
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond molecular structure: critically assessing machine learning for designing organic photovoltaic materials and devices†
Martin
Seifrid‡
 *ab,
Stanley
Lo‡
b,
Dylan G.
Choi
c,
Gary
Tom
bde,
My Linh
Le
f,
Kunyu
Li§
c,
Rahul
Sankar§
c,
Hoai-Thanh
Vuong§
c,
Hiba
Wakidi§
c,
Ahra
Yi§
c,
Ziyue
Zhu§
c,
Nora
Schopp
c,
Aaron
Peng
c,
Benjamin R.
Luginbuhl
c,
Thuc-Quyen
Nguyen
*c and
Alán
Aspuru-Guzik
*bdeghij
*ab,
Stanley
Lo‡
b,
Dylan G.
Choi
c,
Gary
Tom
bde,
My Linh
Le
f,
Kunyu
Li§
c,
Rahul
Sankar§
c,
Hoai-Thanh
Vuong§
c,
Hiba
Wakidi§
c,
Ahra
Yi§
c,
Ziyue
Zhu§
c,
Nora
Schopp
c,
Aaron
Peng
c,
Benjamin R.
Luginbuhl
c,
Thuc-Quyen
Nguyen
*c and
Alán
Aspuru-Guzik
*bdeghij
aDepartment of Materials Science and Engineering, North Carolina State University, Raleigh, North Carolina 27695, USA. E-mail: m_seifrid@ncsu.edu
bDepartment of Chemistry, Chemical Physics Theory Group, University of Toronto, 80 St. George St., Ontario M5S 3H6, Canada. E-mail: aspuru@utoronto.ca
cDepartment of Chemistry and Biochemistry, Center for Polymers and Organic Solids, University of California Santa Barbara, Santa Barbara, California 93106, USA. E-mail: quyen@chem.ucsb.edu
dDepartment of Computer Science, University of Toronto, 40 St George St, Toronto, ON M5S 2E4, Canada
eVector Institute for Artificial Intelligence, 661 University Ave. Suite 710, Toronto, Ontario M5G 1M1, Canada
fMaterials Department, University of California Santa Barbara, Santa Barbara, California 93106, USA
gDepartment of Chemical Engineering & Applied Chemistry, University of Toronto, 200 College St., Ontario M5S 3E5, Canada
hDepartment of Materials Science & Engineering, University of Toronto, 184 College St., Ontario M5S 3E4, Canada
iLebovic Fellow, Canadian Institute for Advanced Research (CIFAR), 661 University Ave., Toronto, Ontario M5G 1M1, Canada
jAcceleration Consortium, University of Toronto, 80 St. George St, Toronto, ON M5S 3H6, Canada
First published on 20th May 2024
Abstract
Our study explores the current state of machine learning (ML) as applied to predicting and designing organic photovoltaic (OPV) devices. We outline key considerations for selecting the method of encoding a molecular structure and selecting the algorithm while also emphasizing important aspects of training and rigorously evaluating ML models. This work presents the first comprehensive dataset of OPV device fabrication data mined from the literature. The top models achieve state-of-the-art predictive performance. In particular, we identify an algorithm that is used less frequently, but may be particularly well suited to similar datasets. However, predictive performance remains modest (R2 ≅ 0.6) overall. An in-depth analysis of the dataset attributes this limitation to challenges relating to the size of the dataset, as well as data quality and sparsity. These aspects are directly tied to difficulties imposed by current reporting and publication practices. Advocating for standardized reporting of OPV device fabrication data reporting in publications emerges as crucial to streamline literature mining and foster ML adoption. This comprehensive investigation emphasizes the critical role of both data quantity and quality, and highlights the need for collective efforts to unlock ML's potential to drive advancements in OPV.
 Martin Seifrid | Martin Seifrid received his PhD from the University of California, Santa Barbara in 2019, where he worked in the Center for Polymers and Organic Solids with Prof. Guillermo C. Bazan. He used computational and experimental methods to study molecular design and structure–processing–property relationships in molecular and polymeric organic semiconductors. He carried out his postdoctoral research at the University of Toronto with Prof. Alán Aspuru-Guzik. There, he developed self-driving laboratories for autonomous molecular design, automated synthesis and characterization, and organic laser materials. In August 2023, Martin Seifrid joined the Department of Materials Science & Engineering at North Carolina State University. |
1 Introduction
Early in the development of OPV devices, it was discovered that bulk heterojunction (BHJ) device active layers led to higher power conversion efficiencies (PCEs) than the simpler planar heterojunction.1 Since then, extensive work has been devoted to optimizing the morphology of BHJ active layers with various processing conditions (e.g., solvent, concentration, solvent additives, thermal annealing, etc.).2–4 Consequently, it is well-understood that the device fabrication procedure (e.g., processing solvent, spin coating speed, annealing temperature) plays an important role in the morphology of the OPV active layer because the process of film formation is kinetically limited.5 The importance of morphology with respect to OPV device performance has been studied extensively,2,3,5 and plays a role in processes including exciton diffusion,6 charge separation and transport,7 and device stability.8,9 However, comprehending the intricate correlations between diverse materials and device fabrication parameters, and their collective influence on the final device performance poses an enduring challenge. This intricacy stands as a formidable barrier, impeding the accurate prediction of OPV device performance and contributing to the laborious and costly nature of the production processes.Given the many complex relationships and processes that ultimately determine OPV device performance, the potential of machine learning (ML) to accelerate the development of OPV materials and devices has emerged as a tantalizing promise. ML techniques could allow scientists to quickly screen potential combinations of donor (D) and acceptor (A) materials, or suggest the most appropriate device fabrication parameters for a given combination of materials. Previous efforts in using ML to predict the PCE of OPV devices have been focused exclusively on the donor and acceptor materials. Notably, the Harvard Clean Energy Project10–12 and others13–19 have used DFT-computed molecular descriptors to predict PCEs. A similar approach has recently incorporated genetic algorithms in an effort to design high-performing non-fullerene acceptor-based (NFA) devices.20–22 Others have generated one-hot encodings based on human intuition regarding molecular substructures.23 However, the most accurate models to date are those that have used high-throughput domain-specific descriptors.17,24–30 Notably, the only device fabrication parameter to have been incorporated so far is the donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor (D:A) ratio.17 Others, which – as discussed above – are known to play an important role in determining PCE, have not been explored yet.
:acceptor (D:A) ratio.17 Others, which – as discussed above – are known to play an important role in determining PCE, have not been explored yet.
In this work, we critically assess the state of ML for predicting PCE of OPV devices. We have curated a dataset of molecular structures, experimental device fabrication parameters and device performance, and evaluate the effect of various structural representations, ML algorithms, and device fabrication parameters on model performance. We also discuss difficulties related to gathering the dataset, as well as the considerations related to using experimental data from the literature, which is applicable to many materials domains outside of OPV.
