
A single-atom iron catalyst on hierarchical N-doped carbon for highly efficient oxygen reduction in Zn–air batteries†
Jun-Fei
Gu
a,
Jichao
Wang
a,
Qing
Wu
a,
Caixia
Wang
*b,
Francis
Verpoort
 *ac and
Somboon
Chaemchuen
*ad
*ac and
Somboon
Chaemchuen
*ad
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luoshi Road, Wuhan 430070, P. R. China. E-mail: francis@whut.edu.cn; sama_che@hotmail.com
bSchool of Civil Engineering and Architecture, Wuhan University of Technology, 122 Luoshi Road, Wuhan 430070, P. R. China. E-mail: caixia.wang@whut.edu.cn
cJoint Institute of Chemical Research (FFMiEN), Peoples Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya Str., 117198 Moscow, Russia
dDepartment of Chemical Engineering, Faculty of Engineering, Mahidol University, Nakhon Pathom 73170, Thailand
First published on 4th June 2024
Abstract
Single-atom iron electrocatalysts have emerged as up-and-coming alternatives to platinum-based catalysts for the oxygen reduction reaction. However, their further development has been impeded by complex fabrication procedures and limitations in long-term stability. This study developed a chemical vapor deposition approach for synthesizing an efficient iron single-atom electrocatalyst denoted as Fe-SA@NC, utilizing vaporized ferrocene to deposit on a hierarchical N-doped carbon derived from ZIF-8. The preparation process maintained the initial pore structure throughout the deposition process by utilizing a two-step pyrolysis, preventing the collapse or deformation of the pore structure and frameworks. The optimized catalyst exhibited an exceptional half-wave potential (0.932 V) and kinetic current density (28.38 mA cm−2 at 0.9 V vs. RHE), along with high turnover frequency (36.37 s−1) and mass activity (5.68 A mg−1), and remarkable long-term stability in an alkaline electrolyte, exceeding those of commercial Pt/C and most previously reported iron-based electrocatalysts. Moreover, it also demonstrated outstanding practicability in both liquid and solid Zn–air batteries. The formation of well-dispersed Fe–N4 with strong interaction on hierarchical N-doped carbon was verified in the correlation of the structural activity and the excellent performance of Fe-SA@NC. This work sheds some light on the facile synthesis of single-atom catalysts with effective efficiency and stability.
Introduction
Developing clean and renewable energy sources is crucial for meeting the worldwide energy demands.1,2 Among the potential candidates for the next generation of energy storage devices, rechargeable Zn–air batteries (ZABs) have emerged as a promising option due to their high energy density, low cost, eco-friendliness, and excellent safety profile.3–8 Despite their potential, the practical application of ZABs has been hampered by the sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode.9 While Pt-based catalysts have been commercialized and exhibit exceptional electrocatalytic performance, they remain limited by their high cost, scarcity, and poor long-term stability.10–12 Consequently, there is a growing need for alternative catalysts to address these shortcomings.In recent years, extensive research efforts have focused on utilizing atomically dispersed non-noble metal–nitrogen–carbon (M–N–C) catalysts for the oxygen reduction reaction (ORR).13–20 These catalysts have demonstrated remarkable electrochemical performance, rivalling or surpassing that of Pt-based catalysts. Among them, single-atom Fe–N–C catalysts with atomically dispersed Fe–N4 active sites have garnered significant interest.21–30 This attention can be attributed to their exceptional intrinsic activity, remarkable stability, abundant availability, 100% atom utilization, and cost-effectiveness, making them a potentially viable alternative to commercial Pt/C.31–40 Nevertheless, it is worth highlighting that the pyrolysis process employed for synthesizing iron single-atom catalysts often leads to the formation of inactive Fe species and agglomerated components like nanoparticles.24,34,41 Consequently, an additional acid etching step becomes necessary, greatly complicating the preparation process, limiting the synthesis scalability, and hindering practical applications.42,43 Hence, an efficient and convenient method to accurately synthesize atomically uniformly dispersed Fe–N–C catalysts is urgently needed. Furthermore, the practical applications of these catalysts should be further explored to fully unleash their potential.
Zeolite-imidazole frameworks (ZIFs) and their derived carbon materials (NC) have been identified as an optimal precursor for generating active sites, resulting in markedly improved ORR activity.21,35,44–47 For example, using ZIF-8 as a precursor with the steric confinement method has been a widely adopted approach for achieving single-atom dispersion.48,49 This method involves the adsorption of an Fe-containing salt solution onto ZIF-8, enclosing a solitary Fe salt molecule within the micropore of ZIF-8, and employing the micropore channel as a molecular cage to separate the Fe salt molecule. The confinement of pore structure limits Fe atom aggregation during pyrolysis, facilitating the synthesis of atomically dispersed Fe–N–C catalysts with enhanced activity. However, the inherent limitations of the steric confinement method hinder its ability to reliably generate single atoms, as pyrolysis-induced pore destruction and skeleton shrinkage often result in the undesired agglomeration of Fe atoms into clusters. Additionally, the conventional pyrolytic ZIF-8 precursor exhibits restricted metal loading due to the consistent content of the adsorbed metal salt solution, thereby limiting the formation of active sites.
