial well-defined state restored. Historically, these three properties have been fulfilled through the use of idealised model systems such as planar single crystals,10 curved single crystals,11 or nanoparticles supported on planar surfaces,12,13 but the applicability of these systems to “real-world” catalysis remains an open topic of debate.3,14–18 Therefore, there is a need for a well-defined, but non-idealised, catalytic material that is robust and that can be reliably regenerated to a consistent initial state for the purposes of benchmarking catalytic activity19 and the identification of active sites.20
Herein, we report a well-defined 2 nm Pt nanoparticle catalyst supported on SiO2 that can be dynamically oxidised and reduced and reliably regenerated back to an initial state with little-to-no sintering of the nanoparticles. Using in situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) we identify two types of sites on the catalyst, well-coordinated terrace-like atoms, and under-coordinated edge and vertex-like atoms. Combining DRIFTS and ex situ Transmission Electron Microscopy (TEM) we find that the catalyst is resistant to CO, O2, and H2 treatments up to 350 °C, and find that no sintering of the nanoparticles occurs during CO oxidation up to 250 °C, but at elevated temperatures the nanoparticles rapidly sinter. Using the Temporal Analysis of Products (TAP) technique21 we find a high batch-to-batch reproducibility of the catalyst, and that it is possible to measure consistent catalytic activity for CO oxidation over metallic and oxidised Pt after over a hundred cycles of oxidation, reduction, and reaction. The key parameters that facilitate this resilience are the low weight loading of the catalyst (<1%) coupled with the mild oxidative, reductive, and reactive treatments. Further, we demonstrate how this catalyst can be used to identify and quantify active sites on the surface using the kinetic site deconvolution technique.22 This catalyst can be synthesised using well-established synthesis techniques, and as such, can act as a benchmarking catalyst, alongside other benchmarking catalysts such as EUROPt-1,23 to understand structure–activity relationships and resolve reaction mechanisms using techniques such as TAP,24 DRIFTS coupled with modulation excitation spectroscopy,25,26 and steady-state isotopic transient kinetic analysis.27 Further, the weak interaction between Pt and SiO2 means that the nanoparticles remain spherical,28,29 which allows for simpler comparison with theoretical simulations over freestanding nanoparticles. Finally, the ability for the catalyst to dynamically regenerate while maintaining its structure allows for in-depth insight into the nature of active sites over both reduced and oxidised Pt.
2. Materials and methods
2.1. Catalyst synthesis
The synthesis procedure and extensive characterisation of the 2 nm Pt/SiO2 catalysts used in this work are outlined in our previous publication,22 but the synthesis procedure is expanded upon here. The synthesis of the 2 nm Pt nanoparticles follows a typical colloidal method,30 and is as follows. First, 100 mg of chloroplatinic acid hydrate (H2PtCl6·xH2O, 99.9%, Sigma-Aldrich), 2.5 ml of sodium hydroxide (NaOH, 1 M), and 20 mg of poly(vinylpyrrolidone) (PVP, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich) were added to 10 ml of ethylene glycol (Sigma-Aldrich) in a 50 ml three-next round-bottom flask. The flask was attached to a Schlenk line and evacuated at 25 °C for 30 minutes while vigorously stirring (400 rpm). The flask was then heated using a heating mantle while under vacuum to 80 °C at a ramp rate of 10 °C min−1, then held at temperature for 30 minutes. Then, the flask was heated from 80 °C to 200 °C at 10 °C min−1, with Ar gas purging introduced at 130 °C during the heating ramp. The flask was then held at 200 °C for 2 hours. After the reaction, the solution was cooled to room temperature while under Ar gas purging. The colloidal suspension was then diluted by adding 50 ml of acetone and was centrifuged at 6500 rpm (2574.3 g, 5.45 cm radius) for 10 minutes, twice, sequentially. Then, the Pt nanoparticles were re-dispersed in 80 ml of ethanol via sonication for 5 minutes at 25 °C. The dispersed Pt solution was then centrifuged at 2000 rpm (243.7 g, 5.45 cm radius) for 10 minutes. The supernatant contains the colloidal 2 nm Pt nanoparticles dispersed in ethanol solvent (Fig. 1a and b).
000, Sigma-Aldrich) were added to 10 ml of ethylene glycol (Sigma-Aldrich) in a 50 ml three-next round-bottom flask. The flask was attached to a Schlenk line and evacuated at 25 °C for 30 minutes while vigorously stirring (400 rpm). The flask was then heated using a heating mantle while under vacuum to 80 °C at a ramp rate of 10 °C min−1, then held at temperature for 30 minutes. Then, the flask was heated from 80 °C to 200 °C at 10 °C min−1, with Ar gas purging introduced at 130 °C during the heating ramp. The flask was then held at 200 °C for 2 hours. After the reaction, the solution was cooled to room temperature while under Ar gas purging. The colloidal suspension was then diluted by adding 50 ml of acetone and was centrifuged at 6500 rpm (2574.3 g, 5.45 cm radius) for 10 minutes, twice, sequentially. Then, the Pt nanoparticles were re-dispersed in 80 ml of ethanol via sonication for 5 minutes at 25 °C. The dispersed Pt solution was then centrifuged at 2000 rpm (243.7 g, 5.45 cm radius) for 10 minutes. The supernatant contains the colloidal 2 nm Pt nanoparticles dispersed in ethanol solvent (Fig. 1a and b).