2 Results & discussion
2.1 Data curation
In this section, we describe some of the most salient aspects of data curation. However, further details are provided in the Methods section. Broadly, the lack of uniform reporting standards and machine-readable data posed a significant challenge to compiling a large and complete dataset of OPV device fabrication and performance.To compile our dataset, we began with the dataset of 565 devices in Wu et al.,23 which only contained donor and acceptor names, short circuit current density (JSC), open circuit voltage (VOC), fill factor (FF), and PCE (Fig. 1). We first evaluated the quality of the data by comparing reported PCE values with values calculated from the product of JSC, VOC, and FF:
| PCEcalculated = JSC × VOC × FF | (1) |
| |PCEreported − PCEcalculated| > 0.01 × PCEcalculated | (2) |
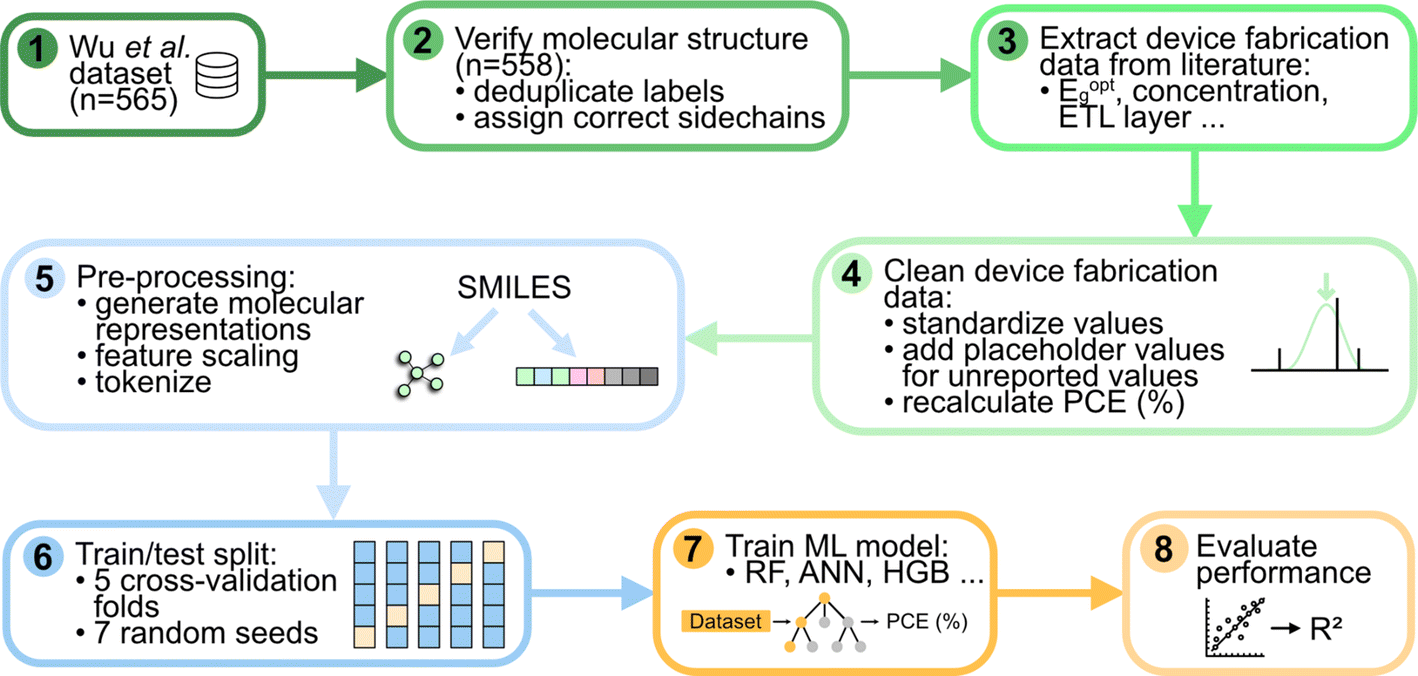 | ||
| Fig. 1 Flowchart describing the steps involved in curating the first dataset of OPV device fabrication data, and testing and validation of representations and models. Eoptg: optical excitation gap energy, ETL: electron transport layer, SMILES: simplified molecular-input line-entry system, RF: random forest, ANN: artificial neural network, HGB: histogram gradient boosting, R2: coefficient of determination. | ||
The threshold of 0.01 was chosen arbitrarily. It reflects that if the reported and calculated PCE values were within 1%, we are relatively certain that any inconsistencies were only due to minor rounding errors. This allowed us to quickly identify data points with incorrect or nonsensical entries. To gather the fabrication data for the 565 devices, we reviewed the 277 original reports from which the dataset was initially constructed and extracted information from the 565 individual devices. To accurately extract the desired parameters, each paper was checked by multiple scientists. We found a number of duplicates, unreliable reports or incorrect references in the original dataset, which were eliminated, leading to the 558 devices in the final dataset.
We associated every donor and acceptor name with a molecular structure in the form of a SMILES (Simplified Molecular Input Line Entry System) string. This proved particularly challenging because of the diversity of polymer and NFA names, as well as frequent overlaps between material names. Due to the lack of a standardized naming convention for conjugated molecules and polymers, there are cases where numerous entries were ambiguously labeled as “P1”, despite having markedly distinct structures. On the other hand, research groups will sometimes use different names for the same molecular structure. For example, IT-F appears as F-ITIC, ITVfIC, ITIC3, and ITIC-F. In these instances, duplicate labels were replaced with a consistent name for the purpose of clarity. Finally, representing molecules with SMILES strings is limited. Polymers cannot be easily represented as the repeating, statistical entities that they actually are. While this is an active area of research,31–34 we simply encoded the structure of the monomer. Additionally, regioregularity – which is a factor for both polymers and NFAs – is not easily represented in SMILES notation. For example, IT-F and other NFAs with mono-substituted end groups are usually obtained as a mixture of three isomers, the populations of which play a role in determining PCE.35–38 However, SMILES forces us to choose a single isomer.
To select film and device fabrication parameters, we considered fundamental processes in OPV, such as light absorption and exciton formation, exciton migration, charge separation, and charge migration to the electrodes. Each of these processes is influenced by specific parameters related to the donor, acceptor, and active layer casting conditions that can be directly controlled by the researcher. The selection of parameters aimed to capture the relevant information for these processes. These parameters included donor and acceptor material properties (HOMO, LUMO, Eoptg), D:A ratio, solvent, total solids concentration, additive (and concentration), active layer thickness, thermal annealing temperature, hole transport layer (HTL), electron transport layer (ETL), hole mobility, and electron mobility. Other well-studied device performance characteristics such as effective mobility, Langevin prefactor, and Vocvs. ln(I) slope were not reported with enough consistency to be included in the dataset.
In extracting material properties (HOMO, LUMO, Eoptg), we observed that multiple values were reported for the same material. This may arise from a variety of factors: differences in purity, dispersity (in the case of polymers), measurement method, or measurement error.39 Using the reported values would result in data points with the same molecular representation (i.e. molecular structure) having multiple different property values. Accounting for such variation in a ML model is nontrivial, and disentangling variations from experimental noise and differences in material composition was not possible. For materials with variation in the reported material properties, the mean was taken as the corresponding feature value. Some materials had identical values reported multiple times. Upon further investigation, this repetition was due to multiple papers citing a single source-often from the literature describing the synthesis of the material. If the repetition is greater than 10 times, only one of the entries is used in the calculation of the mean. Upon further investigation, it was determined that this was due to papers citing a single source (often the material property values reported in the paper describing the initial synthesis). If the value was repeated more than ten times, we automatically discounted nine of the entries when fitting.
