
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbnormal copper coordination obtained by a TiO2 overlayer as the key to enhance photocatalytic hydrogen generation†
Vien-Duong
Quach
 a,
Maria Chiara
Spadaro
bg,
Diana
Dragoe
d,
Marc
Botifoll
b,
Hervé
Vezin
f,
Christophe
Colbeau-Justin
a,
Franck
Dumeignil
e,
Jordi
Arbiol
bc,
Robert
Wojcieszak
e and
Mohamed Nawfal
Ghazzal
*a
a,
Maria Chiara
Spadaro
bg,
Diana
Dragoe
d,
Marc
Botifoll
b,
Hervé
Vezin
f,
Christophe
Colbeau-Justin
a,
Franck
Dumeignil
e,
Jordi
Arbiol
bc,
Robert
Wojcieszak
e and
Mohamed Nawfal
Ghazzal
*a
aUniversité Paris-Saclay, CNRS UMR 8000, Institut de Chimie Physique, F-91405 Orsay, France. E-mail: mohamed-nawfal.ghazzal@universite-paris-saclay.fr
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC, BIST, Campus UAB, Bellaterra, ES-08193 Barcelona, Catalonia, Spain
cICREA, Pg. Lluís Companys 23, ES-08010 Barcelona, Catalonia, Spain
dUniversité Paris-Saclay, CNRS UMR 8182, Institut de Chimie Moléculaire et des Matériaux d'Orsay, F-91405 Orsay, France
eUCCS – Unité de Catalyse et Chimie du Solide, Université de Lille, CNRS UMR 8181, F-59000 Lille, France
fLASIRE – Laboratoire Avancé de Spectroscopie pour les Interactions la Réactivité et l'environnement, Université de Lille, CNRS UMR 8516, F-59000 Lille, France
gDepartment of Physics and Astronomy “Ettore Majorana”, CNR-IMM, University of Catania, via S. Sofia 64, Catania 95123, Italy
First published on 10th June 2024
Abstract
Strong metal–support interaction (SMSI) is a pivotal strategy in thermal catalysis; however, its application in photocatalysis leaves ample area for further development. A method inducing SMSI between earth-abundant metals, such as copper and TiO2, at room temperature, and thus hindering the agglomeration of copper species remains rarely reported. In this work, we achieved SMSI construction of TiO2 overlayers on Cu nanoparticles via a straightforward soft-chemistry method. SMSI coverage is stable even after high-temperature treatment in the air (500 °C), as demonstrated by chemical mapping and surface analysis. The method is more accurate than thermal reduction since it produces a metastable, highly active anatase phase. Interestingly, the TiO2 overlayer induces the formation of four-coordinated copper(II) species surrounded by oxygen atoms, resulting in coexisting CuO2 planes, which were monitored through high-resolution transmission electron microscopy and electron paramagnetic resonance spectroscopy. The stronger interfacial interaction by forming Ti–O–Cu bonding promotes charge carrier separation, producing twice as much H2 than the low interfacial interaction within a conventional photoactive system wherein copper was decorated onto TiO2. Our approach offers a rational design for SMSI materials in photocatalysis, which is extendable to other catalytic reactions.
Introduction
Solar fuel production presents a promising method for converting sunlight into a renewable, sustainable, and environmentally friendly energy source. Hydrogen, a clean fuel generated through solar water splitting, has emerged as a promising solution for climate challenges. H2 can be produced through three distinct approaches: photocatalytic (PC), photoelectrochemical (PEC), and photovoltaic–electrolysis (PV–EC) routes.1–3 Although PEC and PV–EC are recognized as advanced solar-to-hydrogen chemical fuel technologies, their scalability requires significant investment, leading to high-cost implications. In contrast, the photocatalytic process has demonstrated low-cost investment and a promising conversion of solar energy to green hydrogen at the 100 m2 scale.4 Since the discovery of the light-induced water splitting reaction over a titanium dioxide (TiO2) electrode in the 1970s,5 efforts have been devoted to enhancing its efficiency. While the development of novel nanostructures,6–8 construction of heterojunctions,9 induction of oxygen vacancies,10 or coupling TiO2 with a promising 2D graphdiyne carbon allotrope11 have shown effective improvement in photocatalysis, the most widely adopted strategy involves surface coupling with metallic nanoparticles. Noble metals, such as Pt, Pd, and Au,12 generally exhibit the highest photoactivity due to their intrinsic properties. Recent developments have focused on downsizing these cocatalysts from nanoparticles to single atoms, aiming to modify their electronic properties, although this approach has encountered certain bottlenecks.13,14 From this perspective, the design and control of the metal–support interface have emerged as novel factors to enhance the activity in catalytic reactions, leading to the development of the so-called strong metal–support interaction (SMSI) concept.15,16 While SMSI construction is a pivotal strategy in thermal catalysis, its application in photocatalysis leaves ample room for further studies.17,18 The exploration of the aforementioned MSI phenomena should be extended as it can lead to a rearrangement of electrons within both materials,19 favorable separation of photogenerated charge carriers, and modification of the adsorption of H2O or CO2.6,20 Various methods have been proposed to construct a strong metal–support interface, especially under mild conditions, including the mechano-chemical, soft-chemical, and photoinduced reduction of the support,21,22 instead of the conventional reductive high temperature conditions (500–700 °C). However, most reported studies have demonstrated that the SMSI concept works for VIIIB group metals and more recently for IB group metal Au,23,24 with a few studies dedicated to earth-abundant copper metal nanoparticles.25–27In this study, we regulated the interaction between TiO2 and CuO2 for photocatalytic hydrogen generation. Copper and its oxidized forms in general have become one of the most promising alternatives to noble metals for proton photoreduction. Copper exists in various oxidation states, including Cu0, CuI, CuII, and CuIII, and can exist not only in an exclusive state but also in mixed states involving both zero-valent metals and oxides.28 Cu0, Cu(I)2O, and Cu(II)O are typically the most common copper species reported in TiO2-based photocatalysis.29,30 Although less explored in photocatalytic reactions, especially with TiO2-based photocatalysts, CuO2 has been reported for fast charge transport, showing great potential in cuprate superconductor engineering, solid-state physics, and cancer treatment.31–33 Herein, we identify CuO2 as a highly effective candidate for photocatalytic hydrogen generation. Additionally, tuning the interfacial interaction by constructing a metal oxide–support interface facilitates the transport and prolongs the lifetime of charge carriers that play a pivotal role in photon-to-hydrogen conversion.