This study reports the synthesis of atomically dispersed Fe single atoms anchored on a hierarchical N-doped carbon matrix, denoted as Fe-SA@NC. The methodology utilized ZIF-8 as a precursor to derive hierarchical nitrogen-doped porous carbon (NC). Following the step, a strategic iron chemical vapor deposition (CVD) technique was applied to deposit iron single atoms on NC. Notably, the CVD technique offered distinct advantages by implementing a two-step pyrolysis process that bestowed two significant benefits upon the synthesis procedure. Firstly, during the initial stage, the basic pore structure of the carrier was established, which was a crucial component of the overall approach since the pore structure played a vital role in determining the catalyst's performance. Secondly, iron was deposited onto the NC support in the subsequent step without destroying or significantly altering the pore structure or framework. This critical aspect of the method guaranteed that the efficiencies achieved from the initial determination of the pore structure were preserved throughout the deposition process. As a result, utilizing a two-step pyrolysis technique offers a dependable and efficient approach to catalyst design. Moreover, this method significantly enhanced the degree of graphitization in the resulting material, thereby contributing to a noticeable improvement in catalyst stability.
Experimental
Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%), 2-methylimidazole (98%), methanol (99%), and ferrocene (99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals and materials were used without further purification.Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fe-SA@NC-2), 20:3 (Fe-SA@NC-3)) and different pyrolysis temperatures of the NC (i.e., 700 °C (Fe-SA@NC700), 800 °C (Fe-SA@NC800), 900 °C (Fe-SA@NC900), and 1000 °C (Fe-SA@NC)).
:2 (Fe-SA@NC-2), 20:3 (Fe-SA@NC-3)) and different pyrolysis temperatures of the NC (i.e., 700 °C (Fe-SA@NC700), 800 °C (Fe-SA@NC800), 900 °C (Fe-SA@NC900), and 1000 °C (Fe-SA@NC)).
Materials characterization
The Fe-SA@NC catalysts were characterized using a variety of techniques. Firstly, the morphology of the catalysts was examined using a JSM-IT800 scanning electron microscope (SEM). Energy-dispersive X-ray spectroscopy (EDS) element mapping and transmission electron microscopy (TEM) images were obtained using a Talos F200S microscope operating at 200 kV. The presence of a single Fe atom in the Fe-SA@NC catalysts was confirmed using an FEI Titan Themis G2 60-300 spherical aberration-corrected high-angle annular dark-field scanning transmission electron microscope (AC HAADF-STEM). X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance X-ray powder diffractometer with a Cu Kα radiation source (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Fisher ESCALAB 250Xi spectrometer. The Fe loading of Fe-SA@NC was analysed by inductively coupled plasma-optical emission spectrometry (ICP-OES, Prodigy 7), while elemental analysis results were collected on a Vario EL cube. The surface area and pore size of the catalysts were calculated by Brunauer–Emmett–Teller (BET) analysis using N2 adsorption/desorption isotherms at 77 K, taken on a Micromeritics ASAP 2020 instrument. Raman spectra were recorded using a LabRam Odyssey instrument with an excitation wavelength of 532 nm. Finally, the X-ray absorption fine structure spectra (XAFS) data were collected at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF, operated at 2.5 GeV with a maximum current of 200 mA), respectively. The measurements were carried out at the Mo K-edge (20019 eV) and LIII-edge (2525 eV) at room temperature. All samples were pelletized as disks of 13 mm diameter with 1 mm thickness using BN powder as a binder. The ATHENA module implemented in the IFEFFIT software packages was used to reduce and analyse the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data, processed according to standard procedures.
Results and discussion
Catalyst synthesis and characterization
The Fe-SA@NC catalysts were fabricated via a CVD strategy, using a two-step procedure, as illustrated in Fig. 1a. First, the NC was synthesized through pyrolysis of ZIF-8 at 1000 °C under the 5% H2/Ar flow. Subsequently, ferrocene and the achieved NC powders were placed upstream and downstream of the porcelain boat, respectively. When this system was heated above 100 °C, the ferrocene sublimated into the gas phase and deposited in the porous structure of NC. After subsequent heat treatment at a high temperature of 900 °C for 2 h, the iron atoms slipped out of the parallel cyclopentadiene and coordinated with the N vacancies in the NC. By employing this CVD method, gaseous ferrocene was homogeneously dispersed into the carbon matrix without steric limitations of iron aggregation in the NC host, as proved by further characterization. | ||
| Fig. 1 (a) Schematic illustration of the preparation of the atomically dispersed Fe-SA@NC catalysts. (b) XRD patterns, (c) Raman curves (D band: defect sp3 carbon; G band: graphitic sp2 carbon), (d) high-resolution XPS N 1s spectra of the catalysts. | ||
XRD was conducted to evaluate the crystallization of Fe-SA@NC catalysts and NC (Fig. 1b). The XRD pattern indicated two broad peaks at approximately 24° and 44°, attributed to the (002) and (101) planes of graphitic carbon, respectively.51 These two broad peaks suggested that the material's crystallinity was defective and amorphous with a higher degree of graphitization, thus favouring the incorporation of Fe atoms.52 Importantly, no peaks associated with metallic iron, iron compounds, or impurities were observed, indicating that the Fe atoms did not form nanoparticles in the Fe-SA@NC catalysts but provided an excellent atomic dispersion. The Raman spectrum of Fe-SA@NC catalysts, as shown in Fig. 1b, displayed two peaks at 1342 and 1583 cm−1. Notably, the intensity ratio of the D and G bands (ID/IG) for Fe-SA@NC-2 was 1.03, lower than those of Fe-SA@NC-1 (1.05) and Fe-SA@NC-3 (1.08), indicating a higher degree of graphitization in Fe-SA@NC-2 compared to the other catalysts.
