
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando measurement of electrocatalyst potential on particulate photocatalysts for overall water splitting†
Yudai
Kawase
a,
Keisuke
Obata
 a,
Yuu
Shioiri
a,
Tomohiro
Higashi
b and
Kazuhiro
Takanabe
*a
a,
Yuu
Shioiri
a,
Tomohiro
Higashi
b and
Kazuhiro
Takanabe
*a
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: takanabe@chemsys.t.u-tokyo.ac.jp
bInstitute for Tenure Track Promotion, University of Miyazaki, 1-1 Gakuenkibanadai-nishi, Miyazaki 889-2192, Japan
First published on 5th July 2024
Abstract
Photocatalytic water splitting using semiconductor particulates is an effective technique for solar-energy conversion to chemical energy in the form of hydrogen molecules. The photocatalytic reactions proceed via two half reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER). The active sites in these reactions are electrocatalysts that have been deposited on particulate photocatalyst surfaces. It is important to understand the electrocatalyst potential during the photocatalytic reactions to understand the reaction mechanism. Several measuring methods have been developed for photoelectrodes, but it is still challenging to measure the electrocatalyst potential directly during unbiased overall water splitting with particulate photocatalysts. This study presents an inverted photocatalyst structure to evaluate the operating potential of CoOOH electrocatalysts during overall water splitting. The structure consists of Al-doped SrTiO3 (SrTiO3:Al) photocatalyst powder deposited on a CoOOH electrode. In addition, we estimated the fraction of holes that reach the SrTiO3:Al photocatalyst surface after carrier generation in the bulk from the light-intensity dependence of the CoOOH electrocatalyst potential balanced with the hole quasi-Fermi level in the photocatalyst. The hydrogen production rate was found to be controlled by modulating the potential applied to the CoOOH electrode, which perturbs the hole quasi-Fermi level in the SrTiO3:Al photocatalyst.
1 Introduction
For half a century, particulate photocatalysts have been studied to convert solar energy into chemical fuels.1–6 Photocatalytic water splitting is one example of solar-energy conversion into hydrogen.6–8 Although much effort has been devoted to it,6,9–13 it is difficult to achieve high solar-to-hydrogen energy conversion efficiency due to the complexities of photocatalytic processes.The processes are separated into the following events. First, semiconductor photocatalysts absorb photons to produce electron–hole pairs called excitons. After excitons are separated into electrons and holes, these charge carriers are transported to the surface by diffusion and drift. Finally, catalytic reactions are driven at the photocatalyst/water solid/liquid interface, such as the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in the case of water splitting.14 For efficient HER and OER, electrocatalysts (often in the form of nanoparticles) are loaded on particulate photocatalyst surfaces to promote the reactions.6
These electrochemical reactions require multiple electron transfers by holes and electrons transported to the surface. Therefore, the electrochemical potentials of the loaded electrocatalysts in a steady state during photocatalysis determine the overall reaction rates rather than the potential positions of the top of the valence band and the bottom of the conduction band. The electrocatalytic performance has a significant impact on the efficiency of photocatalytic reactions,15–18 so measuring the operating potential of electrocatalysts is necessary to understand the photocatalytic reaction mechanisms and provide strategies to achieve efficient water splitting.
Recently, several studies have measured the electrocatalyst potential or surface potential on photoelectrochemical electrodes, where either reduction or oxidation reactions occur on the electrode surfaces. Pinson used a GaP photoelectrode coated with a 16-nm Au film. The film potential balanced with the electrochemical potential of holes and the GaP photoelectrode potential balanced with the electrochemical potential of electrons were separately measured.19 Boettcher et al. used double working electrodes and measured the potential of surface electrocatalysts (e.g., IrOx and NiOx) separately from the potential of TiO2 photoelectrodes.20,21 They also reported a technique using atomic force microscopy to measure the electrochemical potential of cobalt phosphate electrocatalysts loaded on α-Fe2O3 photoelectrodes during the photoelectrochemical OER.22 The method was applied to an Ni/n-Si photoanode23 and to CoOx/BiVO4 and Pt/BiVO4 particles fixed on FTO substrates.24
Li et al. also used a double working-electrode system with an SrTiO3 photoelectrode to investigate surface potential.25 Osterloh et al. developed a contactless method for measuring the photovoltage of BiVO4 photoelectrodes using Kelvin-probe surface-photovoltage spectroscopy.26 Many important insights into the surface potential of electrocatalysts loaded on photoelectrodes have been obtained, but the studies have used electrode-type photo(electro)catalysts (i.e., photoelectrodes). Non-biased and particulate photocatalyst systems require further investigation.
For particulate photocatalysts, excited charges for reduction and oxidation are balanced in a single particle and act as a closed circuit. Sagara et al. investigated the photogenerated electron potential of TiO2, In2O3, and SnO2 semiconductor powders dispersed in aqueous solutions by measuring the electron transport rates from the oxide semiconductors to metal electrodes immersed in the solution.27 Talin et al. used a single particle of a SrTiO3 photocatalyst that was sandwiched between two gold substrates and measured the difference in their potentials without photocatalytic reactions.28
To obtain in-depth knowledge of electrocatalysts on particulate photocatalysts under operating conditions, direct potential measurement methods during unbiased photocatalytic overall water splitting are strongly needed. However, to the best of our knowledge, there has been no such measurement method. In conventional photocatalyst systems, electrocatalyst nanoparticles are deposited on particulate photocatalysts that are dispersed in aqueous solutions. Selective contact with electrocatalyst nanoparticles is difficult, so measuring their potentials remains a challenge. In 2020, Domen et al. reported efficient overall water splitting using Rh/Cr2O3/CoOOH/SrTiO3:Al (catalyst modified SrTiO3; cSTO) with an apparent quantum efficiency of up to 96% at wavelengths between 350 and 360 nm.29 This efficient photocatalyst provides a strong incentive to investigate how the electrocatalytic potential on the surface shifts during photocatalytic overall water splitting.