 | ||
| Fig. 1 TEM images of (a) and (b) as synthesised 2 nm Pt colloids and (c) the as prepared 2 nm Pt nanoparticles supported on SiO2. Insets show size distribution of nanoparticles. No sintering of the nanoparticles is observed after calcination and reduction. | ||
The synthesis of the supported Pt/SiO2 catalyst is as follows. First, the non-nanoporous 10–20 nm SiO2 powder (Sigma-Aldrich, Product No. 637238) was calcined in air at 700 °C for 1 hour. Then, 300 mg of the calcined SiO2 powder was dissolved in 10 ml of ethanol via sonication for 30 minutes at 25 °C. Then, 3 ml of the colloidal 2 nm Pt nanoparticle solution was added dropwise to the 10 ml ethanol solution with vigorous stirring (400 rpm). The suspension was sonicated for 30 minutes at 25 °C in a 100 ml beaker and was then transferred to a 40 ml vial. The vial was placed under vacuum and the solvent was evaporated via vigorous stirring (600 rpm) overnight. The resulting Pt/SiO2 catalyst was placed into a tube furnace and heated from 25 to 500 °C at a ramp rate of 2 °C min−1 in air, then held at 500 °C for 1 hour to remove the majority of the carbonaceous capping agent. The sample was then cooled to 25 °C. The calcined catalyst was then heated from 25 to 300 °C at a ramp rate of 2 °C min−1 under 10% H2 balanced in Ar flow, leaving the as prepared catalyst. No sintering of the nanoparticles was observed during calcination and reduction (Fig. 1c). It was found that a low metal loading (<1 wt%) was crucial for preventing sintering during the pretreatment process.
2.2. Temporal analysis of products experiments
The temporal analysis of products experiments were performed in a home-built TAP reactor that has been described previously.31 The TAP experiment has been described extensively in the previous literature21,24,31 but is summarised here for clarity. A quartz microreactor (4 mm diameter, 64.1 mm height) is filled with a layer of inert packing (Sand, 50–70 mesh, Sigma-Aldrich, calcined in air at 700 °C overnight) to a height of 29.7 mm, followed by a layer of ∼5.4 mg of Pt/SiO2 catalyst (sieved between 50 and 70 mesh), which is followed by a second layer of inert packing to a total height of 64.1 mm. The amount of catalyst packed in the microreactor is precisely known but varies slightly between each packed bed. A solenoid valve is attached to the entrance of the microreactor, and the exit of the microreactor is attached to an ultra-high vacuum (UHV) system containing a mass spectrometer. The microreactor is then evacuated to a base pressure of 10−10 torr and a short, quantified pulse of gas (∼100 μs, ∼1015 molecules) is sent into the entrance of the packed bed using the solenoid pulse valve. The gas then diffuses through the packed bed via Knudsen diffusion and interacts with the thin layer of the catalyst and eventually diffuses out the packed bed and into the UHV system. At the reactor exit, the flux (or flow) of gas leaving the microreactor is measured using the mass spectrometer for a given collection time (typically 4 s). The temperature of the catalyst is recorded by a type-K thermocouple inserted into the catalyst bed. The quartz microreactor is heated using a hand-woven nichrome heating element, with the temperature controlled using a PID controller connected between the nichrome heating wire and a DC power supply.After being packed in the microreactor, any remaining carbonaceous material is removed from the as prepared Pt/SiO2 catalyst by first performing and oxidation and subsequent reduction of the catalyst. First, the catalyst was oxidised using repeated pulses of O2 (160 μs width) until no more exit flux of CO2 is recorded. Next, the oxidised Pt catalyst was reduced using repeated pulses of H2 (160 μs width) to return the Pt to a metallic state and to remove any remaining carbonaceous material. After the O2 and H2 treatments no production of CO2 (m/z 44) or methane (m/z 16) can be recorded during O2 and H2 pulsing respectively indicating a clean metallic (pristine) surface.22 Between experiments the pristine state of the catalyst is regenerated using a milder version of the pretreatment with smaller pulses of O2 and H2 (∼108 μs).
During the TAP experiment the exit flux of only one m/z value can be recorded at a time, therefore the whole range of m/z values are scanned during sequential pulses and combined together to form a pulse set.31 The exit flux responses are baseline corrected using the last few datapoints of the exit flux response and normalised relative to the integrated signal of the Ar inert tracer gas that is included in each pulse (Ar normalised exit flux). This allows for consistent comparison of datasets as the raw signal recorded by the mass spectrometer will drift over time and is dependent on a number of factors (e.g., multiplier voltage), but the Ar normalised signal for a given gas mixture will remain constant. If the exit flux response does not return to the baseline during the collection time due to slowly desorbing or reacting gases, the signal is baseline corrected using the first few datapoints of the whole experiment (Ar normalised raw exit flux). For the case where there is no baseline drift the Ar normalised exit flux and Ar normalised raw exit flux are equivalent. All gas mixtures reported are balanced in Ar.