The Fe-SA@NC catalysts' pore structure and specific surface area were determined through N2 adsorption–desorption at 77 K (Fig. S1†). The N2 adsorption–desorption isotherms of the samples displayed a steep increase at relatively low pressure (P/P0 < 0.1) mixed with a small hysteresis loop at high relative pressure (P/P0 > 0.8), presenting an obvious combination of type I and IV isotherms, indicating the existence of hierarchical micropores and mesopores. According to the BET results, the specific surface areas of the samples were calculated to be 1348, 1090, and 1006 m2 g−1 on Fe-SA@NC-1, Fe-SA@NC-2, and Fe-SA@NC-3, respectively (Table S1†). Pore-size distribution analysis also confirmed that all the catalysts exhibited mixed pore structures comprising micropores, mesopores, and macropores, consistent with the nitrogen isotherm. This porous structure was responsible for providing the catalysts with a high specific surface area during catalytic processes. The use of CVD to deposit Fe single atoms within the hierarchical pores of NC slightly affected the specific surface area of the resulting material. The increasing Fe loading decreased the specific surface area, implying a successful encapsulation of Fe atoms (Tables S1 and S2†). The increasing density of Fe single atomic sites followed an increase in Fe content, thus leading to further decreases in the specific surface area. Due to the pivotal role of specific surface area and pore structure in mass transfer and active site exposure, a large specific surface area with low active site density would result in poor catalytic performance. Likewise, excess Fe content caused blockage of pores, which inhibited mass transfer and adequate active site exposure, resulting in a reduced active site density. Therefore, a moderate Fe content and specific surface area were required to ensure the optimal improvement of the catalytic activity. Furthermore, XPS was carried out to investigate the composition and chemical states of these catalysts (Fig. 1d and S2†). The high-resolution N 1s spectra of the Fe-SA@NC catalysts depicted the presence of various nitrogen species, including pyridinic-N (398.5 eV), Fe–N (400.2 eV), pyrrolic-N (401.2 eV), graphitic-N (402.6 eV), and oxidized-N (405.6 eV), as shown in Fig. 2c.53 Moreover, in the high-resolution Fe 2p spectra, the peaks at approximately 710.8 and 724.4 eV matched well with the Fe–N, suggesting the existence of Fe–N bonding in Fe-SA@NC catalysts (Fig. S2b†). It has been reported that pyridinic-N played a crucial role in the formation of Fe–Nx, which could readily bond with iron to form Fe–Nx, thus enhancing the intrinsic ORR activity. This signified that a high content of pyridinic-N could boost the catalyst's ORR performance.54 Furthermore, the XPS elemental analysis revealed differences in the atomic content of Fe and N in catalysts with varying Fe loadings. Specifically, the Fe atomic content in Fe-SA@NC-2 was found to be 0.68 atom%, while the N content was 4.74 atom%. Notably, the ratio of pyridine nitrogen in this catalyst was 33.4%, higher than that of the other two catalysts (Tables S3 and S4†). These findings suggested that Fe-SA@NC-2 was poised to exhibit superior catalytic ORR activity.
 | ||
| Fig. 2 (a) TEM image and (b) AC HAADF-STEM enlarged image. (c) HAADF-STEM image and corresponding EDS mapping images for Fe, C, and N. (d) Fe K-edge XANES, (e) FT-EXAFS, (f) R-space FT-EXAFS fitting, and (g) WT-EXAFS contour plots of Fe-SA@NC-2. | ||
Considering that Fe-SA@NC-2 exhibited the highest ORR activity (Fig. 3a), the following morphological and structural characterizations were performed on Fe-SA@NC-2. Despite the shrinkage of the surface of Fe-SA@NC-2, its dodecahedron morphology, as seen in ZIF-8 and NC, was retained, although the framework was destabilized, as indicated in Fig. S3.† The atomic dispersion of Fe-SA@NC-2, as shown in the TEM images in Fig. 2a and S4a,† revealed no detectable nanoparticles or clusters in the porous carbon matrix. Even more significantly, a high-angle annular dark-field scanning transmission electron microscope revealed the uniform dispersion of isolated Fe atoms (bright spots) on the NC support, resulting from the difference in contrast caused by the atomic number, as seen in Fig. 4b. The corresponding enlarged AC HAADF-STEM image confirmed the presence of Fe single atoms as bright spots circled in red, shown in Fig. 2b. Additionally, EDS elemental mapping indicated that Fe, C, and N elements were homogeneously distributed on the carbon matrix, as demonstrated in Fig. 2c.