Extracting active layer and device fabrication details often required digging through the paper's supporting information or references. D:A ratios were not consistently reported, requiring reviewers to refer to the paper's Methods section in order to calculate them from the total solids concentrations of the individual (D and A) solutions. Additionally, some papers reported either active layer thickness or spin coating speed, as they are related. Thus, the reporting of these parameters was inconsistent across papers. For optional fabrication features (solvent additive and concentration, thermal annealing temperature and time) it was assumed that if they were not reported, they were not used.
2.2 Structural representations
Machines cannot directly interpret molecular structure. Therefore, it is necessary to represent molecules and their structure in a machine-readable format. Choosing an appropriate representation is crucial to achieving accurate and interpretable models. We examined several methods for representing molecules in this task (Fig. 2). The simplest such approaches represent a molecule with a binary value (one-hot encoding, OHE) or with values corresponding to material properties. OHE encodes the presence of a molecule in a data point as a binary (true–false) bit in a vector wherein the position of each bit corresponds to a unique molecule, but provides no information about the identity or properties. When representing molecules simply with their material properties, we chose energetic quantities that are known to be relevant in OPV applications: HOMO and LUMO energy levels, the HOMO–LUMO energy gap (EHLg), and the optical excitation gap (Eoptg).40 However, these descriptors still lack any information about molecular structure, which is relevant to solubility, solid-state packing and microstructure. Some information about molecular structure and physical properties can be encoded using Mordred descriptors, a set of computed numerical values describing two- and three-dimensional molecular properties, such as constitutional, topological, and electronic features.41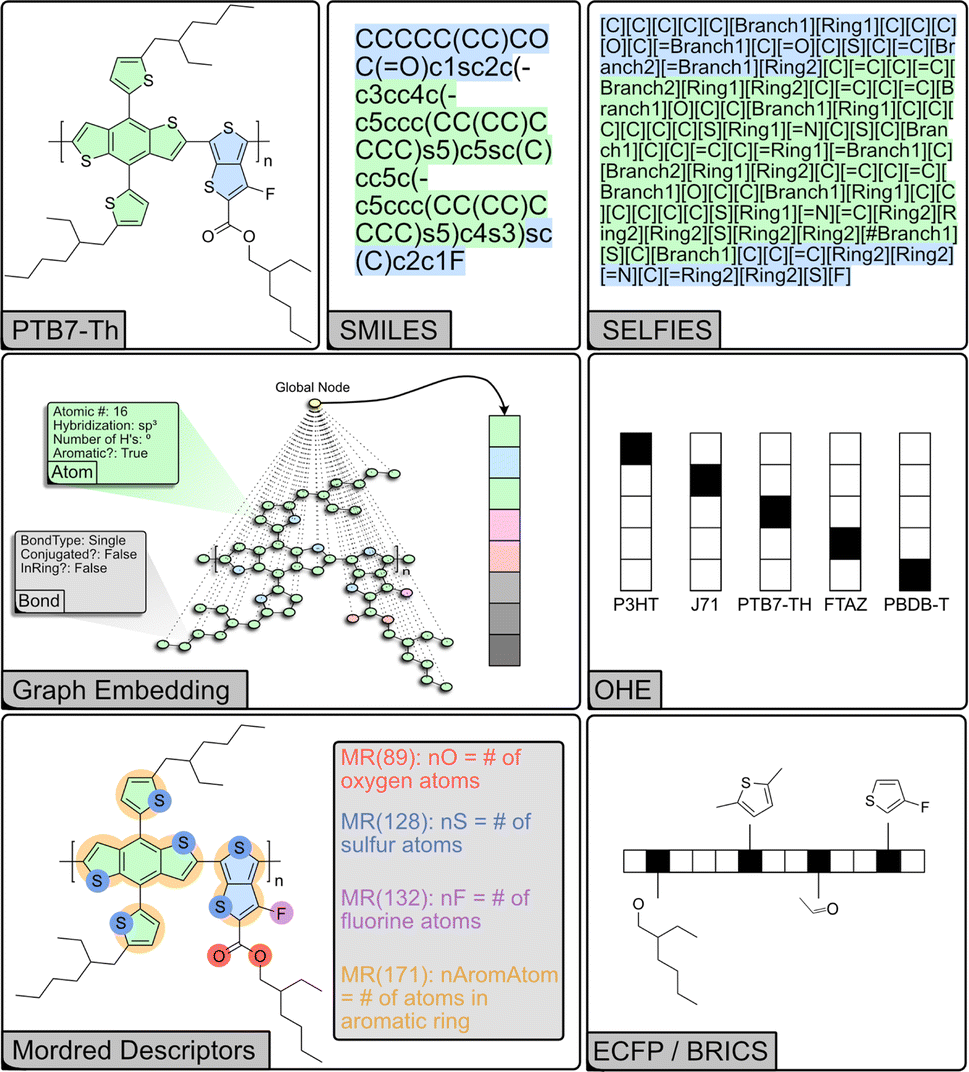 | ||
| Fig. 2 Schematic representation of different structural encodings of the common donor polymer PTB7-Th. | ||
Alternatively, molecules can be decomposed into substructures, or fragments, which are then encoded as arrays. Extended-connectivity fingerprints (ECFP) effectively capture local structural information and are a common cheminformatics tool for modeling structure–activity relationships and calculating structural similarity. ECFP breaks down molecular structures into circular topological fingerprints around each atom up to a set maximum radius and hashes the presence or absence of the substructure into a bit vector of set length – similar to OHE but for substructures. ECFP includes additional atomic characteristics including “heavy” atom count, valence minus the number of hydrogens, atomic number, atomic mass, atomic charge, the number of implicit and explicit hydrogens, and whether the atom is part of a ring.42 BRICS decomposes molecules into fragments of varying size based on common retrosynthetic and drug-like substructures, which are then encoded as arrays of varying length.43 This representation makes it possible to identify common substructures and analyze their impact on the overall molecular properties. Recently, the polymer-unit fingerprint (PUFp) has also been introduced as a way to provide a more explainable fingerprint-based representation for conjugated molecules.44
Molecular structure can naturally be represented by a graph where atoms are the nodes and bonds are the edges. However, encoding the graph is also non-trivial. Several line notations (string-based representations) have been developed for this purpose, wherein atoms are represented by specific characters and the bonds can be either explicit or implicit. SMILES captures both the topology and connectivity of atoms, enabling programmatic comparison and manipulation of molecular structures.45 SELFIES (Self-Referencing Embedded Strings) is a new system aimed at generative models in particular.46 Unlike SMILES which can have many invalid strings, every string of SELFIES characters is inherently valid. Recently, graph neural networks (GNN) have demonstrated state-of-the-art performance on chemical regression tasks.47–50 Molecules are represented as graphs; node and edge features are vectors associated with each atom or bond, describing properties of the node (i.e. atomic number, hybridization, aromaticity etc.) or edge (i.e. bond order, conjugation, stereochemistry). Global features contain information about the entire molecule. The GNN then aggregates the features for each node based on the graph connections into messages, which are then used to learn updated nodes and edges. The global node allows for message-passing between all the nodes in the graph, pooling the edge and node features into global features, and can be thought of as a node that is connected to all other nodes.51 A final aggregation layer takes the updated graph and outputs the final prediction. In addition, the embeddings generated by GNNs can be fed into other model architectures. To generate the graph embeddings, the GNN is trained to predict a relevant set of properties. During training, the GNN generates a learned representation (latent space) that captures information about the relationship between molecular structure and the target property. This representation, which is extracted from the embedding layer of the network, can then be used by another ML model to predict a separate target.48,52,53
2.3 Model selection
Another crucial aspect of building accurate models for predicting the properties of OPV devices is the choice of ML algorithm. Key considerations include whether the algorithm is appropriate for the task at hand (e.g., supervised, unsupervised, or reinforcement learning; classification or regression), the size of the dataset, whether information about the model's uncertainty is required, the algorithm's computational cost, and its interpretability. In this context, we evaluated eleven ML algorithms, which can be classified within five broad categories: non-parametric, linear, kernel-based, tree-based, and deep learning (DL). Each algorithm has its own strengths, limitations, and assumptions. The choice of algorithm depends on the specific characteristics of the dataset, the nature of the problem, and the desired output. Researchers should carefully evaluate and select the most suitable model for their specific application in the context of OPV devices.Model performance can be quantified based on a number of different parameters including R2 score (coefficient of determination), root mean square error (RMSE), and mean absolute error (MAE). These metrics all capture different aspects of model performance. However, we have observed that the correlation coefficient (R) is occasionally reported instead of the R2 score, and as the sole performance metric.23,24,28–30 This is not a robust metric since R only captures the linear correlation between true and predicted values while the R2 score quantifies the deviation of the model prediction from the ground truth. However, while R is less strict in evaluating the model performance, a model with high R score may indicate modest success in training, and can still provide predictions that approximate the ground truth up to some correction term. In order to gain a better statistical understanding of the variability in model performance, cross-validation should be performed. This provides information on the variability of the model with different train and test sets. We achieve this by carrying out 5-fold cross-validation from seven independent random seeds which provide 35 different train and test set combinations. Given that the random seeds are independent, we can determine variability based on the standard error of the mean. Finally, it is also important to carefully apply feature scaling so as not to overestimate the model's performance. The training set features should be scaled first, ensuring a standardized feature space that the models can learn from, and then that scaling should be applied to the test set features. If all features are scaled together, this can result in data leakage between the train and test sets, implicitly encoding information about the train set into the test set, providing overestimates on the model performance.54,55
