
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the nano-scale structure and composition dynamics during the phase transition towards complete separation of CeO2–ZrO2 solid solutions†
Seol Hee
Oh‡
a,
Hyun-Kyu
Kim‡
 b,
Sun-Young
Park‡
c,
Yeong-Cheol
Kim
b,
Deok-Hwang
Kwon
a,
Sungeun
Yang
ad,
Ho-Il
Ji
ad,
Hye Jung
Chang
ad,
Kyung Joong
Yoon
a,
Ji-Won
Son
a and
Jong-Ho
Lee
*ad
b,
Sun-Young
Park‡
c,
Yeong-Cheol
Kim
b,
Deok-Hwang
Kwon
a,
Sungeun
Yang
ad,
Ho-Il
Ji
ad,
Hye Jung
Chang
ad,
Kyung Joong
Yoon
a,
Ji-Won
Son
a and
Jong-Ho
Lee
*ad
aEnergy Materials Research Center, Korea Institute of Science and Technology, Seoul 02792, Korea. E-mail: jongho@kist.re.kr
bSchool of Energy Materials and Chemical Engineering, Korea University of Technology and Education, Cheonan 31253, Korea
cTechnology Support Center, Korea Institute of Science and Technology, Seoul 02792, Korea
dDivision of Nanoscience & Technology, University of Science and Technology, Seoul 02792, Korea
First published on 25th June 2024
Abstract
Over the past few decades, CeO2–ZrO2 (CZO) solid solutions have rapidly replaced conventional CeO2 as a support material in three-way catalysts (TWCs) because of their superior oxygen releasing/storing capability. However, the phase stability of CZO has long been controversial as can be inferred from many conflicting reports on its structural or compositional stability. In a recent experimental observation, it was noted for the first time that even the solid solution can be completely separated into ceria and zirconia. This finding adds a new layer to the existing confusing controversy, thereby constraining the appropriate utilization of this material. Therefore, in order to elucidate the precise cause and mechanism behind the controversial phase evolution phenomena associated with phase instability, we performed a well-defined model experiment with epitaxial Ce0.75Zr0.25O2 thin films and systematically observed the detailed structural and compositional changes at each stage of the evolution process. Furthermore, in order to ascertain the precise driving force and the underlying reaction mechanism that can be used to manipulate the breakdown or onset of such phase evolution, we calculated the grand potentials for not only the initial phase but also all intermediate and final phases that appear during the phase evolution reaction by using ab initio thermodynamics based on DFT.
Introduction
Ceria (CeO2)–zirconia (ZrO2) (CZO) solid solutions have been widely investigated over the past 20 years, given the large impact as three-way catalyst (TWC) promoters in place of conventional CeO2.1–3 The main advantage of CZO over (Zr-free) CeO2 lies in its higher resistance to sintering, leading to longer lifetime of the TWC by retarding the decrease of the active surface area of the catalyst during operation at high temperature.2,3 Among the various compositions, CeO2-rich compositions (around 60–70 mol%) are the most effective materials across a diverse range of chemical processes, not only as TWC promoters but also as fuel cell materials.2,3Another positive effect induced by the insertion of tetravalent Zr (Zr4+) into CeO2 is to improve the oxygen storage capacity (OSC), mobility of lattice oxygen as well as structural stability of the CeO2 lattice.1–3 The most common explanation so far is that the Zr4+ cation, which has a smaller ionic radius (0.84 Å) than Ce4+ (0.94 Å), and favors the 7-fold coordination at room temperature,4 can facilitate the formation of oxygen vacancies5 thereby enhancing the diffusivity of lattice oxygen. The formation of oxygen vacancies can also enhance the structural stability of the ceria lattice by releasing the compressive stress generated by the reduction of Ce4+ (0.94 Å) into the larger Ce3+ (1.04 Å) ion.6 But, there is still uncertainty about whether Zr insertion has a positive impact on the intrinsic stability of the CZO solid solution.7 Due to this ambiguity, it poses challenges in the practical utilization of CZO-based catalyst materials.
Over an extended period, there has been extensive experimental and theoretical investigation on the phase stability of CZO-based materials across a broad compositional range.8–12 However, the findings reveal a multitude of conflicting reports regarding the phase stability of the CZO material, as anticipated given the intricate nature of the CZO phase diagram.13 Even amid this considerable conflict, the most uncontested interpretation is that CZO-based materials exhibit significant phase complexity within the intermediate composition range, specifically when the Ce content falls between 20 and 80 mol%. Unfortunately, this intermediate composition region is commonly employed in the catalyst field.
