
Rational electrolyte design for Li-metal batteries operated under extreme conditions: a combined DFT, COSMO-RS, and machine learning study†
Liang-Ting
Wu
 a,
Yu-Ting
Zhan
a,
Zhong-Lun
Li
a,
Po-Ting
Chen
a,
Bing Joe
Hwang
ab and
Jyh-Chiang
Jiang
*a
a,
Yu-Ting
Zhan
a,
Zhong-Lun
Li
a,
Po-Ting
Chen
a,
Bing Joe
Hwang
ab and
Jyh-Chiang
Jiang
*a
aDepartment of Chemical Engineering, Sustainable Electrochemical Energy Development Center (SEED Center), National Taiwan University of Science and Technology, Taipei 106, Taiwan. E-mail: jcjiang@mail.ntust.edu.tw
bNational Synchrotron Radiation Research Center (NSRRC), Hsinchu 30076, Taiwan
First published on 29th May 2024
Abstract
Developing electrolytes for Li-metal batteries capable of operating under extreme conditions is a significant challenge and is often hindered by the absence of systematic solvent screening studies. In this study, 190 binary mixtures comprising 20 solvents were assessed by calculating the density functional theory (DFT) and conductor-like screening model for realistic solvents (COSMO-RS) to identify electrolytes with a wide liquid temperature range and high LiTFSI solubility. Tetramethylene sulfone (TMS) has emerged as a promising candidate because of its high boiling point and low enthalpy of fusion, which increase the bubble point and reduce the eutectic temperature in mixtures. Utilizing a machine learning model with seven σ-descriptors, Li- and TFSI-ion binding energies were accurately predicted. These binding energies were primarily influenced by strong electrostatic and van der Waals interactions. This integrated approach highlights the effectiveness of the combined DFT, COSMO-RS, and machine learning techniques for guiding electrolyte design.
1. Introduction
Lithium-metal batteries (LMBs) are used extensively in electric vehicles, consumer electronics, and battery energy storage systems (BESS).1 Various battery configurations related to Li-metal anodes have been proposed in recent decades. These include anode-free rechargeable Li-metal batteries (AFLMBs),2,3 Li–S batteries,4–6 and Li batteries with high-voltage cathode materials,7,8 which differ significantly from those of commercial Li-ion batteries (LIBs). Furthermore, the liquid range of the electrolyte is crucial for maintaining optimal electrochemical performance across various scenarios of battery operation, such as extreme weather conditions (for example, heatwaves and cold snaps) that can reduce the real range of electric vehicles,9–12 and unmanned underwater/aerial vehicles13,14 that use batteries to function at low (or high) temperatures. Commercial carbonate-based electrolytes, such as 1.0 M LiPF6 in EC/DMC and EC/EMC,15,16 are no longer the optimal electrolyte formulations for most variants. Therefore, the exploration of new electrolyte formulations that are better suited to the unique characteristics and requirements of emerging Li-metal battery configurations is necessary. For example, Li–S batteries primarily use ether solvents because they can effectively reduce soluble polysulfides during cycling.4–6,17 Meanwhile, preventing the electrolyte from freezing and evaporating during battery operation is essential. However, systematic and effective studies on the screening of binary organic solvents are limited. The choice of solvent often relies on experience and literature, leading to challenges in developing electrolytes with the desired properties for specific applications.Multi-scale modeling and simulation have greatly enhanced our understanding from the molecular to the cellular level.18 Density functional theory (DFT) is helpful for calculating the physicochemical properties of molecules and small clusters, such as the electrochemical windows of solvents,19 reaction kinetics of electrolyte decomposition,20,21 electrodes structural evolution,22 and Li-ion diffusion barriers in solid electrolytes.23,24Ab initio molecular dynamics (AIMD) simulations facilitate the investigation of interfacial reactions between electrolytes and electrodes.25–28 Molecular dynamics simulations based on (reactive) force-field have been widely used to study the solvation sheath of ions,29,30 ion transportation,31–33 SEI formation, and dendrite growth.34,35 The phase-field model can effectively simulate ionic diffusion, stress evolution, and electrodeposition during electrochemical processes.36 Additionally, temperature and current density distribution inside batteries can be simulated using the finite element method (FEM).37–39 Furthermore, machine learning (ML) shows high potential in novel electrolyte discovery and properties prediction,40 it quickly and effectively reveals composition–property relationships of materials.41 Recently, generative artificial intelligence (GAI) has been considered a promising model due to its excellent content-generating capability.42 The presence of ML/AI techniques makes highly efficient material screening possible.
Although ML is promising in materials science, constructing suitable and high-accuracy models remains challenging, particularly regarding data collection and algorithm selection. Shi et al. reported an automatic modeling method that automatically selects algorithms and optimizes hyperparameters.43 Meanwhile, the balance between the number of features and the dataset size must be carefully considered. Redundant descriptors can lead to overfitting when the model is trained on small datasets, resulting in poor predictions for data outside the training set.44 Therefore, feature selection and transformation are frequently used to reduce the number of features and address overfitting.45 Additionally, K-fold cross-validation (K-fold CV) divides the data into K subsets, with one subset assigned as the test set and the remaining subsets as the training set, repeated K times.45 The K-fold CV method ensures that the results are independent of how the training and test data sets are selected.
In this study established a comprehensive thermodynamic database for binary mixtures of 20 commonly used organic solvents in Li batteries using DFT calculations and the conductor-like screening model for realistic solvents (COSMO-RS) model.46–48 The solvents included cyclic carbonates (EC, PC, FEC, VC),49,50 linear carbonates (DMC, DEC, EMC, FEMC),49,51,52 cyclic ester (GBL),49 linear ester (MA, EA, MP),49,53 cyclic ether (DOL, THF),49 linear ether (DME, DEE, FDMB),29,49,54,55 sulfone (TMS),56,57 sultone (PES),58 and amide (DMAC).59 The liquid range of 190 binary mixtures composed of arbitrary pairs of these solvents was examined. Subsequently, the solubility of the lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) salt in the binary mixtures was calculated at the eutectic composition and 298.15 K. Additionally, combined with ML,60–64 the screening surface charge densities (σ-profiles) of 20 solvents and 3 diluents (TTE,65 BTFE,66 TFEO67) were utilized as descriptors to predict the binding energy of the Li- and TFSI-ions with solvents and diluents. The ion binding ability of solvents plays a crucial role in investigating the solvation structure of ions. Additionally, the resulting solvation sheath significantly affects the solid-electrolyte interphase (SEI) formation, Li+ transference number, and overall electrochemical performance of batteries.68 By employing combined DFT, COSMO-RS, and ML techniques, this study significantly enhances the understanding of binary electrolytes and provides computational insights and guidelines for electrolyte design in LMBs operating under extreme conditions.