This study presents an operando measurement system for electrocatalyst potential on particulate photocatalysts by using a novel photocatalyst structure. We fabricated an inverted photocatalyst structure in which photocatalyst powder is deposited on the electrode. Fig. 1 shows the structure and measurement system. The focus was on the kinetically sluggish OER side,30 where the photocatalyst powder was deposited on a CoOOH electrocatalyst fabricated on F-doped tin oxide-coated glass substrates (FTO) to obtain cSTO/CoOOH/FTO.
 | ||
| Fig. 1 Illustration of the measurement system for photocatalytic water splitting using the inverted photocatalyst structure. | ||
We monitored the open circuit potential (OCP) of the CoOOH electrode to monitor the shift of the electrode potential due to photo-generated holes transported from cSTO to CoOOH/FTO while measuring the quantity of produced hydrogen and oxygen gases. We also investigated the dependence of the photocatalytic performance on the amount of CoOOH, light intensity, and CoOOH potential. Since the potential of the electrocatalyst is considered to be balanced with the quasi-Fermi level, which is the electrochemical potential of charge carriers in a semiconductor under light illumination,31 this also leads to quantitative evaluation and understanding of the quasi-Fermi level in the particulate photocatalysts. This study transfers the knowledge of electrocatalysis to particulate photocatalysis providing quantitative description of important parameters in photocatalysis.
2 Experimental
2.1 Synthesis of the Al-doped SrTiO3 photocatalyst
Al-doped SrTiO3 (SrTiO3:Al) was synthesized according to a reported method.29,32 Briefly, SrCl2 (anhydrous, >98.0%, Kanto Chemical Co., Inc.), Al2O3 (<50 nm particle size, Sigma-Aldrich), and SrTiO3 were mixed by grinding in an agate mortar at a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.02:1. The mixture was transferred to an alumina crucible, heated at 1150 °C for 10 h, and cooled to room temperature. The powder was washed with copious ultrapure water using ultrasonication to remove unreacted substances and filtered, which provided a white powder (SrTiO3:Al).
:0.02:1. The mixture was transferred to an alumina crucible, heated at 1150 °C for 10 h, and cooled to room temperature. The powder was washed with copious ultrapure water using ultrasonication to remove unreacted substances and filtered, which provided a white powder (SrTiO3:Al).
2.2 Electrocatalyst loading on the Al-doped SrTiO3 photocatalyst
Rh, Cr2O3, and CoOOH electrocatalysts were loaded on SrTiO3:Al (Rh/Cr2O3/CoOOH/SrTiO3:Al, cSTO) using a reported photodeposition method.29,32 SrTiO3:Al (30 mg) was dispersed in ultrapure water (5 mL), to which 30 μL of an aqueous solution of 0.01 M RhCl3 was added (FUJIFILM Wako Pure Chemical Corporation). LED light (λ = 370 nm, 0.1 W cm−2, Asahi Spectra Co., Ltd, CL-1501) was irradiated to the solution for 10 min with stirring. After that, 30 μL of an aqueous solution of 0.01 M K2CrO4 (≥99.0%, FUJIFILM Wako Pure Chemical Corporation) was added to the suspension, and LED light was applied again for 5 min. Next, 30 μL of an aqueous solution of 85 mM Co(NO3)2·6H2O (99.5%, FUJIFILM Wako Pure Chemical Corporation) was added, and LED light was applied for another 10 min. The suspension was filtered and dried in air to obtain cSTO [Rh (0.1 wt%)/Cr2O3 (0.05 wt%)/CoOOH (0.5 wt%)/SrTiO3:Al]. The wt% of Rh, Cr2O3, and CoOOH was calculated assuming that all precursors dissolved in reaction aqueous solution were loaded on the surface of the SrTiO3:Al photocatalyst via photodeposition.2.3 Photocatalytic reaction
For overall water splitting, 10 mg of cSTO was dispersed in 5 mL of 0.5 M potassium borate buffer solution (K-borate, pH 9.0–9.2). K-borate solution was made from H3BO3 (≥99.5%, Sigma-Aldrich) and KOH (99.99%, Sigma-Aldrich). The suspension was sonicated for 5 min. The glass beaker containing the suspension was transferred to a photocatalyst reactor [see Fig. S1 in the ESI†], and Ar purging at 0.1 L min−1 was conducted.The Ar flow rate was changed to 0.01 L min−1, and LED light (λ = 370 nm, 59 mW cm−2) was applied. The distance from the LED light to the suspension was 6 cm. The produced gas was detected by gas chromatography (GC-8A, SHIMADZU Corp.) with a molecular sieve 5 Å column and a thermal conductivity detector. The apparent quantum yield (AQY) for overall water splitting was calculated using eqn (1):
 | (1) |
2.4 Fabrication of the CoOOH/FTO electrode
The CoOOH OER electrocatalyst was fabricated on FTO substrates based on previous studies.33,34 First, FTO substrates (∼10 Ω sq−1, t = 1.1 mm, n-type, Peccell Technologies, Inc.) were cut into 10 mm × 30 mm and cleaned with acetone (Sigma-Aldrich) and ethanol (FUJIFILM Wako Pure Chemical Corporation) with ultrasonication for 5 min each. After sonication, the FTO substrate was washed with a copious amount of ultrapure water. Cu tape was attached on an area of 10 mm × 5 mm on the FTO to obtain an electrical connection.The FTO was covered with seal tape (Asada Corporation) except for the Cu tape area and a 10 mm × 10 mm area for CoOOH electrodeposition. CoOOH was electrodeposited on the FTO by chronopotentiometry (CP) at 0.5 mA cm−2 for 10 s, 1 min, or 10 min in ∼10 mL of 0.1 M Co(NO3)2·6H2O.