Furthermore, phase transition phenomena induced by such phase instability can manifest differently depending on experimental conditions, leading to various interpretations of their causes and driving forces. Several explanations have been proposed regarding the driving force behind phase evolution phenomena, including differences in surface energy due to particle size discrepancies or between the particle surface and the interphase.14,15 Some researchers have also reported that its stability is governed by the interplay between surface energy and stress induced by the disparity in size among constituent cations.8,11,12 However, despite numerous studies, the driving force behind phase evolution phenomena associated with its phase instability has yet to be clearly elucidated. Recently, we have demonstrated the phase separation phenomena in Ce-rich composition (Ce0.75Zr0.25O2), which had long been known to be stable. What's even more fascinating is that this CZO solid solution is completely separated into the initial states of pure ceria and zirconia phases before mixing.16 While we have previously revealed through density functional theory (DFT) and defect chemical calculations that the phase separation phenomenon is due to the thermodynamically unstable CZO, the precise mechanism remains elusive.
Hence, to provide a rational basis for the design of more efficient CZO-based catalyst materials, we have to understand the exact cause and reaction mechanism which can manipulate the breakdown or onset of phase instability in CZO materials even though their structural and functional complexity makes it more difficult to understand the phenomena.17 In order to experimentally observe the phenomenon while minimizing the influence of complex experimental variables in the test environment, we designed a well-defined model experiment with epitaxial Ce0.75Zr0.25O2 thin films and systematically observed the detailed structural and compositional changes at each stage of phase evolution process by means of surface morphology characterization by scanning electron microscopy, crystallographic data by X-ray diffraction, and element distribution mapping by crystal transmission electron microscopy equipped with energy dispersive spectroscopy. The transformation of phases at each stage throughout the phase evolution process was validated through ab initio thermodynamic calculations.
Results and discussion
The epitaxial relationship between the CZO thin films and YSZ substrates has been investigated using X-ray diffraction. As demonstrated in θ–2θ scans (Fig. S1(a)†) to detect diffraction signals arising from the lattice planes parallel to the sample surface, the preferential growth axis of the CZO thin films deposited on YSZ (001) is identified as (001). The coherent lattice planes that are perpendicular to the sample surface were also confirmed by XRD ϕ scans. According to the ϕ scans in Fig. S1(b)†, the CZO (001) thin films exhibit 4-fold symmetric peaks for the (111) plane. Based on the entirety of XRD results, we confirm that the CZO thin films exhibit strong alignment with the specified directions, displaying no discernible non-epitaxial features.The phase evolution phenomena in an epitaxial CZO film exposed to the reduction condition at 1000 °C for 30, 60, and 120 hours were investigated through XRD analysis (Fig. 1a). According to the XRD results in Fig. 1a, a single characteristic peak positioned at around 33° appeared for pristine CZO films (black colored bottom line) indicating single oriented epitaxial characteristics without any impurities.18 On the other hand, in reduced CZO films, diffraction peaks at around 26.9, 28.6, 29.7, and 36.8° representing the Ce-rich phase appeared where its peak intensity increases with the extended reduction time, signifying the ongoing decomposition of the initial CZO phase and subsequent formation of the Ce-rich phase at the surface with the prolonged reduction time (the full XRD spectrum is shown in Fig. S2† for better understanding). The increasing presence of the Ce-rich phase over time is further supported by surface SEM images of the reduced CZO films (Fig. 1b–e).
 | ||
| Fig. 1 (a) XRD pattern of CZO thin films depending on the reduction time. (b–e) Surface SEM images of the (b) pristine film and reduced CZO thin film for (c) 30 h, (d) 60 h, and (e) 120 h. | ||
To delve into the mechanism governing the phase evolution in the CZO film, we conducted measurements of cross-sectional STEM along with EDS elemental mapping at exposure times of 30, 60, and 120 hours under a reducing atmosphere at 1000 °C. In the EDS mapping images in Fig. 2, the red and green colors indicate cerium (Ce) and zirconium (Zr), respectively. Quantitative compositional analysis was carried out on both the pristine film and the reduced film, focusing on specific regions marked in the mapping images (R1–R5), and the corresponding results are summarized in Table 1.