The modelling of the TAP experiments uses Multi-Zone TAP Reactor Theory32,33 (MZTRT) coupled with our previously published 3-pathway reaction model for CO oxidation over 2 nm Pt nanoparticles.22 The one-dimensional MZTRT model has been proven to be valid for our given reactor setup with a reactor L/R (length/radius) > 3.5, no pre-volume, and an idealised vacuum at the reactor exit.31,34,35 The MATLAB script used to perform the simulation and curve fitting is provided in our previous publication,22 with the model regression performed using the lsqcurve fit function and the 95% confidence intervals estimated using the nlparci function. The data is reported as inlet normalised exit flux, where the exit flux curves are normalised to the amount of reactant (or inert in the case of Ar) pulsed into the reactor. First, the curves are normalised to the tracer Ar and are then divided by the known gas mixture (i.e. 20% O2 in Ar), then corrected using pre-calculated correction factors. The correction factors are calculated by pulsing a known mixture of each gas through an inert bed. The calibration coefficients for O2 and CO2 were found to be 0.977 and 1.220 respectively. The equation for calculating the normalised exit flux is as follows:
 | (1) |
2.3. Diffuse reflectance infrared spectroscopy measurements
The DRIFTS measurements were performed using a Harrick Praying Mantis Low Volume High Temperature Reaction Chamber (LV-HVC) and a Harrick Praying Mantis Low Temperature Reaction Chamber (CHC-CHA-4) reactor cell equipped with ZnSe windows. The DRIFTS cells were mounted into a Fourier-transform infrared spectrometer (FTIR, Bruker INVENIO-R) equipped with a liquid-nitrogen-cooled HgCdTe (MCT) detector using the Harrick Praying Mantis diffuse reflectance accessory. During all experiments the FTIR and Praying Mantis accessory are purged at a flow rate of 5 L m−1 and 14 L m−1 respectively using a purge gas generator (Parker Spectra 15), with the purge gas flown through a gas purifier (Drierite L68GP) before being flown into the system. As a large temperature gradient exists between the thermocouple used to measure the reaction cell temperature and the catalyst surface exposed to the infrared beam36 the sample temperature was calibrated using an optical pyrometer and a bed packed with Al2O3. All temperatures reported are the calibrated values. The sample temperature was maintained using a Harrick ATK Temperature Controller. The sample cups in the DRIFT cells are prepared by first inserting a 250-mesh stainless steel disc to the bottom of the cup, then the cup is packed with 120-grit SiC up to 1 mm from the top, finally the rest of the cup is packed using the Pt/SiO2 catalyst (sieved between 50 and 70 mesh). As adsorbate concentration can be linearly related to the absorbance intensity or Kubelka–Munk37,38 (KM) intensity depending on the relative reflectivity of the DRIFTS features39 we ensure that the relative reflectivity of our DRIFTS features is below the threshold where absorbance is no longer linear,39 and as such, report all spectra in absorbance units. The DRIFTS reaction chambers were connected to an external gas handling system consisting of a set of parallel mass flow controllers that is described in our previous publication and in section 2.5.40After being packed into the DRIFTS sample cup, the remaining carbonaceous material from the as prepared Pt/SiO2 catalyst is removed by first performing an oxidation and subsequent reduction of the catalyst. First, the catalyst was heated from 25 to 350 °C at a ramp rate of 30 °C min−1 in a 20% O2 stream at a total flow rate of 25 sccm, the temperature was then held at 350 °C for 20 minutes. Then, the stream was switched to a 5% H2 stream while at 350 °C and held for an additional 30 minutes. The stream was then switched to one containing Ar and the catalyst was cooled to 25 °C. This generates the initial clean metallic (pristine) catalyst surface. Between experiments, the pristine state of the catalyst surface is regenerated by flowing 5% H2 at 350 °C for 10 minutes. All gas mixtures reported are balanced in Ar.
2.4. Transmission electron microscopy measurements
The ex situ TEM measurements were performed using an aberration-corrected TEM (ARM 200 F, JEOL) at 200 kV using a single tilt holder. For imaging the unsupported particles, 1 ml of the colloidal 2 nm Pt nanoparticle suspension was diluted in 3 ml of ethanol and sonicated for 5 minutes at 25 °C. Then, 10 μl of the diluted Pt colloidal mixture was dropped onto the TEM grid (FCF300-CU-UB, Electron Microscopy Science) three times using a micropipette. For imaging the supported Pt/SiO2 catalysts, the catalyst powder was dispersed in 5 ml of ethanol solvent and sonicated for 20 minutes at 25 °C. Then, the supernatant was dropped at least 5 times onto the TEM grid using a micropipette.2.5. CO and O2 chemisorption experiments
The CO and O2 chemisorption experiments were performed using a home-built rapid transient flow reactor that has been described previously,40 but is summarised here. Two parallel gas streams are generated using 4 mass flow controllers per stream (8 total, Alicat Scientific) which are fed into a two-position four-port microelectric valve (VICI) where one line is sent into the packed bed reactor, and the other is sent through a bypass line. Both streams are isothermal and isobaric. The reactor and bypass lines are both connected to a sampling capillary that is connected to a UHV system containing a mass spectrometer that can sample at rates up to 250 Hz. During the chemisorption experiments, a stream of Ar is flown through the bed over the pristine catalyst. Then, the microelectric valve is actuated and a stream consisting of CO or O2 and a Kr tracer is introduced to the packed bed. By measuring the difference between the normalised tracer signal to the normalised reactant signal (normalised to the steady-state signal) the total amount of CO or O2 adsorbed on the catalyst can be calculated using the known volumetric flow rates and partial pressures of the gas through the following relationship: | (2) |
For the chemisorption experiments 5.73 mg (sieved between 50 and 70 mesh) of the as prepared Pt/SiO2 catalyst is diluted in SiC with a 1:4 volumetric dilution ratio and is loaded into the reactor with a bed height of 2.65 mm. The reactor is then filled up to the top with additional SiC giving a total bed height of 43.30 mm. The as prepared catalyst is then heated to 300 °C and oxidised in a 5% O2 stream at a flow rate of 50 sccm for 30 minutes. The oxidised catalyst is then reduced in a 5% H2 stream for an additional 30 minutes. The catalyst is then cooled to room temperature in Ar. For the CO chemisorption experiments an Ar stream is replaced with a 0.4% CO, 1% Kr stream at 30 °C, with both streams at a total flow rate of 25 sccm. Between chemisorption experiments the adsorbate covered catalyst is heated to 300 °C and reduced in a 5% H2 stream for 30 minutes to regenerate the pristine catalyst. The same process is repeated for the O2 chemisorption experiments, but instead using a 0.4% O2, 1% Kr stream. The experiments are repeated 3 times.