2.5 Electrochemical measurement
Electrochemical measurements were performed using a three-electrode configuration with Hg/Hg2Cl2 (saturated KCl) (BAS Inc.) as a reference electrode, a coiled-Pt wire as a counter electrode, and a potentiostat (VMP3, Bio-Logic Science Instruments). For the activation of the CoOOH/FTO electrode, 10 cycles of cyclic voltammetry (CV) were performed in the range of 1.2–1.8 V versus a reversible hydrogen electrode (vs. RHE) at a scan rate of 10 mV s−1 in 0.5 M K-borate (pH 9.0–9.2). For the evaluation of electrocatalytic performance, CP was conducted at 0.5, 0.1, 0.05, 0.01, 0.005, and 0.5 mA cm−2 for 5 min each. For iR compensation, electrochemical impedance measurement was conducted after CP to obtain the uncompensated resistance.2.6 Fabrication of the inverted photocatalyst structure
To fabricate the photocatalyst structure, 3 mg of cSTO was dispersed in 3 mL of ultra-pure water. The suspension was ultrasonicated for 5 min. 40 μL of the suspension was drop-cast on a CoOOH/FTO electrode on a hot plate at 150 °C. Finally, a cSTO-loaded CoOOH/FTO electrode was obtained (cSTO/CoOOH/FTO). The samples were characterized by UV-vis spectroscopy (V-770, JASCO Corp), scanning electron microscopy (SEM; JSM-IT800, JEOL Ltd), inductively coupled plasma-optical emission spectroscopy (ICP-OES; iCAP PRO XP, Thermo Fisher 226 Scientific K.K.), and X-ray photoelectron spectroscopy (XPS; JPS9030, JEOL Ltd). XPS was performed using Mg Kα radiation, and the binding energy scale was calibrated using the C 1s peak at 285.0 eV.2.7 Electrode potential measurements
The system shown in Fig. 1 was used for measurements. A cSTO/CoOOH/FTO electrode was placed in a glass cell containing ∼60 mL of 0.5 M K-borate. Hg/Hg2Cl2 (saturated KCl) was used as a reference electrode. The potential was converted to an RHE scale (ERHE = EHg/Hg2Cl2 + 0.241 + 0.059 × pH). LED light (λ = 370 nm, 0.01–0.1 W cm−2) and a solar simulator equipped with an AM 1.5G filter (100 mW cm−2, 1 sun; XES-40S2-CE, SAN-EI Electric Co., Ltd) were used as light sources. For the LED light, the r(photons) corresponding to light intensity of 12, 30, 59, and 94 mW cm−2 is 1.3 × 102, 3.3 × 102, 6.7 × 102, and 1.1 × 103 μmol h−1 cm−2, respectively (see Table S1†). The distance from the light source to the electrode was 6 cm for the LED light and 10 cm for the solar simulator, respectively. The cell was connected to the GC setup. The OCP of the CoOOH/FTO electrode and the evolved oxygen and hydrogen gases were detected simultaneously under light illumination.In addition, the electrode potential was controlled by chronoamperometry (CA). A coiled-Pt wire was used as a counter electrode. The reference electrode and the counter electrode were located in a chamber that was separated from the main one with a glass frit. The internal quantum yield (IQY) for overall water splitting was calculated using eqn (2):
 | (2) |
3 Results and discussion
3.1 Photocatalytic performance of cSTO and electrocatalyst performance of CoOOH/FTO
Fig. 2a shows the photocatalytic performance of cSTO photocatalyst powder in overall water splitting using 0.5 M K-borate (pH 9.0–9.2) and LED light (λ = 370 nm, 59 mW cm−2) with the reactor shown in Fig. S1.† Under illumination, the cSTO photocatalyst powder produced H2 and O2 gases. The ratio of production rates of hydrogen and oxygen gases was equal to the theoretical value of 2:1 with an AQY of 15% at the wavelength centered at 370 nm.
 | ||
| Fig. 2 (a) Gas production rate and QY in photocatalytic overall water splitting by Rh (0.1 wt%)/Cr2O3 (0.05 wt%)/CoOOH (0.5 wt%)/SrTiO3:Al (cSTO, 10 mg) in 0.5 M K-borate (5 mL, pH 9.0–9.2). The light source was an LED (λ = 370 nm, 59 mW cm−2). (b) Electrocatalytic performance of the CoOOH/FTO electrode with various Co amounts obtained by CP. The solid lines represent exponential fitting results. WE: CoOOH/FTO or bare FTO, RE: Hg/Hg2Cl2 (sat. KCl), CE: coiled-Pt wire, and electrolyte: 0.5 M K-borate (pH 9.0–9.2). | ||
Three different amounts of CoOOH OER electrocatalyst were prepared on FTO substrates by CP at 0.5 mA cm−2 for 10 s, 1 min or 10 min in 0.1 M Co(NO3)2 aqueous solution. The loading amount of Co was determined to be 0.01, 0.04 and 0.4 μmol cm−2 by ICP-OES (see Fig. S3†). 0.01 μmol of Co was comparable to the Co amount loaded on 0.04 mg of cSTO. The electrocatalytic performance of the CoOOH/FTO electrode was evaluated by CP (Fig. 2b). Scatter plots were obtained from CP and exponential fitting results are shown as solid lines.
The electrode potential to produce 0.05 mA cm−2 shifted to a negative potential with the increase in Co loading (1.68, 1.64, and 1.61 V vs. RHE for 0.01, 0.04, and 0.4 μmol cm−2 Co, respectively). Hydrated CoOx electrocatalysts drive the OER throughout the materials, not just on the surface, so the electrocatalytic performance improved as the amount of Co increased.35
3.2 CoOOH potential measurement on the cSTO/CoOOH/FTO electrode
Fig. S4a and b in the ESI† show the top-view SEM images of CoOOH/FTO and cSTO/CoOOH/FTO. cSTO particles of about 500 nm in diameter were agglomerated and deposited on the CoOOH electrode. For the photocatalytic reaction, sufficient photocatalyst powder is commonly dispersed in aqueous solutions to fully absorb the incident photons. The loading amount of cSTO was varied and the potential of the cSTO/CoOOH electrode was measured (Fig. S5†). Under light illumination, the photo-generated electrons were transported to deposited Rh/Cr2O3 nanoparticles and drove the HER, while the generated holes were transported to the CoOOH/FTO electrode and CoOOH nanoparticles that were pre-loaded on the SrTiO3:Al photocatalyst. Therefore, the positive shift in electrode potential was caused by hole transport through only a small portion of the cSTO photocatalysts in contact with the CoOOH electrode.The most positive potentials that the cSTO/CoOOH electrodes reached were 1.46, 1.48, 1.46, and 1.44 V vs. RHE for 0.02, 0.04, 0.2, and 5 mg of cSTO, respectively. When the loading amount of cSTO is small, the incident photons are not fully absorbed by the cSTO photocatalyst on the electrode. As a result, the potential shift to the positive side becomes smaller. In contrast, for a large amount of cSTO, cSTO particles were aggregated and stacked on the electrode (Fig. S4d†). That probably prevents efficient hole transport to the CoOOH/FTO electrode. To maximize the potential shift, further investigation was conducted with 0.04 mg of cSTO.