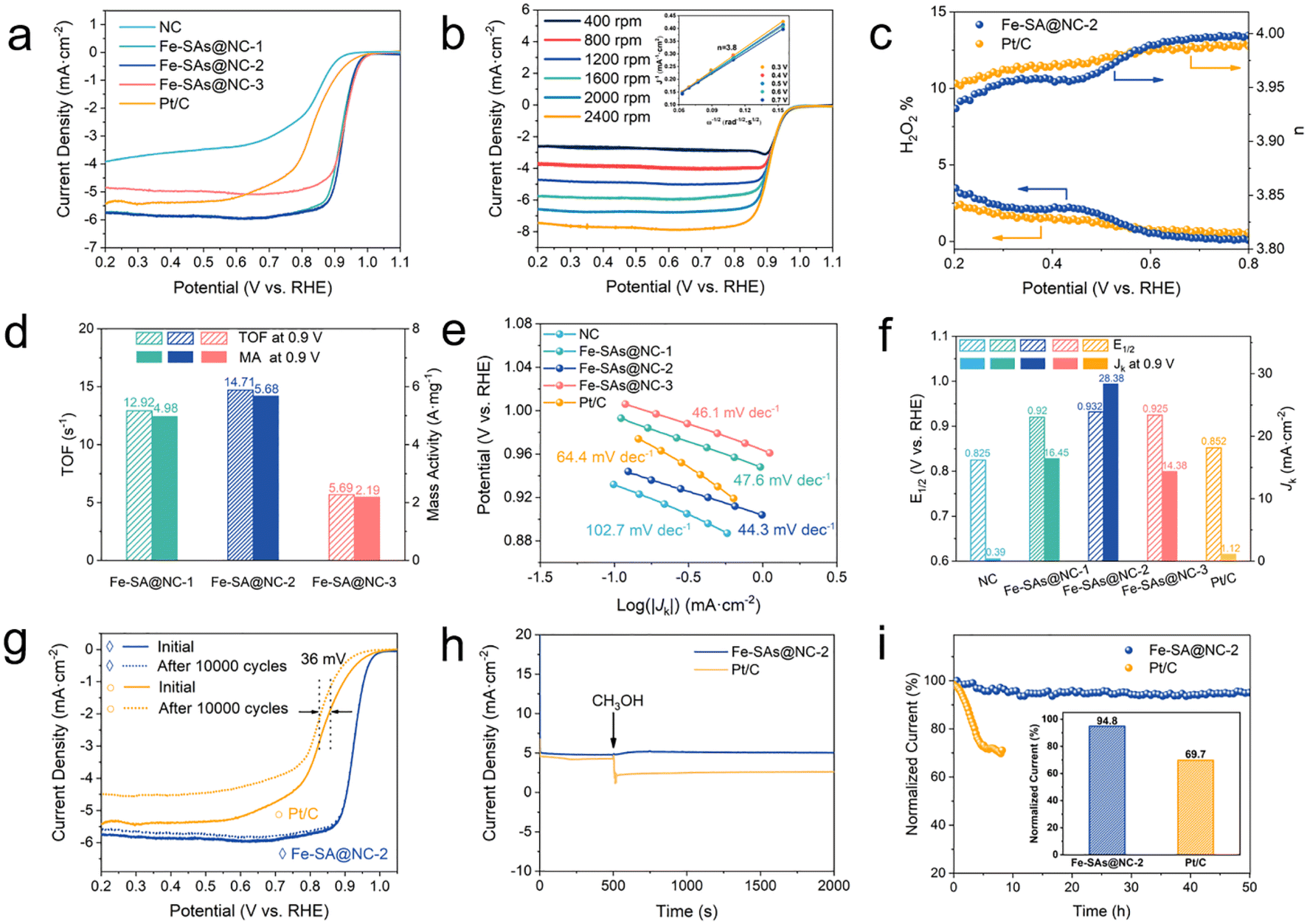 | ||
| Fig. 3 Electrocatalytic ORR performances of Fe-SA@NC catalysts. (a) LSV curves at a rotation rate of 1600 rpm under 10 mV s−1. (b) LSV curves under different rotation rates with the inset showing the K–L plots and electron-transfer number. (c) Electron-transfer number and hydrogen peroxide yield according to an RRDE test. (d) TOF and mass activity values at 0.9 V. (e) Tafel plots at 1600 rpm. (f) Half-wave potential and kinetic current density obtained at 0.9 V vs. RHE. (g) LSV curves before and after 10000 cycles. (h) Methanol tolerance test. (i) Long term i–t chronoamperometric stability test. | ||
 | ||
| Fig. 4 (a) Schematic illustration of liquid ZABs. (b) Open-circuit voltage plots (inserted digital images of output circuit driven by liquid ZABs using Fe-SA@NC-2 and the corresponding LED display). (c) Polarization and power density curves. (d) Specific capacities. (e) Cycling performance of the liquid ZABs using Fe-SA@NC-2 and Pt/C + RuO2 catalysts at a current density of 1 mA cm−2. (f) Open-circuit plots (inserted digital images of the output circuit driven by liquid ZABs using Fe-SA@NC-2 and the corresponding LED display). (g) Polarization and power density curves, and (h) cycling performance of solid ZABs using the Fe-SA@NC-2 + RuO2 catalyst. | ||
XAFS was carried out to decipher the electronic structure and coordination environment of Fe species in Fe-SA@NC-2. Fig. 2d and e display the Fe K-edge XANES and EXAFS of Fe-SA@NC-2 and reference samples. The isolated Fe atoms in Fe K-edge XANES indicated an oxidation state between Fe0 and Fe3+, similar to standard FePc (Fig. 2d). In contrast, the Fourier-transformed k3-weighted EXAFS spectra of the Fe-SA@NC-2 catalyst showed a distinct peak around 1.47 Å, which could not be attributed to Fe–Fe, suggesting that it could be assigned to either Fe–N, Fe–C, or Fe–O coordination (Fig. 2e). Additionally, the R space EXAFS fitting analysis confirmed that the atomically dispersed Fe atom was coordinated with four N atoms with an average bond length of 2.02 Å, which indicated the formation of the Fe–N4 structure in this catalyst (Fig. 2f). The Fe K-edge EXAFS and the curve fit of K space and R space imaginary components are shown in Fig. S5 and S6.† The fitting results agreed well with the Fe–N4 model. The R factor was 0.015, and the fitting parameters are summarized in Table S5.† The wavelet transform (WT) plot of the Fe K-edge EXAFS revealed a single intensity maximum at 3.2 Å−1, which corresponded well to Fe–N in FePc and was slightly smaller than that of Fe–O in Fe2O3 (Fig. 2g). Therefore, the peak should be assigned to Fe–N(O). Moreover, the WT plots of Fe foil and Fe2O3 showed Fe–Fe contributions that were not detected in Fe-SA@NC-2, suggesting the presence of mononuclear Fe centres without an Fe-derived crystal structure. These results provided strong evidence for the nature of the Fe species and demonstrated the unique architecture of Fe-SA@NC-2.