Results and discussion
Microstructure and morphology characterization
The interaction between titania and cuprate oxide was regulated using a soft-chemistry method.6 We impregnated the surface of silica spheres (used as inert support) either with CuCl2 or titania precursor to construct nanostructures: SiO2@TiO2@Cu (TiO2/Cu) and SiO2@Cu@TiO2 (MSI-Cu/TiO2). The details of the synthesis and the aspect of the samples are shown in Fig. S1.† The microstructure and morphology of the as-synthetized photocatalysts were observed using transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1A, S2A and S3A†). The images show the spherical shape of nanostructures scaffolded using an SiO2 core. The diameter of the spheres was estimated to be about 200 nm. The SiO2 microspheres behave as convex nano-lenses, which optimize electromagnetic field enhancement or even lead to an illumination focus point at the front surface of the nanostructure, resulting in optimal light harvesting.34 High-resolution transmission electron microscopy (HRTEM) shows a TiO2 anatase phase-structured shell with a thickness of less than 10 nm for SiO2@TiO2 (Fig. S2B†). The thin TiO2 overlayer was observed to cover copper nanoparticles with a size of 7–10 nm for MSI-Cu/TiO2 (Fig. 1B). The crystalline structure of copper oxide species was assigned to orthorhombic CuO2 and the indexed Fast Fourier Transform (FFT) patterns of the metallic species clearly showed the presence of the {111} and {110} family planes, noting that the observed structure was oriented along its [110] zone axis. For TiO2/Cu, we could observe a homogeneous dispersion of copper species and TiO2, forming a continuous shell around the silica spheres. The HRTEM images in Fig. S3B and C† and their corresponding indexed FFT showed that the studied nanoparticle crystallized in the CuO2 orthorhombic structure with a preferential orientation along the [001] zone axis. From the results, we suspect that the cuprate CuO2 does not exist separately in such a crystalline form. In fact, its coexistence as superconducting CuO2 planes is evidenced by the abnormal coordination of the copper electronic state.35 This will be clarified by EPR and XPS later. | ||
| Fig. 1 Microstructure and morphology of the photocatalyst. (A) TEM image of MSI-Cu/TiO2. (B) High magnification HRTEM image emphasizing the encapsulated cuprate nanoparticle and its indexed power spectrum. (C) EELS-STEM chemical mapping of MSI-Cu/TiO2 and chemical composition: Ti L edge at 456 eV (scarlet red), O K edge at 532 eV (kelly green), Cu L edge at 931 eV (lemon yellow) and Si K edge at 1839 eV (ultramarine blue). (D) Magnified image of EELS-STEM mapping. (E) Extrapolated EELS spectra at different regions on the MSI-Cu/TiO2 surface. (F) Powder XRD patterns noted with Miller indices of SiO2, SiO2@TiO2, MSI-Cu/TiO2, and TiO2/Cu. (G) LEIS spectra measured at an iso-dose of 1 × 1015 He+ ions per cm2 for MSI-Cu/TiO2. | ||
The crystalline structure was further analyzed using X-ray powder diffraction (XRD), as shown in Fig. 1F. Diffractograms exhibited broad peaks at 22°, which correspond to an amorphous SiO2 core. In addition, characteristic peaks at 25.3° were recorded in the diffraction patterns of samples that identify the lattice plane (101) of the TiO2 anatase phase. Other characteristic peaks of TiO2 anatase showed up in the patterns, pointing out the presence of their corresponding lattice planes, including (004), (020), (200), (105), and (204). The patterns displayed no trace of cuprate species due to the small size of the nanoparticles and their low quantity as well.
Scanning transmission electron microscopy coupled with electron energy loss spectroscopy (STEM-EELS) provided the chemical mapping of different elements (Ti, O, Si, and Cu), hence visualizing their spatial distribution within the nanostructures. Fig. 1C, S2D, S3E and F† show that the core of the sphere, mainly consisting of Si and O, was surrounded by a homogeneous TiO2 overlayer. The EELS spectrum obtained within the marked regions in Fig. 1D evidence the presence of both Ti L edges (456 eV) and O K edges (532 eV) EELS signals at the shell (regions 2 and 3) and the core surface (region 4). Remarkably, the Ti signal appears in region 3, where the Cu EELS signal was also observed (Fig. 1E), demonstrating the coverage of the Cu by a TiO2 overlayer. To further investigate the TiO2 overlayer and confirm the nature of the CuO2 coverage, we performed low-energy ion scattering (LEIS) depth profiling spectroscopy. LEIS reveals information about the composition in the depth of a surface (1–5 nm) by bombarding a sample with noble gas ions of well-defined and low energy at 4.1 × 1015 He+ ions per cm2 (Fig. 1G). As the surface is progressively etched by the ion beam, gradients of concentrations from the uppermost layers to the layers below can be inspected. The first recorded signals, at a very low ion dose, were moderate, reflecting the removal of contamination (carbon, hydrogen, etc.) by the ion beam; these species were discarded for analysis. Given the lack of standardization for measurements on nanoparticles distributed over porous support and the initial presence of contamination layers, the translation of the results to a probed depth expressed in nanometre would not be attempted. The presence of Cu atoms in the outer shells of the MSI-Cu/TiO2 sample was evidenced. However, the signal of Ti was dominant compared to the ones of Cu and Si, indicating the low quantity of Cu and Si atoms on the surface. In general, LEIS measurements directly confirm the existence of a Cu enrichment in the outer shells of the photocatalyst. It demonstrates that the TiO2 overlayer partially covers CuO2 nanoparticles or that the thin layer is discontinuous.