 | ||
| Fig. 2 STEM images and simultaneously measured EDS elemental mapping images of reduced CZO thin films for (a and b) 30 h, (c and d) 60 h, and (e and f) 120 h. | ||
| Region | Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr EDX ratio :Zr EDX ratio |
Structure | Space group | M 5/M4 ratio | Ce3+ fraction |
|---|---|---|---|---|---|
| Pristine | 79.30:20.70 |
Cubic fluorite |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
||
| R1 | 78.67:21.33 |
Cubic fluorite |
Fmm |
0.85(1) | 4% |
| R2 | 78.60:21.40 |
Defect fluorite |
Fmm |
0.87(2) | 10% |
| R3 | 70.28:29.72 |
Pyrochlore |
Fdm |
1.04(3) | 71% |
| R4 | 98.51:1.49 |
Cubic |
Ia |
1.12(3) | 100% |
| R5 | 2.29:97.71 |
Tetragonal | P42/nmc | 1.09(5) | 89% |
The pristine CZO thin film has an average cation composition ratio of 79.3:20.7 (Ce/Zr = 3.83) across the entire specimen, which is slightly deviated from the nominal composition ratio of 75:25 (Ce/Zr = 3). Nevertheless, in the early stages of reduction shown in Fig. 2b, certain Ce-rich precipitates start to form near the surface, resulting in slight Ce-deficient (or Zr-rich) regions on the surface. On the other hand, the majority of the internal areas of the film maintain the original composition ratio (marked as R1). In the more advanced intermediate stage of reduction (Fig. 2d), a larger amount of Ce-rich precipitates is generated on the surface, leading to the internal formation of aggregated Zr-rich phases. Interestingly, in the regions where such phase separation occurs, large pores up to 100 nm in size are also formed. This phenomenon is likely the outcome of the rapid migration of larger-sized Ce cations toward the surface, leaving smaller-sized Zr cations behind. In this intermediate stage, it is observed that near the region (R3) where phase separation occurs, there is a slight decrease in the Ce/Zr ratio (70.28:29.72), while in the remaining area (R2), the original composition ratio still appears to be maintained. As the reduction progresses further (Fig. 2f), this phase separation reaction spreads throughout the entire film and is expected to proceed until CZO is fully separated into Ce-rich (R4) and Zr-rich phases (R5).
Interestingly, as revealed by the compositional analysis results in Table 1, the separated phases are identified not just as specific rich phases but as distinct cerium oxide (R4) and zirconium oxide (R5). These findings suggest that similar to the experimental results of thinner CZO films (approximately 25 nm thick) in a previous study,16 the current thicker CZO film (250 nm thick) is also expected to undergo complete separation into cerium oxide (R4) and zirconium oxide (R5) phases with longer reduction times. The results of pure cerium oxide and zirconium oxide, consisting of 99% or more major elements, can be more clearly observed in higher magnification images in Fig. S3.†
To investigate structural changes associated with CZO phase separation, we acquired fast Fourier transformation (FFT) patterns for both the pristine sample and the reduced CZO, with a particular emphasis on the specified regions of R1–R5. The electron diffraction pattern of the pristine CZO in Fig. 3a represents the FFT pattern of the cubic fluorite structure (space group Fmm) with the [110] zone axis. The cubic fluorite structure has a fully disordered cation phase which has 8-fold coordinated oxygen ions as shown in Fig. 4a.
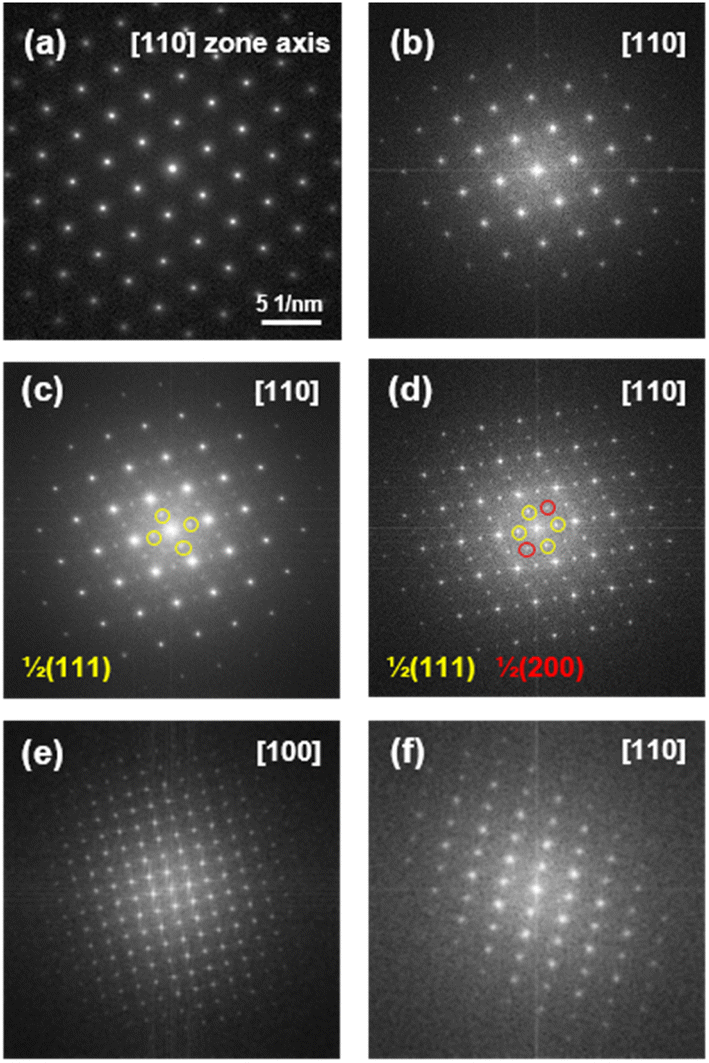 | ||
| Fig. 3 FFT patterns of the (a) pristine CZO thin film, (b) R1, (c) R2, (d) R3, (e) R4 and (f) R5 regions marked in Fig. 2. | ||
 | ||
| Fig. 4 Local structures of (a) cubic fluorite, (b) defect fluorite, and (c) pyrochlore lattice. | ||