3. Results and discussion
3.1. Characterisation of the pristine 2 nm Pt/SiO2 catalyst
The pristine 2 nm Pt nanoparticles supported on SiO2 were found to consist of two distinct types of sites: well-coordinated terrace-like Pt atoms, and under-coordinated edge and vertex Pt atoms (Fig. 2a). The sites were assigned using CO DRIFTS (Fig. 2b) where three distinct spectral features were observed. Feature 1 which has a peak maximum at 2070 cm−1 is ascribed to CO bound linearly on the well-coordinated terrace-like Pt sites. Feature 2 which consists of a shoulder from ∼1960 to 2060 cm−1 is ascribed to CO bound linearly on the under-coordinated edge and vertex sites. Feature 3 which consists of a broad adsorption feature from 1740 to 1860 cm−1 is ascribed to bridge and multi-bound CO on both the well-coordinated and under-coordinated sites. These assignments are consistent with those observed previously over Pt nanoparticles.22,41–44 Quantification of these absorbance features gives an approximate ratio of 80% of the linear bound CO being adsorbed on well-coordinated sites, and 20% of the linear bound CO being adsorbed on under-coordinated sites.22,31 | ||
| Fig. 2 (a) Schematic of a ∼2 nm truncated icosahedron nanoparticle structure with well-coordinated and under-coordinated atoms highlighted in grey and orange respectively. (b) Absorbance spectra taken from the pristine Pt/SiO2 catalyst at 25 °C during 0.1% CO flow at 100 sccm. The three features highlighted correspond to CO adsorbed linearly on well-coordinated sites, CO adsorbed linearly on under-coordinated sites, and multi-bound CO adsorbed on both well-coordinated and under-coordinated sites. | ||
To determine the density of active sites on the catalyst, a series of CO and O2 chemisorption experiments were performed over the pristine 2 nm Pt/SiO2 catalyst (Fig. 3). It was found that an equivalent amount (2.6 μmol gcat−1) of CO and O2 could be adsorbed onto the pristine catalyst, indicating that the saturation coverage of O* was twice that of CO* as O2 readily dissociates over Pt to make 2O*.45 This differs from what has been observed over Pt(111) single crystals, where the saturation coverage of CO at room temperature is expected to be between 0.5 and 0.68 monolayers depending on the pressure,46–48 and the saturation coverage of O* being 0.25 monolayers.49,50 Some minor desorption of CO was found to occur at room temperature after CO chemisorption, indicating a weaker binding affinity for CO over the 2 nm nanoparticles at high coverage than over the Pt(111) surfaces.51 Over stepped Pt(112) single crystal surfaces, the saturation coverage of O* at room temperature was recorded to be as high as 0.54 monolayers,52 showing that O* can accumulate at higher quantities on more defected surfaces. This is further reinforced by recent work performed on a curved Pt single crystal which found higher O* coverages over more vicinal surfaces at room temperature.53 Further, small nanoparticles are known to be easier to oxidise than more bulk-like structures.54 When combined together the decreased binding energy of CO* over the 2 nm nanoparticles and the increased affinity of O* over more under-coordinated surfaces can be used to rationalise the much higher ratio of O* to CO* seen on the nanoparticles, but further study into this effect is required. The 1:2 ratio of CO to O* on the 2 nm nanoparticle catalyst could also be explained by formation of a PtOx species rather than a chemisorbed oxygen, but no evidence of bulk oxidation of Pt could be found at room temperature.22
 | ||
| Fig. 3 (a) CO and (b) O2 chemisorption experiments over the pristine Pt/SiO2 catalyst. The data is a combination of three separate chemisorption experiments. The total amount of CO and O2 adsorbed is calculated by determining the area (highlighted in yellow) between the Kr tracer and the adsorbate. | ||
3.2. Identifying the boundaries under which significant restructuring occurs
The 2 nm Pt/SiO2 catalyst was tested to identify the bounds under which it would significantly restructure. The catalyst is resistant to O2 treatments up to 500 °C and H2 treatments up to 350 °C as they are utilised in the initial pretreatment procedure. The resistance to O2 treatments at elevated temperatures is expected based on previous work over Pt nanoparticles supported on SiO2 single crystals which measured a sintering rate of 0.025 nm−1 min−1 at 650 °C in oxidative environments.29 To test if the catalyst was also resistant to CO, a series of CO adsorption experiments were performed (Fig. 4a). The pristine Pt/SiO2 catalyst was first exposed to CO at 25 °C, showing a similar absorbance spectrum to that measured previously (see Fig. 2). Next, the catalyst was heated to 350 °C in an inert atmosphere, and again exposed to CO. The resulting spectra shows dramatic shifts in absorbance for the linearly bound and multi-bound CO sites, in line with our previous work.22 The large increase in the number of under-coordinated sites vs. well-coordinated sites, coupled with the shift towards lower frequencies suggests the nature of the surface is changing. This phenomenon is being investigated and will be elaborated upon in a future publication. The catalyst was then cooled down in Ar to remove any CO bound to the surface, and the CO adsorption experiment was repeated again on the 350 °C treated catalyst. As can be seen, there is no difference in the adsorption spectra between the pristine and the CO treated catalyst indicating that no irreversible restructuring has occurred. This was confirmed by the ex situ TEM image of the Pt/SiO2 catalyst after the DRIFTS experiments (Fig. 4b) where no sintering of the nanoparticles was observed with an average particle size of 1.96 ± 0.46 nm. | ||
| Fig. 4 (a) Sequential absorbance spectra taken from the Pt/SiO2 catalyst during 0.1% CO flow at 100 sccm over the pristine catalyst at 25 °C (grey), at 350 °C (red), and at 25 °C after the 350 °C CO treatment (black). The features labelled (1), (2), and (3) point to the absorbance features corresponding to linear bound CO on well-coordinated sites, linear bound CO on under-coordinated sites, and multi-bound CO respectively. (b) Representative high resolution TEM image of the Pt/SiO2 catalyst after DRIFTS measurements. Inset shows the size distribution of nanoparticles. | ||
To determine if the Pt/SiO2 catalyst was resistant to reactive environments it was exposed to a stream of 0.27% O2 and 0.13% CO at various temperatures, after which it was reduced in H2, and CO adsorption experiments were performed at room temperature (Fig. 5a). A consistent DRIFTS absorbance spectrum was recorded over the pristine catalyst and after exposure to the reactive environment at 250 °C, indicating that no irreversible restructuring had occurred. However, after the catalyst was exposed to the reactive environment at 350 °C the integrated absorbance decreases, and the distribution of sites also change. The decrease in the integrated absorbance is associated with a decrease in the total number of sites available for CO to adsorb, which indicates sintering has occurred. This was confirmed by ex situ TEM (Fig. 5b) and chemisorption experiments (Fig. 5c and d) where the nanoparticle size had increased from 2 nm to ∼7 nm and the site density had decreased ∼5 fold. The difference in the distribution of peaks in the absorbance spectra also indicates some morphological change of the surface. The sharp feature assigned to linear bound CO on the well-coordinated sites (∼2090 cm−1) has decreased significantly relative to the feature assigned to the linear bound CO on under-coordinated sites (∼2020 cm−1). While not quantitative this indicates that on the large particles, after exposure to the reactive environment, the number of under-coordinated sites relative to the well-coordinated sites increases. This could indicate a roughening of the nanoparticle surface, which has been previously observed on single crystals,55 but requires further analysis. While this irreversible restructuring of the surface is undesirable from a catalytic perspective, the well-defined nature of the initial state of the catalyst means it becomes possible to directly compare pre and post-mortem analysis to determine how more aggressive reactive environments change the catalyst. Further the destructive nature of the CO oxidation experiments means that it should be used with caution when determining catalyst states.
 | ||
| Fig. 5 (a) Room temperature CO DRIFTS spectra taken over the pristine Pt/SiO2 catalyst (grey), and after exposure to a 0.27% O2, 0.13% CO reaction stream for 10 minutes at 200 °C (blue) and 350 °C (red). After exposure to the reactive stream the catalyst was cooled back to room temperature in a 5% H2 stream. The spectra were acquired at room temperature in a 0.1% CO stream. All experiments used a total flow rate of 100 sccm. The features labelled (1), (2), and (3) point to the absorbance features corresponding to linear bound CO on well-coordinated sites, linear bound CO on under-coordinated sites, and multi-bound CO respectively. (b) Representative high resolution TEM image of the Pt/SiO2 catalyst after the 350 °C reactive stream treatment. (c) CO and (d) O2 Chemisorption experiments over the sintered Pt/SiO2 catalyst. The data is a combination of three separate chemisorption experiments. | ||
3.3. Testing the ability to regenerate the well-defined catalyst state
To test how reliably the pristine catalyst state could be generated, a series of Temperature Programmed Oxidation (TPO) experiments were performed in the TAP reactor. First, CO was adsorbed at room temperature to saturation. Then, a gas mixture of 20% O2 was repeatedly pulsed over the CO-covered catalyst while ramping up to 350 °C at a rate of 8 °C min−1 while measuring the temperature dependent Ar normalised CO2 exit flux (Fig. 6). Two methods were utilised to probe the ability to generate a well-defined pristine catalyst state. First, multiple batches of the same catalyst were synthesised from the same batch of colloids. Excellent batch-to-batch reproducibility was observed (Fig. 6a) for the TPO experiments, demonstrating that the initial pristine state of the catalyst can be produced consistently between different catalyst batches. However, swapping out catalysts between each experiment is extremely time and material costly so a series of cycling experiments were performed on the same catalyst to test its longevity. The TPO experiments were repeated on the same catalyst after 1, 15, and 110+ cycles of oxidative, reductive, and reactive treatments (Fig. 6b). The pristine state of the catalyst was regenerated using the oxidative and reductive treatment outlined in section 2.2. After 15 cycles, no difference in the TPO profile could be observed. After 110+ cycles, a marginal decrease the integrated Ar normalised CO2 production was observed, but the overall profile shape remained consistent. The used catalyst from the TAP experiments was analysed via ex situ TEM, and no sintering of the nanoparticles was observed with an average size of 1.86 ± 0.36 nm (Fig. 6c). This demonstrates that the pristine state of the Pt/SiO2 catalyst can be reliably regenerated even after hundreds of oxidative, reductive, and reactive cycles. | ||
| Fig. 6 Temperature programmed oxidation experiments performed using the TAP reactor where O2 is repeatedly pulsed over a CO-covered pristine Pt/SiO2 catalyst while heating from 25 to 350 °C at 8 °C min−1 over (a) three different batches of the Pt/SiO2 catalyst, (b) the same catalyst after 1, 15, and 110+ sequential oxidative, reductive, and reactive treatments. (c) Representative high resolution TEM image of the Pt/SiO2 catalyst after the TAP experiments with the inset showing the size distribution of the nanoparticles. High batch-to-batch reproducibility is found over the Pt/SiO2 catalysts, and no sintering is observed after the TAP experiments. | ||
3.4. Identifying state and site dependent activity for CO oxidation
