
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLateral functionalization of a one-dimensional covalent organic framework for efficient photocatalytic hydrogen evolution from water†
Chao
Liu
ab,
De-Li
Ma
 ab,
Peng-Ju
Tian
b,
Chao
Jia
b,
Qiao-Yan
Qi
b,
Guo-Fang
Jiang
*a and
Xin
Zhao
*b
ab,
Peng-Ju
Tian
b,
Chao
Jia
b,
Qiao-Yan
Qi
b,
Guo-Fang
Jiang
*a and
Xin
Zhao
*b
aAdvanced Catalytic Engineer Research Center of the Ministry of Education, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, Hunan, China. E-mail: gfjiang@hnu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xzhao@sioc.ac.cn
First published on 22nd May 2024
Abstract
Covalent organic frameworks (COFs), a class of porous crystalline organic polymers, have emerged as promising materials for photocatalysis. Structural functionalization of COFs is an effective strategy to improve their photocatalytic performance. However, this approach is mainly limited within interior parts of COFs. Herein we report exterior functionalization of a one-dimensional (1D) COF by introducing terpyridine units on its edges to anchor Pt(II) cations. The as-obtained 1D COF (Pt-Tpy-COF) exhibits high photocatalytic activity for hydrogen evolution from water, with a hydrogen evolution rate up to 7.8 mmol g−1 h−1. Experimental studies and theoretical calculations reveal that the high performance of Pt-Tpy-COF benefits from its distinct 1D framework with readily accessible active sites. This study not only demonstrates the potential of 1D COFs as photocatalysts, but also provides valuable insights for the design and development of highly efficient catalysts for various catalytic applications based on the structural features of this new type of nanoporous crystalline framework materials.
Introduction
The rapid development of modern industry has brought the pressing issues of energy crisis and environmental pollution to the forefront of global concerns.1,2 For decades, fossil fuels such as natural gas, petroleum, and coal have served as the primary sources of energy.3,4 However, the unchecked use of these non-renewable resources has led to severe environmental damage and resource depletion.5,6 In light of these challenges, there is an urgent need for new and sustainable energy sources to ensure harmonious development of human society. Hydrogen, with its exceptionally high calorific value and production of only a clean final product (H2O), stands out as the most promising candidate among all the biological, chemical, and fossil fuels.7,8 Given the abundant natural resources of sunlight and water, a compelling solution to the impending energy crisis is the production of hydrogen through photocatalytic water splitting, a process involving splitting water molecules into hydrogen and oxygen using a photocatalyst.9–11 However, photocatalytic water splitting faces several practical challenges, primarily due to its thermodynamically unfavorable nature.12–14 Efforts to address these challenges have focused on the development of efficient photocatalysts.15–17Photocatalysts are essential for harnessing solar energy to drive chemical reactions. In this context, nanoporous materials with high specific surface areas have garnered significant attention from researchers.18–20 These materials offer a larger active space for reactant adsorption and subsequent catalytic reactions.21–24 In recent years, covalent organic frameworks (COFs), a class of crystalline porous polymers,25–28 have emerged as promising materials for photocatalytic studies.29–32 One of the key advantages of COFs is their capacity for atomically precise integration of organic units, which enables the creation of diverse pre-designed backbones and allows various functionalization.33–35 As a result, their optical and electrical properties can be regulated to meet the criteria for optimal performance. The past few years have witnessed the significant progress on COF-based photocatalysts for water splitting.36–39 However, much research has focused on two-dimensional (2D) and three-dimensional (3D) COFs.40–43 For these two types of COFs, functional moieties are usually located within their interior channels, which might result in limited accessibility of active sites. Compared to them, one-dimensional (1D) COFs (or analogues) are constructed by linearly extending building blocks.44–49 Such a structural feature not only favours their exfoliation dispersion but also enables exterior functionalization at their lateral positions (Scheme 1). However, this advantage has rarely been explored.50,51 In this work, we demonstrate that exterior functionalization of a 1D COF with a metal coordinating unit makes it a highly efficient photocatalyst for hydrogen evolution from water, opening up a way to develop high performance catalysts based on 1D COFs.
 | ||
| Scheme 1 Illustration for the comparison of different distributions of active sites (represented by red balls) in a (a) 3D COF, (b) 2D COF, and (c) 1D COF. | ||
Results and discussion
Structure and property characterization
To realize the above design, we devised a 1D COF (Tpy-COF) in which tripyridyl units are incorporated on its two edges. Subsequently, Pt(II) cations were introduced to bind with the tripyridyl units via post-coordination (Pt-Tpy-COF) (Scheme 2). Tpy-COF was synthesized as yellow powder via the condensation of tetraamine (Pyr-NH2) and dialdehyde (Tpy-CHO) under a solvothermal condition. Then, the as-obtained Tpy-COF was reacted with Pt(COD)Cl2, still under the solvothermal condition to afford Pt-Tpy-COF as brown powder. The chemical structures of the COFs were analyzed by Fourier transform infrared (FT-IR) spectroscopy. The comparison of the FT-IR spectra of the monomers and the COFs showed that the intensity of the N–H stretching vibration peak (∼3346 cm−1) of the amino group in Pyr-NH2, the peak of the carbonyl group (1709 cm−1), and the peak corresponding to O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H (2825 cm−1 and 2734 cm−1) in Tpy-CHO were significantly reduced after the polycondensation reaction, indicating a high degree of polymerization. The peaks at 1627 cm−1 in the FT-IR spectra of Tpy-COF and Pt-Tpy-COF were attributed to the stretching vibration band of imine bond (Fig. 1a), indicating the formation of imine linkages from the condensation of the two monomers. Furthermore, the chemical structure of Tpy-COF at the molecular level was also researched by solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectroscopy. As shown in Fig. 1b, the low-field signal at 155.8 ppm was assigned to the carbon atom of the imine group, while the peak at approximately 153.7 ppm was attributed to the CN bond in the pyridine ring. In addition, other signals at 150.4, 146.7, 140.9, 136.6, 135.0, 132.6, 128.1, 124.7 and 117.5 ppm were assigned to the carbon atoms in the aromatic rings. Those findings matched with the carbon species in the structure of Tpy-COF.