The cubic fluorite structure remains unchanged in the initial stage of phase separation, as evidenced by the FFT pattern for R1 in Fig. 3b. In the middle stage of phase separation, satellite reflections appear at the 1/2〈111〉 and/or 1/2〈200〉 positions marked with yellow and red circles in Fig. 3c and d. It is of interest to note that deficient fluorite-type structures contain short range ordering that gives rise to the satellite reflections in electron diffraction patterns.19,20 Here, the 1/2〈111〉 reflections in the FFT pattern for R2 in Fig. 3c are attributed to oxygen vacancy ordering in a defect fluorite structure,21,22 as illustrated in Fig. 4b (space group Fmm).23 The appearance of additional reflections at 1/2〈200〉 in the FFT pattern for R3 in Fig. 3d represents the pyrochlore structure in which cations are alternately ordered along the 〈110〉 direction as shown in Fig. 4c.24–26 In addition to changes in the crystal structure of CZO, the areas where phase separation occurs exhibit variations in the Ce/Zr ratio within CZO, as observed in the previous EDS analysis results in Table 1. According to the EDS analysis, the Ce/Zr ratio remains unchanged in the defect fluorite structure (R2), while in the pyrochlore structure (R3), there is a relatively increased Zr content. These results indicate that, in the early stages of the phase separation reaction, the reduction leads to a decrease in the Ce/Zr ratio as Ce and O are released from the CZO. Simultaneously, the crystal structure stabilizes towards the pyrochlore structure rather than the fluorite structure due to the reduced content of Ce and O. In the latter part of the phase separation reaction, distinctly separated two phases of ceria and zirconia become evident throughout the entire CZO film. According to the electron diffraction patterns from the fully separated ceria (R4) and zirconia (R5) shown in Fig. 3e and f, it is observed that ceria exhibits a cubic structure with a space group of Ia, featuring a 2 × 2 × 2 supercell derived from the parent fluorite, while zirconia exhibits a tetragonal structure with a space group P42/nmc.
In fact, the transition from the fluorite structure to pyrochlore structure has been frequently reported.24–26 This structural transition is generally influenced not only by oxygen nonstoichiometry but also by the size difference between constituent cations in the pyrochlore structure. Usually, the preference for transitioning from a fluorite structure to a pyrochlore structure becomes more pronounced as the disparity in cation sizes increases.27 Indeed, during the reduction process of CZO initially in the fluorite structure, there is an elevation in the concentration of Zr4+ (0.84 Å), possessing a smaller ionic size compared to Ce4+ (0.94 Å). This increase serves to accentuate the size difference between the two ions. Moreover, the size difference between Zr and Ce ions is anticipated to be further accentuated as Ce4+ (0.94 Å) undergoes reduction to Ce3+ (1.04 Å) in a reducing atmosphere.28,29 This cumulative effect contributes significantly to facilitating the transition to the pyrochlore structure.
The valence state of Ce in the reduced CZO thin film was examined through electron energy loss spectroscopy (EELS). Fig. 5a and b show the EELS spectra of the Ce M5 edge and M4 edge acquired from the R1–R5 and standard samples. The CeO2 and CeF3 powders were used as a reference for Ce4+ and Ce3+, respectively. The Ce-M4,5 edges correspond to the electron transition between the 3d and 4f states of the Ce.30 The shoulder peak in the spectrum of the CeO2 in Fig. 5b is related to the strong covalent hybridization between the Ce 4f and O 2p states.31,32 The Ce valence was quantitatively analyzed by calculating the M5/M4 intensity ratio because the ratio reflects the degree of occupancy of the 4f electron state;33 the 4f electron state of the Ce4+ is empty, while the Ce3+ is occupied with a single electron.34 In this study, the Ce3+ fraction for different positions was estimated in Fig. 5b by calculating a linear interpolation, based on the M5/M4 value for the Ce4+ ions (0.84) and Ce3+ (1.12) obtained via our TEM system. The average M5/M4 ratios of each region are presented in Table 1. The M5/M4 ratio in the R1 is 0.85 which corresponds to the Ce3+ fraction of 4%. It indicates that 4% Ce4+ ions were reduced to Ce3+ due to the oxygen vacancies produced by the Zr addition into ceria. As the reduction time increased, the Ce3+ fraction increased, showing 10% and 71%, respectively, in the defect fluorite (R2) and the pyrochlore structure (R3). Considering that the M5/M4 ratio of the R4 is 1.12, all Ce4+ ions in the precipitated cerium oxide are finally reduced to Ce3+.