Linear methods are the simplest: they assume that there is some linear relationship between the input features and the target value, which can be used to predict the target value based on a linear combination of the input features. One of the greatest advantages to linear regression is the interpretability of the model: linear effects are easy to quantify, and to describe. As expected, multiple linear regression (MLR) is only useable with relatively low dimensional input data (material properties), where it achieves an R2 score of 0.35 ± 0.03. Linear models are not able to effectively handle binary (OHE, ECFP) or sequence-based (SMILES, SELFIES) features.
Non-parametric models such as k-nearest neighbors (KNN) make no assumptions about the distribution of data from which a sample is drawn. The algorithm computes the distances from the nearest number of datapoints (k) around the new datapoint (i.e. test set) to predict the property of the new datapoint. The distance algorithm and k can be user-defined or vary based on the local density of points. KNN is simple and versatile but can be computationally expensive for large datasets.56 While KNN is most often used for classification tasks, it can also be employed for regression. In regression, KNN predicts the average target value of the k nearest points in the training set. Because the KNN algorithm simply predicts the average of the nearest data points, it performs remarkably well across many different representations. In fact, its performance is comparable to or better than much more complex models in the cases of OHE (R2 = 0.31 ± 0.03), material properties (R2 = 0.43 ± 0.03), SMILES and SELFIES (R2 = 0.41 ± 0.03 and 0.41 ± 0.04, respectively), and even graph embeddings (R2 = 0.43 ± 0.03).
Kernel methods apply a non-linear transformation (e.g. radial basis function, polynomial, sigmoid, etc.) to the input features, known as the kernel trick, which can then be used in many different algorithms. Support vector regression (SVR) uses this approach to find an optimal hyperplane in the same or higher dimension that maximizes the margin (distance) between the hyperplane and the nearest data points while also minimizing the prediction error. SVR is effective for handling high-dimensional or non-linear data and outliers. However, it does not scale efficiently for very large datasets. Kernel ridge regression (KRR) is a regularization technique that takes advantage of the kernel trick in order to learn a linear relationship in the kernel space, and a non-linear relationship in the original space similar to SVR. Unlike SVR, KRR uses a squared error loss while SVR calculates the loss from the distance between the test set and the decision boundary of the hyperplane which is determined by the user (i.e. epsilon). Gaussian process regression (GPR) also uses the kernel trick, but it takes a Bayesian approach. GPs are probabilistic models that learn the probability distribution (posterior) over all data points. A prior distribution, typically a Gaussian distribution, over all possible functions to model the data must be specified, with the covariance of the distributions determined by the kernel function. The updated distribution or posterior distribution incorporates information from the prior distribution and the dataset using Bayes' theorem which is then used to make predictions on new, unseen data points. It is particularly useful for small datasets and can capture complex relationships with uncertainty estimates. The biggest disadvantage of GPs is the high computational demand which remains a challenge for high-dimensional and large datasets. In general, KRR and GPR are out-performed by other methods on all representations except for ECFP, where the Tanimoto kernel57 can be employed, leading to good predictive performance (R2 scores of 0.52 ± 0.02 and 0.56 ± 0.02, respectively). On the other hand, SVR is comparable to the top methods for most representations, including R2 scores of 0.53 ± 0.03 with ECFP and 0.56 ± 0.03 with Mordred descriptors.
Decision trees are commonly used for both classification and regression. Decision trees are interpretable and versatile, making them valuable for a wide range of applications in data analysis and prediction. They work by recursively partitioning the data into subsets based on feature values, ultimately leading to a tree-like structure where each leaf node represents a predicted outcome. However, they are prone to overfitting since every data point can be explained with sufficient tree depth. This can be mitigated by using ensemble methods and carefully selecting the model parameters such as tree depth. We evaluate a number of different ensemble tree-based methods including: random forest (RF), gradient boosted trees (XGBoost),58 natural gradient boosting with uncertainty estimation (NGBoost),59 and histogram gradient boosting (HGB), the implementation of which is inspired by LightGBM.60,61 Tree-based methods are able to handle non-linear relationships within the feature space, perform automatic feature selection,62 and can be interpreted by analyzing the decision boundaries established by the trees. RF is an ensemble of decision trees trained on various bootstrapped subsets of the entire dataset, with features randomly selected for branches within the trees. A final prediction and uncertainty is attained by consensus of the ensembles. XGBoost is an optimized implementation of gradient boosting that sequentially combines many weak individual predictive models to form a strong final ensemble predictive model, with each subsequent model trained to correct the errors of the previous models. However, this comes with trade-offs in the form of lack of interpretability, trial-and-error hyperparameter tuning, and higher computational cost. NGBoost is similar to XGBoost, also learning the errors of previous trees through boosting, but predicts the parameters of a distribution rather than the regression value directly. NGBoost is useful when uncertainty estimation is desired and offers better interpretability through the analysis of the predicted distributions. HGB regression is much more efficient by virtue of first binning the input features and building the trees off of bins rather than floating point values. As a result, the number of split candidates and the computational cost are significantly reduced. As an added benefit, one of the bins is reserved for missing values, which is very useful for incomplete datasets as will be discussed below. The tree-based methods discussed above are almost indistinguishable in terms of predictive performance, except for XGBoost which performs slightly worse. Most importantly, tree-based methods perform best with ECFP (between R2 = 0.58 ± 0.02 and 0.59 ± 0.02), and Mordred descriptors (between R2 = 0.56 ± 0.03 and 0.6 ± 0.03) because ECFP and Mordred descriptors extract relevant chemical and molecular features unlike other molecular representations such as SMILES or one-hot encodings.