2. Computational methods
2.1. DFT calculation
The solvent molecule geometries were optimized using DFT calculations performed using the Amsterdam Density Functional (ADF)69,70 module within the Amsterdam Modeling Suite (AMS 2021.102). The Minnesota hybrid meta-generalized gradient M06-2X functional71,72 and the TZP basis set (Triple Zeta Slater type orbitals with a polarization function)73 were employed. To validate the optimized structures, frequency calculations were conducted to ensure they exhibited full positive vibrational frequencies. Subsequently, the input files for the COSMO-RS calculations, denoted as *. coskf files, were generated using DFT calculations in the ADF module.742.2. COSMO-RS calculation
The vapor–liquid equilibria (VLE) and solid–liquid equilibria (SLE) calculations for all binary mixtures, as well as the solubility of LiTFSI, were performed using the COSMO-RS module within AMS.74,75 This computational approach considered the melting point, boiling point, and enthalpy of fusion of the pure solvents, which are listed in Table S1.† For calculating LiTFSI solubility, its melting point, enthalpy of fusion, and density were set at 507.15 K,76 13.205 kJ mol−1,77 1.33 g cm−3,78 respectively. To screen all binary mixtures efficiently, the Python Library for Automating Molecular Simulation (PLAMS)79 was used to automate the process of calling the AMS to conduct a series of calculations and export the results.2.3. Binding energies calculation
The molecular structures of all the compounds were optimized and their full positive vibrational frequencies were verified using DFT calculations in the Gaussian 09 package at the M06-2X/6-311 + g(d,p) level of theory. The binding energies of Li- and TFSI-ions when interacting with a molecule were computed using the following formula:| EB,ion = Ecomplex − Emolecule − Eion |
2.4. Multiple linear regression machine learning models
This study used Scikit-learn, a Python machine learning package, to implement the multiple linear regression model.81 To mitigate overfitting, the σ-descriptors were segmented into seven regions and their sums were computed. All descriptors were standardized to normalize features within the same range. Standardization involves shifting the values by subtracting the mean of the feature and then dividing it by the standard deviation of that feature, as shown in the following equation:where Sij represents the i-th σ-descriptor (Si) of molecule j (i ranges from 1 to 7 and j ranges from 1 to 23), Si,Mean and Si,STD represent the mean value and standard deviation of the Si descriptor across the 23 molecules, and zij represents the z-score of corresponding Sij. For cross validation, a K-fold cross-validation (K-fold CV) strategy was employed, in which the original data were randomly divided into K subsets. One subset was used as the test set to evaluate the performance of the model and the remaining subsets were used for training. This process was repeated K times to ensure that each subset served as the test set. Test predictions from each iteration of the K-fold CV were averaged to assess the overall performance of the model. In this study, the leave-one-out cross-validation (LOOCV) method was implemented, where K is the number of samples (23 in this study). A multiple linear regression model was used, as shown by the following equation:

where EB represents the predicted binding energy, Ci represents the i-th coefficient of regression, zi represents the z-score of i-th σ-descriptor (Si), and the binding energy includes the Li-ion binding energy (EB,Li) and TFSI-ion binding energy (EB,TFSI).

3. Results and discussion
3.1. The phase diagram and liquid range of binary mixtures
To compute the phase diagram of the binary mixtures, the structures of 20 common organic solvents were optimized using the Amsterdam Modeling Suite (AMS); a σ-profile of each molecule was obtained. The optimized structures of these molecules are shown in Fig. S1.† Experimental data, including the melting point, boiling point, and enthalpy of fusion of each solvent, were collected from handbooks, literature sources, and chemical suppliers. These data were used as inputs for the COSMO-RS calculations to ensure highly accurate results. In instances where experimental data were unavailable for certain molecules, the values predicted from the quantitative structure–property relationship (QSPR) model in AMS were utilized. The physicochemical properties of the 20 solvents used as the inputs are listed in Table S1.†To validate the accuracy of the calculated phase diagram, a comparison with experimental data is essential. However, experimental phase diagram data of solvent mixtures used in electrolytes, including both solid–liquid and vapor–liquid equilibria, is still lacking. Therefore, we chose the common solvent mixtures EC/DMC and EC/PC, which are well-known in carbonate-based liquid electrolytes and whose phase diagrams have been extensively studied by Ding and Qian et al.82–84 The calculated phase diagram of the EC/DMC mixture is shown in Fig. 1a. The green solid line represents the solid–liquid equilibrium curve, with the lowest point being the eutectic point.85 The calculated eutectic temperature is 256.54 K, and the eutectic composition consists of 0.287 mole fractions of EC and 0.713 mole fractions of DMC. The region below the green dashed line (T < 256.54 K) represents the solid state. The solid and liquid states coexist in the region between the green solid line and the green dashed line. When the mole fraction of EC is greater than 0.287, EC melts and remains in the liquid state, coexisting with solid-state DMC. Conversely, when the mole fraction of EC is less than 0.287, DMC melts and mixes with solid-state EC. The EC/DMC mixture remains in a liquid state between the green solid line and the red solid line, which is ideal for electrolyte applications due to its high ionic conductivity and safety. In the region between the red and blue solid lines, part of the EC/DMC mixture is in a liquid state, while the other part is in a gaseous state. The red and blue solid lines represent the bubble and dew point curves, respectively. Above the red solid line, all EC and DMC are in the gaseous state. We aim to identify mixtures with an extensive liquid state range, specifically the region between the red and green solid lines. Moreover, the calculated bubble-point temperature of the EC/DMC mixture at eutectic composition was 374.68 K, resulting in a liquid range from 256.54 to 374.68 K. To compare with experimental values, we extracted solid–liquid equilibria data from ref. 82 (green dots) and vapor–liquid equilibria data from ref. 83 (red and blue dots), as represented in Fig. 1a. Our calculated results match the experimental data excellently. For the second example, the calculated phase diagram of the EC/PC mixture is depicted in Fig. 1b. The eutectic and bubble-point temperatures were calculated to be 219.03 K and 515.00 K, respectively, with the eutectic composition consisting of 0.111 mole fraction EC and 0.889 mole fraction PC. In contrast to the EC/DMC mixture, which displays a distinct two-phase region between the bubble and dew point curves, the bubble and dew point curves of EC/PC overlap significantly because of their similar chemical structures and intermolecular interactions. We further compared the phase diagram with data from ref. 84, finding that our calculations were highly consistent with the experimental data. This suggests that COSMO-RS calculations provide reliable results even when only data for pure compounds are available. Subsequently, the phase diagrams of 190 binary mixtures, each composed of arbitrary pairs selected from 20 solvents, were calculated. All phase diagrams are summarized in the archived files in the ESI.†
 | ||
| Fig. 1 The phase diagram shows (a) EC/DMC and (b) EC/PC binary mixtures. The green line represents the solid–liquid equilibria curve, while the red and blue lines denote the bubble and dew point curves, respectively, in vapor–liquid equilibria. The green, red, and blue dots represent experimental data previously published in ref. 82–84. (c) The calculated bubble point temperature in the upper triangular matrix and eutectic temperature in the lower triangular matrix for 190 binary mixtures at their eutectic compositions. | ||
Fig. 1c illustrates the calculated bubble points and eutectic temperatures of the 190 binary mixtures at their respective eutectic compositions, represented by the upper and lower triangular matrices, respectively. The five binary mixtures with the highest bubble-point temperatures were TMS/PES (555.48 K), EC/TMS (550.37 K), PC/TMS (541.88 K), FEC/TMS (538.48 K), and EC/PES (522.31 K). These high bubble-point temperatures were primarily attributed to the high boiling temperatures of TMS (559.15 K), PES (529.75 K), EC (519.15 K), PC (514.75 K), and FEC (485.15 K), because they were the solvents with the highest boiling points. The high boiling points exhibited by sulfone and sultone compounds (TMS and PES) are ideal for extending the bubble-point temperature of binary mixtures. All bubble-point temperatures of TMS-containing mixtures were higher than 360 K. However, linear esters (MA, EA, and MP) and cyclic ethers (DOL and THF) are not suitable solvents for batteries operating in high-temperature environments because of their generally low bubble-point temperatures. Additionally, the boiling points of these pure compounds were lower than those of the other compounds in the solvent set.