Defective surface structure and electronic states
We employed electron paramagnetic resonance spectroscopy (EPR) to analyze the nature of Cu species and hence get information about the oxidation state and coordination of EPR active copper sites (Fig. 2A); the EPR spectrum of SiO2@TiO2 as a reference is illustrated in Fig. S4A.† In detail, Cu2+ (d9) species contain an odd number of electrons assigned to a spin s = 1/2, resulting in an EPR transition. The two principal isotopes of copper (63Cu and 65Cu) have similar nuclear spins of 3/2, leading to the splitting of the Zeeman line into four lines (ml = 3/2, 1/2, (−1)/2, (−3)/2).36–38 However, only the splitting in the z-direction is large enough to be visualized. The Jahn–Teller distortion is most encountered in octahedral complexes, especially six-coordinate copper(II) species surrounded by oxygen atoms.39Fig. 2A compared the EPR spectra of MSI-Cu/TiO2 and TiO2/Cu. In particular, the MSI-Cu/TiO2 photocatalyst exhibited an axial EPR spectrum involving typical parallel and perpendicular edges, revealing two different Cu sites, in agreement with previous studies.40,41 The hyperfine splitting A// of 122 G at a g//-factor of 2.402 can be attributed to the charge of donor atoms coordinating to Cu. The splitting into four peaks unveils the presence of Cu2+ sites coordinated with four oxygen atoms, which attest the existence of CuO2, as revealed by HRTEM. The partial coverage of TiO2 onto cuprate nanoparticles could create heterobimetallic sites via metal–oxygen–metal support bonding, particularly Ti–O–Cu, leading to the extraordinarily abnormal coordination of Cu2+ (crystal structure42 in Fig. 2B). Copper-deposited on TiO2 observed an isotropic spectrum. The outermost copper species on TiO2/Cu are generally bound to polar water molecules, which are most likely to coordinate to Cu2+. As a result, the EPR spectrum of these hydrated Cu species typically displayed the isotropic pattern, which is similar to the EPR spectra of aqueous copper complexes such as [Cu(H2O)6]2+ or [Cu(H2O)5(OH)]+.43,44 Besides, the g-factor of 2.165 estimated from the EPR spectrum of TiO2/Cu is fully consistent with those at about 2.16–2.17 of copper on hydrated zeolite.45,46 In the case of TiO2/Cu, no such bond between cuprate and titania was detected. | ||
| Fig. 2 Characterization of surface electronic properties and catalytic active sites. (A) EPR spectra of MSI-Cu/TiO2 and TiO2/Cu. (B) Crystal structure of cuprate CuO2. XPS spectra: (C) Cu 2p, (D) O 1s, and (E) Ti 2p. | ||
Surface analysis of the photocatalysts was further carried out by X-ray photoelectron spectroscopy (XPS) (Fig. S4B†), recording signals of core levels Cu 2p, O 1s, and Ti 2p. In Fig. 2C, the high-resolution XPS spectra of Cu 2p show the characteristic peaks assigned to Cu 2p3/2 and Cu 2p1/2 transitions with satellite peaks. This indicates the existence of the Cu(II) state within the MSI-Cu/TiO2 sample.47,48 The Cu 2p3/2 can be deconvoluted into three peaks centered at 932.05 eV, 933 eV, and 934.01 eV. The peak at 932.05 eV corresponds to Cu(I) oxide, while these peaks at 933 eV and 934.01 eV are indicative of Cu(II) oxide and Cu(II) hydroxide, respectively.49 The corresponding shake-up satellite peak at 940.5 eV gives rise to Cu(II) oxide, while the others at 942.34 eV and 944.5 eV evidence Cu(II) hydroxide. These Cu(II) 2p transitions shifted toward lower binding energy, and the satellite was much weaker than previous reports.47,49 In addition, the XPS Cu 2p spectrum of the TiO2/Cu sample surprisingly revealed a single peak of Cu 2p3/2 without any shake-up satellite, commonly assigned to the Cu(I) state. The oxidation state change of Cu in TiO2/Cu and the shift towards lower binding energy of Cu(II) in MSI-Cu/TiO2 can be attributed to the impact of electron bombardment in a high vacuum environment.50,51 In fact, the electron bombardment can reduce the copper species dispersed on the photocatalyst surface from Cu(II) to Cu(I). This effect becomes evident in the case of TiO2/Cu, where the TiO2 overlayer did not protect the Cu species and were directly exposed to the electron beam. In general, even though copper species in both MSI and conventional nanostructures were found to exist in the Cu(II) state, the MSI-encapsulated one showed an apparent discrepancy. The combinatory features of Cu(OH)2 and CuO reflect an abnormal coordination of Cu(II) species in MSI-Cu/TiO2, resulting in the coexistence of CuO2 planes, in agreement with the results obtained from HRTEM and EPR.
In the high-resolution XPS spectra of O 1s (Fig. 2D and S4C†), the peaks of lattice oxygen show a shift from ∼530.3 eV for TiO2/Cu and SiO2@TiO2 to 530.07 eV for MSI-Cu/TiO2. In fact, Ti–O–Cu formation creates oxygen vacancies in the interface that negatively charge the surface, hence causing the shift. Moreover, Cu atoms link to electronegative O atoms in Ti–O–Ti to construct Ti–O–Cu. Consequently, the remaining Ti atoms (Ti4+) are reduced to Ti3+, which can be noticed by the intensity of the Ti 2p3/2 shoulder peak at about 457.6 eV (Fig. S4D†).52,53 The high intensity was detected for MSI-Cu/TiO2 (Fig. S4D†). This demonstrates the generation of Ti3+ due to the SMSI construction.
We investigated chemical bonds within our photocatalysts through Fourier-transform infrared spectra, and the results are shown in Fig. S4E.† The broad bands from 3247 cm−1 to 3400 cm−1 are attributed to the stretching vibration of hydroxyl groups in silanol (Si–OH) moiety and adsorbed water molecules.54 The deformation vibration mode of O–H bonds is assigned to the band at 1626 cm−1.54 The bands at about 1109 cm−1 and 806 cm−1 represent the symmetric and asymmetric stretching vibration mode of Si–O–Si bonds, while the band at 953 cm−1 can be due to the stretching vibration of Si–O–Ti bonds. FTIR spectroscopy is not sensitive enough to discriminate between interfacial Ti–O–Cu and Ti–O–Si.
Optical properties and photoelectrochemical photon-to-current conversion
The optical properties were evaluated using UV-visible diffuse reflectance spectroscopy (Fig. S5A and B†). The reference (SiO2@TiO2) exhibited strong absorption at a wavelength of below 400 nm. The absorption bands for MSI-Cu/TiO2 and TiO2/Cu shift toward the visible region. The Cu-mediated photocatalysts absorbed the wavelength from 400 nm to 500 nm, wherein the absorption of MSI-Cu/TiO2 showed a larger extent. This is due to the charge transfer from O 2p of TiO2 to the d-orbitals of Cu.55 The TiO2/Cu samples showed a weak absorption band in the range of 500–600 nm and slightly shifted from 600 nm toward IR, whereas we observed a reverse pattern for MSI-Cu/TiO2. In fact, the band from 500 to 600 nm could be assigned to the interband absorption of Cu2O, while the d–d transition of a single electron among the 3d9 states of Cu2+ species is attributed to the broadband from 600 nm toward IR.51,56 This clearly evidences that Cu2+ species exist in MSI-Cu/TiO2, while a mixed state with a small amount of Cu1+ might be found in TiO2/Cu. We used the Tauc method to estimate the bandgap energy of 3.19 eV, 3.23 eV, and 3.24 eV for MSI-Cu/TiO2, TiO2/Cu, and SiO2@TiO2, respectively.57 The 3.24 eV bandgap energy of SiO2@TiO2 corresponds to the anatase phase of TiO2, which is in agreement with the HRTEM results and previous studies.58,59 A narrower bandgap of the remaining samples can be explained by bandgap alignment between TiO2 and CuOx semiconductor composites, which generates the so-called semiconductor heterojunction. In Fig. S6,† the Mott–Schottky plots of SiO2@TiO2, TiO2/Cu, and MSI-Cu/TiO2 with positive slope intrinsically demonstrated n-type TiO2. Because the Cu content is very low and highly