We also evaluated two common DL model architectures: multi-layer perceptron (MLP) neural networks (NN) and GNNs. MLPs are versatile and powerful models that have been shown to be capable of learning complex relationships between the input features and target variables. These models are made up of multiple interconnected layers of neurons with non-linear activation functions (e.g. ReLU, sigmoid, tanh). The weights and biases of the neurons are optimized over many iterations using gradient descent and backpropagation via the connections between neurons to minimize the loss function (mean squared error). However, they require careful architecture design, parameter tuning, and large quantities of training data. GNNs are neural network architectures designed to handle data that can be represented in the form of a graph. GNNs can effectively learn relationships and patterns in molecular structures because local features can be influenced by distant atoms and bonds through message-passing.47,50,63 Here we utilize the GraphNets architecture for GNN prediction, and the ChemProp message passing networks for generating the molecular embeddings.48–50,64,65
The tree-based models (RF, HGB, XGB, NGB) significantly outperform the DL models (i.e. MLP and GNN) because they are insensitive to irrelevant features and can learn irregular functions.62 Given the small size of our dataset, the limitations of DL models are pronounced. DL models can easily overfit to a small training set by learning from spurious correlations of irrelevant features and predict inaccurate output values.
The best DL model (MLP trained on ECFP, R2 = 0.46 ± 0.04) performs well because the ECFP representation extracts relevant features from the molecular structure,42 making the inherently challenging task of learning the most relevant features from sparse and low quantity data less difficult for the MLP algorithm. In addition, DL models are biased towards smooth functions which make it challenging for the model to learn from tabular data and discrete data, as is the case with this dataset.62 Interestingly, while the GNN regressor struggled due to the size of the dataset, the graph embeddings extracted from the GNN embedder combined with a tree-based or kernel-based prediction model outperformed the direct prediction of PCE from the graph.
Independent of model choice, the best representations are found to be material properties, ECFP, Mordred descriptors, and graph embeddings (model performance in Table S1†). OHE is a good baseline representation in that if the same model performs worse with a new representation, it is likely that the new representation is uninformative. OHE performs poorly across all of the models because none of the chemical, substructural, or atomic characteristics are captured. We find that most representations perform significantly better than OHE. While the PUFp representation provides model performance comparable to the material properties representation (Table S2†), it suffers from a lack of generalizability since the PUFp workflow creates a fingerprint that is specific to the dataset.44 However, when only including fabrication conditions, the nature of which will be detailed in Section 2.4, the difference is very small. This suggests that fabrication conditions alone are not very informative. None of the models that were evaluated were able to take advantage of the sequence information in SMILES and SELFIES, explaining their poor performance. Models such as long short-term memory (LSTM) networks may be useful. However, the dataset is likely too small for the LSTM network to accurately learn the syntax and grammar of string-based representations in addition to accurately learning the complex relationships between the representation and properties.66,67
Despite the modest R2 scores, we find that our top-performing models (RF and HGB) either match or surpass the state of the art for other similar OPV datasets (Fig. S2†).22,29 In particular, we achieve better predictive performance (R2 = 0.5 ± 0.03) using only ECFP and the material properties available in the dataset in comparison to the performance (R2 = 0.4) achieved when using computed descriptors.22
We also explored the ability of multi-output models for predicting PCE. While the most common approach is to predict PCE as the single output, PCE can also be predicted as the product of its components – JSC, VOC, and FF (eqn (1)). We selected three models: two that natively support multi-output regression in scikit-learn (RF, HGB), and an artificial neural network (ANN) made in PyTorch, which is similar to the MLP described above (further details available in Methods). The models were trained concurrently on PCE, JSC, VOC, and FF. Model scores for PCE prediction were calculated using eqn (1) from the predicted JSC, VOC, and FF (Fig. S3†). Ultimately, model performance was not significantly different between directly predicting PCE (Fig. 3) and predicting PCE from JSC, VOC, and FF. However, we observed that all models were able to predict JSC much better than VOC, despite heuristics suggesting a strong correlation between VOC and LUMOA–HOMOD,68 which suggests that this problem is worth further investigation.