Next, the loading amount of CoOOH on the FTO substrate was changed. Fig. 3a shows the OCP of 0.4 μmol cm−2 CoOOH/FTO (without cSTO), cSTO/0.01 μmol cm−2 CoOOH/FTO, cSTO/0.04 μmol cm−2 CoOOH/FTO, and cSTO/0.4 μmol cm−2 CoOOH/FTO electrodes as a function of time. No potential shift was observed for the CoOOH/FTO electrode without loading of cSTO under illumination (dotted line in Fig. 3a). It confirms that the positive potential shift was caused by hole transport from cSTO to the CoOOH electrode.
 | ||
| Fig. 3 (a) OCP and (b) gas production rate of the cSTO/CoOOH/FTO electrode in 0.5 M K-borate (pH 9.0–9.2) under LED illumination (λ = 370 nm, 59 mW cm−2). The production rate was obtained by GC detection, and by estimation from OCP under illumination and the j–E curve of CoOOH/FTO in the dark. WE: cSTO/CoOOH/FTO electrode and RE: Hg/Hg2Cl2 (sat. KCl). | ||
For cSTO/0.04 μmol cm−2 CoOOH/FTO and cSTO/0.4 μmol cm−2 CoOOH/FTO, the measured potential was shifted to a positive potential under illumination and reached stable values, 1.59 and 1.56 V vs. RHE, respectively, after 7 h. The potential is sufficient for driving the OER with the CoOOH/FTO electrode (see Fig. 2b). The potential reached after 7 h of irradiation was stable. In the case of cSTO/0.04 μmol cm−2 CoOOH/FTO, OCP was stable at 1.59 V vs. RHE from 7 h to 11 h (Fig. S6†). To reach steady-state OCP values, 7 h of illumination were needed. This long time to activate CoOOH was due to the low flux of excited holes. As shown in Fig. S7,† the oxidation peaks of Co species were observed in the range of 1.3–1.5 V vs. RHE, requiring a large amount of charge before reaching the steady state. On the other hand, for cSTO/0.01 μmol cm−2 CoOOH/FTO, the potential reached 1.40 V vs. RHE after 7 h. 0.01 μmol cm−2 of CoOOH was probably deposited non-uniformly due to low Co content, so the contact between cSTO and CoOOH was not sufficient for efficient hole transport, which may have resulted in an insufficient potential shift to drive the OER.
Rh (0.1 wt%)/Cr2O3 (0.05 wt%)/SrTiO3:Al (without CoOOH pre-loading) on the CoOOH electrode was also prepared to measure the OCP under LED light illumination (Fig. S8†). Even after 7 h, the electrode potential reached 1.48 V vs. RHE, which is more negative than the electrode potential of the cSTO (with CoOOH pre-loading)/CoOOH electrode, 1.56–1.59 V vs. RHE. The pre-loading of CoOOH on SrTiO3:Al by photodeposition probably promotes charge separation and carrier transport to the SrTiO3:Al surface, which results in a maximized electrode potential shift. To measure the maximized electrode potential shift, CoOOH was loaded not only on the FTO electrode but also on the SrTiO3:Al photocatalyst surface.
The gas production rates [r(H2) and r(O2)] of cSTO/0.04 μmol cm−2 CoOOH/FTO and cSTO/0.4 μmol cm−2 CoOOH/FTO were detected by GC. As shown in Fig. S9,†r(H2) was 2.5 and 2.7 μmol h−1 cm−2 after 30 min of illumination, which corresponded to an AQY at 370 nm of 0.7 and 0.8% for cSTO/0.04 μmol cm−2 CoOOH/FTO and cSTO/0.4 μmol cm−2 CoOOH/FTO, respectively. The low AQY value is due to the small amount of cSTO of 0.04 mg.
To estimate the internal quantum yield (IQY), the transmittance (T) and reflectance (R) of 0.04 mg of cSTO were measured (Fig. S10†). For the transmission spectrum, 0.04 mg of cSTO was loaded on SiO2 substrates. In addition, SiO2 substrates coated with blackbody spray were employed for the reflection spectrum. T and R were 0.84 and 0.09 at λ = 370 nm. The absorbance (A) was calculated using 1 − (T + R), and the IQY was 11 and 12% for cSTO/0.04 and 0.4 μmol cm−2 CoOOH/FTO according to eqn (2). The QY of cSTO/CoOOH/FTO reached 70–80% of the QY of the powder system (15%; see Fig. 2a) when assuming that the A of cSTO in the powder system is 1. The calculated quantum yields are summarized in Table S2.†
Bar graphs in Fig. 3b exhibit the average values of r(H2) and r(O2) after the electrode potentials were stabilized (see Fig. 3a). The average r(H2)/r(O2) values were 0.58/0.25 and 0.55/0.25 μmol h−1 cm−2 for cSTO/0.04 μmol cm−2 CoOOH/FTO and cSTO/0.4 μmol cm−2 CoOOH/FTO, respectively. The gas production rates were almost the same between the two electrodes with a r(H2)/r(O2) of 2. As the CoOOH amount decreased from 0.4 to 0.04 μmol cm−2, the cSTO photocatalyst shifted the CoOOH electrocatalyst's potential toward a more positive value (from 1.56 to 1.59 V vs. RHE) to achieve the same level of gas production rates.