The five binary mixtures with the lowest eutectic temperatures were THF/TMS (147.44 K), MA/TMS (147.91 K), DOL/TMS (148.02 K), MA/THF (151.18 K), and DOL/THF (151.24 K). This was observed because THF, MA, and DOL have low melting points (164.77 K, 174.90 K, and 175.94 K, respectively), whereas TMS has the lowest enthalpy of fusion at 1.372 kJ mol−1. In solid–liquid equilibria, a solid-state compound dissolves into a liquid-state compound; therefore, the enthalpy of fusion of the solid-state compound is crucial in a eutectic system. TMS, which has the lowest enthalpy of fusion, is likely highly soluble in other solvents, resulting in a very low eutectic temperature despite its relatively high melting point (301.60 K). All eutectic temperatures of TMS-containing mixtures are lower than 243 K. This suggests that TMS, DOL, and MA are ideal candidates for reducing the eutectic temperature of binary mixtures because of their low enthalpies of fusion (<8 kJ mol−1).
3.2. The solubility of LiTFSI salt in binary mixtures
An ideal solvent mixture must exhibit excellent solubility of Li salts, with commercial electrolytes typically dissolving a 1 M salt in binary solvent mixtures. To further enhance electrochemical performance, the salt concentration must be maximized in (localized) high-concentration electrolytes.86 Therefore, the LiTFSI salt was selected as a representative because of its widespread use in LMBs; the solubility of the LiTFSI salt in 190 binary mixtures was calculated. The solubility calculation is based on the following equation:where xLiTFSI and γLiTFSI represent the molecule fraction and activity coefficient of LiTFSI dissolved in the electrolyte mixture. The activity coefficient of LiTFSI can be solved by the COSMO-RS model, which is dependent on temperature, pressure, and composition. ΔfusHLiTFSI represent the enthalpy of fusion of LiTFSI; Tm,LiTFSI represent the melting point of LiTFSI. Based on this equation, LiTFSI exhibits higher solubility when its activity coefficient in the solvent mixture is lower. This implies that the chemical potential of LiTFSI in the mixture should be minimized, and the interaction between LiTFSI and the solvent mixture should be maximized. The activity coefficient of LiTFSI in mixtures can be calculated using the COSMO-RS model, which can then be used to calculate the solubilities. The melting point, enthalpy of fusion, and density of LiTFSI were 507.15 K,76 13.205 kJ mol−1,77 and 1.33 g cm−3,78 respectively. These values are essential for computing the solubility of the solid solute. The solubility of the salt can be influenced by temperature and the composition of the binary mixture. To simplify the comparison, the temperature at 298.15 K and the eutectic composition of each mixture was adopted as the examination condition. Fig. 2a summarizes the calculated LiTFSI solubility and mole fraction of solvent A, which represents the component in the column index, in the binary mixtures at the eutectic composition; the dark red and blue colors indicate that the binary mixture is close to the component in the row and column index, respectively. The EC/FEC, FEC/VC, and FEC/PES mixtures exhibited low LiTFSI solubilities (<1 M); therefore, they were unsuitable as high-concentration electrolytes. Similarly, FEMC and FDMB exhibited weak LiTFSI dissolution abilities, as evidenced by the 52.6% FEMC- and FDMB-containing mixtures with LiTFSI solubility lower than 2 M. This can be attributed to weak interactions between the fluorinated solvents. In contrast, the cyclic ethers displayed excellent LiTFSI dissolution abilities, as all DOL- and THF-containing mixtures had solubilities exceeding 2.5 M.