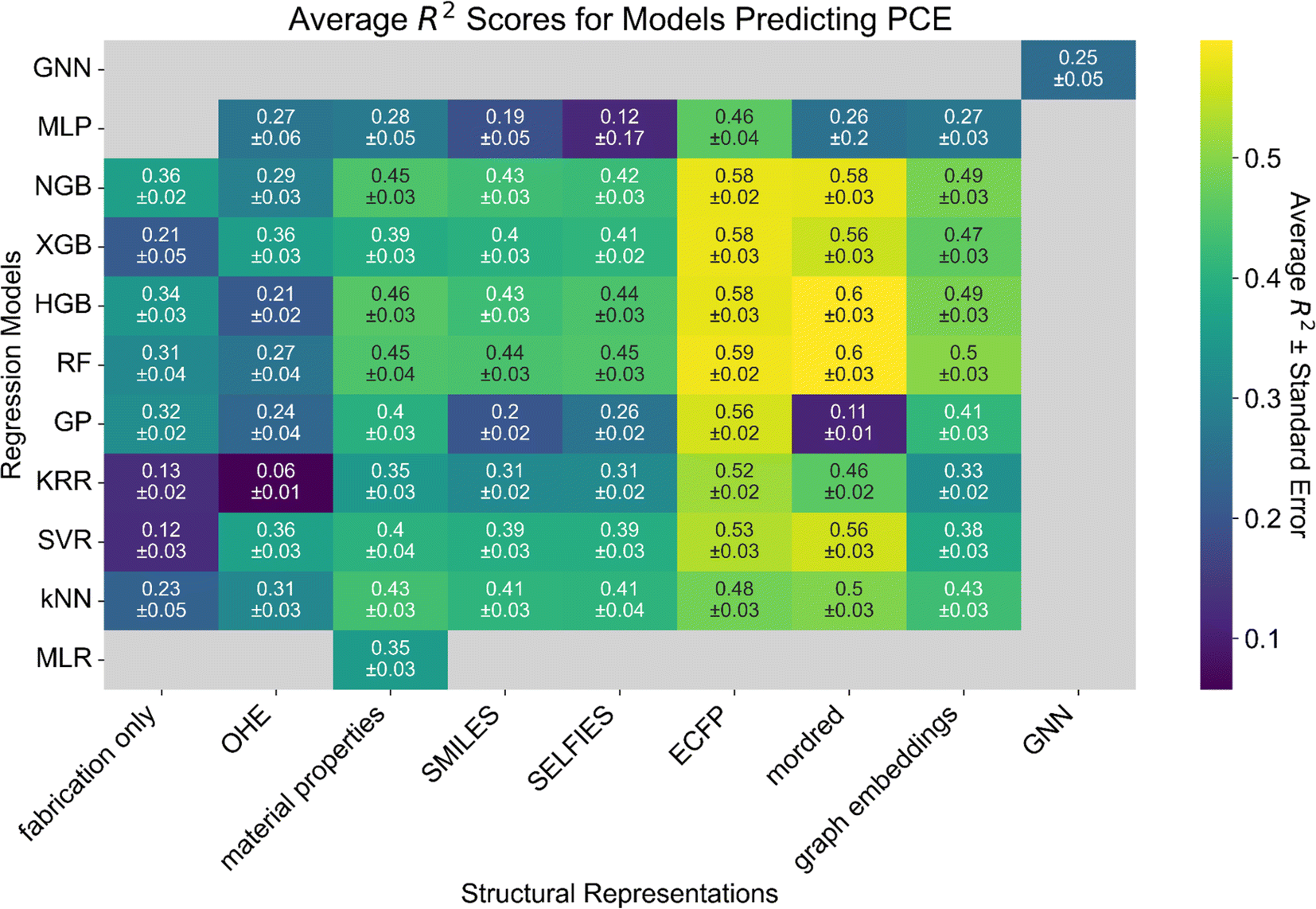 | ||
| Fig. 3 Heatmap of model performance for predicting PCE from the molecular structure of the donor and acceptor materials as measured by the R2 score. Tree-based models (RF, XGBoost, HGB, and NGBoost) perform best on a dataset of this size (558 points). Structural representations based on ECFP and Mordred descriptors perform best across the board. Numbers in each cell correspond to the average R2 score of the model over seven independent five-fold cross-validations ± the standard error of the mean. Gray cells correspond to models that were not tested or failed to yield reasonable results (0 ≤ R2 ≤ 1). Heatmaps for the RMSE, MAE and R (for comparison to other published works only) scores are presented in Fig. S1.† | ||
2.4 Introducing fabrication features
Empirically, device fabrication parameters are known to play a role in determining the PCE of OPV devices.2,3 The same can be observed in our dataset. The most common donor and acceptor combination is PBDB-T and ITIC with 11 occurrences. The PCEs of the PBDB-T:ITIC devices range from 7.3% to 11.2%, a significant difference for top-performing devices in different papers.Encoding scalar device fabrication features such as energy levels, annealing temperature or the D:A ratio is straightforward. However, encoding categorical features is more challenging. In the case of OPV device fabrication, these are (i) solvent, (ii) solvent additive (if used), (iii) the hole transport layer, HTL, and (iv) the electron transport layer, ETL. Similarly to the donor and acceptor materials, these categorical features could be encoded using one-hot encoding or as labels. However, as seen with OHE of the active layer materials, these encodings are not informative for a ML model. The HTL and ETL were encoded with their energy levels, as determined from literature reports (Table S3†). The solvent and solvent additive were encoded with physicochemical descriptors obtained from the HSPiP database (Table S4†).69
The features beyond molecular structure were split into four broad categories: material properties (as in Fig. 3), film fabrication parameters (fabrication), device architecture, and electrical characterization. As discussed in greater detail below, a small number of features (total solids concentration and spin coating speed in particular) were missing from many of the data points (Fig. 4a). These features were excluded in order to maximize the size of the dataset on which the models were trained (Fig. 4b and S4†). Data points with missing values were only provided to the HGBR models, while those data points were removed for all other models. Consequently, the datasets provided to all algorithms except HGBR are ca. 95% of the size of the HGBR model's dataset (Fig. 4b), which we do not regard as being a significant difference when comparing the HGBR models to all others.
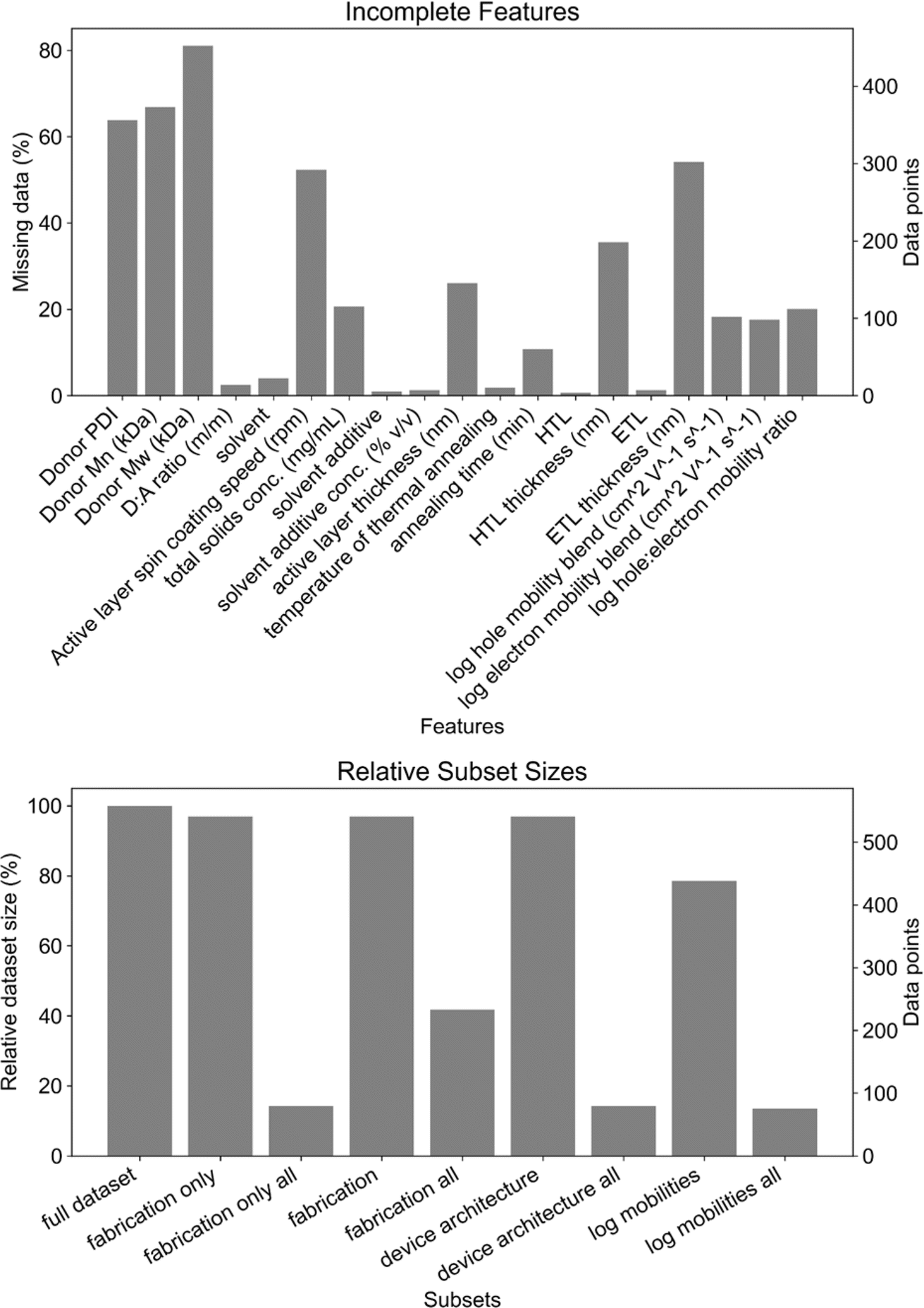 | ||
| Fig. 4 Top: percentage (left) and amount of data (right) missing from key categories. Bottom: relative (left) and absolute (right) dataset sizes of subsets of processing parameters. | ||
The correlation between each set of features and the output variables were evaluated using both Pearson's correlation coefficient (R) and Spearman's rank correlation coefficient (ρ). R is a measure of the extent to which two variables are linearly correlated, while ρ is a measure of rank correlation (i.e. the extent to which two variables are correlated through some monotonic function). Feature correlations (R and ρ) with PCE are relatively low (Fig. S5–S8†). Only some of the material property features show moderate correlation with PCE (|R| > 0.3 or |ρ| > 0.3): donor HOMO and LUMO, and all acceptor properties (HOMO, LUMO, HOMO–LUMO gap, and optical gap). These stronger correlations are expected from what is known about the function and design of OPV devices. Of the processing and device architecture features only active layer thickness and spin coating speed show weak correlation with PCE (|R| > 0.15 or |ρ| > 0.15). There is also only moderate correlation at best (|R| ≤ 0.28 and |ρ| ≤ 0.25) between the selection of solvent and solvent additive with PCE. There are two potential explanations for this. First, the identity and physical properties of solvents and solvent additives are known to strongly influence the performance of OPV devices through a number of factors (e.g., material solubility and rate of evaporation).4,5 Second, it is possible that there is also a degree of spurious correlation between solvent or solvent additive and PCE because higher-performing materials are more often dissolved in particular solvents.