 | ||
| Fig. 5 EELS spectra of the Ce M edge obtained from (a) the R1–R5 marked in Fig. 2 and (b) CeO2 and CeF3 standard powders. (c) Scatter plot showing the M5/M4 EELS ratio (left axis) and Ce3+ fraction (right axis) with respect to Ce concentration. | ||
Based on all the above analysis results, the structural deformation that occurs during the phase separation process of the CZO thin films consists of three reaction steps as shown in Fig. 6a; (1) transformation into the defect fluorite with formation of oxygen vacancies, (2) transformation into the pyrochlore structure with precipitation of ceria, and (3) complete separation of the phase into C-type Ce2O3 and tetragonal ZrO2. The relevant reaction equations for the three-stage evolution process are:
| Ce0.784+Zr0.224+O2 → Ce0.083+Ce0.704+Zr0.224+O1.96 | (1) |
| Ce0.083+Ce0.704+Zr0.224+O1.96 → 0.733Ce0.53+Ce0.24+Zr0.34+O1.75 + 0.267CeO1.5 | (2) |
| Ce0.53+Ce0.24+Zr0.34+O1.75 → 0.7CeO1.5 + 0.3ZrO2 | (3) |
 | ||
| Fig. 6 (a) Phase evolution, (b) relative grand potential (ΔΩ) of the four phases at T = 1300 K and pO2 = 10−16 atm, and (c) relative grand potential (ΔΩ) of two ceria phases as a function of T at pO2 = 10−16 atm. The ceria phase is changed from CeO2 to Ce2O3 at T = 1100 K. | ||
Through DFT calculations, we could validate that this three-step evolution process aligns with the transition towards a thermodynamically more stable phase as reduction of CZO proceeds. Fig. 6b shows the relative grand potential of the four phases (CZO, rCZO, pyro-CZO, and ceria + zirconia) at T = 1300 K and pO2 = 10−16 atm. As depicted in Fig. 6b, when the CZO phase is subjected to a high-temperature reducing atmosphere, it undergoes gradual reduction, transitioning into intermediate phases like rCZO and pyro-CZO phases, until it eventually fully segregates into thermodynamically stable ceria and zirconia phases. These thermodynamic calculations effectively explain the phase separation phenomenon observed in our experimental results. Furthermore, the relative grand potential results depicted in Fig. 6c for the two ceria phases (CeO2 and Ce2O3) corroborate our findings that the resultant ceria phase after reduction was Ce2O3.
Experimental
Sample preparation and characterization
Ce0.75Zr0.25O2 (CZO) powders were synthesized by the glycine-nitrate process (GNP). Individual chemical compounds of Ce(NO3)36H2O (Aldrich, 99.99%), and ZrO(NO3)2·xH2O (Aldrich, 99.99%) were used as starting materials. These materials were dissolved in 200 mL of distilled water, and mixed with glycine (H2NCH2COOH) with the glycine to nitrate (G/N) ratio of 0.55. The solution was heated on a hot plate to evaporate the water, initiating self-combustion. Subsequently, the resulting powder from the reaction chamber underwent ball milling and was heat-treated at 600 °C for 3 hours to achieve complete single-phase formation. The calcined CZO powder was pelletized using uniaxial press and cold isostatic press apparatus and sintered at 1500 °C for 5 hours. The CZO pellet was used for a pulsed laser deposition (PLD) target. Epitaxial CZO thin films were deposited on YSZ (001) substrates by using PLD. Before the deposition, all the substrates were cleaned in order to prevent an artificial contribution, causing degradation of the crystal quality, resulting from ultrasonic cleaning with acetone, methyl alcohol, and distilled water. The deposition was performed at a constant temperature of 750 °C under oxygen partial pressure of 50 mTorr with a laser fluence of 2 J cm−2 using a KrF excimer laser. The epitaxial growth along the [001] direction was examined by X-ray diffraction θ–2θ and φ scan with a RIGAKU Smart Lap X-ray diffractometer equipped