The scatter plots in Fig. 3b show the gas production rates from CoOOH/FTO electrodes estimated from the OCP measurement (Fig. 3a) and j–E curve (Fig. 2b). Both 0.04 and 0.4 μmol cm−2 CoOOH/FTO electrodes should produce an OER current of 7 μA cm−2 at their measured potentials (1.59 and 1.56 V vs. RHE), which corresponds to a r(O2) of 0.07 μmol h−1 cm−2. The estimated values correspond to 28% of the detected r(O2) values for cSTO/CoOOH/FTO.
There are several possible reasons for the difference between the estimated and detected values. First, the estimated values are for O2 produced on the CoOOH/FTO electrode; however the OER is also driven on CoOOH nanoparticles pre-loaded on SrTiO3:Al photocatalytic particles. Next, the non-uniform distribution of electrochemical potential may be another reason. The measured CoOOH/FTO potentials are average values. The electrochemical potential near cSTO could be more positive than average, and if so, the measured O2 production rate would be higher than estimated. While further investigation is needed to reveal the potential distribution, the measured average potentials of CoOOH/FTO electrocatalysts shifted to a sufficient positive potential to drive the OER by photo-generated holes transported from cSTO to CoOOH/FTO. It can be concluded that the method, which enables the measurement of surface electrocatalyst potentials during photocatalytic overall water splitting, has been established.
Chen et al. studied the surface hole quasi-Fermi level (Ef,h) of a SrTiO3 photoanode using the dual-working electrode technique.25 The obtained photocurrent and the electrode potential balanced with Ef,h are compared with previously reported values (Table S3†). In the previous study using a SrTiO3 photoanode, Ef,h did not reach 1.23 V vs. RHE under open circuit conditions and the photocurrent was negligible. The SrTiO3 photoelectrode requires an applied voltage to generate a photocurrent. In contrast, the present study successfully measured Ef,h during overall water splitting under open circuit conditions by using a SrTiO3 particulate photocatalyst and an inverted structure. This is the biggest advantage of the proposed system.
One remaining problem is stability. The gas production rate decreased with time (Fig. S9†). As shown in Fig. S4c,† after the reaction, the number of agglomerated particles was reduced, indicating the detachment of the cSTO photocatalyst from CoOOH/FTO electrodes. The XPS spectra of the cSTO/0.4 μmol cm−2 CoOOH/FTO electrode were obtained before and after the reaction (Fig. S11†). While Rh- and Cr-derived peaks were not clearly observed due to very small amounts of loading on the cSTO photocatalyst, Co 2p1/2 and Co 2p3/2 peaks centered at the binding energies of 796 and 781 eV were observed. Since Co on cSTO as well as Rh and Cr are not expected to be seen, the Co 2p peaks are considered to originate from exposed CoOOH/FTO electrodes. As the intensity and position of Co peaks did not change significantly, CoOOH loaded on FTO substrates was not degraded during the reaction. In contrast, SrTiO3:Al photocatalyst derived peaks in Sr 3d and Ti 2p spectra diminished after the reaction. It is due to the detachment of cSTO from CoOOH/FTO electrodes, which is in good agreement with the SEM images of cSTO/CoOOH/FTO after the reaction (Fig. S4c†). One strategy to prevent the detachment of cSTO is annealing and necking to strengthen the adhesion. Another strategy is using the particle transfer method36,37 to fabricate cSTO/CoOOH/FTO electrodes. This method, which creates a strongly attached single-particle layer with the substrate, is expected to create a uniform and strongly adherent cSTO layer.
In addition, the OCP of cSTO (0.02, 0.04, and 0.2 mg)/0.4 μmol cm−2 CoOOH/FTO electrodes was measured under simulated AM 1.5G solar illumination. As shown in Fig. S12,† the most positive OCP of 1.41 V vs. RHE was obtained when using a cSTO of 0.04 mg loaded on the 0.4 μmol cm−2 CoOOH/FTO electrode. The measured OCP was negative compared to that under LED light illumination (1.56–1.59 V vs. RHE). The intensity of light available to the cSTO photocatalyst (λ < 387.5 nm) is 4 mW cm−2 under AM 1.5G irradiation and 59 mW cm−2 under LED irradiation. The smaller light intensity caused a smaller OCP shift in the case of AM 1.5G irradiation.
3.3 Light intensity dependence of electrocatalyst potential and photocatalytic activity
After illumination at 59 mW cm−2 on cSTO/0.4 μmol cm−2 CoOOH/FTO for 7.5 h, light intensity was varied in the range of 12–94 mW cm−2. Fig. 4a shows the CoOOH potentials in cSTO/0.4 μmol cm−2 CoOOH/FTO under various light intensities. When the light intensity decreased from 59 to 30 and 12 mW cm−2, the CoOOH potential responded to the light-intensity change and shifted to a negative potential. This is due to a decrease in the number of holes transported from cSTO to CoOOH because the photogenerated charge carriers decrease as the photon number decreases. | ||
| Fig. 4 (a) OCP and (b) gas production rate of the cSTO/0.4 μmol cm−2 CoOOH/FTO electrode in 0.5 M K-borate (pH 9.0–9.2) under LED light illumination (λ = 370 nm, 12–94 mW cm−2). WE: cSTO/0.4 μmol cm−2 CoOOH/FTO electrode and RE: Hg/Hg2Cl2 (sat. KCl). (c) Gas production rate and (d) CoOOH electrocatalyst potential as a function of photon numbers obtained from (a) and (b). | ||
When the light intensity returned to 59 mW cm−2, the CoOOH potential also returned to 1.57 V vs. RHE, while at higher light intensities (94 mW cm−2), the potential did not change significantly (1.57 V vs. RHE). This is probably due to the increase in the carrier recombination rate by the high photon number, resulting in no increase in the number of holes transported from cSTO to the CoOOH electrocatalyst. This phenomenon is similar to that in time-resolved spectroscopy, where a high photon number promotes recombination, resulting in lower carrier mobility in semiconductors.38,39
Fig. 4b shows the gas production rate when measuring the CoOOH potential. Similar to the CoOOH potential changes, the gas production rate changed as the light intensity increased or decreased. The average values are summarized in Fig. 4c as a function of the photon number. r(H2)/r(O2) was 0.26/0.12, 0.39/0.16, 0.46/0.24, and 0.52/0.22 μmol h−1 cm−2 for photon numbers (light intensity) of 8.0 × 1019 (12 mW cm−2), 2.0 × 1020 (30 mW cm−2), 4.0 × 1020 (59 mW cm−2), and 6.4 × 1020 photons h−1 cm−2 (94 mW cm−2), respectively. Based on the results, the gas production rate was proportional to the 0.4th power of the incident photon number.