Although the KRR and GPR models with ECFP structural representations, which used the Tanimoto kernel, performed well, we were not able to evaluate their performance when device fabrication features are included. Although mixing different kernels is possible, it is unclear whether it would be meaningful and how the weights would be learned.52,57 The Tanimoto kernel calculates distance based on all 4096 ECFP features while device fabrication feature distances would be calculated one at a time. As such, assigning correct weights would be non-trivial.
Instead, we evaluate top-performing algorithms with ECFP and Mordred representations (the best molecular representations from the previous step): SVR and the tree-based algorithms (RF, HGB, XGBoost, NGBoost). When combined with device fabrication parameters, Mordred descriptors slightly outperform ECFP for encoding molecular structure of the donor and acceptor materials on average (Fig. 5), however the difference is not statistically significant. For example, the performance of RF models increases from R2 = 0.58 ± 0.03 to R2 = 0.60 ± 0.02.
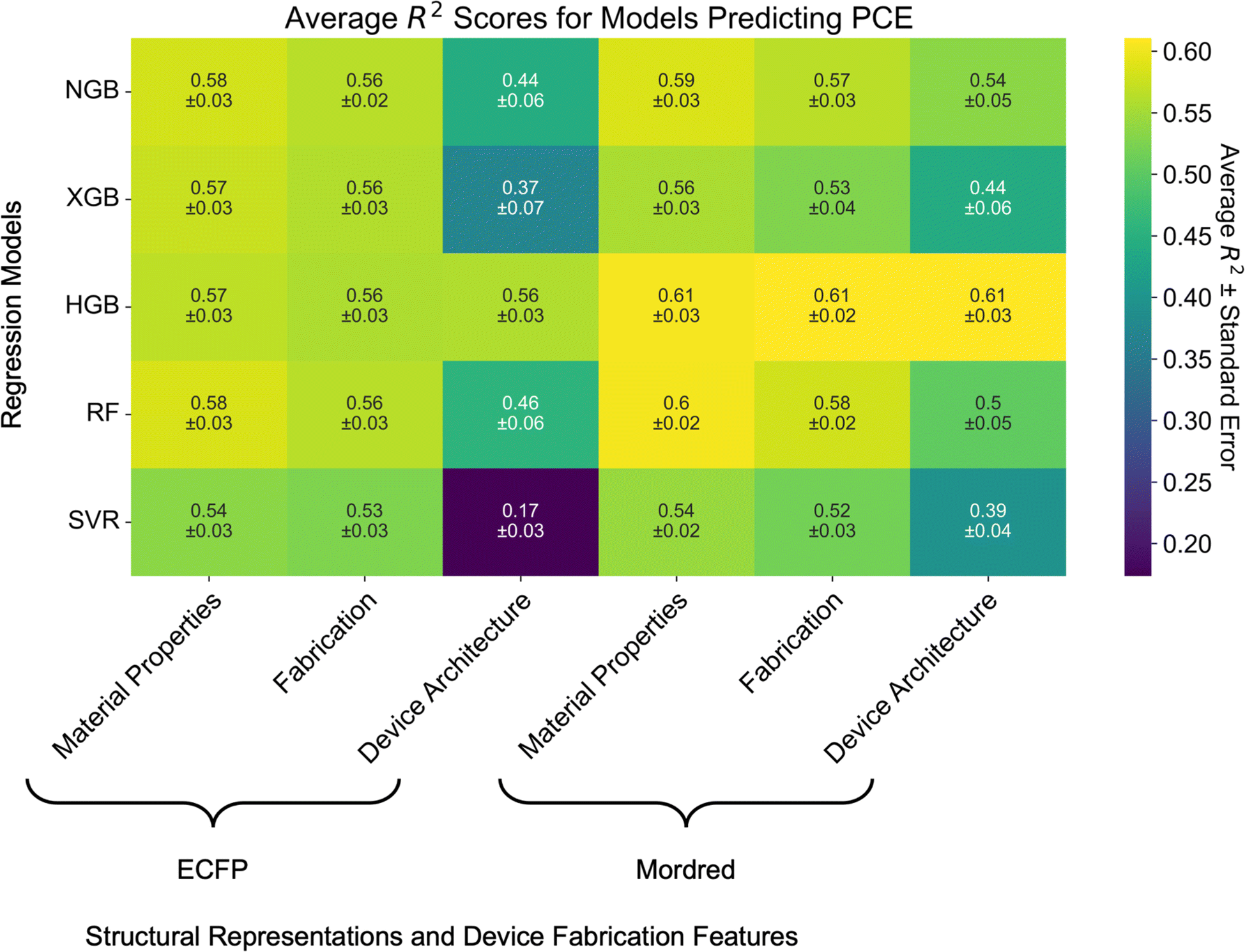 | ||
| Fig. 5 Heatmap of model performance as measured by the R2 score for predicting PCE from molecular structure and various subsets of OPV device fabrication parameters. Numbers in each cell correspond to the average R2 score of the model over seven independent five-fold cross-validations ± the standard error of the mean. Heatmaps for the RMSE and MAE scores are presented in Fig. S9.† | ||
Additional features do not significantly increase performance over the models simply trained on molecular structure. Performance of all tree-based models except HGB degrades by ca. 0.01–0.02 between models trained on molecular structure and material properties compared to models trained on molecular structure and the fabrication subset of features. This can be attributed to the fact that the material properties features do not contain missing data and are closely tied to molecular structure. That is not the case for the other features in the fabrication and device architecture subsets. Furthermore, adding the device architecture subset of features significantly decreases model performance for all but HGB across both ECFP and Mordred molecular descriptors. This may be related to the imbalanced selection of HTLs and ETLs in OPV device fabrication.