with a rotating anode using monochromatic copper radiation (Kα1 = 0.154056 nm). The epitaxial CZO thin films were annealed at 1000 °C under air or 4% hydrogen mixed argon gas for different annealing times using a tube furnace. Surface morphologies of the reduced CZO thin films were investigated by field emission scanning electron microscopy (Inspect 50, FEI). Cross-sectional element mapping data of the reduced CZO thin films were obtained using scanning transmission electron microscopy (STEM, Talos F200X) with energy dispersive spectroscopy. Prior to immersion, a focused ion beam (FIB, Helios NanoLab 600) system was used to prepare STEM samples.Computational details
The 2 × 2 × 2 fluorite cubic CeO2 supercell (Fig. 7a) that contains 32 Ce and 64 O atoms is used to form Ce0.75Zr0.25O2 (CZO, Fig. 7b) by randomly replacing 8 Ce atoms with 8 Zr atoms. Reduction of CZO was implemented by removing 6 O atoms in the O sublattice (reduced CZO: rCZO, Fig. 7c). In such cases, a vast array of structures emerges for CZO and rCZO. To address this, 20 random structures were generated for each phase, and their average internal electronic energy was calculated. | ||
| Fig. 7 Typical cells of (a) CeO2, (b) CZO, (c) reduced CZO (rCZO), and (d) pyro-CZO. CeO2 is the 2 × 2 × 2 fluorite cubic supercell that contains 32 Ce and 64 O atoms. CZO is formed by replacing 8 Ce atoms with 8 Zr atoms, and rCZO is formed by adding 6 VO's in CZO. The replacement and addition are all conducted with randomness. Pyro-CZO contains 8 VO's. | ||
The pyro-CZO with the Ce:Zr ratio of 75:25 is derived from the typical pyrochlore structured CZO with the Ce:Zr ratio of 50:50 that is already well-known as a catalyst support for TWC.35–37 The introduction of this distinct pyro-CZO structure, characterized by a Ce:Zr ratio of 75:25, can be attributed to the experimental analysis of the composition and crystal structure conducted in this study. The superstructure of this pyro-CZO is formed with an ordering of 8 Zr atoms and 8 O vacancies in the metal and O sublattices, respectively. In this pyro-CZO structure, each Zr atom is neighbored by two O vacancies.
Generally, the formation energy (Eform) of phase A from phase B is defined as:
| Eform = EA − EB | (4) |
| Ωi = Ei − TSi − μONO − ΩM | (5) |
| Si = kBlnω | (6) |
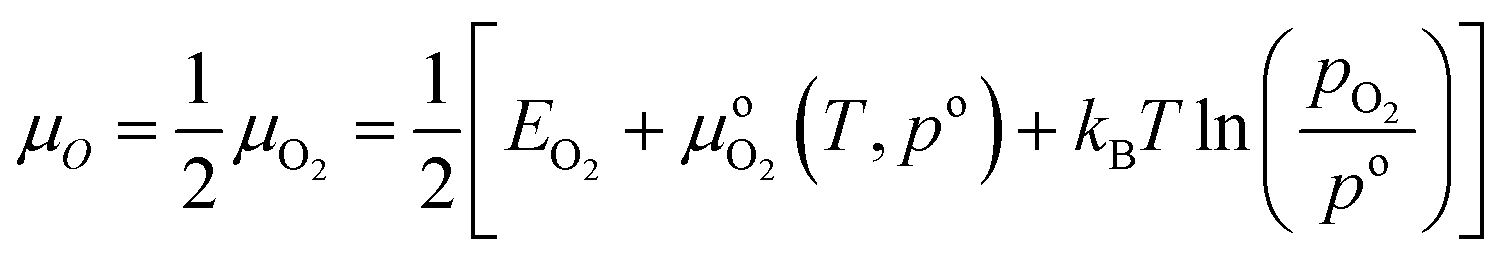 | (7) |
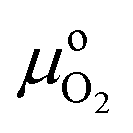 (T, po) is the difference in the chemical potential of O2 between T = 0 K and the temperature of interest at the reference pressure (po, 1 atm), and pO2 is the oxygen partial pressure.
(T, po) is the difference in the chemical potential of O2 between T = 0 K and the temperature of interest at the reference pressure (po, 1 atm), and pO2 is the oxygen partial pressure.  (T, po) can be obtained from the NIST-JANAF thermochemical tables.38,39
(T, po) can be obtained from the NIST-JANAF thermochemical tables.38,39
All DFT calculations are performed by using the Vienna ab initio simulation package (VASP) code.40,41 The electron wave functions are described using the projector augmented wave (PAW) method of Blochl, which is implemented in the VASP code by Kresse and Joubert.42,43 The exchange correlation energy is described via the generalized gradient approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE).44 The GGA + U method is employed to consider strongly localized Ce 4f electrons with effective Hubbard U parameters (U = 5 and J = 0).45–47 The cut-off energy is 500 eV, and the k-point mesh is 2 × 2 × 2 Monkhorst Pack.48 The ionic and electronic optimizations are converged when the total energy difference between successive calculation steps is less than 10−2 and 10−3 eV, respectively.