Tabata et al. examined the light-intensity dependence of water splitting efficiency on a K4Nb6O17 photocatalyst and reported that r(H2) was proportional to the square root of light intensity in the range of 1–100 mW cm−2.40 Hisatomi et al. reported a transition from proportional dependence to square-root dependence of r(H2) and r(O2) on the Rh2−yCryO3/(Ga1−xZnx)(N1−xOx) photocatalyst above 1 × 1022 photons h−1.41 The obtained light-intensity dependence of photocatalytic activity in the present study is consistent with such behavior under high light intensities.
Fig. 4d shows the measured potential of CoOOH as a function of the photon number. In the range of 0.8–4.0 × 1020 photons h−1 cm−2, a linear relationship was observed, and the slope was 15 mV dec−1. The hole density (p) is described as eqn (3) with the carrier density in an intrinsic semiconductor (ni), elementary change (q), Boltzmann constant (k), absolute temperature (T), the Fermi level in an intrinsic semiconductor (Ei), and the hole quasi-Fermi level (Ef,h).
 | (3) |
Therefore, Ef,h should be shifted by 60 mV when the hole density (p) changes by one order (60 mV dec−1) according to eqn (4).
 | (4) |
The discrepancy between the measured and estimated slopes originated from carrier transport losses. The slope measurement takes into account the carrier loss in the processes of photon absorption, charge separation, and carrier transport. The hole density change (Δp) on the SrTiO3:Al photocatalyst surface calculated using eqn (5) was 1.8 when the photon number changed by one order (ΔEf,h = 0.015 V).
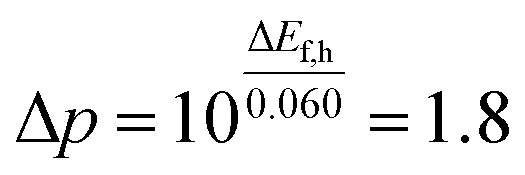 | (5) |
18% of the holes were transported to the SrTiO3:Al photocatalyst surface after charge carrier generation in the bulk. The hole transport rate was in close agreement with the measured IQY values. It can be concluded that the proposed method is able to measure the Ef,h in the SrTiO3:Al photocatalyst corresponding to the quantum efficiency in photocatalytic water splitting.
3.4 Control of the hole quasi-Fermi level in SrTiO3:Al
Fig. S13 in the ESI† shows the time course of the electrode potential and produced current when the electrode potential was controlled by CA. In the beginning, cSTO/0.4 μmol cm−2 CoOOH/FTO was illuminated for 17 h under open circuit conditions to stabilize the electrode potential. It took a longer time compared to the result in Fig. 3a, which was most likely due to the different contact conditions between cSTO and CoOOH/FTO. This resulted in lower r(H2) and r(O2) relative to Fig. 3b.The electrode potential reached 1.53 V vs. RHE and produced H2 and O2 with a r(H2)/r(O2) of 0.18/0.08 μmol h−1 cm−2 (Fig. 5a). The band diagram at OCP is shown in the middle panel in Fig. 5b. The CoOOH/FTO electrode potential was balanced with the Ef,h in the SrTiO3:Al photocatalyst and had a value of 1.53 V vs. RHE to drive the OER. The potential of Rh/Cr2O3 nanoparticles loaded on the SrTiO3:Al photocatalyst should be more negative than 0 V vs. RHE to drive the HER.
 | ||
| Fig. 5 (a) Gas production rate of the cSTO/0.4 μmol cm−2 CoOOH/FTO electrode at varied cSTO/CoOOH/FTO electrode potentials. WE: cSTO/0.4 μmol cm−2 CoOOH/FTO electrode, RE: Hg/Hg2Cl2 (sat. KCl), CE: Pt wire, electrolyte: 0.5 M K-borate (pH 9.0–9.2), and light source: LED light illumination (λ = 370 nm, 59 mW cm−2). (b) Plausible band diagram when Ef,h was more negative, equivalent, or more positive relative to OCP (1.53 V vs. RHE). | ||
After OCP measurement, CA was conducted at 1.46, 1.48, 1.50, 1.54, 1.56, and 1.58 V vs. RHE under illumination, and the OCP was measured again. Finally, CA was performed under dark conditions. Since the reference and counter electrodes were in a different chamber from the one containing cSTO/0.4 μmol cm−2 CoOOH/FTO (see Fig. 1), the detected gas was produced only by cSTO/0.4 μmol cm−2 CoOOH/FTO.
Fig. 5a shows r(H2) and r(O2) at various electrode potentials. Bar graphs with deep color exhibit detected gas production rates under light illumination, and bar graphs with pale color show the gas production rates under dark conditions. The estimated values from the current density during CA in the dark are also shown as scatter plots.
Below OCP (i.e., at 1.46, 1.48, and 1.50 V vs. RHE), r(H2)/r(O2) was 0.22/0.09, 0.22/0.08, and 0.19/0.07 μmol h−1 cm−2, respectively. r(H2) increased as the potential decreased from OCP, while r(O2) was almost the same with respect to OCP. A plausible band diagram is depicted in the left panel of Fig. 5b. The controlled CoOOH potential was more negative than the potential driving the OER. Therefore, photo-generated holes were used only for the oxidation of Con+ to Con+1.