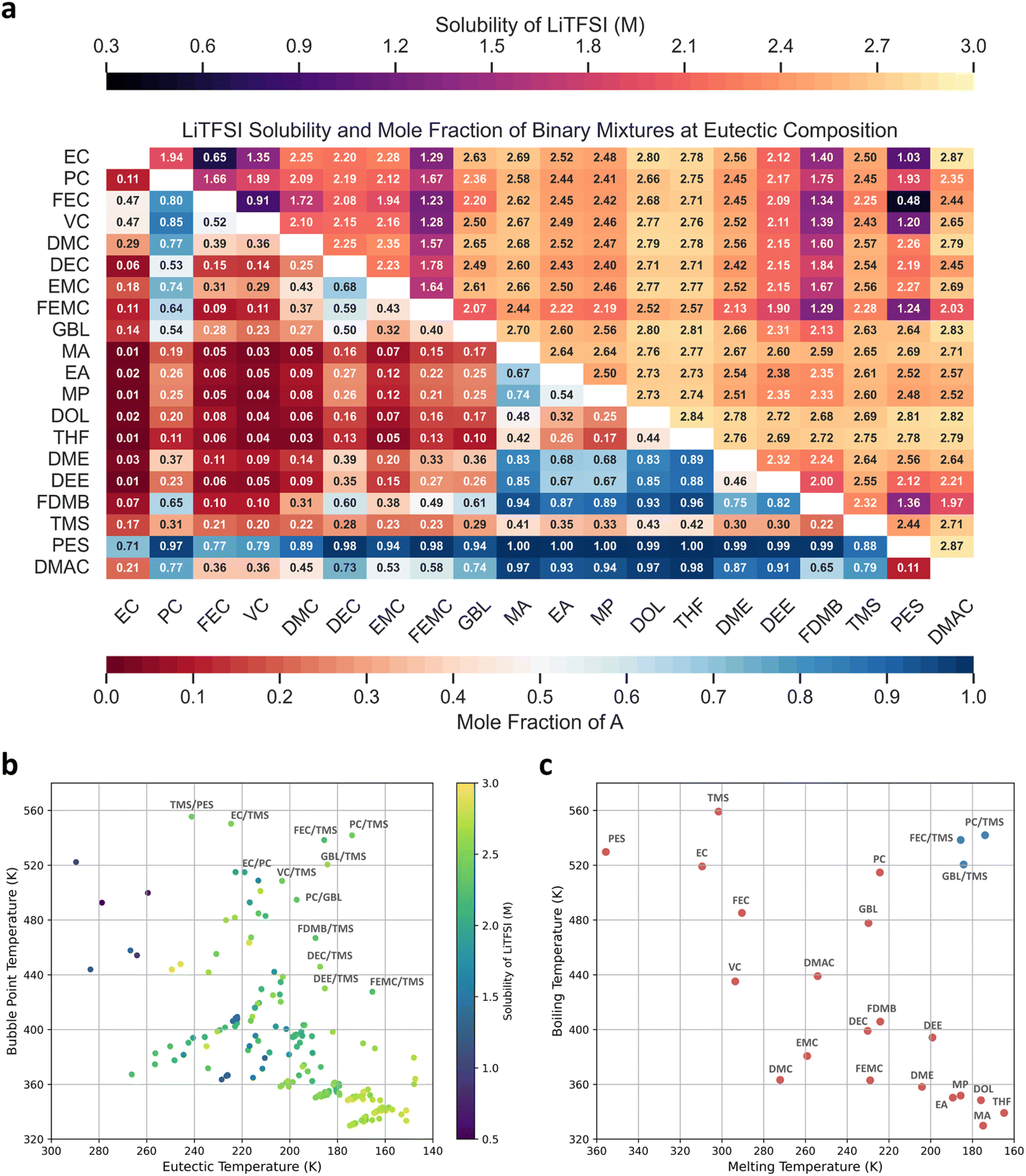 | ||
| Fig. 2 (a) The calculated LiTFSI solubility (upper triangular matrix) and mole fraction of solvent A (lower triangular matrix) for 190 binary mixtures at eutectic composition. (b) The eutectic and bubble point temperature of 190 binary mixtures. The color of each point represents the solubility of LiTFSI at 298.15 K and the eutectic composition. (c) The melting and boiling temperatures of 20 organic solvents, with three selected binary mixtures showing a wide liquid range, are included for comparison. | ||
To further screen for an electrolyte that can maintain the liquid phase under extreme conditions and exhibit excellent LiTFSI solubility, all relevant thermodynamic information was integrated into Fig. 2b. Each point on the graph represents a binary mixture. The PC/TMS mixture is observable in the upper-right corner of Fig. 2b owing to its high bubble-point temperature (541.88 K), low eutectic temperature (173.93 K), and high LiTFSI solubility (2.45 M). The mole fractions of PC and TMS at eutectic compositions were 0.308 and 0.692, respectively. Compared with its pure components (Fig. 2c), the bubble-point temperature of the mixture was between the boiling points of PC (514.75 K) and TMS (559.15 K). However, the eutectic temperature of the mixture was lower than those of PC (224.35 K) and TMS (301.60 K), indicating that PC/TMS was a representative eutectic system. Additionally, FEC/TMS and GBL/TMS exhibited an extensive liquid range, from 185.61 to 538.48 K and 184.18 to 520.54 K, respectively. They exhibited relatively high LiTFSI solubilities of 2.25 M (FEC/TMS) and 2.63 M (GML/TMS), highlighting their suitability for extreme temperature applications. Therefore, the following electrolyte design strategies were proposed for applications in extreme conditions: for electrolytes that need to operate at high temperatures, consider adding compounds with high boiling temperatures, such as TMS and PES. For electrolytes required to operate at low temperatures, compounds with low enthalpies of fusion, such as TMS, form eutectic systems and extend the liquid range. Adding nonvolatile Li salt to the binary mixture increased the bubble point temperature and reduced the eutectic temperature. Consequently, a wider liquid range was expected in the electrolytes than in the solvent mixtures. However, extending the liquid range in electrolytes is beyond the scope of this study, and assessing this extension using the current COSMO-RS method remains challenging.
3.3. Sigma-profile analysis
The σ-profiles of compounds included the charge density distribution information of molecules. As reported by Alkhatib et al., the intensity of a specific σ interval indicates the surface area of the molecule with that strength of screening charge density.87 Different σ intervals correspond to various interactions, such as van der Waals forces, electrostatic interactions, and hydrogen bonding. Therefore, analyzing the σ-profile can provide insights into the properties of a specific compound. We first examine the dependence of σ-profiles on the bin size of screening charges, as shown in Fig. S2.† Taking EC as an example, its σ-profiles were constructed using 11, 31, 51, 71, and 91 bins. With 11 points, only two broad peaks are present in the positive and negative regions, respectively, and the profile details are not visible. With 31 points, the peak around −0.005 e Å−2 splits into two peaks, revealing more features of the molecule. The σ-profiles gradually converge as the number of points increases to 51 and 71, suggesting that 51 data points are sufficient to represent the screening charge of the molecule. If the number of points is further increased to 91, the profile becomes overly complex and difficult to interpret. Therefore, we adopt the default AMS output format, which uses 51 points from 0.025 to −0.025 e Å−2. Fig. 3 shows the σ-profiles of 20 solvents, complemented by 3 diluent molecules commonly found in localized high-concentration electrolytes. The COSMO surface exhibited a screening charge density, which was opposite to that of the molecule. In general, electrons are predominantly distributed on O atoms in solvents, resulting in negative charge accumulation, whereas H atoms on C carry a positive charge. Regarding the COSMO surface and σ-profiles, O atoms were surrounded by a positive screening charge, whereas H atoms were encircled by a negative screening charge. Fig. 3a–c show the σ-profiles of carbonate and ester molecules, which exhibited a peak at approximately 0.008–0.012 e Å−2. This was indicative of the characteristic O of carbonyl groups. In contrast, the ether molecules exhibited a peak at approximately 0.015 e Å−2 (Fig. 3d and e), indicating a higher charge density on the O than on the carbonyl group. Additionally, sulfone (TMS) and sultone (PES) exhibited distinct peaks at approximately 0.010–0.013 e Å−2 (Fig. 3e), which were similar to the profiles of the carbonate and ester molecules. PES exhibited a downshift in the screening charge scale compared with that of TMS, which can be attributed to the presence of three O atoms sharing a positive screening charge, whereas TMS had only two oxygen atoms. Additionally, DMAC exhibited a distinct peak at approximately 0.015 e Å−2, which extended to 0.020 e Å−2, indicating that DMAC had the highest screening charge density in the amide group compared with the other solvents. | ||
| Fig. 3 The σ-profiles and the COSMO surface of (a–e) 20 common solvents and (f) 3 diluents. The COSMO surface is representing by red (positive) and blue (negative) colors, corresponding to the screening charge density in σ-profiles. The sign of the screening charge density is opposite to the molecular charge density, where positive screening charge surrounds oxygen atoms and negative screening charge surrounds hydrogen atoms. | ||
The diluents exhibited a prominent peak at approximately 0.004 e Å−2 (Fig. 3f), which was attributed to the presence of fluorine atoms. In general, the involvement of these highly fluorinated molecules in the Li-ion solvation sheath is minimal, as shown by the σ-profiles of the diluents. However, F atoms dominated the charge-density distribution, overshadowing the solvation ability of ethereal O, resulting in a weaker Li-ion solvation capacity. Other fluorinated molecules, such as FEC, FEMC, and FDMB, exhibited a characteristic peak corresponding to F atoms in their σ-profiles.