Conclusions
Despite CZO's widespread adoption in three-way catalysts (TWCs) due to its superior oxygen storing/releasing properties, its phase stability remains contentious, as evidenced by many conflicting reports. Our recent experimental findings have revealed the unprecedented separation of CZO into its constituent components, ceria and zirconia, intensifying the ongoing debate and challenging its practical applications. To address this, we conducted well-defined model experiments using epitaxial Ce0.75Zr0.25O2 thin films, meticulously tracking structural and compositional alterations at each evolutionary stage.According to the structural and compositional analysis, the phase separation of CZO thin films involves a structured process delineated into three key reaction steps:
(1) Transformation into defect fluorite: initially, the CZO thin films undergo a transformation into defect fluorite, accompanied by the generation of oxygen vacancies.
(2) Conversion into the pyrochlore structure: subsequently, there is a transition into the pyrochlore structure facilitated by the precipitation of ceria within the thin films.
(3) Complete separation into Ce2O3 and ZrO2 phases: finally, the phase fully separates into distinct entities, forming C-type Ce2O3 and tetragonal ZrO2 phases, thus concluding the phase separation process.
Additionally, by leveraging ab initio thermodynamics based on Density Functional Theory (DFT), we calculated grand potentials for all phases involved in the evolution, elucidating the driving forces and reaction mechanisms governing CZO's phase instability. This comprehensive approach sheds light on the intricate dynamics underlying CZO's phase evolution, paving the way for informed strategies to modulate its stability and optimize its utility in catalytic applications.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Seol Hee Oh conceived the experiments and analyses. Seol Hee Oh and Sun-Young Park designed and participated in all the experimental work. Hyun-Kyu Kim performed DFT calculations. Sungeun Yang provided suggestions for the experimental setup, Yeong-Cheol Kim helped analyze the simulation results and commented on the manuscript. Hye Jung Chang and Deok-Hwang Kwon assisted in characterizing the structure and composition of samples. Ho-Il Ji and Kyung Joong Yoon provided suggestions for mechanistic analysis of phase evolution. Ji-Won Son provided suggestions for fabricating thin film samples. Jong-Ho Lee supervised the whole research. Jong-Ho Lee and Sun-Young Park prepared and edited the manuscript with input from all authors. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT (MSIT), grant number: NRF-2022M3J1A1065570) and partially supported by the KIST institutional program (2E33263).References
- X. Liang, X. Wang, Y. Zhuang, B. Xu, S. Kuang and Y. Li, J. Am. Chem. Soc., 2008, 130, 2736 CrossRef CAS PubMed.
- M. D. Krcha and M. J. Janik, Catal. Sci. Technol., 2014, 4, 3278 RSC.
- G. Balducci, P. Fornasiero, R. Di Monte, J. Kaspar, S. Meriani and M. Graziani, Catal. Lett., 1995, 33, 193 CrossRef CAS.
- D. A. Daramola, M. Muthuvel and G. G. Botte, J. Phys. Chem. B, 2010, 114, 9323 CrossRef CAS PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752 CrossRef CAS PubMed.
- Z.-K. Han, L. Zhang, M. Liu, M. V. Ganduglia-Pirovano and Y. Gao, Front. Chem., 2019, 7, 436 CrossRef CAS PubMed.
- R. Dziembaj, M. Molenda and L. Chmielarz, Catalysts, 2023, 13, 1165 CrossRef CAS.
- L. Lan, S. Chen, Y. Cao, M. Zhao, M. Gong and Y. Chen, J. Colloid Interface Sci., 2015, 450, 404 CrossRef CAS PubMed.
- G. Colon, F. Valdivieso, M. Pijolat, R. T. Baker, J. Calvino and S. Bernal, Catal. Today, 1999, 50, 271 CrossRef CAS.
- A. Martínez-Arias, M. Fernández-García, A.-B. Hungría, J. C. Conesa and G. Munuera, J. Phys. Chem. B, 2003, 107, 2667 CrossRef.
- K. Kenevey, F. Valdivieso, M. Soustelle and M. Pijolat, Appl. Catal., B, 2001, 29, 93 CrossRef CAS.
- C. C. Chuang, H. I. Hsiang, C. C. Chen, F. S. Yen and M. Yoshimura, J. Am. Ceram. Soc., 2013, 96, 1629 CrossRef CAS.
- J. Kašpar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419 CrossRef.
- S. Pengpanich, V. Meeyoo, T. Rirksomboon and K. Bunyakiat, Appl. Catal., A, 2002, 234, 221 CrossRef CAS.
- D. H. Prasad, S. Park, H.-I. Ji, H.-R. Kim, J.-W. Son, B.-K. Kim, H.-W. Lee and J.-H. Lee, J. Phys. Chem. C, 2012, 116, 3467 CrossRef CAS.