The potentiostat introduced electrons to reduce Con+1 to Con+. The measured cathodic current during CA under illumination is shown with blue lines in Fig. S13† and supported the introduced electrons from the potentiostat. The Ef,h was negative compared to OCP, so the electron quasi-Fermi level should become more negative, resulting in a large overpotential for the HER. Therefore, r(H2) increased as the CoOOH/FTO electrode potential decreased.
This condition is similar to the HER with an electron donor that has a redox potential of 1.46, 1.48, and 1.50 V vs. RHE pinning the Ef,h there. Although the Ef,h was controlled below the onset potential of the OER, we detected O2 that originated from cSTO that was not in contact with CoOOH/FTO. The cSTO particles probably drive overall water splitting regardless of the potential.
Above OCP (i.e., at 1.54, 1.56, and 1.58 V vs. RHE), the detected r(H2)/r(O2) was 0.16/0.10, 0.14/0.10, and 0.13/0.11 μmol h−1 cm−2, respectively. While r(H2) decreased with the electrode potential, r(O2) increased. As the electrode was set at a more positive potential than OCP, cSTO/CoOOH/FTO drove the electrochemical OER regardless of illumination. The r(O2) produced under dark conditions was 0.04, 0.04, and 0.06 μmol h−1 cm−2 at 1.54, 1.56, and 1.58 V vs. RHE, respectively (pale-colored bar graphs in Fig. 5a). These values are consistent with estimated values from the current density during CA under dark conditions (scatter plots in Fig. 5a). The measured current density during CA is also shown in Fig. S13 in the ESI.†
The right panel in Fig. 5b displays a plausible band diagram at electrode potentials above OCP. Since the OER overpotential increased, r(O2) increased with the electrode potential. Accordingly, the electron quasi-Fermi level moved toward a positive potential, so the HER overpotential decreased, which resulted in a decrease in r(H2).
Fig. S14† shows the ratio of r(H2) to r(O2) at various electrode potentials. While r(H2)/r(O2) was 2 at OCP, the ratios were 2.5, 2.7, and 2.9 at 1.46, 1.48, and 1.50 V vs. RHE, respectively. In contrast, the ratios at 1.54, 1.56, and 1.58 V vs. RHE were 1.6, 1.4, and 1.1, respectively. Thus, in the proposed method, r(H2) and r(H2)/r(O2) can be controlled by modulating the Ef,h in the SrTiO3:Al photocatalyst.
3.5 Pt potential measurement on the CrOx/cSTO/Pt electrode
We also attempted to measure the potential of HER electrocatalysts during overall water splitting using CrOx-coated, cSTO-loaded Pt foil (CrOx/cSTO/Pt). Details of the sample preparation are given under Fig. S15 in the ESI.† Fig. S15a† shows the OCP of the CrOx/cSTO/Pt electrode as a function of time. The measured potential was shifted to a negative potential under illumination, but the electrode potential only reached 0.34 V vs. RHE, which is substantially more positive than 0 V vs. RHE.When a metal with a large work function such as Pt is in contact with cSTO, a Schottky barrier will be formed at the interface, inhibiting efficient electron transport.36 Another previous study suggests that Pt may trap holes and act as a recombination site.42 As shown in Fig. S15b,† the lower initial r(H2) of 1.3 μmol h−1 cm−2 compared to cSTO/CoOOH/FTO (2.5–2.7 μmol h−1 cm−2) supports that the cSTO/Pt foil interface acts as a recombination site and efficient electron transport is inhibited.
A previous study reported that surface Pt electrocatalyst potential shifted to a negative potential during the photocatalytic HER probably due to the surface condition change.42 Following this report, the OCP of CrOx/cSTO/Pt was measured after CP at −0.5 mA cm−2 (Fig. S16†). Although electrode potential was negative relative to 0 V vs. RHE during CP, OCP reached 0.6 V vs. RHE. Because of the Schottky junction formed at the interface between the n-type semiconductor photocatalysts and the HER electrocatalysts, it was found difficult to measure the potential of the HER electrocatalyst during overall water splitting. This also suggests that the barrier for photocatalysis resides in the HER catalyst interface on the n-type semiconductor. This type of OCP measurement can be further extended to improve the photocatalytic performance.
4 Conclusions
This study developed an operando measurement system for electrocatalyst potential on particulate photocatalysts. cSTO photocatalyst powder was loaded on a CoOOH/FTO electrode to obtain a novel inverted photocatalyst structure. Under illumination, photo-generated holes were transported from cSTO to the CoOOH/FTO electrode, which shifted the electrode potential to a more positive potential to drive the OER. The potential or quasi-Fermi level applied by photoexcited holes of cSTO to the CoOOH/FTO electrode was successfully measured in this system (i.e., 1.56–1.59 V vs. RHE). The produced hydrogen and oxygen gases were also quantified, confirming the operando potential measurement during overall water splitting with an IQY of 12%. The CoOOH potential was positively shifted by 15 mV when the light intensity increased by one order. The results indicated that 18% of photo-generated holes were transported from the cSTO bulk to CoOOH/FTO. The proposed method was able to measure the Ef,h in the CoOOH/SrTiO3:Al photocatalyst corresponding to the quantum efficiency in photocatalytic water splitting. Finally, the varied CoOOH/FTO electrode potential led to an increase in r(H2) as the potential moved to a negative potential. r(H2) was controlled by modulating the Ef,h in SrTiO3:Al photocatalysts because the electron quasi-Fermi level consistently shifted with it. These methodologies could be further utilized for the quantitative understanding of particulate photocatalysts.Author contributions
Y. K. designed the concept of this study and fabricated the photocatalysts and electrodes. Y. K., K. O., Y. S., and T. H. performed the electrochemical measurements and data analysis. K. T. designed the concept of this study, and planned and supervised this project. All the authors wrote the manuscript.Conflicts of interest
There is no conflict of interest.Acknowledgements
This work was financially supported by the Mohammed bin Salman Center for Future Science and Technology for Saudi-Japan Vision 2030 at The University of Tokyo (MbSC2030). A Grant-in-Aid for JSPS Fellows (No. 22KJ0969) and MERIT-WINGS also supported this work.References
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- P. W. Huang and M. C. Hatzell, Nat. Commun., 2022, 13, 7908 CrossRef CAS PubMed.