To further construct an ML model, data collection and preprocessing are crucial. Recently, Jain et al. reported a method for extracting data from research papers using large language models (LLMs).88 Pande et al. introduced MoleculeNet, a large-scale benchmark for molecular ML that incorporates multiple public datasets.89 Shi et al. introduced FFMDFPA, a framework for extracting, transforming, and sharing data, which greatly improves data accessibility, interoperability, and findability.90 Given that we aim to examine the potential of σ-profiles used in molecular ML, the dataset was constructed with only 23 molecules in this work. The original σ-profiles consist of 51 data points within the range of 0.025 to −0.025 e Å−2, providing 51 descriptors for constructing the ML model. However, this can lead to overfitting since the dataset contains only 23 molecules. Therefore, we divided the σ-profile into seven characteristic regions within the range of −0.021 to 0.020 e Å−2 (intervals of 0.005 e Å−2) to reduce the dimensionality of the feature space and further train the ML model.87 These σ-descriptors, denoted as S1 to S7, represent the sum of the intensity of σ-profiles within each characteristic region. The coverage ranges of the screening charge density were as follows: S1 (−0.021 to −0.016), S2 (−0.015 to −0.010), S3 (−0.009 to −0.004), S4 (−0.003 to 0.002), S5 (0.003 to 0.008), S6 (0.009 to 0.014), and S7 (0.015 to 0.020). The seven σ-descriptors for the 23 molecules are listed in Table 1. Descriptors S1–S2 represent the strong electrostatic interactions with anions, whereas descriptors S6–S7 represent electrostatic interactions with cations. In contrast, descriptors S3–S5 characterize weak interactions because they cover regions of low screening charge density. The σ-descriptors were used to interpret the solubility of LiTFSI. Mixtures containing ethers and amides, such as DOL, THF, DME, DEE, and DMAC, exhibit excellent solubility in LiTFSI. This was observed via their high S7 values ranging from 4.37 to 12.00, which was conducive to solvated Li-ions. In contrast, fluorinated solvents, such as FEMC and FDMB, exhibited low S2, S6, and S7 values, indicating weak ion stabilization abilities.
| Compounds | S 1 | S 2 | S 3 | S 4 | S 5 | S 6 | S 7 |
|---|---|---|---|---|---|---|---|
| EC | 0.00 | 1.60 | 49.87 | 14.93 | 17.33 | 26.50 | 0.00 |
| PC | 0.00 | 0.86 | 54.69 | 29.24 | 16.50 | 26.90 | 0.00 |
| FEC | 0.00 | 22.25 | 23.81 | 14.15 | 39.15 | 17.93 | 0.00 |
| VC | 0.00 | 14.28 | 19.63 | 30.26 | 23.76 | 16.74 | 0.00 |
| DMC | 0.00 | 0.00 | 49.00 | 36.20 | 14.58 | 24.17 | 0.16 |
| DEC | 0.00 | 0.00 | 59.90 | 67.43 | 14.27 | 25.05 | 0.32 |
| EMC | 0.00 | 0.00 | 55.19 | 50.73 | 14.34 | 24.73 | 0.31 |
| FEMC | 0.00 | 11.64 | 40.50 | 45.07 | 51.67 | 17.40 | 0.00 |
| GBL | 0.00 | 0.00 | 53.44 | 30.37 | 8.65 | 24.04 | 2.14 |
| MA | 0.00 | 0.00 | 43.29 | 39.89 | 9.82 | 19.67 | 1.62 |
| EA | 0.00 | 0.00 | 47.47 | 56.16 | 11.52 | 18.89 | 1.88 |
| MP | 0.00 | 0.00 | 46.44 | 52.93 | 13.63 | 18.21 | 2.17 |
| DOL | 0.00 | 0.00 | 47.04 | 28.57 | 9.99 | 15.27 | 4.37 |
| THF | 0.00 | 0.00 | 30.49 | 65.04 | 6.49 | 5.58 | 5.95 |
| DME | 0.00 | 0.00 | 50.59 | 62.28 | 11.42 | 11.48 | 7.08 |
| DEE | 0.00 | 0.00 | 53.96 | 96.01 | 18.03 | 9.98 | 8.26 |
| FDMB | 0.00 | 2.00 | 69.49 | 51.16 | 61.42 | 14.73 | 2.51 |
| TMS | 0.00 | 0.67 | 63.83 | 33.19 | 6.87 | 34.60 | 0.02 |
| PES | 0.00 | 17.26 | 37.27 | 23.02 | 16.64 | 34.64 | 0.00 |
| DMAC | 0.00 | 0.00 | 48.02 | 58.57 | 7.81 | 6.41 | 12.00 |
| TTE | 0.51 | 28.48 | 15.73 | 47.97 | 108.24 | 0.04 | 0.00 |
| BTFE | 0.00 | 21.72 | 23.61 | 44.00 | 81.77 | 3.88 | 0.00 |
| TFEO | 0.00 | 29.79 | 34.88 | 73.13 | 114.32 | 7.71 | 0.00 |
3.4. Prediction of Li+ and TFSI− binding energies by machine learning
Although the thermodynamic properties of the binary mixtures were evaluated and several ideal candidates with a wide liquid range and high LiTFSI solubility were identified, the solvation structure of the ions in the electrolyte remains unknown. Further investigation of these aspects is required to comprehensively understand their suitability for practical applications in LMBs. The interaction between Li-ions and solvents is crucial in determining key aspects of battery performance, such as the solvation structures of Li-ions, Li-ion transport within the electrolyte, SEI formation at the electrodes, and the morphology of the plated Li-metal. A recent study examined the interaction between anions and polymers in gel-polymer electrolytes, which significantly affects salt dissociation and the Li-ion transference number.68 However, assessing the binding energies between solvents and ions requires time-consuming DFT calculations, particularly for large ion clusters and intricate solvation structures. Moreover, interpreting trends in the calculated results can be challenging when relying solely on a few physicochemical properties. Hence, the σ-descriptors were leveraged along with ML techniques to predict the binding energy between Li-/TFSI-ions and molecules. The binding energies were determined using DFT calculations. Fig. 4a shows a schematic of the ion-binding energy prediction strategy. The seven σ-descriptors (S1 to S7) of the 23 molecules served as the independent variables, whereas the calculated binding energies of Li- and TFSI-ions served as dependent variables in the multiple linear regression ML models. The Li-ion binding energy of DMAC (−1.012 eV) was higher than those of the other molecules (Fig. 4b), corresponding to the highest S7 value (12.00). This was followed by ethers, sulfones, and esters, showcasing a similar trend to that of the σ-profiles (Fig. 3). Carbonate solvents exhibit relatively low binding energies because of their low donor numbers.63 In particular, the electron density of the carbonyl O was reduced and delocalized by two connected ethereal O atoms in the carbonate group. In contrast, TFEO exhibited the highest TFSI-ion binding energy (−0.380 eV), which was attributed to its high S5 value (114.32). This was followed by TTE, which exhibited a S5 value of 108.24 and a TFSI-ion binding energy of −0.297 eV. This was observed because TFSI is an ionic liquid anion with a highly delocalized negative charge and the large surface area of TFEO/TTE provides strong van der Waals and moderate electrostatic interactions. | ||
| Fig. 4 (a) A schematic illustration of the strategy for predicting Li- and TFSI-ions binding energies using ML. (b) The calculated Li- and TFSI-ions binding energy of 20 solvents and 3 diluents. (c) The coefficient of the seven σ-descriptors in multiple linear regression. (d) A comparison between the binding energies of Li- and TFSI-ions calculated by DFT and predicted by the multiple linear regression ML model. | ||
To enhance the understanding of the ion binding energies between ions and solvents, multiple linear regression machine-learning models trained on cross-validated folds were utilized. The K-fold cross-validation strategy was adopted to investigate the relationship between σ-descriptors and binding energies. Fig. 4c depicts the corresponding coefficients of the seven σ-descriptors; the mean values and standard deviations are listed in Table 2. For Li-ions, the mean coefficient of S1–S3 was positive (0.035, 0.112, and 0.0485, respectively). Given that the binding energies were negative, this implied that higher values of S1–S3 were unfavorable for the Li-ion binding energy. The low coefficient of S4 (−0.0006) indicates that the influence of the S4 descriptor is negligible for predicting the Li-ion binding energies. However, the coefficients for S5 (−0.1025), S6 (−0.0686), and S7 (−0.0942) were negative, implying that higher values of the S5–S7 descriptors were associated with higher Li-ion binding energies. The S5–S7 descriptors were indicative of screening charge densities ranging from 0.003–0.020 e Å−2. Intensities within this range significantly influenced the binding energies of the Li-ions. All positive screening charges—negative charges within the molecule—contribute to strong electrostatic and dative interactions. With respect to the TFSI-ion binding energies, the coefficients of the σ-descriptors, excluding S5, were relatively low. This may be due to a compromise in the regression model. However, the coefficient for S5, −0.063, indicates that higher S5 values lead to higher TFSI-ion binding energies because higher S5 values signify a molecule with a larger van der Waals surface. The results indicate that strong electrostatic interactions between the TFSI anion and the partial positive charge density region of the solvent molecule were not observed; however, weak van der Waals interactions were dominant. Additionally, the negative coefficients of S5–S7 confirmed the electrostatic interaction between the Li cation and the high-electron-density region of the molecule. The model was validated by comparing the binding energies predicted using the ML model with those calculated by DFT (Fig. 4d). The gray diagonal line represents the ideal scenario, where the predictions perfectly match the DFT calculations. Overall, the model successfully predicted the binding energies of Li- and TFSI-ions with low average standard deviations for Li (0.00873 eV) and TFSI (0.00492 eV). This level of accuracy is sufficient for early-stage evaluations of the binding abilities. Highly accurate potential molecules can be further calculated using DFT at a higher level of theory, if necessary.
| σ-descriptors | C Li,Mean | C Li,STD | C TFSI,Mean | C TFSI,STD |
|---|---|---|---|---|
| S 1 | 0.035 | 0.0094 | 0.0107 | 0.003 |
| S 2 | 0.112 | 0.0226 | −0.0179 | 0.0091 |
| S 3 | 0.0485 | 0.0218 | 0.0056 | 0.0085 |
| S 4 | −0.0006 | 0.0072 | 0.0233 | 0.0026 |
| S 5 | −0.1025 | 0.0344 | −0.063 | 0.0127 |
| S 6 | −0.0686 | 0.0295 | −0.0275 | 0.0105 |
| S 7 | −0.0942 | 0.0227 | −0.0173 | 0.0059 |
Moreover, this combined approach using DFT, COSMO-RS, and ML demonstrates high potential for evaluating phase diagrams, salt solubility, and ion binding energies, all of which are important properties of electrolytes. Our goal is to apply this computational protocol to other metal-ion battery fields, such as Na-ion, Mg-ion, Ca-ion, and Zn-ion batteries, to inspire relevant studies in those areas.
4. Conclusion
A thermodynamic database comprising 190 binary mixtures was established and their liquid ranges and LiTFSI solubilities were analyzed using DFT and COSMO-RS calculations. These results emphasize the exceptional properties of PC/TMS, FEC/TMS, and GBL/TMS, which exhibited wide liquid ranges and high LiTFSI solubilities in their eutectic compositions. To enhance the bubble-point temperature, solvents with high boiling points should be considered. Conversely, to lower the eutectic temperature, the selection of compounds with low enthalpies of fusion is ideal for the formation of a eutectic binary system. Furthermore, seven σ-descriptors (S1 to S7), derived from the screening charge density of molecules, in conjunction with ML techniques were employed to predict the binding energies of Li- and TFSI-ions. The multiple linear regression machine-learning models were trained using data from 23 molecules. Regression analysis showed that a high screening charge density predominantly influenced the binding energy of the Li-ions, whereas the binding energy of the TFSI-ions was primarily determined by the van der Waals surface area. The model exhibited a low average standard deviation (<0.01 eV) that meets the required accuracy for early stage assessment of solvent solvation abilities. This study significantly advances the understanding of the thermodynamics of binary mixtures and streamlines the screening process for promising electrolytes that are suitable for extreme operating conditions. Further evaluation of the electrochemical and interfacial properties is essential to complement the electrolyte design guidelines.Data availability
Data supporting the findings of this study are available from the corresponding author upon request.Author contributions
Liang-Ting Wu: methodology, visualization, data curation, writing-original draft, review & editing. Yu-Ting Zhan: methodology, software, investigation, data curation. Zhong-Lun Li: software, investigation, data curation. Po-Ting Chen: data curation, visualization. Bing Joe Hwang: conceptualization, funding acquisition, writing-review & editing. Jyh-Chiang Jiang: conceptualization, supervision, funding acquisition, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science and Technology Council, Taiwan (NSTC 112-2923-M-011-002, 112-2923-E-011-005, 112-2639-E-011-001-ASP), Sustainable Electrochemical Energy Development Center (SEED Center) supported by the Ministry of Education (MOE), Taiwan. The authors thank the Taiwan National Center of High-Performance Computing (NCHC) for computing resources.References
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- C.-J. Huang, B. Thirumalraj, H.-C. Tao, K. N. Shitaw, H. Sutiono, T. T. Hagos, T. T. Beyene, L.-M. Kuo, C.-C. Wang, S.-H. Wu, W.-N. Su and B. J. Hwang, Nat. Commun., 2021, 12, 1452 CrossRef CAS PubMed.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- J. J. Zak, S. S. Kim, F. A. L. Laskowski and K. A. See, J. Am. Chem. Soc., 2022, 144, 10119–10132 CrossRef CAS PubMed.
- Y. Xia, J. Zheng, C. Wang and M. Gu, Nano Energy, 2018, 49, 434–452 CrossRef CAS.
- Q. Li, D. Ning, D. Wong, K. An, Y. Tang, D. Zhou, G. Schuck, Z. Chen, N. Zhang and X. Liu, Nat. Commun., 2022, 13, 1123 CrossRef CAS PubMed.
- C.-Y. Wang, G. Zhang, S. Ge, T. Xu, Y. Ji, X.-G. Yang and Y. Leng, Nature, 2016, 529, 515–518 CrossRef CAS PubMed.
- Y. Ji, Y. Zhang and C.-Y. Wang, J. Electrochem. Soc., 2013, 160, A636 CrossRef CAS.
- J. R. M. D. Reyes, R. V. Parsons and R. Hoemsen, IEEE Trans. Veh. Technol., 2016, 65, 4016–4022 Search PubMed.
- J. Lindgren and P. D. Lund, J. Power Sources, 2016, 328, 37–45 CrossRef CAS.
- S. W. Paek, S. Kim and C. V. R. Rayappan, 2019.
- Z. Luan, Y. Qin, B. Hu, W. Zhao and C. Wang, J. Energy Storage, 2023, 59, 106479 CrossRef.
- D. Guyomard and J. M. Tarascon, J. Electrochem. Soc., 1993, 140, 3071 CrossRef CAS.
- S. S. Zhang, T. R. Jow, K. Amine and G. L. Henriksen, J. Power Sources, 2002, 107, 18–23 CrossRef CAS.
- J. Shim, K. A. Striebel and E. J. Cairns, J. Electrochem. Soc., 2002, 149, A1321 CrossRef CAS.
- S. Shi, J. Gao, Y. Liu, Y. Zhao, Q. Wu, W. Ju, C. Ouyang and R. Xiao, Chin. Phys. B, 2016, 25, 018212 CrossRef.
- D. Wang, T. He, A. Wang, K. Guo, M. Avdeev, C. Ouyang, L. Chen and S. Shi, Adv. Funct. Mater., 2023, 33, 2212342 CrossRef CAS.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708–11718 CrossRef CAS PubMed.
- R. C. Longo, F. T. Kong, S. Kc, M. S. Park, J. Yoon, D. H. Yeon, J. H. Park, S. G. Doo and K. Cho, Phys. Chem. Chem. Phys., 2014, 16, 11233–11242 RSC.
- X. He, Y. Zhu and Y. Mo, Nat. Commun., 2017, 8, 15893 CrossRef CAS PubMed.
- W. D. Richards, Y. Wang, L. J. Miara, J. C. Kim and G. Ceder, Energy Environ. Sci., 2016, 9, 3272–3278 RSC.
- E. K. W. Andersson, L.-T. Wu, L. Bertoli, Y.-C. Weng, D. Friesen, K. Elbouazzaoui, S. Bloch, R. Ovsyannikov, E. Giangrisostomi, D. Brandell, J. Mindemark, J.-C. Jiang and M. Hahlin, J. Mater. Chem. A, 2024, 12, 9184–9199 RSC.
- L.-T. Wu, S. Nachimuthu, D. Brandell, C.-N. Tsai, P.-H. Wang, Y.-W. Li and J.-C. Jiang, Mater. Today Phys., 2023, 38, 101253 CrossRef CAS.
- L.-T. Wu, E. K. W. Andersson, M. Hahlin, J. Mindemark, D. Brandell and J.-C. Jiang, Sci. Rep., 2023, 13, 9060 CrossRef CAS PubMed.
- L.-T. Wu, S. Nachimuthu, D. Brandell and J.-C. Jiang, Batteries Supercaps, 2022, 5, e202200088 CrossRef CAS.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- Y. Lin, Z. Yu, W. Yu, S.-L. Liao, E. Zhang, X. Guo, Z. Huang, Y. Chen, J. Qin, Y. Cui and Z. Bao, J. Mater. Chem. A, 2024, 12, 2986–2993 RSC.
- M. M. Islam, A. Ostadhossein, O. Borodin, A. T. Yeates, W. W. Tipton, R. G. Hennig, N. Kumar and A. C. T. van Duin, Phys. Chem. Chem. Phys., 2015, 17, 3383–3393 RSC.
- K. D. Fong, J. Self, B. D. McCloskey and K. A. Persson, Macromol, 2021, 54, 2575–2591 CrossRef CAS.
- Y. Shao, H. Gudla, J. Mindemark, D. Brandell and C. Zhang, Acc. Chem. Res., 2024, 57, 1123–1134 CrossRef CAS PubMed.
- H. G. Lee, S. Y. Kim and J. S. Lee, npj Comput. Mater., 2022, 8, 103 CrossRef CAS.
- M. M. Islam and A. C. T. van Duin, J. Phys. Chem. C, 2016, 120, 27128–27134 CrossRef CAS.
- Q. Wang, G. Zhang, Y. Li, Z. Hong, D. Wang and S. Shi, npj Comput. Mater., 2020, 6, 176 CrossRef CAS.
- G. Guo, B. Long, B. Cheng, S. Zhou, P. Xu and B. Cao, J. Power Sources, 2010, 195, 2393–2398 CrossRef CAS.
- V. R. Subramanian, V. D. Diwakar and D. Tapriyal, J. Electrochem. Soc., 2005, 152, A2002 CrossRef CAS.
- L. Weilong, L. Wang, L. Chenglin, L. Yong, Y. Bing and S. Zhenao, Modeling of current distribution in a lithium-ion battery, 2014 IEEE Conference and Expo Transportation Electrification Asia-Pacific (ITEC Asia-Pacific), Beijing, China, 2014, pp. 1–6, DOI:10.1109/ITEC-AP.2014.6940677.
- C. Ling, npj Comput. Mater., 2022, 8, 33 CrossRef.
- Y. Liu, T. Zhao, W. Ju and S. Shi, J. Materiomics, 2017, 3, 159–177 CrossRef.
- Y. Liu, Z. Yang, Z. Yu, Z. Liu, D. Liu, H. Lin, M. Li, S. Ma, M. Avdeev and S. Shi, J. Materiomics, 2023, 9, 798–816 CrossRef.
- Y. Liu, S. Wang, Z. Yang, M. Avdeev and S. Shi, Sci. Bull., 2023, 68, 1259–1270 CrossRef CAS PubMed.
- J. Lever, M. Krzywinski and N. Altman, Nat. Methods, 2016, 13, 703–704 CrossRef CAS.
- Y. Liu, Z. Yang, X. Zou, S. Ma, D. Liu, M. Avdeev and S. Shi, Natl. Sci. Rev., 2023, 10(7), nwad125 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- A. Klamt and F. Eckert, Fluid Phase Equilib., 2000, 172, 43–72 CrossRef CAS.
- A. Klamt, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 699–709 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149–8159 CrossRef CAS.
- J. Wu, X. Wang, Q. Liu, S. Wang, D. Zhou, F. Kang, D. Shanmukaraj, M. Armand, T. Rojo, B. Li and G. Wang, Nat. Commun., 2021, 12, 5746 CrossRef CAS PubMed.
- C.-C. Su, M. He, J. Shi, R. Amine, J. Zhang, J. Guo and K. Amine, Nano Energy, 2021, 89, 106299 CrossRef CAS.
- X. Ma, R. S. Arumugam, L. Ma, E. Logan, E. Tonita, J. Xia, R. Petibon, S. Kohn and J. R. Dahn, J. Electrochem. Soc., 2017, 164, A3556 CrossRef CAS.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- L. Kim, T. Jang and H. R. Byon, J. Power Sources, 2023, 576, 233237 CrossRef CAS.
- H. Jia, Y. Xu, L. Zou, P. Gao, X. Zhang, B. Taing, B. E. Matthews, M. H. Engelhard, S. D. Burton, K. S. Han, L. Zhong, C. Wang and W. Xu, J. Power Sources, 2022, 527, 231171 CrossRef CAS.
- F. Wu, Q. Zhu, L. Li, R. Chen and S. Chen, J. Mater. Chem. A, 2013, 1, 3659–3666 RSC.
- J. Self, D. S. Hall, L. Madec and J. R. Dahn, J. Power Sources, 2015, 298, 369–378 CrossRef CAS.
- M. Xu, L. Hao, Y. Liu, W. Li, L. Xing and B. Li, J. Phys. Chem. C, 2011, 115, 6085–6094 CrossRef CAS.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- Y. Wu, Q. Hu, H. Liang, A. Wang, H. Xu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2300259 CrossRef CAS.
- A. Ishikawa, K. Sodeyama, Y. Igarashi, T. Nakayama, Y. Tateyama and M. Okada, Phys. Chem. Chem. Phys., 2019, 21, 26399–26405 RSC.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- X. Cao, L. Zou, B. E. Matthews, L. Zhang, X. He, X. Ren, M. H. Engelhard, S. D. Burton, P. Z. El-Khoury, H.-S. Lim, C. Niu, H. Lee, C. Wang, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Energy Storage Mater., 2021, 34, 76–84 CrossRef.
- Y.-H. Lin, L.-T. Wu, Y.-T. Zhan, J.-C. Jiang, Y.-L. Lee, J.-S. Jan and H. Teng, Energy Storage Mater., 2023, 61, 102868 CrossRef.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- ADF 2021.102. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125(19), 194101 CrossRef PubMed.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- AMS 2023.1 COSMO-RS. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- M. J. Marczewski, B. Stanje, I. Hanzu, M. Wilkening and P. Johansson, Phys. Chem. Chem. Phys., 2014, 16, 12341–12349 RSC.
- C. Labrèche, I. Lévesque and J. Prud'homme, Macromol, 1996, 29, 7795–7801 CrossRef.
- W. Lee, C. K. Lyon, J.-H. Seo, R. Lopez-Hallman, Y. Leng, C.-Y. Wang, M. A. Hickner, C. A. Randall and E. D. Gomez, Adv. Funct. Mater., 2019, 29, 1807872 CrossRef.
- M. Handzlik, B. v. Beek, P. Melix, R. Rüger, T. Trnka, L. Ridder and F. Zapata, PLAMS. SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com, https://github.com/SCM-NV/PLAMS Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- M. S. Ding, J. Chem. Eng. Data, 2004, 49, 276–282 CrossRef CAS.
- Y.-J. Fang and J.-M. Qian, J. Chem. Eng. Data, 2005, 50, 340–343 CrossRef CAS.
- M. S. Ding, J. Electrochem. Soc., 2003, 150, A455 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- I. I. I. Alkhatib, C. G. Albà, A. S. Darwish, F. Llovell and L. F. Vega, Ind. Eng. Chem. Res., 2022, 61, 7414–7429 CrossRef CAS PubMed.
- J. Dagdelen, A. Dunn, S. Lee, N. Walker, A. S. Rosen, G. Ceder, K. A. Persson and A. Jain, Nat. Commun., 2024, 15, 1418 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC.
- B. He, Z. Gong, M. Avdeev and S. Shi, J. Chem. Inf. Model., 2023, 63, 4986–4994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03026e |
| This journal is © The Royal Society of Chemistry 2024 |