dispersed on the surface of nanostructures, we could not witness any effect of p-type CuOx in these Mott–Schottky plots. We then estimated the flat-band potential, which is the Fermi level as no band bending occurs for our photocatalyst samples. Under UV-visible irradiation, electrons are excited from the valence bands to the conduction bands within both the semiconductors. These photogenerated electrons are injected among TiO2 and cuprate clusters through photo-induced interfacial charge transfer (IFCT). This resulted in the rising of the Fermi level, from −0.683 V vs. RHE for pure TiO2 to −0.725 V vs. RHE and −0.875 V vs. RHE for TiO2/Cu and MSI-Cu/TiO2 (Fig. S6†). Obviously, the metal oxide–support interaction could favor interfacial charge transfer between cuprate and TiO2, evidenced by the largest anodic shift of the flat-band potential. The shift also demonstrates the increasing extent of interfacial band bending, which optimizes charge carrier separation.60 To examine the effect of encapsulation of CuO2 nanoparticles on the photocatalytic hydrogen evolution reaction (HER), we carried out the electrochemical performance of the HER and OER in K2SO4 electrolyte buffered at pH 7 in the presence of argon bubbles. Fig. 3A shows that the current density is markedly improved by the encapsulation of Cu and its abnormal coordination. More interestingly, the encapsulation of Cu increases the current density not only in HER but also in oxygen evolution reaction (OER), where MSI-Cu/TiO2 could be utilized as a photoanode in photoelectrochemical water splitting cell (Fig. 3B). The results demonstrate that the intrinsic HER activity can be significantly improved by covering copper by a TiO2 overlayer. | ||
| Fig. 3 Photoelectrochemical and charge carrier dynamics properties of SiO2@TiO2, MSI-Cu/TiO2, and TiO2/Cu: (A and B) electrochemical activity tests on different photocatalysts in phosphate-buffered K2SO4 0.5 M electrolyte (pH 7) for the (A) HER and (B) OER through linear sweep voltammetry at a scan rate of 0.05 V s−1. (C) Incident photon-to-current efficiency (IPCE) spectra in the ultraviolet range at a fixed bias voltage of 0.6 V vs. Ag/AgCl 3 M (∼1.23 V vs. RHE) in Na2SO3 0.1 M buffered at pH = 7 under intermittent illumination triggered from a AM 1.5G solar simulator. (D) Time-resolved microwave conductivity (TRMC) signals of the samples triggered via laser irradiation (Iex = 6.623 mJ cm−2 at λ = 420 nm). (E) Electrochemical impedance spectroscopy (EIS) Nyquist plots of the front-contact photoelectrodes recorded in the dark and under AM 1.5G illumination in K2SO4 0.5 M in the range from 10−2 Hz to 100k Hz. (F) Normalized open-circuit potential (OCP) decay curves after switching off the AM 1.5G solar irradiation. | ||
Amperometric photocurrent measurement provided valuable insights into charge carrier mobilities and their trapping within the photocatalysts by examining photocurrent responses (current rise, current decay, etc.). After the light was switched on, photogenerated electron–hole pairs were separated rapidly, charging space charge capacitance. Typically, the photocurrent instantaneously rises to a peak value before decaying to a steady state due to electron–hole recombination, accumulation of electrons in the bulk or holes on the surface, and the trapping of electrons or holes at the surface defects.61 We employed sulfite (SO32−) in electrolyte solution to optimize light capture and charge transport. In our case, holding accumulation can be ruled out as a main recombination mechanism thanks to adding sulfite ions as hole scavengers. The higher photocurrent response of TiO2/Cu reveals the superior separation of photogenerated charge carriers to those in MSI-Cu/TiO2 and SiO2@TiO2 (Fig. S5C†). While SiO2@TiO2 outputted a relatively steady-state photocurrent response, MSI-Cu/TiO2 and TiO2/Cu exhibited gradually rising photocurrent curves with no spike. This could be explained by larger depletion layers near the surface or interface of the semiconductor-liquid, leading to effective charge carrier separation.
Photocurrent action spectra, including IPCE and APCE under UV and visible illumination (Fig. 3C and S5D–F†), further examine the photocurrent as a function of the wavelength at an applied bias of 1.23 V vs. RHE. About 1% of the incident photon at λ = 320 nm was absorbed by the SiO2@TiO2 photoelectrode to contribute to the photocurrent on the outer circuit. The other 99% was lost due to charge carrier recombination within the material. The external quantum efficiency (IPCE, Fig. 3C) was much greater at 2.5% for MSI-Cu/TiO2 and nearly 7% for TiO2/Cu. When considering only charge carrier recombination as a main effect on photocurrent response, we estimated the internal quantum efficiency (APCE, Fig. S5E†) at over 5% for MSI-Cu/TiO2 and 9% for TiO2/Cu. Cu-modified photocatalysts converted visible photons to current despite a negligible extent, while SiO2@TiO2 was unactive in the visible range (Fig. S5D and F†). These experimental outputs were consistent with the one of UV-vis DRS and amperometric photocurrent response measurements.
We investigated charge carrier dynamics at microsecond and nanosecond timescales by time-resolved microwave conductivity (TRMC) and time-resolved photoluminescence spectroscopy (TRPL). Fig. S7A† displays the TRMC signal of three photocatalysts triggered by an ultraviolet laser at 360 nm, where TiO2 is activated only. In detail, the signal of SiO2@TiO2 observed the highest maximum intensity and a sharp decay, while MSI-Cu/TiO2 outputted two times smaller maximum intensity but a long-lasting signal. In fact, electrons, the primary mobile charge carriers in TMRC,61 were generated in bulk and then transferred to the surface of TiO2. The sharp TRMC decay of SiO2@TiO2 reflects the disappearance of electrons, which could be attributed to electron–hole recombination or surface trapping. Coupling TiO2 with cuprate nanoparticles induces surface defects, including oxygen vacancies and Ti3+, which are the trapping sites.61 Although the trapping hinders electron transport within the crystalline structure, it precludes electron–hole recombination, leading to a longer charge carrier lifetime. Surprisingly, we recorded the TMRC signal of TiO2/Cu at a much lower maximum intensity. These results suggested that less surface defects are created in this system. We recorded the TRMC signal under visible laser excitation (λex = 420 nm), activating only cuprate oxide. As a result, in Fig. 3D, the TRMC signal of SiO2@TiO2 displayed flat fluctuation at 0 mV, while those of the composites showed higher charge density and an unexpectedly longer lifetime. The visible laser generates charge carriers, mainly electrons, within the CuO2 band structure. These electrons are injected into the conduction band of TiO2, while holes are accumulated into the valence band of CuO2. This inhibits charge carrier recombination and thus prolongs charge carrier lifetime, as evidenced by non-decayed TRMC signals. TRPL spectroscopy further elucidated charge carrier dynamics at nanosecond timescale by inspecting the photons released from the relaxation of photogenerated charges. Time-resolved photoluminescence (TRPL) in Fig. S7B† exhibited the sharpest decay with respect to MSI-Cu/TiO2, followed in order by the one of TiO2/Cu and SiO2@TiO2. The high probability of electron–hole recombination within SiO2@TiO2 would be responsible for its slighter TRPL decay. In the case of TiO2/Cu and MSI-Cu/TiO2, non-irradiated relaxation of electrons, such as electron trapping and transfer, are more favorable, leading to sharper TRPL decay. These experimental results are consistent with the TRMC analysis.