- D. Bahnemann, P. Robertson, C. Wang, W. Choi, H. Daly, M. Danish, H. de Lasa, S. Escobedo, C. Hardacre, T. H. Jeon, B. Kim, H. Kisch, W. Li, M. Long, M. Muneer, N. Skillen and J. Zhang, JPhys Energy, 2023, 5, 012004 CrossRef.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- T. Hisatomi and K. Domen, Next Energy, 2023, 1, 100006 CrossRef.
- X. Xin, Y. Li, Y. Zhang, Y. Wang, X. Chi, Y. Wei, C. Diao, J. Su, R. Wang, P. Guo, J. Yu, J. Zhang, A. J. Sobrido, M.-M. Titirici and X. Li, Nat. Commun., 2024, 15, 337 CrossRef CAS PubMed.
- L. Lin, Y. Ma, J. J. M. Vequizo, M. Nakabayashi, C. Gu, X. Tao, H. Yoshida, Y. Pihosh, Y. Nishina, A. Yamakata, N. Shibata, T. Hisatomi, T. Takata and K. Domen, Nat. Commun., 2024, 15, 397 CrossRef CAS PubMed.
- J. Xiao, M. Nakabayashi, T. Hisatomi, J. J. M. Vequizo, W. Li, K. Chen, X. Tao, A. Yamakata, N. Shibata, T. Takata, Y. Inoue and K. Domen, Nat. Commun., 2023, 14, 8030 CrossRef CAS PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- L. Lin, P. Kaewdee, V. Nandal, R. Shoji, H. Matsuzaki, K. Seki, M. Nakabayashi, N. Shibata, X. Tao, X. Liang, Y. Ma, T. Hisatomi, T. Takata and K. Domen, Angew. Chem., Int. Ed., 2023, 62, e202310607 CrossRef CAS PubMed.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- A. T. Garcia-Esparza and K. Takanabe, J. Mater. Chem. A, 2016, 4, 2894–2908 RSC.
- E. Nurlaela, S. Ould-Chikh, I. Llorens, J.-L. Hazemann and K. Takanabe, Chem. Mater., 2015, 27, 5685–5694 CrossRef CAS.
- Y. Kawase, T. Higashi, M. Katayama, K. Domen and K. Takanabe, ACS Appl. Mater. Interfaces, 2021, 13, 16317–16325 CrossRef CAS PubMed.
- Y. Kawase, T. Higashi, K. Obata, Y. Sasaki, M. Katayama, K. Domen and K. Takanabe, ACS Sustainable Chem. Eng., 2022, 10, 14705–14714 CrossRef CAS.
- W. E. Pinson, Nature, 1977, 269, 316–318 CrossRef CAS.
- F. Lin and S. W. Boettcher, Nat. Mater., 2014, 13, 81–86 CrossRef CAS PubMed.
- M. R. Nellist, F. A. Laskowski, F. Lin, T. J. Mills and S. W. Boettcher, Acc. Chem. Res., 2016, 49, 733–740 CrossRef CAS PubMed.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2017, 3, 46–52 CrossRef.
- F. A. L. Laskowski, S. Z. Oener, M. R. Nellist, A. M. Gordon, D. C. Bain, J. L. Fehrs and S. W. Boettcher, Nat. Mater., 2019, 19, 69–76 CrossRef PubMed.
- M. Shen, A. J. Kaufman, J. Huang, C. Price and S. W. Boettcher, Nano Lett., 2022, 22, 9493–9499 CrossRef CAS PubMed.
- R. Chen, D. Zhang, Z. Wang, D. Li, L. Zhang, X. Wang, F. Fan and C. Li, J. Am. Chem. Soc., 2023, 145, 4667–4674 CrossRef CAS PubMed.
- S. Daemi, A. Kundmann, K. Becker, P. Cendula and F. E. Osterloh, Energy Environ. Sci., 2023, 16, 4530–4538 RSC.
- T. Sagara, Y. Aikawa and M. Sukigara, J. Phys. Chem., 1987, 91, 1173–1177 CrossRef CAS.
- B. Zutter, Z. Chen, L. Barrera, W. Gaieck, A. S. Lapp, K. Watanabe, A. Kudo, D. V. Esposito, R. Bala Chandran, S. Ardo and A. A. Talin, ACS Nano, 2023, 17, 9405–9414 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- F. A. L. Laskowski, M. R. Nellist, J. Qiu and S. W. Boettcher, J. Am. Chem. Soc., 2019, 141, 1394–1405 CrossRef CAS PubMed.
- T. Suguro, F. Kishimoto, N. Kariya, T. Fukui, M. Nakabayashi, N. Shibata, T. Takata, K. Domen and K. Takanabe, Nat. Commun., 2022, 13, 5698 CrossRef CAS PubMed.
- Z. Jin and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12651–12656 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS PubMed.
- Y. Ham, T. Minegishi, T. Hisatomi and K. Domen, Chem. Commun., 2016, 52, 5011–5014 RSC.
- T. Hisatomi, T. Yamamoto, Q. Wang, T. Nakanishi, T. Higashi, M. Katayama, T. Minegishi and K. Domen, Catal. Sci. Technol., 2018, 8, 3918–3925 RSC.
- T. J. Savenije, A. Huijser, M. J. W. Vermeulen and R. Katoh, Chem. Phys. Lett., 2008, 461, 93–96 CrossRef CAS.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS.
- S. Tabata, H. Ohnishi, E. Yagasaki, M. Ippommatsu and K. Domen, Catal. Lett., 1994, 28, 417–422 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef CAS.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218–13219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Illustration of the reactor setup and additional experimental results of ICP-OES, SEM, electrochemical measurements, gas quantification, and UV-vis. See DOI: https://doi.org/10.1039/d4ta02571g |
| This journal is © The Royal Society of Chemistry 2024 |