ORR activity and stability
The ORR activity of the catalysts was evaluated using cyclic voltammetry (CV) and linear sweep voltammograms (LSVs) in a 0.1 M KOH electrolyte with a rotating disk electrode (RDE). In order to study the oxygen reduction process, CV measurements were performed in both O2 and Ar-saturated 0.1 M KOH electrolytes. Fig. S7† shows that in the O2-saturated electrolyte, cathodic peaks were observed for both Fe-SA@NC catalysts and Pt/C. Fe-SA@NC-2 exhibited the highest oxygen reduction peak of 0.873 V (vs. RHE), indicating its outstanding ORR activity. LSV measurements in an O2-saturated 0.1 M KOH electrolyte were then carried out to further investigate the ORR activity of the catalysts. The half-wave potentials (E1/2) of Fe-SA@NC-1, Fe-SA@NC-2 and Fe-SA@NC-3 were found to be 0.920 V, 0.932 V and 0.925 V (vs. RHE), respectively, with all values being higher than that of Pt/C (E1/2 = 0.852 V) (Fig. 3a and f). These results suggested that the hierarchical nitrogen doped porous carbon (NC) derived from ZIF-8 played a significant role in the ORR activity. The catalytic activity of Fe-SA@NC catalysts was superior to that of NC, which showed a half-wave potential at 0.825 V, proving the function of Fe–N4 as the ORR active sites. Furthermore, since the Fe-SA@NC catalysts were prepared with ZIF-8 derived NC as a precursor, NC was synthesized with different pyrolysis temperatures as a carrier for CVD to investigate its ORR performance (i.e., Fe-SA@NC700-2, Fe-SA@NC800-2, Fe-SA@NC900-2 and Fe-SA@NC-2). As shown in Fig. S8,† the half-wave potential of all the samples gradually increased with the rise of pyrolysis temperature, indicating an improvement in ORR performance. Notably, the half-wave potentials of all the Fe-SA@NC catalysts were much higher than that of Pt/C, regardless of the pyrolysis temperature, showing superior catalytic activity. These findings suggested that the pyrolysis temperature of ZIF-8 was a crucial factor in constructing NC, which could influence the activity of the catalysts.To assess the ORR kinetics of Fe-SA@NC-2, RDE tests were conducted at different rotating speeds ranging from 400 to 2400 rpm (Fig. 3b). Based on the Koutecky–Levich (K–L) plot, the electron transfer number of Fe-SA@NC-2 was calculated to be four, indicating a dominant four-electron pathway for the ORR over Fe-SA@NC-2 in alkaline media. Remarkably, RRDE measurements showed a significantly low H2O2 yield of less than 2.5%, with an average electron transfer number of 4.00 (Fig. 3c), which was nearly identical to the n value based on the K–L equation. This suggested a high-efficiency selectivity of the four-electron ORR pathway. Accordingly, the turnover frequency (TOF) and mass activity (MA) of the Fe-SA@NC catalysts were calculated according to the current density at 0.9 V vs. RHE to confirm the contribution of each active metal atom to the ORR process. The intrinsic TOF of Fe-SA@NC-1 was up to 36.37 s−1, which was higher than the 26.92 s−1 of Fe-SA@NC-2 and nearly 2 times that of Fe-SA@NC-3 (17.68 s−1). Besides, the mass activity of Fe-SA@NC-2 was determined to be 5.68 A mg−1, which was higher than the 4.98 A mg−1 for Fe-SA@NC-1 and 2.19 A mg−1 for Fe-SA@NC-3. The above results implied that the intrinsic activity of Fe-SA@NC catalysts was excellent, outperforming those for the reported Fe–N–C electrocatalysts (Table S6†).
The calculated ORR Tafel slope for Fe-SA@NC-2 was found to be 44.3 mV dec−1, while the corresponding values for Fe-SA@NC-1, Fe-SA@NC-3, and Pt/C catalysts were 47.6 mV dec−1, 46.1 mV dec−1, and 64.4 mV dec−1, respectively (Fig. 3e). Therefore, Fe-SA@NC-2 exhibited excellent ORR kinetics performance. The kinetic current density of Fe-SA@NC-2 was 28.38 mA cm−2 at 0.9 V, which was considerably higher than those of NC (0.39 mA cm−2) and commercial Pt/C (1.12 mA cm−2), almost 28 times that of Pt/C (Fig. 3f). To explore the reason for this high performance, the double-layer capacitance (Cdl) was calculated to investigate the electrochemical surface area (ECSA) of the catalysts (Fig. S9†). Fe-SA@NC-2 showed the largest Cdl value, i.e., 48.8 mF cm−2, which was much higher than those of Fe-SA@NC-1 (23.9 mF cm−2), Fe-SA@NC-3 (14.6 mF cm−2), and Pt/C (2.3 mF cm−2), indicating that Fe-SA@NC-2 had more accessible Fe–N4 active sites. Furthermore, the ECSA of the catalysts exhibited the same trend, with Fe-SA@NC-2 having the highest ECSA of 1220 cm2 mg−1.
In order to investigate the durability of the Fe-SA@NC-2 catalyst, the sweep rate of 10 mV s−1 in O2-saturated 0.1 M KOH was used. Fig. 3g demonstrated that the E1/2 of Fe-SA@NC-2 was not shifted after 10000 cycles, whereas the E1/2 of Pt decreased by 36 mV, indicating that Fe-SA@NC-2 exhibited excellent electrochemical stability in the alkaline electrolyte. Moreover, the catalyst showed robust methanol resistance in the alkaline medium, as no change was observed after the methanol injection, while the current density of Pt/C dropped abruptly (Fig. 3h). Furthermore, in the long-term i–t chronoamperometric stability test, Fe-SA@NC-2 showed 94.8% retention after scanning for 50 hours, while Pt/C retained only 69.7% under the same conditions and within the same time frame, as shown in Fig. 3i. The results suggested that Fe-SA@NC-2 had both long-term stability and robust methanol resistance, indicating its potential application in practical situations as an ORR catalyst. The remarkable activity, selectivity, and durability of Fe-SA@NC-2 were mainly attributed to its unique structural properties. The hierarchical porous N-doped carbon materials, serving as a carrier, possessed a high specific surface area and abundant surface anchoring sites. The atomic dispersion of Fe on the hierarchical porous N-doped carbon matrix created numerous and highly uniform Fe–N4 active sites, thereby optimizing the electronic environment for efficient O2 adsorption and reduction. Moreover, the Fe–N4 coordination ensured optimal binding energies for intermediates, thereby enhancing reaction kinetics and promoting the preferred four-electron reduction pathway to water.55 This precise atomic structure, combined with the stabilizing effect of the support, effectively prevented Fe atom agglomeration, ensuring long-term stability. The potential of Fe-SA@NC-2 as the air cathode catalyst was further explored through the fabrication of liquid and solid rechargeable ZABs. In the liquid ZABs illustrated in Fig. 4a, a zinc plate served as the anode and Fe-SA@NC-2 + RuO2-loaded carbon paper as the air cathode. Various techniques were carried out to evaluate the performance of the home-made batteries. The results showed that the Fe-SA@NC-2 + RuO2 based battery exhibited a higher open-circuit voltage (1.45 V) as compared to the commercial Pt/C + RuO2 based one (1.36 V), which closely matched the open-circuit voltage measured by a voltmeter of 1.49 V. Furthermore, it was observed that the Fe-SA@NC-2 + RuO2 based battery was able to power a “WHUT” LED display (Fig. 4b). The discharge polarization curves further demonstrated that the open-circuit voltage of the Fe-SA@NC-2 + RuO2 based battery surpassed that of the Pt/C + RuO2 based one (1.45 V vs. 1.36 V). Additionally, the maximum power density of the Fe-SA@NC-2 + RuO2 battery was found to be 141 mW cm−2, surpassing the capacity of the Pt/C + RuO2 based battery (78 mW cm−2) (Fig. 4c). Moreover, Fe-SA@NC-2 + RuO2 showed a higher discharge specific capacity (710 mA h g−1) compared to Pt/C + RuO2 (640 mA h g−1) at a discharging current density of 7.8 mA cm−2 and normalized to the mass of the zinc plate (Fig. 4d). Regarding long-term stability, the Fe-SA@NC-2 + RuO2 based battery maintained its initial discharging–charging voltage gap of 0.8 V over a 60-hour cycle test, whereas the gap for Pt/C + RuO2 exceeded that of Fe-SA@NC-2 + RuO2 after only 20 hours. This indicated that the Fe-SA@NC-2 + RuO2 based battery exhibited superior long-term stability compared to the Pt/C + RuO2 based battery (Fig. 4e). The performance test results of the solid-state ZABs were equally promising, showcasing an open circuit voltage of 1.42 V, a maximum power density of 62 mW cm−2, and a cycle test of 3.3 h. Moreover, the solid battery also successfully powered an LED display (Fig. 4f–h).
Conclusions
A single-atom Fe-SA@NC catalyst on hierarchical N-doped porous carbon was successfully synthesized using the CVD strategy. The catalyst characterization indicated a uniform dispersion of Fe–N4 active sites on the Fe-SA@NC catalyst. This strategy simplified the process of tuning the content of a single iron atom by solely requiring the adjustment of ferrocene mass to increase the Fe loading. The atomic dispersion and confinement of Fe species within the carbon lattice and their structural coordination to the Fe–N4 moiety were confirmed through EXAFS and AC HAADF-STEM. Significantly, the well-dispersed active iron atom species deposited and strongly interacted with nitrogen-functionalized carbon. Integrating the hierarchical structure of the Fe-SA@NC catalyst was a crucial component of the overall approach compared to earlier reported Fe–N–C catalysts. Accordingly, the best-performing catalyst, Fe-SA@NC-2, exhibited not only exceptional ORR performance with a half-wave potential of 0.932 V vs. RHE, a kinetic current density of 28.38 mA cm−2, and a Tafel slope of 44.3 mV dec−1 but also remarkable long-term stability with no activity decay after 10000 cycles in alkaline electrolytes. Furthermore, it demonstrated outstanding practicability for both liquid and solid Zn–air batteries. The formation of well-dispersed Fe–N4 with strong interaction on hierarchical N-doped carbon was verified in the correlation of the structural activity and the excellent performance of Fe-SA@NC. Furthermore, when utilized as the air cathode for ZABs, Fe-SA@NC demonstrated superior performance compared to commercial Pt/C in terms of open-circuit voltage, power density, specific capacity, and stability. This work sheds some light on the facile synthesis of single-atom catalysts with effective efficiency and stability.
Author contributions
Jun-Fei Gu: investigation, methodology, data curation, formal analysis, writing – original draft; Jichao Wang: investigation, methodology; Qing Wu: formal analysis; Caixia Wang: conceptualization, funding acquisition, project administration, resources, writing – review & editing; Francis Verpoort: conceptualization, data curation, formal analysis, resources, supervision, writing – review & editing; Somboon Chaemchuen: conceptualization, methodology, data curation, visualization, formal analysis, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the State Key Lab of Advanced Technology for Materials Synthesis and Processing for financial support (Wuhan University of Technology).References
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- Y. Li, M. Gong, Y. Liang, J. Feng, J.-E. Kim, H. Wang, G. Hong, B. Zhang and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- H.-F. Wang, C. Tang and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1803329 CrossRef.
- H. Li, L. Ma, C. Han, Z. Wang, Z. Liu, Z. Tang and C. Zhi, Nano Energy, 2019, 62, 550–587 CrossRef CAS.
- T. Zhou, N. Zhang, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC.
- W. Zhou, Y. Liu, D. Wu, P. Zhang, G. Zhang, K. Sun, B. Li and J. Jiang, Sustainable Energy Fuels, 2022, 6, 791–799 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS.
- K. Zeng, X. Zheng, C. Li, J. Yan, J.-H. Tian, C. Jin, P. Strasser and R. Yang, Adv. Funct. Mater., 2020, 30, 2000503 CrossRef CAS.
- S. Zhang, M. Chen, X. Zhao, J. Cai, W. Yan, J. C. Yen, S. Chen, Y. Yu and J. Zhang, Electrochem. Energy Rev., 2021, 4, 336–381 CrossRef CAS.
- N. Karmodak and J. K. Norskov, Angew. Chem., Int. Ed., 2023, 62, e202311113 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- L. Ma, S. Chen, Z. Pei, Y. Huang, G. Liang, F. Mo, Q. Yang, J. Su, Y. Gao, J. A. Zapien and C. Zhi, ACS Nano, 2018, 12, 1949–1958 CrossRef CAS PubMed.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 CrossRef CAS PubMed.
- X. Zhong, W. Yi, Y. Qu, L. Zhang, H. Bai, Y. Zhu, J. Wan, S. Chen, M. Yang, L. Huang, M. Gu, H. Pan and B. Xu, Appl. Catal., B, 2020, 260, 118188 CrossRef CAS.
- Z. Yang, B. Chen, W. Chen, Y. Qu, F. Zhou, C. Zhao, Q. Xu, Q. Zhang, X. Duan and Y. Wu, Nat. Commun., 2019, 10, 3734 CrossRef PubMed.
- Z. Li, S. Ji, C. Wang, H. Liu, L. Leng, L. Du, J. Gao, M. Qiao, J. H. Horton and Y. Wang, Adv. Mater., 2023, 35, 2300905 CrossRef CAS PubMed.
- M. Tong, F. Sun, G. Xing, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314933 CrossRef CAS PubMed.
- X. Li, X. Yang, L. Liu, H. Zhao, Y. Li, H. Zhu, Y. Chen, S. Guo, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2021, 11, 7450–7459 CrossRef CAS.
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Nano Energy, 2016, 25, 110–119 CrossRef CAS.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Adv. Mater., 2020, 32(8), 1907399 CrossRef CAS PubMed.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- S. Liu, M. Wang, X. Yang, Q. Shi, Z. Qiao, M. Lucero, Q. Ma, K. L. More, D. A. Cullen, Z. Feng and G. Wu, Angew. Chem., Int. Ed., 2020, 59, 21698–21705 CrossRef CAS PubMed.