The charge transport capability in the CuO2/TiO2 interface was studied by electrochemical impedance spectroscopy (EIS). The data was fitted to an equivalent circuit model (inset Fig. 3E) involving two parallel branches (Rct1 and CPE1) derived from the interfacial contact between FTO and semiconductive materials. Rct2 and CPE2 represent charge transfer resistance and double-layer capacitance, respectively, at the electrode and electrolyte interface, and Rs stands for ohmic resistance. To address the electrochemical phenomena on a non-uniform surface, it is essential to incorporate constant phase elements (CPE) with an ideality factor (n) to characterize double-layer capacitance and/or coating capacitance rather than relying on ideal capacitors. Nyquist plots were obtained from electrochemical impedance spectroscopy (EIS) measurements in both dark and under UV-visible irradiation, employing a K2SO4 0.5 M electrolyte solution. The first compact semicircles (inset Fig. 3E) might be due to the interfacial contact between FTO glass electrode and our photocatalysts. The second larger semi-circle typically translates the charge transfer impedance between the photoelectrode and the electrolyte. SiO2@TiO2 registered the largest values of charge transfer resistance, followed by TiO2/Cu, and the smallest values belong to MSI-Cu/TiO2 (Table 1). The result reveals that charge transport is more favorable for CuO2/TiO2 composites. Moreover, the low value of the charge transfer resistance on MSI-Cu/TiO2 demonstrates that strong metal oxide–support interaction efficiently contributes to the charge carriers' transfer and separation. Upon illumination condition, the composites became more conductive, evidenced by the smaller diameter of the Nyquist semi-circles. This can be ascribed to the abnormal Cu coordination that facilitates charge transfer.
| Samples | R ct2 (Ω) | E fb (VRHE) | k r (s−1) | |
|---|---|---|---|---|
| a R s, Rint, and Rct were estimated via EIS plot fitting. b E fb values were determined from Fig. S6. c k r was obtained by fitting OCP curves in Fig. S7C. | ||||
| SiO2@TiO2 | Light | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 722 722 |
−0.683 | 1 × 10−6 |
| Dark | 14510 |
|||
| TiO2/Cu | Light | 8465.8 | −0.725 | 2.5 × 10−7 |
| Dark | 13022 |
|||
| MSI-Cu/TiO2 | Light | 6826.6 | −0.875 | 1.9 × 10−8 |
| Dark | 11175 |
|||
Open-circuit potential (OCP) decay analysis offers information about surface recombination between trapped electrons and reaction intermediates. In fact, the charge carrier recombination occurs at the nanosecond or microsecond timescale, while OCP decay takes several minutes. Open-circuit voltage (VOC) is the difference in the Fermi level between a photoelectrode and a counter electrode. Upon illumination, photogenerated electrons fill up the Fermi region and shift the Fermi level toward anodic potentials. Following this, the recombination of photogenerated charge carriers occurs and maintains equilibrium with their accumulation at the interface, resulting in a steady state of VOC. When switching the light off, VOC decays upon recombination process due to a lack of charge carrier generation. The normalized transient decay profiles of OCP are illustrated in Fig. 3F. The results demonstrate that MSI-Cu/TiO2 exhibited the most gradual OCP decay, indicating low surface recombination of charge. An average recombination rate constant kr can be estimated by fitting the OCP decay profile with the pseudo-first-order kinetic model equation:62

Photocatalytic hydrogen production
The photocatalytic activity of photocatalysts was evaluated following the accumulated hydrogen produced under UV-visible light illumination from methanol/water solution (1:3 v/v). We employed X-ray fluorescence spectrophotometry (XRF) to quantify the mass percentage of either elements (Si, Ti, O, Cu) or oxides (SiO2, TiO2, CuOx) in our powder samples. Notably, the insulating SiO2 occupies approximately 85 wt% while the remaining active phase (TiO2 and CuOx), which is involved in photocatalytic reactions, accounts for 15% wt. As a result, we refined the photocatalytic H2 evolution rate based on the mass percentage of the active phase. As shown in Fig. 4A, MSI-Cu/TiO2 exhibited the highest yield of photocatalytic hydrogen production, reaching nearly 60 mmol g−1 after 5 hours of UV-vis irradiation. The amount of H2 generated was approximately twice lower (35 mmol g−1) for TiO2/Cu, and only a negligible amount of H2 was obtained in the sample without CuO2. Undoubtedly, the synergistic combination of TiO2 and CuO2 significantly enhanced the photocatalytic activity for the H2 generation reaction. This is in line with the enhancement of charge carrier separation and lifetime, as corroborated by the PEC results, and despite TiO2/Cu demonstrating superior absorbed photon-to-current efficiency, evidenced by IPCE and APCE, MSI-Cu/TiO2 outperformed others in terms of the overall photocatalytic efficiency. This superiority is closely linked to the charge carrier dynamics within the nanostructures, which are pivotal in improving the photocatalytic activity. The observed color change during the photocatalytic reaction of MSI-Cu/TiO2 from greenish to dark red testifies to a change in the oxidation state of Cu (Fig. S8B†). Indeed, some photogenerated electrons could be promoted from TiO2 to unoccupied d9 orbitals of CuII, resulting in a fulfilled d10 state of CuI. More interestingly, the MSI-Cu/TiO2 turned back to its original greenish color, without air exposure when illumination was cut off, reflecting the release of the photoelectrons from cuprate to TiO2. However, TiO2/Cu displayed no color change, demonstrating a less effective change in the oxidation state of Cu. The partial coverage of TiO2 onto CuO2, showing strong metal oxide–support interaction, generates a significant number of the active sites for photocatalytic reactions, thereby enhancing the overall reaction yield. A 20 hours photocatalytic cycling experiment was performed to evaluate the stability of the MSI encapsulation layer and TiO2/Cu sample. As shown in Fig. S8A,† a decrease in the amount of H2 produced is observed for both the samples. H2 generation decreased by 15% for both systems in the second cycle. The decrease is mainly due to the loss of part of the sample during the recovery by centrifugation. Indeed, after the second cycle, a decrease in H2 generation is still observed for SMSI (∼10%), while TiO2/Cu shows relatively good stability. After the third cycle, the SMSI system shows relatively good stability and remains more efficient in H2 generation than TiO2/Cu (Fig. S8A†).
 | ||
| Fig. 4 Photocatalytic activity of SiO2@TiO2, MSI-Cu/TiO2, and TiO2/Cu: (A) graphical illustration of photocatalytic reactions occurring on TiO2/Cu and MSI-Cu/TiO2 photocatalysts. (B) Hydrogen production investigated using gas chromatography. (C) Formaldehyde and acetaldehyde production investigated via high performance liquid chromatography. Reaction conditions: 10 mg/10 mL photocatalyst, 25 °C, H2O:CH3OH = 3:1 v/v, UV-visible irradiation mercury lamp 150 W. | ||
The photoactivation mechanism and H2 generation over the MSI-Cu/TiO2 sample is depicted in Fig. 5. Under light excitation, electrons and holes are generated in the conduction and valence bands of TiO2, respectively. Photogenerated electrons are transferred to copper, resulting in the reduction of Cu(II) to Cu(I), as evidenced by the change of the sample's color from green to rust color (dark red) (Fig. S8B†). The protonation at the TiO2 overlayer active sites consumes electrons and induces a building field that induces the diffusion of scavenged electrons at the d-orbitals of Cu back to the TiO2 conduction band. When UV-visible illumination was cut off, the electrons at the Cu sites were released back to ensure charge balance within the band structure; this was observed by the reversion to the original sample color as well as the oxidation of Cu(I) to Cu(II). In the case of holes, they are scavenged by the sacrificial electron donor molecules (methanol).