C–H (2825 cm−1 and 2734 cm−1) in Tpy-CHO were significantly reduced after the polycondensation reaction, indicating a high degree of polymerization. The peaks at 1627 cm−1 in the FT-IR spectra of Tpy-COF and Pt-Tpy-COF were attributed to the stretching vibration band of imine bond (Fig. 1a), indicating the formation of imine linkages from the condensation of the two monomers. Furthermore, the chemical structure of Tpy-COF at the molecular level was also researched by solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectroscopy. As shown in Fig. 1b, the low-field signal at 155.8 ppm was assigned to the carbon atom of the imine group, while the peak at approximately 153.7 ppm was attributed to the CN bond in the pyridine ring. In addition, other signals at 150.4, 146.7, 140.9, 136.6, 135.0, 132.6, 128.1, 124.7 and 117.5 ppm were assigned to the carbon atoms in the aromatic rings. Those findings matched with the carbon species in the structure of Tpy-COF.
 | ||
| Scheme 2 The preparation of Tpy-COF and Pt-Tpy-COF. | ||
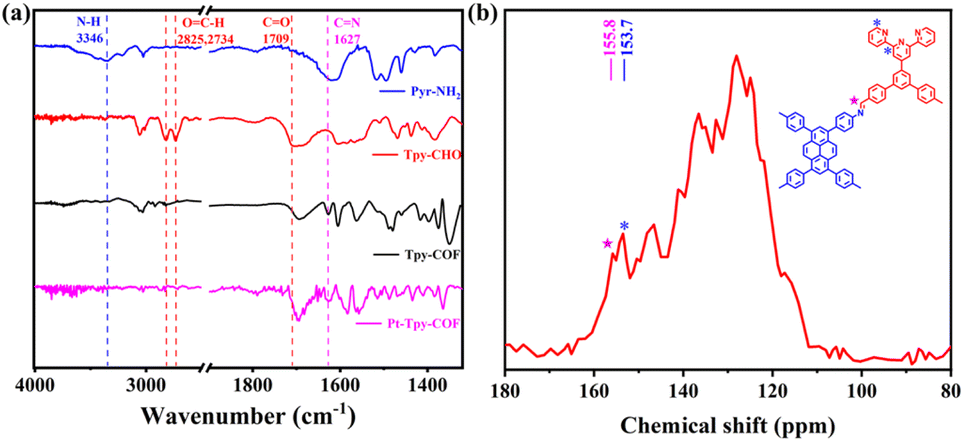 | ||
| Fig. 1 (a) FT-IR spectra of Pyr-NH2, Tpy-CHO, Tpy-COF, and Pt-Tpy-COF. (b) Solid-state 13C CP-MAS NMR spectrum of Tpy-COF. | ||
The crystalline structures of the COFs were elucidated based on the analysis on the experimental and simulated powder X-ray diffraction (PXRD) patterns. For Tpy-COF, a series of diffraction peaks could be observed due to its high crystallinity (Fig. 2a), which provides abundant crystallographic information and thus the peaks could be directly indexed to derive the cell parameters. Two high-intensity diffraction peaks were observed at 3.25° and 4.02°, corresponding to the (100) and (010) crystal planes, respectively. Additionally, six weak-intensity diffraction peaks were observed at 6.07°, 7.32°, 9.78°, 12.28°, 15.99°, and 23.86°, which were attributed to (2–10), (1–20), (300), (220), (4–40), and (8–30) facets. The comparison shows that the experimental PXRD data match well with the theoretical PXRD pattern simulated for the one-dimensional COF with AA stacking (Fig. 2b). In contrast, the theoretical powder diffraction pattern simulated for the 1D structure with AB stacking significantly deviates from the experimental data (Fig. 2c), ruling out the AB stacking structure. The cell parameters of Tpy-COF were obtained base on Pawley refinement, affording a = 29.83 Å, b = 24.11 Å, c = 4.14 Å, α = β = 90°, γ = 114°, with Rp = 3.33% and Rwp = 4.98%.
 | ||
| Fig. 2 (a) Experimental (black) and Pawley refined (red) PXRD patterns of Tpy-COF, simulated PXRD patterns for Tpy-COF with AA stacking (blue) and AB stacking (pink), and the difference between the experimental and the refined PXRD patterns (gray line). (b) Top and side views of the AA stacking structure of Tpy-COF. (c) Top and side views of the AB stacking structure of Tpy-COF. | ||
The comparison between the PXRD patterns of Pt-Tpy-COF and Tpy-COF, indicated that their primary peaks were almost same (Fig. 3a), suggesting that the backbone integrity was preserved after Pt(II) ions were loaded. It also should be noted that several of the fine peaks experienced minor alterations, with some being displaced, some vanishing, and new peaks emerging. All of these observations suggest that the Pt(II) cations were effectively deposited onto the COF framework. Similarly, Pt-Tpy-COF also exhibits high crystallinity. Its 11 diffraction peaks facilitate directly indexing and structure elucidation (Fig. 3b). Two high-intensity diffraction peaks appear at 3.25° and 3.87°, along with nine weak-intensity diffraction peaks at 6.12°, 7.08°, 7.72°, 8.96°, 9.77°, 12.21°, 13.56°, 15.13°, and 18.54°, corresponding to the diffractions of (100), (010), (2–10), (1–20), (020), (3–10), (3–20), (4–20), (4–30), (5–10), and (4–50) planes, respectively. The comparison of the PXRD patterns shows good agreement between the experimental data and the simulated PXRD pattern of the 1D structure with AA stacking (Fig. 3c and d). The difference plot displayed high consistency between them, indicating that the as-prepared polymer conforms to the predicted structure. Pawley refinement gave cell parameters to be a = 29.71 Å, b = 25.02 Å, c = 3.72 Å, α = β = 90°, γ = 113°, with Rp = 4.08%, and Rwp = 5.52%.
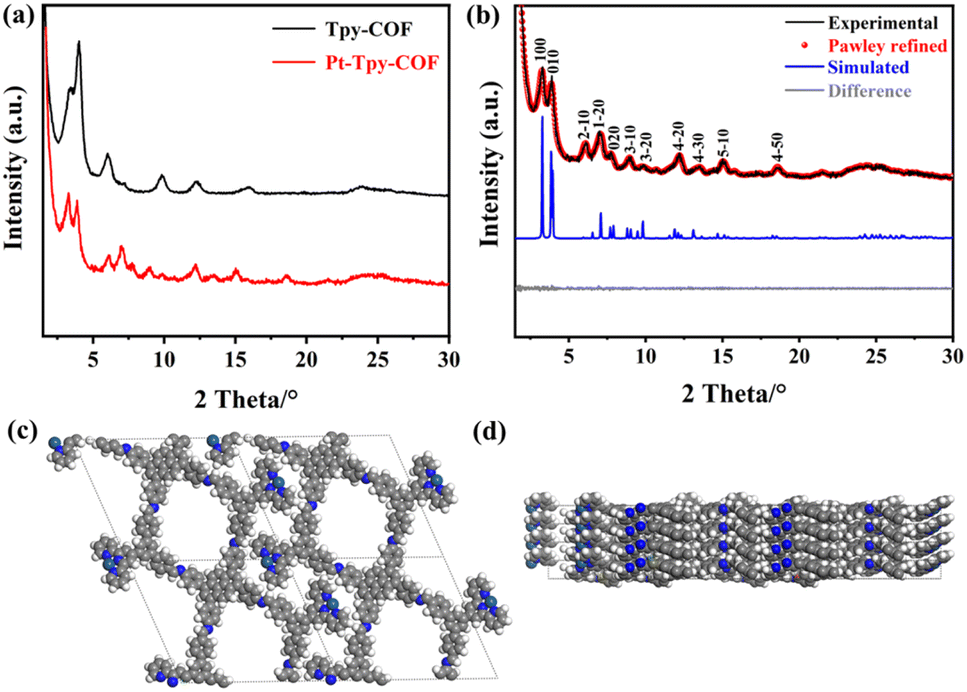 | ||
| Fig. 3 (a) Comparison of the experimental PXRD patterns of Tpy-COF and Pt-Tpy-COF. (b) PXRD profiles of Pt-Tpy-COF: the experimental (black), refined (red), and simulated (blue, based on AA stacking) PXRD patterns, and difference plot between the experimental and refined PXRD patterns (gray). (c) Top and (d) side view of structural models of Pt-Tpy-COF, respectively. | ||
The Pt elemental content in Pt-Tpy-COF was revealed by elemental analysis to be 8.44% (Table S1†), which is just half of the theoretical value (17.91%, Fig. S1†), suggesting that only part of the terpyridine units in Pt-Tpy-COF successfully coordinated with Pt(II). Moreover, the structural features of Pt-Tpy-COF were further determined by X-ray photoelectron spectroscopy (XPS) analysis. The XPS survey spectrum of Pt-Tpy-COF indicates that C, N, O, Cl, and Pt elements are present (Fig. 4a). The C 1s spectrum shows the predominance of C–C, C–H, and C–N bonds in Pt-Tpy-COF (Fig. 4b). From the N 1s spectrum, it is evident that NC, N–H, and N–Pt bonds are predominantly present in Pt-Tpy-COF (Fig. 4c), suggesting that Pt(II) coordinates with the N atom of pyridine. The Pt(II)–N coordination was also evidenced by the Pt 4f spectrum, which shows Pt–N and Pt–Cl bonds largely presenting in Pt-Tpy-COF (Fig. 4d).52 Surprisingly, a small amount of Pt(0) was observed in this spectrum, which might be due to the reduction of partial Pt(II) to Pt(0) during the loading of Pt(II). This could contribute to the subsequent photocatalytic hydrogen evolution process.
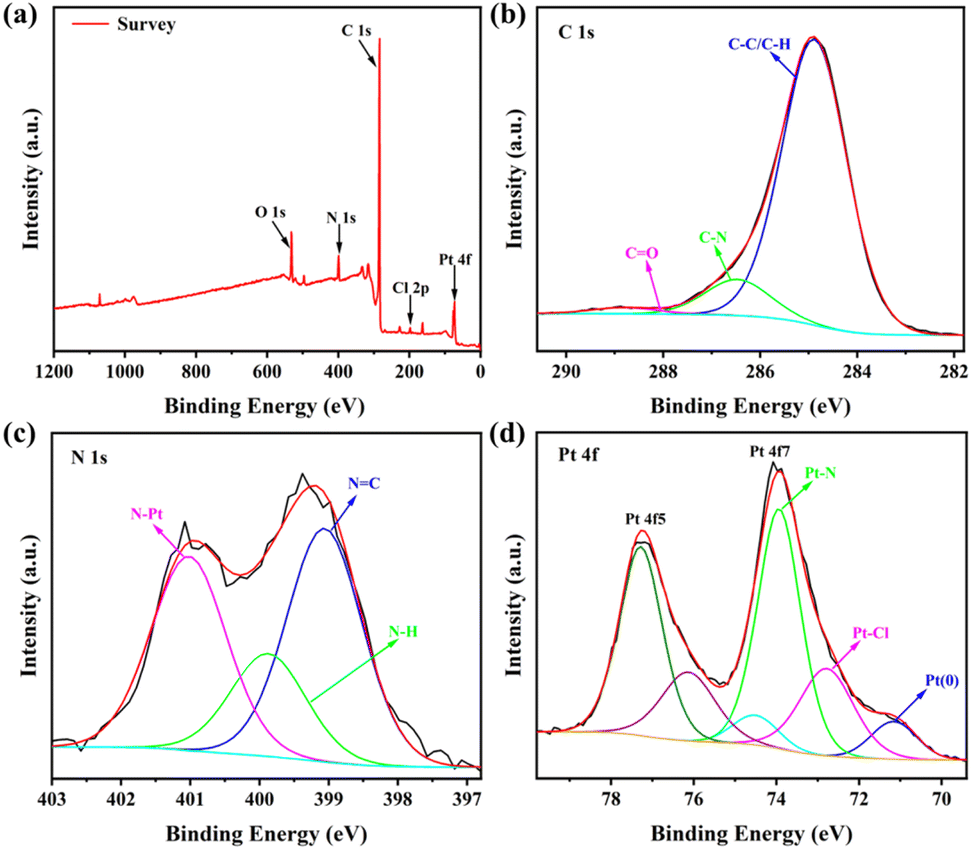 | ||
| Fig. 4 The XPS spectra of Pt-Tpy-COF: (a) survey spectrum, (b) C 1s, (c) N 1s, and (d) Pt 4f. | ||
The porosity of the COFs was assessed by nitrogen adsorption–desorption measurements (Fig. S2†). On the basis of the analysis of N2 adsorption data at low pressure region (P/P0 = 0.01–0.10), the Brunauer–Emmett–Teller (BET) surface areas of Tpy-COF and Pt-Tpy-COF were determined to be 513 and 583 m2 g−1, respectively (Fig. S3†). Their pore size distributions (PSDs) were estimated using the nonlocal density functional theory (NLDFT) fitting, which gave a major PSD of ∼10.1 Å for Tpy-COF and approximately 9.7 Å for Pt-Tpy-COF (Fig. S4†). The values agree well with the theoretical pore size of the COFs (11.8 Å) (Fig. S5†). The almost no change before and after the Pt(II) coordination suggests that the introduction of Pt(II) did not alter the original pore structure of the COF, in accordance with the design of the exterior functionalization. Morphological characterization of the two COFs with transmission electron microscope (TEM) indicates that Tpy-COF and Pt-Tpy-COF both exhibit a nanosheet morphology (Fig. S6†). In the TEM image of Pt-Tpy-COF some black spots could be observed, which was ascribed to the presence of Pt.53 The thermal stability of Pt-Tpy-COF was examined with thermogravimetric analysis (TGA). It showed that weight loss occurred only 5% upon the temperature increasing from 25 to 492 °C (Fig. S7†), suggesting that it possesses a high thermal stability.
Excellent light absorption is prerequisite for photocatalytic applications.54 The ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) of Tpy-COF and Pt-Tpy-COF showed they both exhibit strong light absorption in the visible region. The spectra of Tpy-COF and Pt-Tpy-COF showed wide light absorption up to ca. 600 nm and 650 nm, respectively (Fig. 5a). This red-shift absorption can be attributed to the anchor of Pt(II) cations in Pt-Tpy-COF. Based on the Tauc plot analysis, the optical band gap energies (Eg) of Tpy-COF and Pt-Tpy-COF were calculated to be 2.25 eV and 2.16 eV, respectively (Fig. 5b). Furthermore, Mott–Schottky measurements were conducted (Fig. S8†), on the basis of which the values of conduction band (ECB) of Tpy-COF and Pt-Tpy-COF to were evaluated to be −0.06 V and −0.59 V vs. NHE, respectively. Tpy-COF and Pt-Tpy-COF exhibited positive slopes in the Mott–Schottky plots, suggesting that they were typical n-type semiconductors.55 Afterward, the valence band values (EVB) were calculated via the equation: EVB = ECB + Eg, giving the values of 2.19 V and 1.57 V for Tpy-COF and Pt-Tpy-COF, respectively (Fig. 5c). Since the ECB values of them are more negative than the standard potential of H+/H2, Tpy-COF and Pt-Tpy-COF are promising candidates for the thermodynamic transfer of photoexcited electrons to H+ to facilitate the evolution of hydrogen.
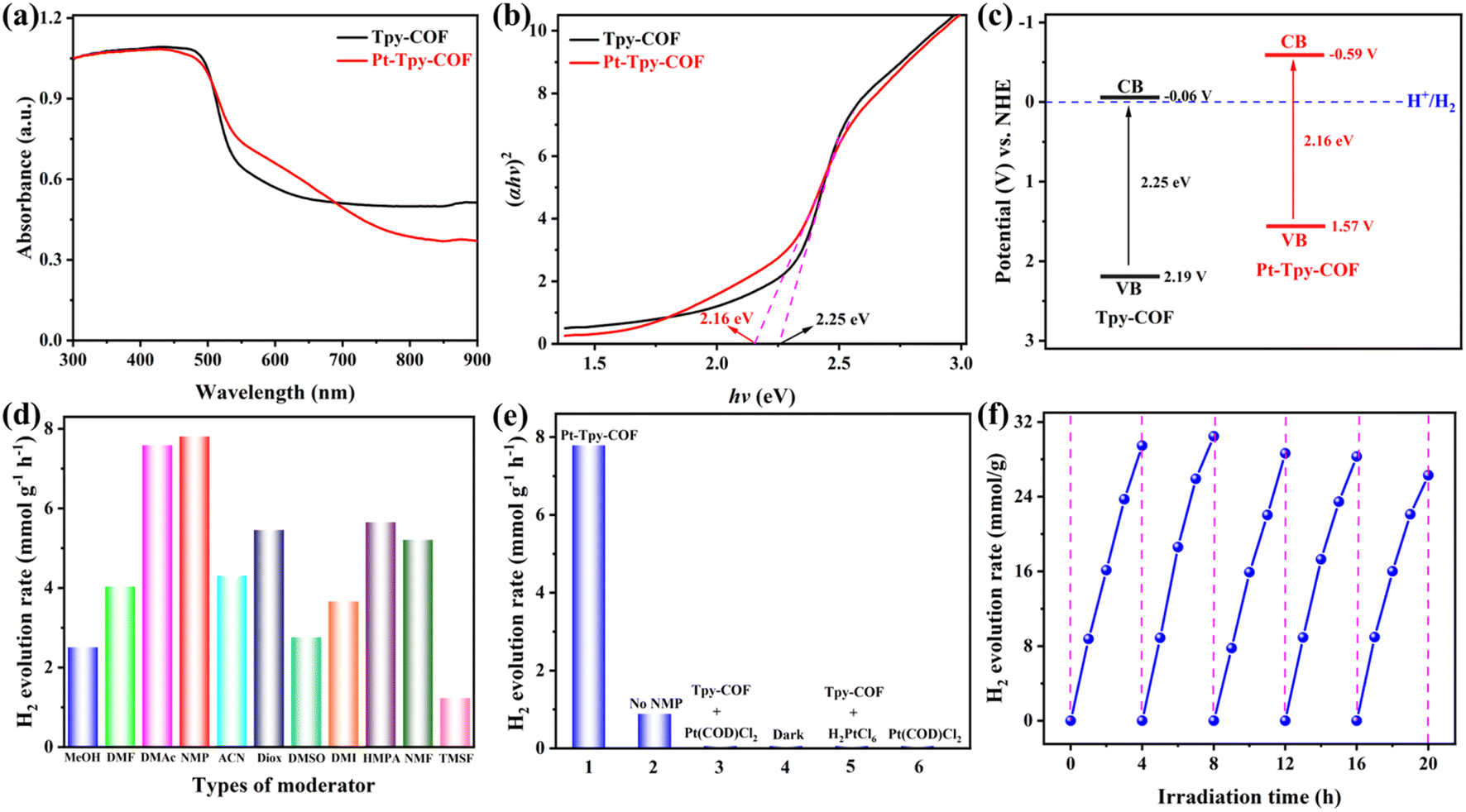 | ||
| Fig. 5 (a) UV-vis DRS spectra, (b) Tauc plots, and (c) band position and band gap of Tpy-COF and Pt-Tpy-COF. (d) Comparison of photocatalytic hydrogen evolution rates for Pt-Tpy-COF with the addition of different moderator species. (e) Comparison of hydrogen evolution rates catalyzed by Pt-Tpy-COF and other catalytic systems under visible light irradiation (λ > 400 nm). (f) Long-term hydrogen production test for Pt-Tpy-COF under visible light irradiation (λ > 400 nm). | ||
Photocatalytic hydrogen evolution
Encouraged by the above characterization results, photocatalytic hydrogen evolution performance of Tpy-COF and Pt-Tpy-COF was examined. Under a condition of visible light irradiation (λ > 400 nm, 300 W Xe lamp), ascorbic acid (AA) as a sacrificial electron donor (SED), and absence of a co-catalyst, Tpy-COF did not exhibit any noticeable activity in hydrogen production. In contrast, Pt-Tpy-COF showed a considerable hydrogen evolution rate (HER) with an average rate of 883.5 μmol g−1 h−1 under such a condition (Fig. S9†). Through further optimization of the conditions, we discovered that the hydrogen production activity of Pt-Tpy-COF was greatly influenced by the types of SEDs and moderator used. For instance, the effect of different SED, such as AA, triethanolamine (TEOA), and lactic acid (LA) was investigated. The results showed that only ascorbic acid worked for hydrogen evolution (Fig. S10†). Therefore, the concentration of ascorbic acid in water was further optimized, which revealed that a concentration of 0.3 M gave the highest HER (7234.8 μmol g−1 h−1) among the five concentrations (0.1–0.5 M) examined (Fig. S11†). In addition, different moderators were also explored, including methanol (MeOH), N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMAc), N-methylpyrrolidone (NMP), acetonitrile (ACN), 1,4-dioxane (Diox), dimethyl sulfoxide (DMSO), 1,3-dimethyl-2-imidazolidinone (DMI), hexamethylphosphoramide (HMPA), N-methylformamide (NMF), and tetramethylene sulfone (TMSF) for which HERs more than 1000 μmol g−1 h−1 was obtained. Among them, NMP was found to be the best by significantly increasing HER to 7801.6 μmol g−1 h−1, while the use of DMAc, Diox, HMPA, and NMF also result in HERs exceeding 5000 μmol g−1 h−1 (Fig. 5d). The control experiment has demonstrated that the behavior of photocatalytic hydrogen evolution is significantly influenced by the light source and catalyst (Fig. 5e).Hydrogen production was not observed in the absence of light irradiation. Substituting the photocatalyst Pt-Tpy-COF with other catalysts, such as Tpy-COF with Pt(COD)Cl2, or Tpy-COF with H2PtCl6, or Pt(COD)Cl2 alone, also did not result in hydrogen evolution. The experimental results demonstrated that hydrogen was produced in the system only when both the photocatalyst Pt-Tpy-COF and light conditions are present. Cycling experiments were conducted to further evaluate the stability of Pt-Tpy-COF in photocatalytic hydrogen evolution. There was no significant decrease in HER and the linear relationship was maintained after five cycles (Fig. 5f). Additionally, the PXRD pattern and FT-IR spectra indicate that the structure of Pt-Tpy-COF remained largely unchanged after the photocatalytic reaction (Fig. S12 and 13†), indicating outstanding reusability of Pt-Tpy-COF.
Optical and electronic properties
In order to study the difference in photocatalytic activity between Tpy-COF and Pt-Tpy-COF for hydrogen evolution, a series of photoelectric experiments were performed. It is well known that efficient generation, separation, and transport of photogenerated charge carriers in a semiconductor photocatalyst are crucial for improving photocatalytic activity. We further investigated the electronic transfer performance of the COFs through electrochemical impedance spectroscopy (EIS) and photocurrent (i–T) tests.56,57 A cathodic photocurrent was obtained for Pt-Tpy-COF at 0 V versus reversible hydrogen electrode under visible light irradiation (λ > 400 nm), which is about 5 times higher than that of Tpy-COF, indicating the high mobility and separation of charge carriers in Pt-Tpy-COF (Fig. 6a). Furthermore, Pt-Tpy-COF also showed enhanced charge carrier transport efficiency and higher electron–hole separation rate, as demonstrated by its smaller semicircle radius than that of Tpy-COF in the EIS (Fig. 6b). A similar phenomenon was observed for photo-luminescence (PL) characterization, which can reflect the electron–hole recombination of the materials. It was found that the fluorescence intensity of Pt-Tpy-COF was lower than that of Tpy-COF, indicating a lower electron–hole recombination rate (Fig. 6c). | ||
| Fig. 6 (a) Transient photocurrent spectra, (b) EIS Nyquist plots, and (c) steady-state PL spectra of Tpy-COF and Pt-Tpy-COF. (d) Schematic diagram for the mechanism of hydrogen production catalyzed by Pt-Tpy-COF. | ||
Mechanism of photocatalytic hydrogen evolution
To gain a better understanding of the process of hydrogen evolution in the photolytic water splitting of Pt-Tpy-COF, we conducted density functional theory (DFT) calculations using the Dmol3 software in Materials Studio 7.0. Electrostatic potential surfaces of the model systems for Tpy-COF, Tpy-COF@H2O, Pt-Tpy-COF, Pt-Tpy-COF@H2O, and H2O were calculated (Fig. S14†). The electrostatic potential surfaces showed that the electron-rich areas of Tpy-COF and Pt-Tpy-COF were centered near the terpyridine portion. Additionally, Pt-Tpy-COF has a more negative electrostatic potential than Tpy-COF, possibly due to the presence of two chlorine ions on the Pt(II) after it is anchored to the terpyridine moiety. The high electronegativity of chloride ions allows their formation of hydrogen bonds with water molecules, resulting in effective adsorption of water molecules to the framework. To evaluate the water molecule adsorption ability of the photocatalyst, physical measure of adsorption energy (ΔEads) was used as a ref. 58. The ΔEads was calculated using the formula: ΔEads = ECOF@H2O − (ECOF + EH2O), where ECOF@H2O represents the free energy of the COF after adsorption of water molecules, ECOF represents the free energy of the COF before adsorption of water molecules, and EH2O refers to the free energy of water molecules. A higher adsorption energy indicates that the COF can more easily adsorb water molecules and form hydrogen bonds. The calculations indicate that the water molecules adsorption energies of Tpy-COF and Pt-Tpy-COF were −14.78 kcal mol−1 and −26.78 kcal mol−1, respectively (Fig. S15 and 16†). The adsorption energy of Pt-Tpy-COF is approximately double the adsorption energy of Tpy-COF. This suggests that water molecules are more readily adsorbed onto Pt-Tpy-COF, promoting electron transfer and ultimately resulting in the evolution of hydrogen.The efficient photocatalytic hydrogen production mechanism of Pt-Tpy-COF is summarized based on experimental results and theoretical calculations (Fig. 6d). As a result of its excellent visible light absorption and dispersion, the electrons of Pt-Tpy-COF were excited from the valence band (VB) to the conduction band (CB) under visible light irradiation. Additionally, the introduction of Pt(II) enhances the electrical conductivity of the framework, which facilitates the photogenerated electrons to transfer from the COF to the electron-deficient platinum, improving the separation of photogenerated electrons and holes. On the other hand, Pt-Tpy-COF exhibits a high adsorption energy for water molecules, which gather around the skeleton through intermolecular hydrogen bonding. The platinum species anchoring on the terpyridine site act as an electron transport bridge, which can transfer enriched electrons to hydrogen atoms of water molecules, resulting in the evolution of hydrogen. Simultaneously, the sacrificial electron donor consumed the holes on the VB of Pt-Tpy-COF, enabling the photocatalytic process to persist. The photogenerated electrons produced through this migration pathway significantly boosted the activity of Pt-Tpy-COF. The Pt(II) cations precisely distributing on the edges of Pt-Tpy-COF enhance the efficiency of the photocatalyst compared to previous photocatalysts with a random distribution of other Pt co-catalysts.41,59,60 A comparison of the photocatalytic hydrogen production performance of Pt-Tpy-COF with that of some COF-based photocatalysts reported in literature indicates that the former is at a moderate level (Table S2†).
Conclusion
In summary, a 1D COF (Pt-Tpy-COF) containing Pt(II) cations was successfully constructed with an external functionalization design, which was further employed as a photocatalyst to achieve a stable and efficient decomposition of water into hydrogen. Experimental and theoretical studies have demonstrated that the introduction of terpyridine units on its edges to anchor Pt(II) cations not only improves the visible light absorption, but also enhances the photoelectric capability for better carrier transport and separation. Furthermore, the 1D structure of Pt-Tpy-COF promotes interactions between the photocatalyst and water molecules. These distinctive structural characteristics result in its high activity in photocatalytic hydrogen evolution. The external functionalization design provides new insights in the design of COF-based photocatalysts at the molecular level, based on which highly efficient hydrogen evolution or other types of photocatalytic reactions may be realized.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We thank the National Natural Science Foundation of China (21725404) and State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology) (2023-KF-6) for financial support.Notes and references
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- K. Prakash, B. Mishra, D. D. Díaz, C. M. Nagaraja and P. Pachfule, J. Mater. Chem. A, 2023, 11, 14489–14538 RSC.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- K. Rogdakis, N. Karakostas and E. Kymakis, Energy Environ. Sci., 2021, 14, 3352–3392 RSC.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 128, 1824–1828 CrossRef PubMed.
- Z. Qian, Z. J. Wang and K. A. I. Zhang, Chem. Mater., 2021, 33, 1909–1926 CrossRef CAS.
- X. Huang and Y.-B. Zhang, Chem. Lett., 2021, 50, 676–686 CrossRef CAS.
- C. Feng, Z.-P. Wu, K.-W. Huang, J. Ye and H. Zhang, Adv. Mater., 2022, 34, 2200180 CrossRef CAS PubMed.
- W.-K. Qin, C.-H. Tung and L.-Z. Wu, J. Mater. Chem. A, 2023, 11, 12521–12538 RSC.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2020, 6, 168–190 CrossRef.
- C.-C. Gu, F.-H. Xu, W.-K. Zhu, R.-J. Wu, L. Deng, J. Zou, B.-C. Weng and R.-L. Zhu, Chem. Commun., 2023, 59, 7302–7320 RSC.
- G. Yuan, L. Tan, P. Wang, Y. Wang, C. Wang, H. Yan and Y.-Y. Wang, Cryst. Growth Des., 2021, 22, 893–908 CrossRef.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef CAS PubMed.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- N. Romero, R. Bofill, L. Francàs, J. García-Antón and X. Sala, Catalysts, 2021, 11, 754 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- J. You, Y. Zhao, L. Wang and W. Bao, J. Cleaner Prod., 2021, 291, 125822 CrossRef CAS.
- W.-T. Chung, I. M. A. Mekhemer, M. G. Mohamed, A. M. Elewa, A. F. M. El-Mahdy, H.-H. Chou, S.-W. Kuo and K. C.-W. Wu, Coord. Chem. Rev., 2023, 483, 215066 CrossRef CAS.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261 RSC.
- X.-H. Han, J.-Q. Chu, W.-Z. Wang, Q.-Y. Qi and X. Zhao, Chin. Chem. Lett., 2022, 33, 2464–2468 CrossRef CAS.
- X. Liu, X. Yang, X. Ding, H. Wang, W. Cao, Y. Jin, B. Yu and J. Jiang, Chin. Chem. Lett., 2023, 34, 108148 CrossRef CAS.
- N. Arora, C. Flores, M. C. Senarathna, C. M. Thompson and R. A. Smaldone, CCS Chem., 2024, 6, 57–68 CrossRef CAS.
- Y. Xu, G. Ren, D. Zhang, L. Sun and Y. Zhao, Chin. J. Chem., 2023, 41, 3447–3472 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- Z. Lin and J. Guo, Macromol. Rapid Commun., 2023, 44, 2200719 CrossRef CAS PubMed.
- Y. Li, X. Song, G. Zhang, L. Wang, Y. Liu, W. Chen and L. Chen, ChemSusChem, 2022, 15, e202200901 CrossRef CAS PubMed.
- N. A. Khan, C. S. Azad, M. Luo, J. Chen, T. Kesharwani, A. Badshah and D. Wang, Energies, 2023, 16, 5888 CrossRef CAS.
- B. Mishra, A. Alam, B. Kumbhakar, D. Díaz Díaz and P. Pachfule, Cryst. Growth Des., 2023, 23, 4701–4719 CrossRef CAS.
- C. Ji, C. Kang, B. C. Patra and D. Zhao, CCS Chem., 2024, 6, 856–881 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- Y. Chen, X. Luo, J. Zhang, L. Hu, T. Xu, W. Li, L. Chen, M. Shen, S.-B. Ren, D.-M. Han, G.-H. Ning and D. Li, J. Mater. Chem. A, 2022, 10, 24620–24627 RSC.
- Y. Zhong, W. Dong, S. Ren and L. Li, Adv. Mater., 2024, 36, 2308251 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomacker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- H. He, R. Shen, P. Zhang, G. Liang and X. Li, J. Mater. Chem. A, 2024, 12, 227–232 RSC.
- Y. Wan, P. Sun, L. Shi, X. Yan and X. Zhang, J. Phys. Chem. Lett., 2023, 14, 7411–7420 CrossRef CAS PubMed.
- M. Lu, S.-B. Zhang, M.-Y. Yang, Y.-F. Liu, J. P. Liao, P. Huang, M. Zhang, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202307632 CrossRef PubMed.
- H. L. Nguyen, C. Gropp and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 2771–2776 CrossRef CAS PubMed.
- Z. Chen, K. Wang, X. Hu, P. Shi, Z. Guo and H. Zhan, ACS Appl. Mater. Interfaces, 2021, 13, 1145–1151 CrossRef CAS PubMed.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- Z. Chen, K. Wang, Y. Tang, L. Li, X. Hu, M. Han, Z. Guo, H. Zhan and B. Chen, Angew. Chem., Int. Ed., 2023, 62, e202213268 CrossRef CAS PubMed.
- Y. Hu, N. Dunlap, H. Long, H. Chen, L. J. Wayment, M. Ortiz, Y. Jin, A. Nijamudheen, J. L. Mendoza-Cortes, S.-h. Lee and W. Zhang, CCS Chem., 2021, 3, 2762–2770 CrossRef CAS.
- Y. Hu, S. J. Teat, W. Gong, Z. Zhou, Y. Jin, H. Chen, J. Wu, Y. Cui, T. Jiang, X. Cheng and W. Zhang, Nat. Chem., 2021, 13, 660–665 CrossRef CAS PubMed.
- C. Jia, A. Duan, C. Liu, W.-Z. Wang, S.-X. Gan, Q.-Y. Qi, Y. Li, X. Huang and X. Zhao, Small, 2023, 19, 2300518 CrossRef CAS PubMed.
- J. Li, J. Lan, R. Cao, J. Sun, X. Ding, X. Liu, L. Yuan and W. Shi, ACS Appl. Mater. Interfaces, 2023, 15, 59544–59551 CrossRef CAS PubMed.
- M. Wang, Z. Wang, M. Shan, J. Wang, Z. Qiu, J. Song and Z. Li, Chem. Mater., 2023, 35, 5368–5377 CrossRef CAS.
- Y. Li, L. Yang, H. He, L. Sun, H. Wang, X. Fang, Y. Zhao, D. Zheng, Y. Qi, Z. Li and W. Deng, Nat. Commun., 2022, 13, 1355 CrossRef CAS PubMed.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS.
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, Q. Li, G. Wu, H. Xia, S.-W. Yang and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208919 CrossRef CAS PubMed.
- W. Dong, Y. Xiao, Z. Qin, B. Qiao and L. Li, J. Mater. Chem. A, 2023, 11, 14760–14767 RSC.
- X. Guan, Y. Qian, X. Zhang and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202306135 CrossRef PubMed.
- Y. Liu, W.-K. Han, W. Chi, J.-X. Fu, Y. Mao, X. Yan, J.-X. Shao, Y. Jiang and Z.-G. Gu, Appl. Catal., B, 2023, 338, 123074 CrossRef CAS.
- C. Yang, C. Qian, M. Yu and Y. Liao, Chem. Eng. J., 2023, 454, 140341 CrossRef CAS.
- X. Wu, M. Zhang, Y. Xia, C. Ru, P. Chen, H. Zhao, L. Zhou, C. Gong, J. Wu and X. Pan, J. Mater. Chem. A, 2022, 10, 17691–17698 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02453b |
| This journal is © The Royal Society of Chemistry 2024 |