- J. S. Bates, J. J. Martinez, M. N. Hall, A. A. Al-Omari, E. Murphy, Y. Zeng, F. Luo, M. Primbs, D. Menga, N. Bibent, M. T. Sougrati, F. E. Wagner, P. Atanassov, G. Wu, P. Strasser, T.-P. Fellinger, F. Jaouen, T. W. Root and S. S. Stahl, J. Am. Chem. Soc., 2023, 145, 26222–26237 CrossRef CAS PubMed.
- R. Jiang, Z. Qiao, H. Xu and D. Cao, Chin. J. Catal., 2023, 48, 224–234 CrossRef CAS.
- Y. Li, Y. Ding, B. Zhang, Y. Huang, H. Qi, P. Das, L. Zhang, X. Wang, Z.-S. Wu and X. Bao, Energy Environ. Sci., 2023, 16, 2629–2636 RSC.
- L. Wu, R. Zhao, G. Du, H. Wang, M. Hou, W. Zhang, P. Sun and T. Chen, Green Energy Environ., 2023, 8, 1693–1702 CrossRef CAS.
- Y. Zhao, H.-C. Chen, X. Ma, J. Li, Q. Yuan, P. Zhang, M. Wang, J. Li, M. Li, S. Wang, H. Guo, R. Hu, K.-H. Tu, W. Zhu, X. Li, X. Yang and Y. Pan, Adv. Mater., 2023, 36, 2308243 CrossRef PubMed.
- G. Chen, R. Lu, C. Li, J. Yu, X. Li, L. Ni, Q. Zhang, G. Zhu, S. Liu, J. Zhang, U. I. I. Kramm, Y. Zhao, G. Wu, J. Xie and X. Feng, Adv. Mater., 2023, 35, 2300907 CrossRef CAS PubMed.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- Y.-C. Wang, Y.-J. Lai, L. Song, Z.-Y. Zhou, J.-G. Liu, Q. Wang, X.-D. Yang, C. Chen, W. Shi, Y.-P. Zheng, M. Rauf and S.-G. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed.
- S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Adv. Energy Mater., 2017, 7, 1700363 CrossRef.
- J. Zhang, M. Zhang, Y. Zeng, J. Chen, L. Qiu, H. Zhou, C. Sun, Y. Yu, C. Zhu and Z. Zhu, Small, 2019, 15, 1900307 CrossRef.
- X. Zhang, X. Han, Z. Jiang, J. Xu, L. Chen, Y. Xue, A. Nie, Z. Xie, Q. Kuang and L. Zheng, Nano Energy, 2020, 71, 104547 CrossRef CAS.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y.-N. Liu, Y. Zhang, X. Ouyang and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201007 CrossRef CAS.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS.
- L. Peng, J. Yang, Y. Yang, F. Qian, Q. Wang, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- X. Wang, J. Du, Q. Zhang, L. Gu, L. Cao and H.-P. Liang, Carbon, 2020, 157, 614–621 CrossRef CAS.
- C. Hu, L. Gong, Y. Xiao, Y. Yuan, N. M. Bedford, Z. Xia, L. Ma, T. Wu, Y. Lin, J. W. Connell, R. Shahbazian-Yassar, J. Lu, K. Amine and L. Dai, Adv. Mater., 2020, 32, 1907436 CrossRef CAS PubMed.
- Q. Lai, L. Zheng, Y. Liang, J. He, J. Zhao and J. Chen, ACS Catal., 2017, 7, 1655–1663 CrossRef CAS.
- H. Jin, H. Zhou, D. He, Z. Wang, Q. Wu, Q. Liang, S. Liu and S. Mu, Appl. Catal., B, 2019, 250, 143–149 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385 CrossRef CAS PubMed.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS PubMed.
- J.-F. Gu, C. Chen, Z.-H. Zheng, J. Hang, W. Sang, J.-C. Wang, Y. Yuan, S. Chaemchuen and F. Verpoort, J. Catal., 2023, 417, 202–212 CrossRef CAS.
- T. Wu, L. Sun, F. Xu and D. Cai, J. Mater. Sci. Technol. Res., 2018, 34, 2384–2391 CrossRef CAS.
- S. Zhou, C. Chen, J. Xia, L. Li, X. Qian, F. Yin, G. Dai, G. He, Q. Chen and H. Chen, J. Mater. Sci. Technol. Res., 2024, 181, 82–90 CrossRef.
- Y. Liu, L. Zhou, S. Liu, S. Li, J. Zhou, X. Li, X. Chen, K. Sun, B. Li, J. Jiang and H. Pang, Angew. Chem., Int. Ed., 2024, 63, e202319983 CrossRef CAS PubMed.
- H. Ji, M. Wang, S. Liu, H. Sun, J. Liu, T. Qian and C. Yan, Electrochim. Acta, 2020, 334, 135562 CrossRef CAS.
- J.-W. Chen, Z. Zhang, H.-M. Yan, G.-J. Xia, H. Cao and Y.-G. Wang, Nat. Commun., 2022, 13, 1734 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03039g |
| This journal is © The Royal Society of Chemistry 2024 |