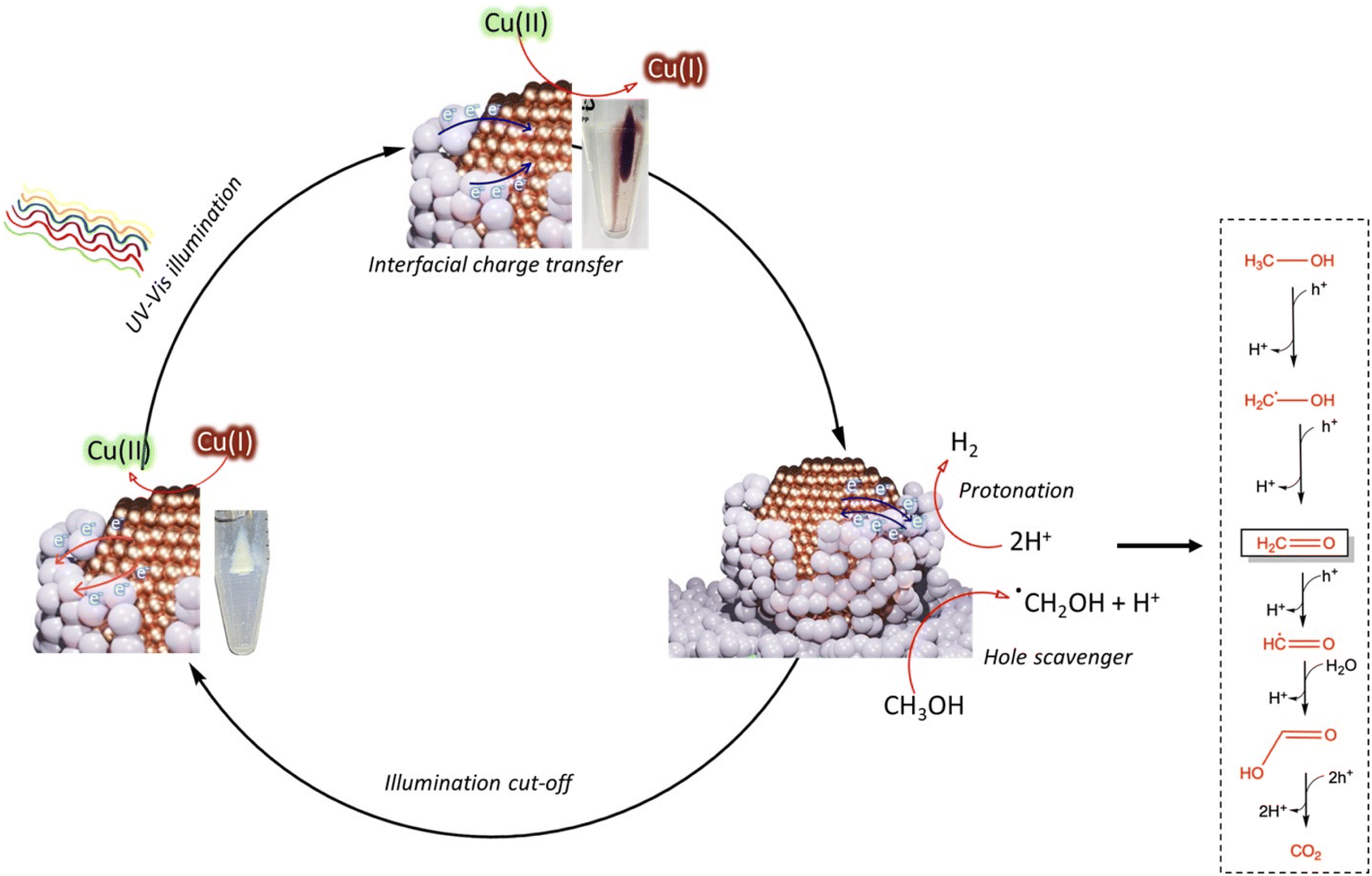 | ||
| Fig. 5 A schematic illustration of the photocatalytic hydrogen production mechanism with electron transfer pathways and the change in the valence state of copper on MSI-Cu/TiO2. | ||
To explore the photocatalytic H2 generation pathway, we extended our investigation to trace intermediates produced during illumination in the liquid phase. Following five hours of illumination, the reaction solutions were extracted and injected into a high-performance liquid chromatography (HPLC)–UV-visible spectrophotometry detector. Formaldehyde (HCHO) and acetaldehyde (CH3CHO) were detected in the liquid phase; HCHO was identified as a primary product from photocatalytic methanol reforming reactions. The production yields of these intermediates are visualized in Fig. 4C, where SiO2@TiO2 exhibited the smallest production yield of HCHO compared to the cuprate-mediated samples. This result points out the low kinetic conversion of CH3OH to HCHO. TiO2/Cu exhibited a slightly greater extent of the HCHO/CH3CHO ratio than MSI-Cu/TiO2. The dissociative adsorption of MeOH, right after the e−/h+ generation, leads to the formation of HCHO, followed by the release of H+. In particular, CH3OH molecules convert to methyl hydroxyl radicals (˙CH2OH) through C–H activation. Then, radicals react with oxidative holes and produce formaldehyde (HCHO). Hydrogen ions generated during reactive transformation would capture photogenerated electrons and combine to release H2 molecules. MSI-Cu/TiO2 generated the higher production of CH3CHO than TiO2/Cu, probably due to the more likely accessibility of Ovac and Ti3+ on the surface in the TiO2 surface facilitated by the abnormal Cu coordination that hastened photocharge transfer. Although converting CH3OH to HCHO is more efficient for TiO2/Cu than MSI-Cu/TiO2, the H2 evolution yield for MSI-Cu/TiO2 was remarkably greater. In the case of TiO2/Cu, the active sites are likely in the vicinity of the CuO2–TiO2 interface, which is typically associated with steric hindrance and low charge transfer. To address this limitation and enhance the kinetic of HCHO oxidation reactions, the position of CuO2 and TiO2 could be switched, promoting strong oxide–support interaction through partial encapsulation.
Conclusions
In this study, we present a soft-chemistry approach to construct composite photocatalysts, wherein a TiOx shell encapsulates highly dispersed CuO2 nanoclusters on an SiO2 core. The fine structure was characterized by transmission electron microscopy (TEM, HRTEM, STEM), in combination with EELS chemical mapping and LEIS, which confirmed the partial coverage of TiO2 onto copper metallic species. Surface analysis through EPR and XPS highlighted the existence of Cu sites coordinated with four oxygen in the case of MSI-Cu/TiO2. Compared to conventional TiO2/Cu, it exhibited a superior photocatalytic yield in the hydrogen evolution reaction. We demonstrated optimal charge carrier dynamics and photon-to-current conversion efficiencies within the SMSI-like nanostructure MSI-Cu/TiO2 using photoelectrochemical measurements and time-resolved spectroscopy techniques. In addition, the results underscore the dominance of charge carrier dynamics in driving photocatalytic enhancement.Experimental
Photocatalyst preparation
(Photo)electrochemical measurements
Photoelectrochemical measurements and electrochemical impedance analysis, including amperometry transient photocurrent (TPC), electrochemical impedance spectroscopy (EIS), Mott–Schottky, and open-circuit voltage decay, were carried out in a three-electrode electrochemical cell with/without illuminating by an AM 1.5G solar simulator. Photoelectrodes were fabricated by drop-casting 100 μL of an ultrasonicated solution (2 mg mL−1) of our samples on fluorine-doped tin oxide-coated glass slides of 25 × 25 × 1 mm2 (FTO glass) before drying overnight at room temperature. The electrochemical setup involved a platinum disk counter electrode and an Ag/AgCl (saturated KCl) reference electrode. The values of potential vs. Ag/AgCl were converted to those versus RHE through the following equation:| ER.H.E = EAg/AgCl + 0.197 + 0.059 × pH |
Experiments were manipulated by a PGSTAT101 Metrohm Autolab potentiostat. An open-circuit voltage decay fitting was executed using the inverse method on Matlab. When the numeric and experimental curves show superimposition, the fitting is reliable for estimating important parameters such as the recombination rate constant.
Photocatalytic hydrogen evolution assessment
The photocatalytic activity of our photocatalyst was evaluated via a hydrogen evolution reaction. The reaction conditions remained identical: the amount of photocatalyst at 10 mg/10 mL, room temperature, and testing duration of five hours under UV-visible irradiation triggered by a mercury lamp (150 W). Reactors were degassed by argon for 20 min to remove the dissolved oxygen completely before illumination. Gaseous products were quantified by an Agilent 990 micro gas chromatography (GC) system facilitated with a thermal conductive detector (TCD). Liquid phase products were detected by high-performance liquid chromatography (HPLC) on an Agilent 1260 Infinity with a UV-visible detector.Data availability
The data that support the findings of this study are openly available at https://zenodo.org/ at http://doi.org/10.5281/zenodo.11489226.Author contributions
V. D. Quach carried out and set up the design of the experiments and investigation process, data curation, wrote the initial draft, and reviewed and edited the final manuscript. M. C. Spadaro, M. Botifoll, and J. Arbiol performed the electron microscopy analyses and discussed their results. D. Dragoe performed XPS measurements and discussed the results. R. Wojcieszak, H. Vezin, and F. Dumeignil carried out LEIS and EPR; R. Wojcieszak discussed the LEIS result. C. Colbeau-Justin instructed V. D. Quach in TRMC. M. N. Ghazzal conceptualized and conceived the idea, acquired financing, supervised the work, contributed to the manuscript skeleton, and reviewed and edited the final manuscript. The manuscript was written with the help of all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the public grant overseen by the French National Research Agency (ANR) through the INGENCAT project (ANR-20-CE43-0014). V. D. Quach and M. N. Ghazzal appreciated Mrs Audrey Gayral and Prof. Marie Erard (Institut de Chimie Physique, Université Paris-Saclay, CNRS UMR 8000, Orsay, France) for the access to TCSPC laser system. V. D. Quach acknowledges Ms. NGUYEN Thi-Ha-Xuyen (Centre d’Innovation Matériaux et Procédés, Institut Mines Telécom, Université de Lille, Lille, France) for the assistance of fitting open-circuit voltage decay by Matlab processing. ICN2 acknowledges funding from Generalitat de Catalunya 2021SGR00457. This study is part of the Advanced Materials program and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17. I1) and by Generalitat de Catalunya. The authors thank the support from the project NANOGEN (PID2020-116093RB-C43), funded by MCIN/AEI/10.13039/501100011033/and by “ERDF A way of making Europe”, by the “European Union.” ICN2 is supported by the Severo Ochoa program from Spanish MCIN/AEI (Grant No. CEX2021-001214-S) and is funded by the CERCA Programme/Generalitat de Catalunya. Part of the present work has been performed in the Universitat Autònoma de Barcelona Materials Science PhD program framework. M. Botifoll acknowledges support from SUR Generalitat de Catalunya and the EU Social Fund; project ref. 2020 FI 00103. ICN2 is founding member of e-DREAM.64References
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- G. D. Gesesse, C. Wang, B. K. Chang, S.-H. Tai, P. Beaunier, R. Wojcieszak, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Nanoscale, 2020, 12, 7011–7023 RSC.
- X. Sun, S. Jiang, H. Huang, H. Li, B. Jia and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202204880 CrossRef CAS PubMed.
- Y. Liu, C. Zhang, J. Feng, X. Wang, Z. Ding, L. He, Q. Zhang, J. Chen and Y. Yin, Angew. Chem., Int. Ed., 2023, 62, e202308930 CrossRef CAS PubMed.
- L. Guo, Z. Yang, K. Marcus, Z. Li, B. Luo, L. Zhou, X. Wang, Y. Du and Y. Yang, Energy Environ. Sci., 2018, 11, 106–114 RSC.
- M. A. Rahman, S. Bazargan, S. Srivastava, X. Wang, M. Abd-Ellah, J. P. Thomas, N. F. Heinig, D. Pradhan and K. T. Leung, Energy Environ. Sci., 2015, 8, 3363–3373 RSC.
- J. Li, A. Slassi, X. Han, D. Cornil, M. Ha-Thi, T. Pino, D. P. Debecker, C. Colbeau-Justin, J. Arbiol, J. Cornil and M. N. Ghazzal, Adv. Funct. Mater., 2021, 31(29), 2100994 CrossRef CAS.
- C. Wang, J. Li, E. Paineau, H. Remita and M. N. Ghazzal, Sol. RRL, 2023, 7(5), 2200929 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS.
- L. Piccolo, P. Afanasiev, F. Morfin, T. Len, C. Dessal, J. L. Rousset, M. Aouine, F. Bourgain, A. Aguilar-Tapia, O. Proux, Y. Chen, L. Soler and J. Llorca, ACS Catal., 2020, 10, 12696–12705 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS PubMed.
- V. D. Quach, R. Wojcieszak and M. N. Ghazzal, ChemNanoMat, 2023, 9(11), e202300329 CrossRef CAS.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- G. Pacchioni and H.-J. Freund, Chem. Soc. Rev., 2018, 47, 8474–8502 RSC.
- S. Rhatigan and M. Nolan, J. Mater. Chem. A, 2018, 6, 9139–9152 RSC.
- M. Li, T. Zhang, S.-Z. Yang, Y. Sun, J. Zhang, F. Polo-Garzon, K. M. Siniard, X. Yu, Z. Wu, D. M. Driscoll, A. S. Ivanov, H. Chen, Z. Yang and S. Dai, ACS Catal., 2023, 13, 6114–6125 CrossRef CAS.
- F. Polo-Garzon, T. F. Blum, V. Fung, Z. Bao, H. Chen, Z. Huang, S. M. Mahurin, S. Dai, M. Chi and Z. Wu, ACS Catal., 2020, 10, 8515–8523 CrossRef CAS.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang and M. Haruta, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- S. Liu, H. Qi, J. Zhou, W. Xu, Y. Niu, B. Zhang, Y. Zhao, W. Liu, Z. Ao, Z. Kuang, L. Li, M. Wang and J. Wang, ACS Catal., 2021, 11, 6081–6090 CrossRef CAS.
- R. Belgamwar, R. Verma, T. Das, S. Chakraborty, P. Sarawade and V. Polshettiwar, J. Am. Chem. Soc., 2023, 145, 8634–8646 CrossRef CAS PubMed.
- C. Zhang, L. Wang, U. J. Etim, Y. Song, O. M. Gazit and Z. Zhong, J. Catal., 2022, 413, 284–296 CrossRef CAS.
- J. Yu, X. Sun, X. Tong, J. Zhang, J. Li, S. Li, Y. Liu, N. Tsubaki, T. Abe and J. Sun, Nat. Commun., 2021, 12, 7209 CrossRef CAS PubMed.
- W. Levason and M. D. Spicer, Coord. Chem. Rev., 1987, 76, 45–120 CrossRef CAS.
- M. Janczarek, Z. Wei, M. Endo, B. Ohtani and E. Kowalska, J. Photonics Energy, 2016, 7, 012008 CrossRef.
- S. J. A. Moniz and J. Tang, ChemCatChem, 2015, 7, 1659–1667 CrossRef CAS.
- W. Tabis, Y. Li, M. L. Tacon, L. Braicovich, A. Kreyssig, M. Minola, G. Dellea, E. Weschke, M. J. Veit, M. Ramazanoglu, A. I. Goldman, T. Schmitt, G. Ghiringhelli, N. Barišić, M. K. Chan, C. J. Dorow, G. Yu, X. Zhao, B. Keimer and M. Greven, Nat. Commun., 2014, 5, 5875 CrossRef CAS PubMed.
- R. Zhang, G. Jiang, Q. Gao, X. Wang, Y. Wang, X. Xu, W. Yan and H. Shen, Nanoscale, 2021, 13, 15937–15951 RSC.
- F.-T. Cheng, Y.-D. Geng, Y.-X. Liu, X. Nie, X.-G. Zhang, Z.-L. Chen, L.-Q. Tang, L.-H. Wang, Y.-Z. You and L. Zhang, Nanoscale Adv., 2023, 5, 3336–3347 RSC.
- G. D. Gesesse, T. Le Neel, Z. Cui, G. Bachelier, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Nanoscale, 2018, 10, 20140–20146 RSC.
- I. Vishik, Science, 2020, 369, 775–776 CrossRef CAS PubMed.
- G. Palmer, in Oxidation and Phosphorylation, ed. M. E. P. Ronald and W. Estabrook, Academic Press, New York, 1967, vol. 10, pp. 594–609 Search PubMed.
- G. W. Brudvig, in Biochemical Spectroscopy, ed. Kenneth S., Academic Press, New York, 1995, vol. 246, pp. 536–554 Search PubMed.
- W. R. Hagen, Dalton Trans., 2006, 4415 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
- N. Sakaguchi Miyamoto, R. Miyamoto, E. Giamello, T. Kurisaki and H. Wakita, Res. Chem. Intermed., 2018, 44, 4563–4575 CrossRef CAS.
- F. A. Taiwo, Spectroscopy, 2003, 17, 53–63 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- B. Kozlevčar and P. Šegedin, Croat. Chem. Acta, 2008, 81, 369–379 Search PubMed.
- J. A. Aramburu, A. Bhowmik, J. M. Garcia-Lastra, P. García-Fernández and M. Moreno, J. Phys. Chem. C, 2019, 123, 3088–3101 CrossRef CAS.
- A. V. Kucherov, J. L. Gerlock, H.-W. Jen and M. Shelef, J. Phys. Chem., 1994, 98, 4892–4894 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- C. Wang, J. Li, E. Paineau, A. Laachachi, C. Colbeau-Justin, H. Remita and M. N. Ghazzal, J. Mater. Chem. A, 2020, 8, 10779–10786 RSC.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. A. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CrossRef CAS.
- X. Qiu, M. Miyauchi, K. Sunada, M. Minoshima, M. Liu, Y. Lu, D. Li, Y. Shimodaira, Y. Hosogi, Y. Kuroda and K. Hashimoto, ACS Nano, 2012, 6, 1609–1618 CrossRef CAS PubMed.
- J. Zhang, C. Y. Toe, P. Kumar, J. Scott and R. Amal, Appl. Catal., B, 2023, 333, 122765 CrossRef CAS.
- H. Chen, Z. Yang, X. Wang, F. Polo-Garzon, P. W. Halstenberg, T. Wang, X. Suo, S. Z. Yang, H. M. Meyer, Z. Wu and S. Dai, J. Am. Chem. Soc., 2021, 143, 8521–8526 CrossRef CAS PubMed.
- M. N. Ghazzal, M. Joseph, H. Kebaili, J. De Coninck and E. M. Gaigneaux, J. Mater. Chem., 2012, 22, 22526 RSC.
- H. Irie, S. Miura, K. Kamiya and K. Hashimoto, Chem. Phys. Lett., 2008, 457, 202–205 CrossRef CAS.
- B. Choudhury, M. Dey and A. Choudhury, Appl. Nanosci., 2014, 4, 499–506 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- C. Colbeau-Justin, M. Kunst and D. Huguenin, J. Mater. Sci., 2003, 38, 2429–2437 CrossRef CAS.
- A. Zaban, M. Greenshtein and J. Bisquert, ChemPhysChem, 2003, 4, 859–864 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- R. Ciancio, R. E. Dunin-Borkowski, E. Snoeck, M. Kociak, R. Holmestad, J. Verbeeck, A. I. Kirkland, G. Kothleitner and J. Arbiol, Microsc. Microanal., 2022, 28, 2900–2902 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03072a |
| This journal is © The Royal Society of Chemistry 2024 |