The ability for the 2 nm Pt/SiO2 catalyst to be dynamically modified while also being able to reliably reproduce an initial catalyst state means that it becomes possible to resolve state and site dependent reactivity for Pt catalysts. A set of TPO experiments were performed where CO was adsorbed over a pristine and oxidised version of the catalyst. The oxidised PtOx catalyst was generated by sending large pulses of O2 at 350 °C over the catalyst for 30 minutes, similar to the initial oxidation treatment performed in section 2.2. Much higher activity was reported at lower temperatures for the PtOx catalyst than the CO covered pristine catalyst during the TPO experiments (Fig. 7a). This matches with previous work showing that a “hyperactive” phase for CO oxidation is present on oxidised Pt group metal nanoparticles.56,57 Further, the sharp peak indicates that a single active site is responsible for the oxidation of CO over PtOx, though further work is required to confirm. The CO covered Pt catalyst shows two features, a sharp peak at approximately 75 °C followed by a broad shoulder up to 350 °C. Our previous work has shown these two features correspond to the oxidation of CO over under-coordinated sites being dominant at low temperature, and the oxidation of CO over well-coordinated sites being dominant at higher temperatures.22 A similar effect is observed when a mixture of 6.6% CO and 13.4% O2 is pulsed over a Pt, CO covered Pt, and CO covered PtOx catalyst while heating from 25 to 350 °C (Fig. 7b) where higher activity is measured at low temperatures over the PtOx catalyst. This ability is to dynamically switch between catalyst states allows for rigorous and reproducible characterisation of the catalyst without irreversibly restructuring the Pt nanoparticles. | ||
| Fig. 7 (a) Temperature programmed oxidation experiments performed using the TAP reactor where O2 is repeatedly pulsed over a CO covered pristine Pt and CO covered PtOx catalyst while heating from 25 to 350 °C at 8 °C min−1. (b) Temperature programmed reaction experiments where a 6.6% CO 13.4% O2 mixture was repeatedly pulsed over the pristine, CO covered pristine, and CO covered PtOx catalyst while heating from 25 to 350 °C at 8 °C min−1. Higher activity is observed at low temperatures over the PtOx catalysts. | ||
The well-defined nature of the catalyst allows for the simultaneous calculation of site-specific intrinsic rate constants and reaction stoichiometry using the kinetic site deconvolution method developed in our previous work.22 An isothermal titration experiment was performed over the CO covered pristine catalyst at 125 °C where O2 was repeatedly pulsed until no more CO2 could be detected (Fig. 8a). As there are two distinct types of sites on the Pt catalyst, it is possible to build a simple site-specific model for CO oxidation, where O2 can react with CO adsorbed on well-coordinated and under-coordinated sites to form CO2, and where O2 can irreversibly adsorb to empty sites on the catalyst without reacting further.22 This reaction model can be combined with Multi-Zone TAP Reactor Theory (see section 2.2) to simulate exit flux response curves which can be regressed to the experimentally measured exit flux curves to calculate the five rate constants in the model (Fig. 8a and b). The first three rate constants  ,
, 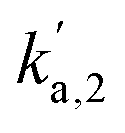 ,
, 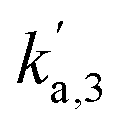 relate to the probability of O2 to react with CO adsorbed on the under-coordinated sites, CO adsorbed on the well-coordinated sites, and with empty sites respectively. The final two rate constants kr,1, kr,2 relate to the intrinsic rate constant for the surface reaction between CO* and O* over the under-coordinated and well-coordinated sites respectively. As the model is regressed to each pulse set individually, an independent set of 5 rate constants is calculated for each pulse set in the experiment (Fig. 8c). Using the reaction probability rate constants, it is possible to determine how much oxygen is consumed by each site:
relate to the probability of O2 to react with CO adsorbed on the under-coordinated sites, CO adsorbed on the well-coordinated sites, and with empty sites respectively. The final two rate constants kr,1, kr,2 relate to the intrinsic rate constant for the surface reaction between CO* and O* over the under-coordinated and well-coordinated sites respectively. As the model is regressed to each pulse set individually, an independent set of 5 rate constants is calculated for each pulse set in the experiment (Fig. 8c). Using the reaction probability rate constants, it is possible to determine how much oxygen is consumed by each site:
 | (3) |
 is the calculated reaction probability constant for site n, L is the reactor length, and D is the diffusivity of the reactant.
is the calculated reaction probability constant for site n, L is the reactor length, and D is the diffusivity of the reactant.
 | ||
| Fig. 8 Identification of reaction pathways from an isothermal titration experiment at 125 °C where O2 is repeatedly pulsed over a CO covered pristine Pt catalyst until no more CO2 production is recorded. (a) Integrated inlet normalised exit flux for O2 and CO2 during the titration experiment and MZTRT model fit. (b) Representative pulse set (60) showing the inlet normalised exit flux with MZTRT model fit. (c) Calculated rate constants from the three-pathway model regression with 95% confidence intervals. (d) Deconvoluted pathways for O2 conversion calculated from MZTRT model. The surface reaction constants as a function of the relative coverage of CO for (e) the well-coordinated sites, and (f) the under-coordinated sites. The CO coverage is estimated by assuming a 1:1 stoichiometry for CO* to CO2. | ||
The total amount of O2 converted during the experiments can be calculated using the integrated inlet normalised exit flux of O2 (Fig. 8a), which when combined with the pathway deconvolution method in eqn (3) allows for the site stoichiometry to be calculated (Fig. 8d). The total amount of O2 consumed during the experiment is equivalent to 76.6 pulses, of which 7% (5.0 pulses) is consumed by CO adsorbed on under-coordinated sites, 30% (23.4 pulses) is consumed by CO adsorbed on well-coordinated sites, and 63% (48.2 pulses) is consumed by empty sites on the catalyst, which corresponds well with the 1:2 ratio of CO* to O* site density on the nanoparticles calculated in the chemisorption experiments (Fig. 3). Alongside the site stoichiometry, the site-specific intrinsic kinetics of the surface reaction between CO* and O* are also calculated (Fig. 8e and f). Similar to our previous work,22,31 the intrinsic rate constant for the reaction over well-coordinated sites shows a linear decrease as a function of CO coverage, whereas the intrinsic rate constant over under-coordinated sites is largely coverage independent.
4. Conclusion
The 2 nm Pt/SiO2 catalyst developed as part of this work has a well-defined structure, can be reliably regenerated without sintering, and can be dynamically modified between an oxidised and a reduced state. The catalyst is resistant to reductive treatments up to 350 °C, oxidative treatments up to 500 °C, but is shown to be susceptible to irreversible restructuring in redox (CO + O2) environments at milder conditions. The high susceptibility of the Pt catalyst to the reactive environment indicates that CO oxidation may not be an ideal probe reaction for studying catalyst structure and should be used with caution. However, it is possible to overcome this susceptibility by using experiments such as TAP which limit exposure to the reactive mixture. This combination of properties makes it an excellent system for surface-science experiments, where a precise molecular understanding of catalytic reactions is possible. We demonstrate how this catalyst can be used to resolve state specific activity, and when coupled with kinetic modelling allows for the simultaneous characterisation and quantification of active sites. In future work, it is hoped that this catalyst can be extended beyond model reactions (CO oxidation) towards more industrially relevant reactions such as propane dehydrogenation. The well-defined nature of this catalyst also means it can be used for benchmarking large-scale machine learning enhanced molecular dynamics simulations of nanoparticles.58 Further, this catalyst can be used as a reference point for systems with increased complexity such as different sized nanoparticles or different supports.Data availability
The raw TEM images are uploaded as part of the ESI.† The Multi-Zone TAP Reactor Theory code used to perform the kinetic site deconvolution is available in our previous publication.22Author contributions
Taek-Seung Kim: investigation, visualization, writing – original draft, writing – review & editing. Christopher R. O'Connor: investigation, visualization, writing – original draft, writing – review & editing. Samantha Le: writing – review & editing, investigation, validation. Christian Reece: conceptualization, funding acquisition, methodology, project administration, software, supervision, visualization, writing – original draft, writing – review & editing. T. S. K. performed the catalyst synthesis, TEM, and the TAP experiments. C. R. O. performed the DRIFTS and chemisorption experiments. S. L. assisted with collection of the TAP, DRIFTS, and chemisorption measurements. C. R. developed the TAP model and supervised the project. C. R. wrote the original draft with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. R. gratefully acknowledges the Rowland Fellowship through the Rowland Institute at Harvard. C. R. and S. L. acknowledge S. L.'s faculty advisor Prof. Eric High from Tufts University.References
- V. Tulus, J. Pérez-Ramírez and G. Guillén-Gosálbez, Green Chem., 2021, 23, 9881–9893 RSC.
- S. Mitchell, A. J. Martín and J. Pérez-Ramírez, Nat. Chem. Eng., 2024, 1, 13–15 CrossRef.
- A. Beck, V. Paunović and J. A. Van Bokhoven, Nat. Catal., 2023, 6, 873–884 CrossRef CAS.
- G. A. Somorjai, Catal. Lett., 1992, 12, 17–34 CrossRef CAS.
- P. Flynn, J. Catal., 1975, 37, 432–448 CrossRef CAS.
- G. A. Somorjai and J. Carrazza, Ind. Eng. Chem. Fundam., 1986, 25, 63–69 CrossRef CAS.
- H. Frey, A. Beck, X. Huang, J. A. Van Bokhoven and M. G. Willinger, Science, 2022, 376, 982–987 CrossRef CAS PubMed.
- C. Vogt and B. M. Weckhuysen, Nat. Rev. Chem., 2022, 6, 89–111 CrossRef PubMed.
- Addressing Rigor and Reproducibility in Thermal, Heterogeneous Catalysis, ed. N. M. Schweitzer, R. Gounder and R. M. Rioux, Zenodo, 2023 Search PubMed.
- G. Ertl, Surf. Sci., 1994, 299–300, 742–754 CrossRef CAS.
- S. V. Auras and L. B. F. Juurlink, Prog. Surf. Sci., 2021, 96, 100627 CrossRef CAS.
- J. Libuda and H. J. Freund, Surf. Sci. Rep., 2005, 57, 157–298 CrossRef CAS.
- G. A. Somorjai and J. Y. Park, J. Chem. Phys., 2008, 128, 182504 CrossRef PubMed.
- C. Reece and R. J. Madix, ACS Catal., 2021, 11, 3048–3066 CrossRef CAS.
- R. Imbihl, R. J. Behm and R. Schlögl, Phys. Chem. Chem. Phys., 2007, 9, 3459 RSC.
- R. Schloegl, R. C. Schoonmaker, M. Muhler and G. Ertl, Catal. Lett., 1988, 1, 237–241 CrossRef CAS.
- P. Stoltze and J. K. Nørskov, Phys. Rev. Lett., 1985, 55, 2502–2505 CrossRef CAS PubMed.
- H. P. Bonzel, Surf. Sci., 1977, 68, 236–258 CrossRef CAS.
- T. Bligaard, R. M. Bullock, C. T. Campbell, J. G. Chen, B. C. Gates, R. J. Gorte, C. W. Jones, W. D. Jones, J. R. Kitchin and S. L. Scott, ACS Catal., 2016, 6, 2590–2602 CrossRef CAS.
- C. J. Wrasman, A. T. Bell, B. D. Chandler, J. W. Harris, S. Kwon, M. R. Ball, S. H. Krishna, S. J. Khatib, P. Bollini, Y. Román-Leshkov, A. Bean Getsoian, R. S. Weber, J. A. Lercher, D. Liu, D. E. Resasco, J. S. Bates, J. N. Hall, E. A. Lebrón-Rodríguez, L. Paz Herrera, J. M. Notestein and J. A. Schaidle, J. Catal., 2024, 433, 115451 CrossRef CAS.
- J. T. Gleaves, G. S. Yablonskii, P. Phanawadee and Y. Schuurman, Appl. Catal., A, 1997, 160, 55–88 CrossRef CAS.
- T.-S. Kim, C. R. O'Connor and C. Reece, Nat. Commun., 2024, 15, 2074 CrossRef CAS PubMed.
- G. C. Bond, F. Garin and G. Maire, Appl. Catal., 1988, 41, 313–335 CrossRef CAS.
- R. Fushimi, in Springer Handbook of Advanced Catalyst Characterization, Springer, 2023, pp. 899–934 Search PubMed.
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- P. D. Srinivasan, B. S. Patil, H. Zhu and J. J. Bravo-Suárez, React. Chem. Eng., 2019, 4, 862–883 RSC.
- C. Ledesma, J. Yang, D. Chen and A. Holmen, ACS Catal., 2014, 4, 4527–4547 CrossRef CAS.
- M. Ahmadi, J. Timoshenko, F. Behafarid and B. Roldan Cuenya, J. Phys. Chem. C, 2019, 123, 10666–10676 CrossRef CAS PubMed.
- S. B. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, J. Catal., 2011, 281, 147–155 CrossRef CAS.
- H. C. Song, S. Oh, S. H. Kim, S. W. Lee, S. Y. Moon, H. Choi, S.-H. Kim, Y. Kim, J. Oh and J. Y. Park, Chem. Commun., 2019, 55, 9503–9506 RSC.
- L. Brandão, E. A. High, T.-S. Kim and C. Reece, Chem. Eng. J., 2023, 478, 147489 CrossRef.
- D. Constales, G. S. Yablonsky, G. B. Marin and J. T. Gleaves, Chem. Eng. Sci., 2001, 56, 133–149 CrossRef CAS.
- D. Constales, G. S. Yablonsky, G. B. Marin and J. T. Gleaves, Chem. Eng. Sci., 2004, 59, 3725–3736 CrossRef CAS.
- D. Constales, S. O. Shekhtman, G. S. Yablonsky, G. B. Marin and J. T. Gleaves, Chem. Eng. Sci., 2006, 61, 1878–1891 CrossRef CAS.
- D. Constales, G. S. Yablonsky, G. B. Marin and J. T. Gleaves, Chem. Eng. Sci., 2001, 56, 1913–1923 CrossRef CAS.
- F. C. Meunier, React. Chem. Eng., 2016, 1, 134–141 RSC.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 259–274 Search PubMed.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38, 448 CrossRef CAS PubMed.
- J. Sirita, S. Phanichphant and F. C. Meunier, Anal. Chem., 2007, 79, 3912–3918 CrossRef CAS PubMed.
- E. A. High, E. Lee and C. Reece, Rev. Sci. Instrum., 2023, 94, 054101 CrossRef CAS PubMed.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef CAS PubMed.
- A. Sangnier, E. Genty, M. Iachella, P. Sautet, P. Raybaud, M. Matrat, C. Dujardin and C. Chizallet, ACS Catal., 2021, 11, 13280–13293 CrossRef CAS.
- R. Lewis and R. Gomer, Surf. Sci., 1968, 12, 157–176 CrossRef CAS.
- P. R. Norton, J. A. Davies and T. E. Jackman, Surf. Sci. Lett., 1982, 122, L593–L600 CrossRef CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, K. Suzuki, K. Amemiya, K. Mase, B. Simon Mun and H. Kondoh, Phys. Chem. Chem. Phys., 2014, 16, 23564–23567 RSC.
- S. R. Longwitz, J. Schnadt, E. K. Vestergaard, R. T. Vang, I. Stensgaard, H. Brune and F. Besenbacher, J. Phys. Chem. B, 2004, 108, 14497–14502 CrossRef CAS.
- H. Steininger, S. Lehwald and H. Ibach, Surf. Sci., 1982, 123, 1–17 CrossRef CAS.
- N. Materer, U. Starke, A. Barbieri, R. Döll, K. Heinz, M. A. Van Hove and G. A. Somorjai, Surf. Sci., 1995, 325, 207–222 CrossRef CAS.
- G. Ertl, M. Neumann and K. M. Streit, Surf. Sci., 1977, 64, 393–410 CrossRef CAS.
- A. Winkler, X. Guo, H. R. Siddiqui, P. L. Hagans and J. T. Yates, Surf. Sci., 1988, 201, 419–443 CrossRef CAS.
- F. García-Martínez, E. Turco, F. Schiller and J. E. Ortega, ACS Catal., 2024, 6319–6327 CrossRef PubMed.
- S. Penner, P. Bera, S. Pedersen, L. T. Ngo, J. J. W. Harris and C. T. Campbell, J. Phys. Chem. B, 2006, 110, 24577–24584 CrossRef CAS PubMed.
- M. A. Van Spronsen, J. W. M. Frenken and I. M. N. Groot, Chem. Soc. Rev., 2017, 46, 4347–4374 RSC.
- M. S. Chen, Y. Cai, Z. Yan, K. K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS.
- E. M. C. Alayon, J. Singh, M. Nachtegaal, M. Harfouche and J. A. Van Bokhoven, J. Catal., 2009, 263, 228–238 CrossRef CAS.
- C. Zhou, H. T. Ngan, J. S. Lim, Z. Darbari, A. Lewandowski, D. J. Stacchiola, B. Kozinsky, P. Sautet and J. A. Boscoboinik, J. Am. Chem. Soc., 2022, 144, 15132–15142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03106g |
| This journal is © The Royal Society of Chemistry 2024 |