- S. H. Oh, H.-K. Kim, J. Kim, Y.-C. Kim, S.-Y. Park, S. Yang, H. I. Ji, K. J. Yoon, J. W. Son and J. H. Lee, J. Phys.: Energy, 2022, 4, 045004 CAS.
- R. Grau-Crespo, N. H. de Leeuw, S. Hamad and U. V. Waghmare, Proc. R. Soc. A, 2011, 467, 1925 CrossRef CAS.
- T. M. Onn, X. Mao, C. Lin, C. Wang and R. J. Gorte, Inorganics, 2017, 5, 69 CrossRef.
- T. A. Schaedler, W. Francillon, A. S. Gandhi, C. P. Grey, S. Sampath and C. G. Levi, Acta Mater., 2005, 53, 2957 CrossRef CAS.
- I. Kaiser-Bischoff, H. Boysen, F. Frey, J.-U. Hoffmann, D. Hohlwein and M. Lerch, J. Appl. Crystallogr., 2005, 38, 139 CrossRef.
- R. Withers, J. Thompson, P. Barlow and J. Barry, Aust. J. Chem., 1992, 45, 1375 CrossRef CAS.
- J. Allpress and H. Rossell, J. Solid State Chem., 1975, 15, 68 CrossRef CAS.
- S. N. Achary, S. K. Sali, N. K. Kulkarni, P. S. R. Krishna, A. B. Shinde and A. K. Tyagi, Chem. Mater., 2009, 21, 5848 CrossRef CAS.
- K. R. Whittle, L. M. Cranswick, S. A. Redfern, I. P. Swainson and G. R. Lumpkin, J. Solid State Chem., 2009, 182, 442 CrossRef CAS.
- M. de los Reyes, K. R. Whittle, Z. Zhang, S. E. Ashbrook, M. R. Mitchell, L.-Y. Jang and G. R. Lumpkin, RSC Adv., 2013, 3, 5090 RSC.
- Y. Liu, R. L. Withers and L. Norén, J. Solid State Chem., 2004, 177, 4404 CrossRef CAS.
- N. M. Cepeda-Sánchez, J. A. Díaz-Guillén, M. Maczka, U. Amador and A. F. Fuentes, J. Mater. Sci., 2018, 53, 13513 CrossRef.
- D. Prusty, A. Pathak, A. Chintha, B. Mukherjee and A. Chowdhury, J. Am. Ceram. Soc., 2014, 97, 718 CrossRef CAS.
- B. Paul, K. Singh, T. Jaroń, A. Roy and A. Chowdhury, J. Alloys Compd., 2016, 686, 130 CrossRef CAS.
- Y. Ding, Y. Chen, K. C. Pradel, M. Liu and Z. Lin Wang, J. Appl. Phys., 2016, 120, 214302 CrossRef.
- M. FinazziHatano, F. de Groot Hatano, A.-M. D. Hatano, J.-P. Kappler, O. S. Hatano, W. F. Hatano and G. K. Hatano, Proceedings of the 11th International Conference on Vacuum Ultraviolet Radiation Physics, 1996, p. 221 Search PubMed.
- P. Gao, Z. Kang, W. Fu, W. Wang, X. Bai and E. Wang, J. Am. Chem. Soc., 2010, 132, 4197 CrossRef CAS PubMed.
- H. Hojo, T. Mizoguchi, H. Ohta, S. D. Findlay, N. Shibata, T. Yamamoto and Y. Ikuhara, Nano Lett., 2010, 10, 4668 CrossRef CAS PubMed.
- A. Pfau and K. Schierbaum, Surf. Sci., 1994, 321, 71 CrossRef CAS.
- P. R. Shah, T. Kim, G. Zhou, P. Fornasiero and R. J. Gorte, Chem. Mater., 2006, 18, 5363 CrossRef CAS.
- A. Gupta, A. Kumar, U. Waghmare and M. Hegde, J. Phys. Chem. C, 2017, 121, 1803 CrossRef CAS.
- J. Wu, A. E. O'Neill, C.-H. Li, J. R. Jinschek and G. Cavataio, Appl. Catal., B, 2021, 280, 119450 CrossRef CAS.
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, 2009 Search PubMed.
- V. Botu, R. Ramprasad and A. B. Mhadeshwar, Surf. Sci., 2014, 619, 49 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217 CrossRef CAS.
- T. Zacherle, A. Schriever, R. De Souza and M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134104 CrossRef.
- L. Shi, E. Vathonne, V. Oison, M. Freyss and R. Hayn, Phys. Rev. B, 2016, 94, 115132 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02923b |
| ‡ SH Oh, H-K Kim, and S-Y Park: Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |