
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon black structural effect within kraft black liquor-based poly(HIPE): enhanced hydrogen storage and electro-capacitive properties†
Romain
Poupart
abc,
Ronan
Invernizzi
bc,
Hervé
Deleuze
 *a,
Liliane
Guerlou-Demourgues
bd,
Jacob
Olchowka
*bd,
David
Talaga
a,
Laurent
Servant
a,
Nicolas
Penin
b,
Jean-Louis
Bobet
b and
Rénal
Backov
c
*a,
Liliane
Guerlou-Demourgues
bd,
Jacob
Olchowka
*bd,
David
Talaga
a,
Laurent
Servant
a,
Nicolas
Penin
b,
Jean-Louis
Bobet
b and
Rénal
Backov
c
aUniv. Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, F-33400 Talence, France. E-mail: herve.deleuze@u-bordeaux.fr
bUniv. Bordeaux, CNRS, Bordeaux INP, ICMCB, UMR 5026, F-33600 Pessac, France. E-mail: jacob.olchowka@icmcb.cnrs.fr
cUniv. Bordeaux, CNRS, CRPP, UMR 5031, F-33600 Pessac, France
dRS2E, Réseau Français sur le Stockage Electrochimique de l'Energie, FR CNRS, 3459, France
First published on 19th July 2024
Abstract
A biopolymer derived from Kraft Black Liquor (KBL), a byproduct of the paper industry composed mainly of lignin and hemicellulose, has been successfully filled with three different types of carbon black (CB) that differ in their specific surface areas, sizes, shapes and surface heteroatoms. These composite CB–KBL dispersions have been subsequently employed to generate porous monoliths through an emulsion-polymerization templating process. After carbonization, the fillers' influence over the resulting carbon monolith structures and textures is investigated. In particular, beyond XRD, Raman spectroscopy demonstrates improved sample structuration through CB filler addition while nitrogen sorption measurements reveal the influence of the fillers over the final composite's porosities. Considering their properties and effectiveness, hydrogen storage at 77 K reveals that some materials offer up to 1.4 wt% of H2 storage capacity, being higher than that of some commercial carbon materials (with the same specific surface) offering 1.2 wt% hydrogen retention. When addressing their electrochemical energy storage properties, some of these electrode materials deliver extremely promising specific capacities and rate capabilities, with values up to 47 mA h g−1 at 1 A g−1 in alkaline electrolyte, higher than those of the well-known “YP-80F” commercial material tested under the same conditions. These enhanced energy storage properties, while employing a high tonnage paper industry by-product as a carbonaceous source and carbon blacks as structural and textural modifiers, render these materials realistic candidates favoring a sustainable energy transition.
1 Introduction
In the recent decade, interest in porous materials has increased drastically among all the fields of chemistry of materials (inorganic, hybrid and organic ones). While inorganic porous materials are mostly silica-based1 or zeolites,2 hybrid organic–inorganic ones rely essentially on MOFs,3 and organic materials are essentially emerging from polymer chemistry.4 When addressing the latter set of materials, various processes can trigger desired morphologies.4,5 Among all processes, the emulsion templating approach appears to be a straightforward technique to achieve interconnected macroporous materials, called polyHIPEs (HIPE being the acronym for High Internal Phase Emulsion).6 Since the process was patented by Unilever,7,8 various types of emulsion have been employed, especially with the recent endeavour of making use of more bio-related synthon or raw materials9,10 under the current trend of reducing fossil-based products. Regarding the particular context of bio-related poly(HIPE), Foulet et al.11 pioneered the generation of a porous HIPE using a byproduct of the paper milling industry i.e. kraft black liquor (shortened hereafter as KBL), an alkaline aqueous suspension composed mainly of lignin and hemicellulose. This by-product material is known to produce porous carbon-based materials after an appropriate thermal treatment (and are so-called carboHIPEs).12Porous carbons are known to address hydrogen storage13 and for their specificities as electrode materials for electrochemical energy storage,14 related to their intrinsic microporosity/small mesoporosity standing as the criteria of choice.13 Therefore, to be produced at the industrial high-scale level, functional carbon-based materials need to be both abundant and cheap. Through a realistic approach, KBL is appears to be of crucial interest as being widely available; the paper industry generates annual KBL tonnage ranging from 500 millions15 to 1.3 billion tons per year.16 However, bare carbon materials produced from KBL-based monoliths tend to be mainly amorphous, bearing only small amounts of partially graphitized domains. Graphite,17–20 even in the form of (nano)-21fibers,22 is known to efficiently adsorb hydrogen while enhancing the charge transport properties. Considering hydrogen storage properties, other techniques, namely H2 compression or solid storage through hydrides, require harsh implementation conditions such as high energy or high temperature and pressure to absorb hydrogen.23 Adsorption on carbon is easier as it does not need tremendous requirements unlike other current technologies mentioned before. Increasing the hydrogen storage uptake of carbons would be of crucial interest. Considering supercapacitors and battery applications, addition of CBs in electrode formulation is well known to enhance charge transport effectiveness. Here, CBs will be embedded within a continuous carbonaceous network favouring both a self-supported character and graphitic domain extension while maintaining a certain degree of micro- and mesoporosity of the carbonaceous backbone.
In this paper, we investigate different carbon black (CB) inclusions24,25 (from 0.5 wt% to 5 wt%) regarding the HIPE preparations. We evaluate their effect over the carbonaceous KBL-based-carboHIPE backbone structure and texture while addressing both their enhanced hydrogen sorption and charge storage properties. As such, three CBs varying in their morphologies, specific surface area and graphitization levels are employed. CB modified KBL-based monoliths were finally carbonized, leading to carboHIPE samples. Several characterization techniques have been implemented. First, Raman spectroscopy and XRD have depicted the impact of the CBs' addition over the carbonaceous backbone structure. Then, nitrogen sorption measurements were employed for the carbon surface area and adsorption property assessments. Through these characterization studies, we show that the presence of CB influences the KBL-based carbon monolith porosities at various length scales while optimizing the graphitization degree. Moreover, the presence of CB almost doubles the amount of hydrogen adsorbed at 77 K compared to the CB free monolith. Finally, when employed as electrode materials for supercapacitors, the specific capacity increases up to 47 mA h g−1 at 1 A g−1 in alkaline electrolyte versus 35 mA h g−1 for bare KBL materials.
2 Materials and methods
2.1 Materials
Kraft black liquor (with an average residual dry mass of about 45 wt%) was kindly gifted by Smurfit Kappa Cellulose du Pin (Biganos, France) and has been concentrated using a rotavapor to a 50%-amount of dry matter prior to being used for experiments. (±)-Epichlorhydrin (purum, ≥99%), Pluronic® F-108, and 1,2-dichloroethane (ACS reagent, ≥99.0%) were supplied by Sigma-Aldrich/Merck. Isopropanol (≥98%, Technical) and diethyl ether (≥99.7%) have been provided by VWR. PBX 55 (CBA), Vulcan XC-72 (CBB) and PBX 51 (CBC) have all been supplied by Cabot Corporations. Except for KBL, all reagents have been used without further purification.The as-received kraft black liquor comes as a viscous black liquid. Its main physico-chemical properties are: dynamic viscosity μ = 7000 mPa s at 23 °C; pH = 14; density ρ = 1.3 g mL−1. It has been concentrated with a rotavap up to ≈50 wt%. Phenol group content and total hydroxyl group content of black liquor were determined to be 0.5 mmol g−1 and 0.8 mmol g−1, respectively, according to an already published analysis.26
2.2 Preparation of the KBL–CB composite monoliths
Self-standing macrocellular materials have been obtained following a protocol previously published,27 while additionally dispersing CB into the hydrophilic alkaline KBL phases. Briefly, a mixture of KBL (20 g), Pluronic® F-108 (0.9 g) and CB (either 0.5; 1; 3 or 5 wt% of PBX 51; 55 or VXC 72, respectively) has been gently mixed (100 rpm) together at room temperature until surfactant and CBs are homogeneously mixed. Following this dispersion step, epichlorhydrin (2.2 g) was added dropwise and mixed for 10 minutes. The as-prepared continuous phase was inserted into a 60 mL syringe as part of the emulsification system. In the meantime, 1,2-dichloroethane was added in a second 60 mL syringe. Emulsification occurred during 80 minutes in a laboratory-made emulsifier system composed of two syringes (60 mL, internal diameter in mm) connected through a small section of a nylon tube (external diameter = 11 mm; internal diameter = 4 mm; length = 15 mm) as described in Fig. S1.†After the emulsification process, the native emulsion was removed from the syringes and poured into a PTFE cylindrical mold (d = 45 mm and h = 15 mm) pinched between two Teflon plates. The mold has been placed in an oven (Thermo Scientific Heraeus Vacutherm VT6060) heated at 60 °C for 24 h in order to allow an epichlorhydrin reaction with the lignin and hemicellulose alcohol moieties. After crosslinking, the resulting polymerized monolith was rinsed for two days with isopropanol and one day with diethyl ether before being dried for two days at 85 °C under vacuum. No further washing with deionized water has been applied at that stage.
2.3 Carbonization procedure
Approximately 200 mg of the monolith has been carbonized in a tubular alumina oven to obtain carboHIPEs, using a slightly adapted protocol from the literature.12 Typically, a sample is degassed under an Argon flux (0.3 L min−1) for 30 minutes, and then, (still under the Ar flux) the temperature is increased to 900 °C with a heat rate of 5 °C min−1 along with plateaus at 300, 600 and 900 °C for 1 h each. Cooling down was uncontrolled and led by the oven inertia. Carbonized samples, named KBLc, have been finally washed for 6 hours with deionized water and then dried overnight in a stove set at 100 °C in order to remove the embedded salts. It was shown that the presence of salt decreases the porosity and lowers the energy storage performance.272.4 Hydrogen storage experiment
Newly synthesized KBLc samples have been directly used for hydrogen adsorption. Typically, about 100 mg of materials have been placed in the sample holder of a controlled Sieverts' type apparatus. Prior to measurements, the apparatus was degassed under vacuum for 5 minutes, followed by an injection of 4 bar of argon, then further degassed for 10 min followed by an injection of Ar and finally degassed for 20 more minutes before being used. The desired hydrogen pressure was pre-loaded inside the experimental setup. Then, the desired pressure was permitted to expand in the sample holder at the temperature of liquid nitrogen. In order to ascertain reaching mechanic equilibrium (constant pressure), a 15-minute rest ensuring thermodynamic stabilization was imposed. After each adsorption, the sample was degassed at room temperature. The experiments were performed step-by-step at different pressures ranging from 0.1 bar to 60 bar (step of approximately 10 bars). High purity hydrogen has been used for this set of experiments.2.5 Capacitive charge storage
For each material, an electrode was prepared by mixing in a mortar the (washed) active material with carbon black (Alfa Aesar) and polytetrafluoroethylene (PTFE) in a weight ratio of 80/15/5. These mixtures were thoroughly ground and became self-supported electrodes. Then, disks of 16 mm diameter are cut and pressed at 5 bars on a nickel foam, which is used as a current collector. The active mass loading of each electrode was around 5 mg cm−2. Electrochemical measurements were carried out employing a 5 M KOH alkaline solution at 25 °C in a 3-electrode system with a Hg/HgO electrode (1 M NaOH) and a platinum wire being the reference and counter electrodes respectively. Then, for the most interesting materials, symmetric supercapacitors (Swagelok type) were built using two identical electrodes separated using a glass fiber spacer (260 micrometre thickness) impregnated with 5 M KOH.We also attempted to test the monolith as an electrode without reshaping or additional formulation. However, this brittle material could not maintain its self-standing structure during cell assembly. The pressure exerted when closing a Swagelok-type cell or using a crocodile clip during testing in a beaker cell caused it to crumble.
2.6 Characterization techniques
Zeta-potential measurements have been performed on the pure CBs by adding 50 mg of the CB in 10 mL of deionized water. The suspensions have been sonicated for 5 minutes prior to being analyzed on a Malvern Zetasizer Nano ZS.Scanning electron microscopy (SEM) studies have been performed on a Hitachi TM-1000 apparatus using 15 kV as accelerating voltage. Samples have been coated with a thin Pt–metal layer beforehand.
Porosity of the different materials has been investigated by Mercury Intrusion Porosimetry (MIP) using an Autopore IV 9500 porosimeter (Micromeritics) with the following parameters: contact angle = 155°, Hg surface tension = 485 mN m−1 and maximum intrusion pressure fixed at 124 MPa. The determination of the pore size distributions was based on the Washburn equation.
The sample specific surface areas were determined by recording nitrogen sorption isotherms (77 K) with a 3Flex gas sorption analyzer (Micromeritics) after degassing the materials at 250 °C under vacuum for 10 h. The specific surface areas were evaluated using the Brunauer, Emmett and Teller (BET) method.28
Raman spectra were recorded in backscattering configuration using a Labram HR (Horiba Group, France) confocal microspectrometer equipped with a confocal microscope. Spectra were recorded using the 514.5 nm (2.41 eV) wavelength generated from an Ar ion laser. The spectral resolution was 6 cm−1 through a confocal hole aperture of 150 μm and a 600 grooves per mm diffraction grating. The laser was focused on the sample by means of a 50× objective (0.75 numerical aperture). The beam power at the sample surface was around 200 μW avoiding overheating effects due to laser irradiation. Spectra were recorded with an acquisition time of 60 s and 4 accumulations. We proceeded with the spectral hand-made baseline corrections using LabSpec 5 software before deconvolutions. Spectra have been recorded from 500 to 3500 cm−1. Deconvolutions have been performed using two different fitting methods: first with a common 2-band method (D and G; with D situated at 1350 and G at 1600 cm−1, respectively)12 and then with a 5-band method, as developed by Sadezky and coworkers (D1, D2, D3, D4 and G situated at 1360, 1620, 1500, 1180 and 1580 cm−1, respectively). The Raman spectra were fitted using a Voigt function (which combines Gaussian and Lorentzian functions).29
X-ray diffraction measurements were performed with a Philips PANalytical X'Pert Pro equipped with a copper source (λCu Kα1 = 0.15405 nm and λCu Kα2 = 0.15443 nm) between 2θ = 10 and 80° with a 0.02° (2θ) step size and a 2.022° (2θ) active width in the detector.
3 Results and discussion
3.1 Materials preparation
Three different types of CB have been selected as fillers (Fig. S2†) based on their microstructure (Fig. S3–S5,† respectively) and their specific surface area (see Table S1 and Fig. S6†). PBX 55; VXC 72 and PBX 51 exhibit specific surface areas of 75, 250 and 1500 m2 g−1 respectively, while also offering different states of carbon organization. PBX 51, having the highest specific area, also has micropores as well as pores of 10 nm, according to NLDFT treatment of the data, while the two other carbon blacks have a low amount of pores smaller than 60 nm. The specific surface areas were determined using nitrogen adsorption isotherm experiments while carbon organization has been probed by Raman spectroscopy. Initial materials obtained from bare KBL have already been described and characterized.27 Several preparations have been performed in order to compare the carbon black filler influence on the final materials' porosity and graphitic character (Table 1).| Carbon black (CB) in use | Sample's labelling | CB amount (wt%) |
|---|---|---|
| None | KBLc | 0 |
| PBX 55 | CBA-0.5 | 0.5 |
| PBX 55 | CBA-1 | 1 |
| PBX 55 | CBA-3 | 3 |
| PBX 55 | CBA-5 | 5 |
| Vulcan XC 72 | CBB-0.5 | 0.5 |
| Vulcan XC 72 | CBB-1 | 1 |
| Vulcan XC 72 | CBB-3 | 3 |
| Vulcan XC 72 | CBB-5 | 5 |
| PBX 51 | CBC-0.5 | 0.5 |
| PBX 51 | CBC-1 | 1 |
| PBX 51 | CBC-3 | 3 |
| PBX 51 | CBC-5 | 5 |
CBs are easily dispersed into the KBL initial suspensions at that stage, due to their negative ζ-potentials (Table S1†) and the KBL solution alkaline character. The negative ζ-potential values may indicate that CBs possess carboxylic acid moieties over their surfaces.30,31 KBL being an alkaline sol (pH = 14), the presence of pending carboxylic acids on the CB's external surface may favor their dispersion states. Besides we do not have to omit a synergistic scenario where the KBL alkaline medium (containing Na2CO3, for instance) may so-called “activate” the CBs by changing the surface moieties.32 This specificity of the alkaline KBL medium is thereby very important as surfactant molecules are here unnecessary to disperse the CB particles, allowing the generation of homogeneous CB/KBL suspensions with ease. The emulsification process has been chosen over direct crosslinking of the KBL for mechanical reasons. The material produced via emulsions is more self-standing after the curing process than the one obtained via direct crosslinking. Besides, DCE has been chosen over other oils (such as castor oil) due to its better separation process with the alcohol used for the washing step, through distillation, for example, leading to a lower environmental impact as assessed by a life-cycle analysis (LCA).33 Emulsification was achieved using a double-syringe translating device, where the two non-miscible phases are sheared to generate the direct concentrated oil-in-water emulsion (Fig. S1†). After pouring into PTFE molds and cured in an oven (60 °C for 24 hours), the native self-standing polymerized materials (the polyHIPEs) are washed, dried and cut into smaller pieces to fit the carbonization-tubular setup diameter (Fig. S7†). Following this, scanning electron microscopy (SEM) and mercury intrusion porosimetry (MIP) investigations have been performed.
Through SEM investigations (Fig. 1), we can evidence that final materials are still porous, independent of the nature or concentration of CBs. Compared to a pristine (i.e. CB-free) HIPE material obtained from KBL,27 only the CBA-loaded sample at 0.5 wt% (Fig. 1A) shows the clear pattern of a KBL-based polyHIPE. Others, such as CBA – 5 (Fig. 1D), CBC – 0.5 (Fig. 1I) or CBC – 3 (Fig. 1K), demonstrate only partial emulsion-templated-domains. Despite this random phase separation scenario, the final materials are rather porous, which was the main objective of this emulsion templating process.
 | ||
| Fig. 1 Scanning Electron Microscopy (SEM) micrographs of the different KBL-based monoliths loaded with carbon blacks. Samples loaded with CBA: pictures (A) to (D) with 0.5; 1; 3 & 5 wt% CBA in them, respectively. With CBB: pictures (E) to (H) with 0.5; 1; 3 & 5 wt% CBB in them, respectively. With CBC: pictures (I) to (L) with 0.5; 1; 3 & 5 wt% CBC in them, respectively. The scale bar represents 30 μm. | ||
Beyond SEM qualitative observations, more quantitative insights regarding the samples' macroscopic texture have been reached through MIP measurements. As mentioned previously,27 pure KBL-based carbon exhibits a Gaussian-type pattern, centred at 0.4 μm. In the case of a porous interconnected monolith, the pore distribution represents the diameters of the connecting pore windows (and the pore diameters themselves). Here, the results of MIP experiments show that most of the materials present intrusion volume occurrences revealing a bimodal character of Hg intrusion peaks in rather spread ranges between 0.1–2 and 8–200 μm (Fig. 2). This indicates that the addition of CB influences the macroporosity compared to the reference pure KBL-based carbon. This is especially the case for CBA-1 and CBB-1 samples as well as all low CB 0.5 wt%-loaded samples. As the connecting windows are emerging from the emulsion coalescence phenomenon, we cannot exclude the fact that the CBA CB nanoparticles contribute toward the emulsion instability while favoring the coalescence process and thereby the induced connecting windows' higher diameters.
 | ||
| Fig. 2 Mercury Intrusion Porosimetry (MIP) profiles of the (A) CBA-; (B) CBB- & (C) CBC-loaded samples. We have to underline that the geometries assessed under Hg porosimetry are the ones minimizing Hg impregnation; this is to say the connecting pore windows of adjacent cells and not the cells themselves in the present cases. | ||
As summarized in Table S2,† the porosity value is centered at 57 ± 5% between all the synthesized samples. In the series, densities tend to reduce when CBs with the highest specific areas (namely CBB and CBC) are incorporated. Besides, the bulk density slightly diminishes from 0.60 ± 0.02 g mL−1 to 0.49 ± 0.02 g mL−1, when the amount of CBB increases from 0.5 wt% to 5 wt%. The same trend is observed when considering the decreasing skeletal density from 1.36 ± 0.02 g mL−1 to 1.32 ± 0.02 g mL−1.
The most interesting result is that only CBA samples (except CBA-0.5) do not evidence huge intrusion values at 3 μm or higher, contrary to other fillers. One potential explanation is to attribute these values to an emulsion dephasing scenario (SEM in Fig. S8†). Indeed, this behavior is mostly linked to CBs bearing the highest surface areas. The surfactant is implied to interact preferentially with the CBs as PEO-containing amphiphilic surfactants are known to interact with CBs.30,34 As such, the Pluronic entities anchored at the CB external surfaces cannot be part of the oil-in-water droplet stabilization anymore, leading to an exacerbated coalescence scenario. This phenomenon, not detrimental to the material self-standing character integrity, enhances both the emulsion coalescence and final macrocellular connectivity.
3.2 Raman spectroscopy investigations
After KBL-based sample carbonization, the resulting carboHIPE monoliths (Fig. S9†) have been studied by Raman spectroscopy in order to highlight the impact of the CB addition over the carbonaceous backbone structuration. Typical Raman spectra are provided in Fig. 3 and exhibit two main peaks: the D band, located at 1350 cm−1 and the G band, situated at 1600 cm−1. Briefly, the D-band is attributed to the carbon defect sites associated with vacancies and/or grain boundaries35 whereas the G-band is related to the in-plane vibrations of carbon atoms in graphene sheets with E2g2 symmetry; typical of graphitic domains.36 | ||
| Fig. 3 Examples of (A) 2-peak fitting and (B) 5-peak fitting on a Raman spectrum obtained for a pristine KBL-based carbon monolith sample. | ||
At first, a 2-peak fitting procedure was employed in the 500–2000 cm−1 range. An example of such a fitting is given in Fig. 3A. Based on these two broad band intensities, we first evaluated the R1 ratio, which is correlated with the number of graphitized domains: the lower the R1, the more graphitized domains are present (eqn (1)).
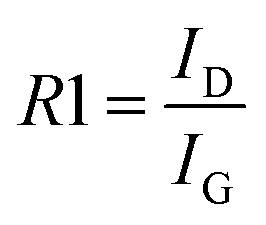 | (1) |
In order to address higher insight, another ratio was employed, R2,36 representing a disorder parameter defined as (eqn (2)):37
 | (2) |
The R2 values obtained for all samples are summarized in Table S3.† Both ratios are supposed to increase with the presence of defects (or reversely decrease with higher graphitization), as expressed in eqn (1) and (2). The results reported in Table S3† evidence a slight decrease of R2, like R1, while adding the CB. It is worth pointing out that all CBs lead to a similar behavior. However, only the lowest amounts of CB (i.e. either 0.5 or 1 wt%) lead to the strongest decrease of those ratios compared to the initial KBL material without carbon blacks (e.g. R1 = 4.19; R2 = 0.81). One hypothesis, already suggested by Always-Cooper et al.,38 is that addition of carbon black in dilute concentrations (lower than 1 wt%) leads to a strong graphitic domain expansion. The transfer of this behavior to the present materials is not to be excluded, as Ravikumar et al.24 noticed that adding carbon black into a phenolic resin (such as lignin) improved the arrangements of carbon basal planes. Also, Zhang et al.39 noticed that phenolic resin can be trapped onto carbon black fillers while being involved in the carbonization process. This trapped resin can account for broader organized carbon domains, with smaller amounts of CBs leading to larger domains that are allowed to grow. We can summarize this CB-induced effect as pseudo epitaxial growth where the graphitic backbone of the CB is transferred toward the surrounding carbonaceous matrix. When increasing the amount of CB, this pseudo epitaxial growth is still acting but the geometric proximity of the concentrated CB (at high concentrations) is promoting dislocation domains where sp2 configuration is relaxing the constraints in sp3 configuration, increasing the sp3 defects while minimizing the sp2 graphitic order.
In order to support our view deduced from the 2-peak fitting procedure, we performed a 5-peak fitting, following a method developed by Sadezky et al.29 and involving five bands in order to describe more accurately the Raman spectra: D1, D2, D3, D4 and G situated at ∼1360, ∼1620, ∼1500, ∼1180 and ∼1580 cm−1, respectively, using a Voigt function (combining Gaussian and Lorentzian functions). Through this fitting method, each defect is treated independently: D1 is associated with defects generated by heteroatoms; D2 is related to the disorder inside the graphite lattice with E2g2 symmetry; D3 infers the out of plane vibrations due to defects and/or heteroatoms while D4 addresses the –CH species in aliphatic hydrocarbon chains.36 Moreover, the graphite band (as observed in the 2-band fitting method) is deconvoluted into a new G-band and a defect band, “D2”, for the sake of higher accuracy.
Examples of this 5-peak fitting method are provided in Fig. 3B. Ratios are given following the equations (eqn (3)):w
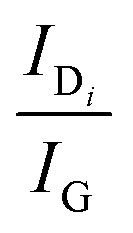 | (3) |
 | (4) |
All ratio variations are presented in Table S4†. Similarly, for the 2-peak fitting, the ID1/IG ratio tends to decrease with the addition of carbon black, again with the lowest values for the two lowest CB amounts (0.5 and 1 wt%). While the initial KBL material possesses a ID1/IG ratio of 4.14, adding 0.5 wt% of CBC can help reach a ratio of 2.25 and 0.5 wt% of CBA leads to a value of 2.60. Concerning the other ratios, namely ID2/IG; ID3/IG and ID4/IG, we decided not to focus on them as their variations appear more erratic. These erratic variations are also noticed by Sadezky and coworkers in their paper29 and should be taken less into account. On the other hand, the ratio IG/Iall increases by addition of the CB filler, as expected. The optimum values are obtained for compositions of 0.5- and 1 wt%-CB added, regardless of their nature. For the carbonized sample of pure KBL the ratio is as low as 0.14 with the highest being for CBA 0.5 and 1 wt% added to the initial mixture. CBB and CBC, for their part, lead to values of 0.20.
In summary, ID1/IG and IG/Iall ratios give the same conclusion as the 2-peak fitting method: adding a low amount of CBs (in wt%) into the KBL-based materials leads to a better ordering of the material carbonaceous backbones, where, here, again a balance between graphitization domains and dislocation zones has to be taken into account. This balance relies on the amount of CB introduced acting as nucleation sites through the affinity (π–π staking and van der Waals) between the sp2 carbon backbone of the CB particles and the aromatic carbon rising from the KBL phenolic moieties. Following the heterogeneous nucleation, pseudo-epitaxial growth of graphitic ordered domains will occur until reaching dislocation appearance, at higher CB contents. Native dislocations lead to carbon backbone sp3 disordering enhancement. Moreover, the “low” amount of CBs (0.5 or 1 wt%) can be misinterpreted, as this amount is calculated from the whole black liquor weight (i.e. the liquid suspension) while being concentrated at 50 wt% (dry extract). Additionally, CBs harness a low bulk density. According to the MSDS sheets of VXC 72, its bulk density is comprised between 0.2 and 0.680 g cm3,42 meaning that even small gravimetric contents represent volumetric contents at the milliliter scale.
3.3 XRD diffraction patterns
XRD experiments have been performed on the initial carbonized KBL reference sample (CB free) and on the ones with CBA, B and C fillers added. For each sample, the load concentrations of filler were set to 0.5 and 5 wt%. XRD investigations on bare carbon blacks' powders (Fig. S3–S5†) have also been performed. From the CB's XRD patterns, CBA presents more organized, crystallized carbon as the diffraction peaks are rather narrow. CBB presents broader peaks but is still recognizable. CBC, on the opposite, possesses broader and less defined peaks indicating carbon containing exalted amorphous domains with defects.As can be observed on the diffractogram (Fig. 4), the initial carbonized KBL material exhibits two large diffraction peaks centered at 2θ = 26° and 44°. These large peaks are typical of a poorly crystallized material. Adding a small fraction of CBs seems to improve slightly the sample crystallinity, in agreement with the Raman conclusion: for instance, the intensity of the peak at 26° appears enhanced upon addition of CBs. Adding 5 wt% of filler renders the peak clearly distinguishable, as can be observed for CBA, leading to an even better crystallinity. The same trend can be applied for CBB and CBC (Fig. 4B). While pure CBC is mainly amorphous, its carbonization when embedded in the KBL-matrix exalts the carbon structural diffraction peaks, thus strengthening the hypothesis of graphitic domains' growth.
 | ||
| Fig. 4 XRD diffractograms (A) a comparison between the KBLc material with two different CBA-loaded materials and (B) the three different CB-loaded materials at 5 wt%. XRD patterns of the CBA, CBB, and CBC are presented in Fig. S3–S5† for the sake of comparison. | ||
3.4 Nitrogen physisorption characterization
To further investigate the textural properties, nitrogen physisorption (Fig. S10†) experiments have been performed to evaluate the samples' specific surface area (SSA) (Table 2). All the obtained carbonaceous foams present activated carbon profiles with the physical artefact visible from the desorption curve, at medium relative pressure. These types of carbon have both micro- and mesoporosity.27 One can notice that the hysteresis loops (i.e. the physical artefact induced through nitrogen capillary condensation) between the adsorption and desorption curves occurring at medium relative pressure are more pronounced when CBs are added, especially for CBB-5 and CBC-5. Interestingly enough, these two samples are the ones with the lower ratio of microporosity over the total volume of pores. This indicates, overall, a higher degree of small mesoporosity.| Samples name | Specific surface area (m2 g−1) | BJH adsorption pore volume (cm3 g−1) | BJH desorption pore volume (cm3 g−1) | Micropore volume/total volume ratio (%) | Micropore specific area/total specific area ratio (%) |
|---|---|---|---|---|---|
| KBLc | 640 | 0.056 | 0.068 | 84.8 | 97.6 |
| Pure CBA | 75 | 0.22 | 0.23 | 3.37 | 16.0 |
| CBA-0.5 | 410 | 0.040 | 0.048 | 81.5 | 95.2 |
| CBA-3 | 600 | 0.060 | 0.074 | 82.9 | 97.2 |
| CBA-5 | 515 | 0.063 | 0.077 | 78.6 | 96.1 |
| Pure CBB | 250 | 0.69 | 0.73 | 9.19 | 48.9 |
| CBB-0.5 | 460 | 0.055 | 0.067 | 79.1 | 96.5 |
| CBB-5 | 570 | 0.097 | 0.13 | 70.9 | 94.6 |
| Pure CBC | 1500 | 1.2 | 1.3 | 28.7 | 76.9 |
| CBC-0.5 | 690 | 0.066 | 0.083 | 81.9 | 96.3 |
| CBC-5 | 455 | 0.094 | 0.11 | 69.3 | 95.4 |
Overall, the specific surface areas range between 400 and 700 m2 g−1. Compared to the reference material (specific surface area of the CB-free sample is 640 m2 g−1),27 the addition of CB leads to a constant specific surface area, independent of their own initial specific surface area. On the opposite, using the BJH (from Barrett–Joyner–Halenda) model to determine the pores' volume of the prepared materials, with and without carbon black, leads to an increase in this volume over all the materials containing CB as a filler. The reference KBLc material exhibits a BJH volume of 0.056 and 0.068 cm3 g−1 for the adsorption and desorption, respectively. Other values of the micropore volumes obtained for the materials bearing CB are higher, except for CBA-0.5 (which is far lower addressing a volume of 0.040 and 0.048 cm3 g−1 for the adsorption and desorption respectively) and for CBB-0.5, on par with KBLc. It is important to note that the highest amount of CB leads to the highest BJH volume and that the largest values are reached for CBB-5 and CBC-5 compounds. Overall, all the carbons obtained present a microporosity centred at around 1.25 nm as well as another pore population with size below 0.6 nm (∼0.55 nm) as seen in Fig. S11.†
As for the CBs, low specific surface areas and pore volumes tend to be correlated with low Raman R1 and R2 ratios. High R1 and R2 ratios tend to promote higher microscopic volumes. Under this scenario involving larger microporous volumes, these CBs/carbo(HIPE) composite materials have been further tested first toward hydrogen storage capabilities.
3.5 Hydrogen storage assessment
Hydrogen adsorption experiments have been performed at the liquid nitrogen temperature (77 K). As H2 adsorption relies here on weak van der Waals interactions,43 adsorption can be disrupted under the slightest amount of energy. In order to minimize this phenomenon, the experiment is performed at the lowest reachable temperature. As liquid nitrogen is widely available, 77 K can be obtained with ease. Considering Fig. 5, KBLc (CB free) adsorbs the lowest H2 amount, only retaining a maximum of 0.8 H2 wt%. The inclusion of carbon black charges promotes enhanced H2 adsorption capabilities. CBA-loaded materials exhibit different isotherm profiles, without a real plateau as it still adsorbs H2 at pressures higher than 70 bar. CBB- and CBC-based materials store the highest hydrogen contents. The CBB-loaded sample can store up to 1.2 wt% while CBC-loaded samples can retain as high as 1.4 H2 wt%. These results match the literature, as Panella and coworkers described a master curve for H2 adsorption of different carbons at 77 K.44 In their work, a carbon retaining between 1 and 1.5 wt% at liquid nitrogen temperature harnesses a specific surface area around 500 m2 g−1 (according to the error bars) and a micropore volume spread between 0.05 and 0.2 cm3 g−1, with a similar master curve also presented by Thomas while considering micropores.45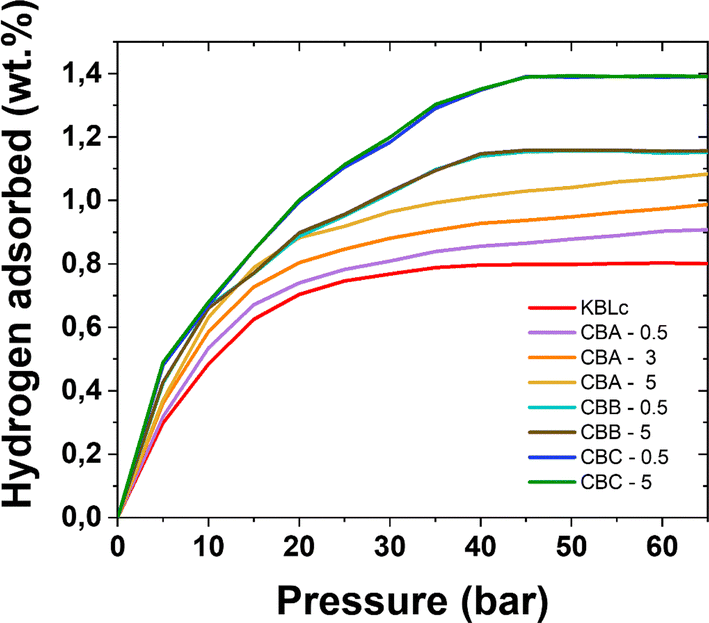 | ||
| Fig. 5 Hydrogen adsorption isotherms at 77 K for the different obtained samples. | ||
Indeed, the results concerning CBC-based carbons are outstanding. As such, regarding their H2 adsorption properties, they are indeed superior to another carbon emerging from biomass such as the AC500 described by Toprak.46 Toprak's carbon, arising from cherry laurel, possesses a higher specific surface area, 809 m2 g−1. Thus, the CBC-carbo(HIPEs) are promising materials when dedicated toward hydrogen adsorption, bearing specific areas lower than 690 and 455 m2 g−1 for CBC-0.5 and CBC-5, respectively. At pressure higher than 30 bar, CBCs are on par with AC35, commercial activated carbon from CECA storing 1.4 wt% at 30 bar and higher, described by Weinberger and Darkrim Lamari.47 CBC-carbo(HIPE) materials are also on par with the commercially available reference carbon from Norit (specific surface area of 970 m2 g−1), addressing a retention of only 1.5 wt% at 77 K.48 In comparison to other commercial carbons, CBC-0.5 and CBC-5 are better than SA20 and Filtrasorb400 (from, respectively, Ingevity and Chemviron) that retain 1.2 wt% at 60 bar and similar to SA1500 and TH90I (from, respectively, Ingevity and Silcarbon), all four carbons addressing specific surface areas ranging from 983 to 2200 m2 g−1.49 CBC-based materials, being partly ordered materials, offer a good compromise between graphite-like and amorphous materials, thus describing an intermediary behavior. One hypothesis is that due to their intermediary behavior, they exhibit better H2 storage properties, harnessing the best of two worlds: the specific area, coming from all the defects and also the multiple spread graphite domains, giving sites for adsorption.
In comparison, activated carbons show, by far, higher H2 adsorption. For instance, in the work of Panella et al., their activated carbon harnessing more than 2500 m2 g−1 can retain up to 4.5 wt% at 77 K.44 More recently, Stock et al. describe the use of an activated carbon achieving more than 3000 m2 g−1 that can retain close to 5.8 wt% of H2 at 77 K and 37 bars of dihydrogen pressure, with measurement made at more than 50 bars showing a decrease in the H2 adsorption.50 These impressive results were shown with activated carbons, possessing specific area by far higher than 1000 m2 g−1.
Besides, we have to underline the fact that the self-standing CB-carbo(HIPE) composite materials presented here are employed while bearing a monolithic character; this is to say that they are not nano-powdered materials inducing undoubtedly high flowability and are not activated carbons. This morphological characteristic is indeed of first importance providing additional modular value to their intrinsic H2 enhanced hydrogen storage properties when compared to the literature.
3.6 Charge storage in supercapacitors
Unlike experiments conducted for hydrogen storage, the monolith could not be directly used as an electrode for supercapacitors. Instead, it was ground and tested as an active material in a conventional electrode formulation (see the Capacitive charge storage section). The carbonaceous materials obtained with 0.5% and 5 wt% of CB have been tested as electrode materials for supercapacitors. Their electrochemical performance was evaluated in a 5 M KOH aqueous electrolyte using cyclic voltammetry and galvanostatic charge/discharge techniques, and compared to that of KBLc. The voltammogram of each material, measured at 5 mV s−1, displays a rectangular-like shape typical of capacitive storage, whereas the higher area of the envelops for CBA-0.5%, CBB-0.5%, CBC-0.5% and CBA-5% compared to KBLc suggests a higher charge storage capacity for these materials (Fig. 6A). This result was further confirmed by the galvanostatic charge/discharge measurements at various current densities, as illustrated in Fig. 6B. CBA-0.5%, CBB-0.5%, CBC-0.5% and CBA-5% clearly exhibit higher specific capacities and rate capabilities than KBLc, CBB-5% and CBC-5% and even outperformed the commercial electrode material YP-80F tested under the same conditions. YP-80F delivers a specific capacity close to 45 mA h g−1 at 0.5 A g−1 while CBA-0.5% and CBC-0.5% present a capacity of 51 mA h g−1 at the same current density. Moreover, they exhibit a good capacity retention at higher current densities, with a capacity of 41 to 42 mA h g−1 at 10 A g−1 while commercial Kuraray YP-80F only delivers a capacity of 36 mA h g−1 at 10 A g−1. It is important to note that these results seem to be correlated with the graphitization degree indicated by the ratio ID1/IG: the higher the graphitization, the higher the specific capacity (Fig. 6C). On the other hand, as shown in Fig. 6D, the specific capacity does not appear to be directly correlated with the surface area (determined by nitrogen adsorption), even if porosity and pore size distribution play a crucial role in the charge storage performance.51,52 As exemplified for birnessite materials exhibiting a discrepancy in specific surface areas, the electrochemically active surface appears to be different from the specific surface.53 In addition, to reach the most efficient energy storage performance, it is important to obtain a balance between porosity, high specific surface areas and more importantly a high degree of accessibility. Indeed, increasing specific surface areas also leads to increased pore volumes and trapped air within, which can impede solute imbibition into the porosity due to Laplace pressure. This “balance” criterion is particularly evidenced when comparing YP-80F with CBA-0.5, where the former possesses a specific surface area three times larger than that of CBA-0.5 but exhibits lower specific capacity (Fig. 6D). Among these materials, CBA-0.5% stands out with a specific capacity reaching 48 mA h g−1 at 1 A g−1versus 36 mA h g−1 for KBLc. Following these results, some of the most promising materials (CBA-0.5, CBA-5 and CBB-0.5) have been tested in symmetrical cells, as displayed in Fig. 6E, and compared to the commercial material YP-80F. Once again, CBA-0.5 and CBB-0.5 exhibited a CV envelop larger than that of YP-80F, suggesting that they are able to deliver a higher specific capacity upon cycling. Besides, CBA-0.5 demonstrated a stable capacity of around 15 mA h g−1 at 2 A g−1 upon 5000 cycles, higher than that of YP-80F (Fig. 6F), with 13 mA h g−1 at 2 A g−1.54 All these results underscore the benefits of engineering the carbon material graphitization degree and porosity through introduction of small amounts of carbon black before carbonization, thereby enhancing its electrochemical energy storage performance. | ||
| Fig. 6 (A) Cyclic voltammetry performed in 5 M KOH at 5 mV s−1, (B) specific capacities measured through galvanostatic charges/discharges at different current densities in 5 M KOH, (C) comparison of the specific capacity measured at 1 A g−1versus the ID1/IG ratio and (D) comparison of the specific capacity measured at 1 A g−1versus the specific surface area determined by nitrogen adsorption. (E) Cyclic voltammetry performed in 5 M KOH at 5 mV s−1 in symmetrical cells. (F) Galvanostatic charge–discharge of symmetrical cells at 2 A g−1 in 5 M KOH. | ||
4 Conclusion
We have successfully dispersed different carbon blacks into a black liquor-based-sol, a paper industry byproduct. After polymerization and subsequent carbonization, CB inclusions are shown to tune both the final materials' porosity and associated pore volume. At the microscopic length scale, two different Raman deconvolution analyses, 2-peak or 5-peak fitting, led to the conclusion that addition of less than 1 wt% of CBs tends to generate materials bearing extended graphitic domains in agreement with XRD investigations.The increase in the pore volumes (specifically micropores) fosters these materials suitability towards energy storage applications, especially hydrogen adsorption and electrochemical energy storage. Considering hydrogen retention, experiments performed at 77 K demonstrate that the addition of CBs increased the stored H2 quantities. In particular, CBC loaded materials retained 1.4 wt% H2, capabilities being similar, if not better, than those of commercial carbons. Moreover, the CB-based carbo(HIPE) intrinsic self-standing nature is of additional value as being both non-flowable and modular regarding their external shapes. Additionally, when employed as electrode materials for supercapacitors, the higher carbonaceous backbone organization, induced through a low amount of CB addition, strongly enhances the specific capacity in alkaline electrolyte, leading to capacity and rate capabilities higher than those for a traditional commercial carbon electrode material (YP-80F). However, for supercapacitor applications, the monolith could not be directly used as an electrode due to its friable nature, causing it to crumble under pressure during cell assembly. This issue necessitates further development in cell assembly or design, or in monolith synthesis, to fully take advantage of carbon's self-standing nature.
Data availability
My co-authors and I could state that data are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to the grant “Grands Programmes de Recherche, Post Petroleum Materials –PPM” of Bordeaux University. The authors thank Isabelle Ly for the TEM images and Lucas Fourier for the zeta-potential experiments. We wish to thank Dr Marie-Anne Dourges for acquiring the nitrogen physisorption isotherms and for the MIP experiments. Finally, the authors also thank Smurfit Kappa for providing the raft black liquor.References
- R. Brady, B. Woonton, M. L. Gee and A. J. O'Connor, Innovative Food Sci. Emerging Technol., 2008, 9, 243–248 CrossRef CAS.
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev., 2003, 45, 297–319 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- R. Poupart, D. Grande, B. Carbonnier and B. Le Droumaguet, Prog. Polym. Sci., 2019, 96, 21–42 CrossRef CAS.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- D. Barby and Z. Haq, Low Density Porous Cross-Linked Polymeric Materials and Their Preparation and Use as Carriers for Included Liquids, US Pat., US4522953A, 1985 Search PubMed.
- D. Barby and Z. Haq, Low Density Porous Cross-Linked Polymeric Materials and Their Preparation, European Pat., EP0060138B1, 1986 Search PubMed.
- H. Zhang, R. Zhao, M. Pan, J. Deng and Y. Wu, Ind. Eng. Chem. Res., 2019, 58, 5533–5542 CrossRef CAS.
- S. Kramer, N. Skušek and P. Krajnc, Polym. Chem., 2023, 14, 1330–1338 RSC.
- A. Foulet, M. Birot, G. Sonnemann and H. Deleuze, React. Funct. Polym., 2015, 90, 15–20 CrossRef CAS.
- A. Foulet, M. Birot, R. Backov, G. Sonnemann and H. Deleuze, Mater. Today Commun., 2016, 7, 108–116 CrossRef CAS.
- Y. Xia, Z. Yang and Y. Zhu, J. Mater. Chem. A, 2013, 1, 9365–9381 RSC.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293–1302 CrossRef CAS.
- N. S. Kevlich, M. L. Shofner and S. Nair, Sep. Sci. Technol., 2017, 52, 1070–1094 CrossRef CAS.
- L. Pola, S. Collado, P. Oulego and M. Díaz, Chem. Eng. J., 2022, 448, 137728 CrossRef CAS.
- M. S. Dresselhaus, K. A. Williams and P. C. Eklund, MRS Bull., 1999, 24, 45–50 CrossRef CAS.
- A. D. Lueking, L. Pan, D. L. Narayanan and C. E. B. Clifford, J. Phys. Chem. B, 2005, 109, 12710–12717 CrossRef CAS PubMed.
- S. Orimo, G. Majer, T. Fukunaga, A. Züttel, L. Schlapbach and H. Fujii, Appl. Phys. Lett., 1999, 75, 3093–3095 CrossRef CAS.
- S. Orimo, A. Züttel, L. Schlapbach, G. Majer, T. Fukunaga and H. Fujii, J. Alloys Compd., 2003, 356–357, 716–719 CrossRef CAS.
- C.-W. Huang, H.-C. Wu and Y.-Y. Li, Sep. Purif. Technol., 2007, 58, 219–223 CrossRef CAS.
- J. S. Im, S.-J. Park and Y.-S. Lee, Mater. Res. Bull., 2009, 44, 1871–1878 CrossRef CAS.
- M. Mohan, V. K. Sharma, E. A. Kumar and V. Gayathri, J. Energy Storage, 2019, 1, e35 CrossRef CAS.
- N. L. Ravikumar, K. K. Kar, S. Sarkar and D. Sathiyamoorthy, Polym. Compos., 2010, 31, 2069–2078 CrossRef CAS.
- A. Verma, K. Baurai, M. R. Sanjay and S. Siengchin, Polym. Compos., 2020, 41, 338–349 CrossRef CAS.
- C. Forgacz, M. Birot and H. Deleuze, J. Appl. Polym. Sci., 2013, 129, 2606–2613 CrossRef CAS.
- R. Poupart, R. Invernizzi, L. Guerlou-Demourgues, J. Olchowka, M.-A. Dourges, J.-L. Bobet, H. Deleuze and R. Backov, Langmuir, 2023, 39, 16385–16394 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- H. Sis and M. Birinci, Colloids Surf., A, 2009, 341, 60–67 CrossRef CAS.
- M. Youssry, F. Z. Kamand, M. I. Magzoub and M. S. Nasser, RSC Adv., 2018, 8, 32119–32131 RSC.
- C. Ishizaki and I. Martí, Carbon, 1981, 19, 409–412 CrossRef CAS.
- A. Foulet, M. Birot, G. Sonnemann and H. Deleuze, J. Clean. Prod., 2015, 91, 180–186 CrossRef CAS.
- H. Ridaoui, A. Jada, L. Vidal and J.-B. Donnet, Colloids Surf., A, 2006, 278, 149–159 CrossRef CAS.
- S.-G. Kim, O.-K. Park, J. H. Lee and B.-C. Ku, Carbon Lett., 2013, 14, 247–250 CrossRef.
- D. G. Henry, I. Jarvis, G. Gillmore and M. Stephenson, Earth Sci. Rev., 2019, 198, 102936 CrossRef CAS.
- E. Fuentes-Quezada, E. de la Llave, E. Halac, M. Jobbágy, F. A. Viva, M. M. Bruno and H. R. Corti, Chem. Eng. J., 2019, 360, 631–644 CrossRef CAS.
- R. M. Alway-Cooper, D. P. Anderson and A. A. Ogale, Carbon, 2013, 59, 40–48 CrossRef CAS.
- X. Zhang, S. Khor, D. Gao and E. Sum, Mater. Chem. Phys., 2012, 131, 735–742 CrossRef CAS.
- Y. Ban, Y. Wang, N. Li, R. He, K. Zhi and Q. Liu, R. Soc. Open Sci., 2018, 5, 180717 CrossRef CAS.
- C. Sheng, Fuel, 2007, 86, 2316–2324 CrossRef CAS.
- VULCAN® XC72 (VXC72), CAS 1333-86-4, 16080001, Fuel Cell Store, Bryan, Texas, USA, 2012, https://www.fuelcellstore.com/msds-sheets/vulcan-xc72r-msds.pdf, accessed 2024-01-20.
- M. Rzepka, P. Lamp and M. A. de la Casa-Lillo, J. Phys. Chem. B, 1998, 102, 10894–10898 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209–2214 CrossRef CAS.
- K. M. Thomas, Catal. Today, 2007, 120, 389–398 CrossRef CAS.
- A. Toprak, Biomass Convers. Biorefin., 2020, 10, 977–986 CrossRef CAS.
- B. Weinberger and F. D. Lamari, Int. J. Hydrogen Energy, 2009, 34, 3058–3064 CrossRef CAS.
- J. Jagiello, A. Ansón and M. T. Martínez, J. Phys. Chem. B, 2006, 110, 4531–4534 CrossRef CAS PubMed.
- P. Ramirez-Vidal, R. L. S. Canevesi, G. Sdanghi, S. Schaefer, G. Maranzana, A. Celzard and V. Fierro, ACS Appl. Mater. Interfaces, 2021, 13, 12562–12574 CrossRef CAS.
- S. Stock, N. Kostoglou, J. Selinger, S. Spirk, C. Tampaxis, G. Charalambopoulou, T. Steriotis, C. Rebholz, C. Mitterer and O. Paris, ACS Appl. Energy Mater., 2022, 5, 10915–10926 CrossRef CAS.
- Q. Dou and H. S. Park, Energy Environ. Mater., 2020, 3, 286–305 CrossRef CAS.
- E. H. Lahrar, P. Simon and C. Merlet, J. Chem. Phys., 2021, 155, 184703 CrossRef CAS.
- A. Lemoine, R. Invernizzi, G. Salvato Vallverdu, J. Olchowka, L. Guerlou-Demourgues, I. Baraille and D. Flahaut, ACS Appl. Energy Mater., 2022, 5, 12359–12372 CrossRef CAS.
- B. Karamanova, E. Mladenova, M. Thomas, N. Rey-Raap, A. Arenillas, F. Lufrano and A. Stoyanova, Gels, 2023, 9, 983 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02097a |
| This journal is © The Royal Society of Chemistry 2024 |