Empirically, device fabrication parameters are known to be important factors in OPV device performance. The lack of improvement in model performance when including these features suggests that it may be due to the quality of the training data. The top-performing model is HGB trained on Mordred descriptors and any of the device fabrication feature subsets (R2 = 0.61 ± 0.03). This may be due to the fact that HGB can handle missing data points, and therefore is capable of learning input feature–target relationships even from data points with missing features (e.g. total solids concentration and spin coating speed).
The top ML model architecture, HGB, using Mordred descriptors for molecular structure and trained on all available features achieves an R2 score of 0.61 ± 0.03. The model predictions are poor for data points where the true PCE is below 5%. Predictions in this range are as high as ca. 12%. This is to be expected because this range is under-represented in the dataset and therefore the model sees fewer examples on average. A similar phenomenon can be observed in the high-PCE tail of the distribution. In both cases, predictive errors tend toward the points with the highest density in the dataset (ca. 10% PCE). Additionally, there are no predictions near the maximum PCE (15.71%). The same phenomena are observed for models with different molecular representations and data (Fig. S10†).
Notably, a model trained on Mordred descriptors and the device architecture subset of features (Fig. 6) is not significantly better than the same model trained on only Mordred descriptors, which achieves a R2 score of 0.6 ± 0.03 (Fig. 3). This suggests that the models' performance is limited by the quality of the underlying data.
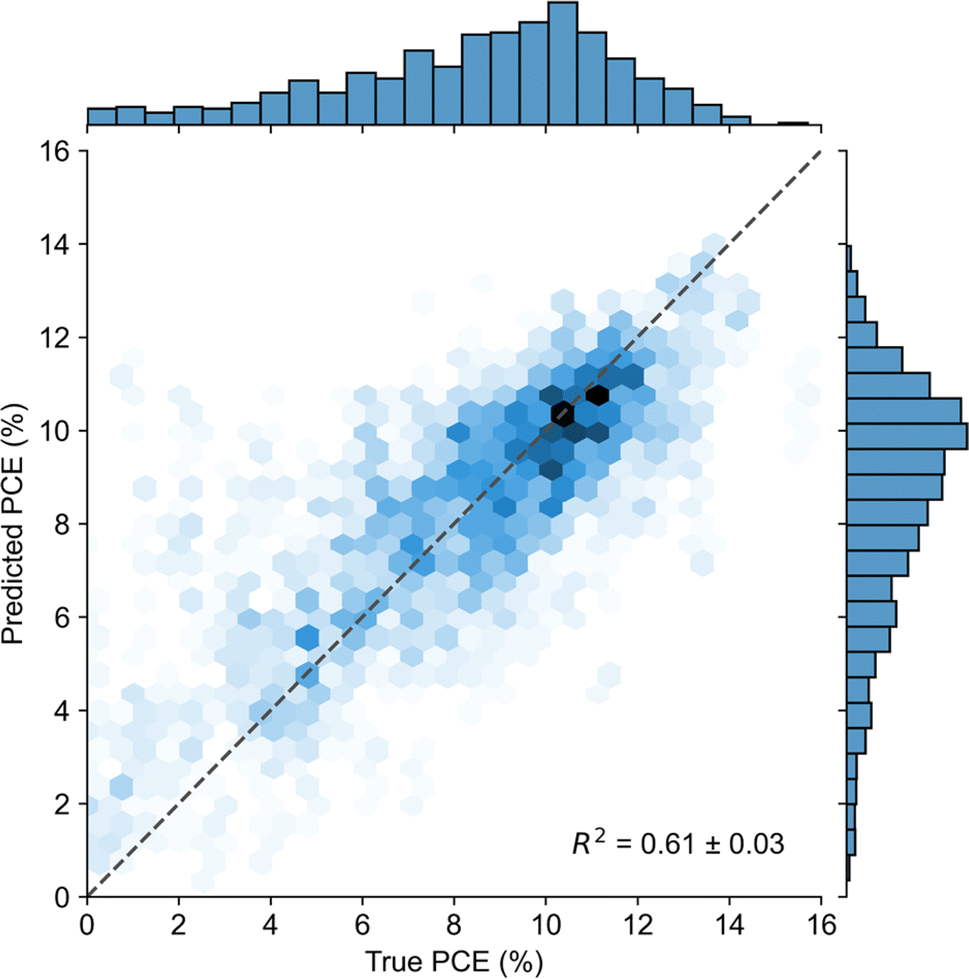 | ||
| Fig. 6 Scatter plot of predicted PCE from the top model (HGB trained on the device architecture subset of features). Data is aggregated over all five folds of each of the seven randomly seeded cross-validation splits. The opacity of the hex-binned cells corresponds to the count of predictions within each cell. The dashed line represents ideal predictions. | ||
Finally, we evaluated the performance of the HGB model when including features related to charge carrier mobility: log(hole mobility) and log(electron mobility) from space charge-limited current (SCLC) diode measurements, as well as the ratio of the two. The resulting model performance is significantly better (R2 = 0.68 ± 0.02, Fig. S11†) than anything else reported in the literature. However, including these values does not make a lot of sense from a practical perspective. In order to predict the PCE of a device, one would already need to have measured the hole and electron mobilities. This involves fabricating a device with a modified electrode architecture, and is therefore less efficient than simply measuring the device's performance experimentally.
2.5 Handling missing values in real-world data
Datasets gathered from historical or literature sources are often imperfect. One of the most common drawbacks is incomplete reporting of values, or missing values. Our datasets face a similar challenge in that most of the processing and device fabrication features are missing data. As seen in Fig. 4, the missing values are spread among a large percentage of the data points such that only 14% of datapoints79 have all of the features that were considered (i.e., excluding polymer molecular weights and charge carrier mobilities). While roughly half of the features are not missing many values (less than 4%), total solids concentration, spin coating speed, and thickness features (active layer, HTL, ETL) are missing significant amounts of data. The lack of active layer, HTL, and ETL thickness data is understandable given the difficulty of measuring thickness on the tens to hundreds of nanometers scale. However, not reporting total solids concentration (21% missing, 115 data points) and spin coating speed (52% missing, 292 data points) cannot be easily excused as these are fundamental and easily obtained experimental parameters when fabricating OPV devices.To address the issue of missing values, we can try to impute data. Imputing can help to fill in missing data based on various statistical treatments and assumptions about its distribution. We explored imputing data using some of the most common imputation techniques: mean, median, most-frequent, uniform- and distance-KNN, and iterative. The simplest imputing strategies are mean, median and most-frequent, which replace missing values with the mean, median and mode of the available data, respectively. Mean imputing is most useful when the standard deviation of the distribution is relatively small and data is missing at random. On the other hand, median imputing is more suitable for distributions where there are outliers because it is less sensitive to them. Finally, most-frequent (or mode) imputing is not suitable for imbalanced distributions, as is the case for some of the features in our dataset. In KNN-based imputing strategies, missing values are completed using the mean of the k (usually five) nearest samples in the dataset. They leverage the similarity between instances and are effective when data points with similar attributes tend to have similar outcomes. The distance between samples can be measured in two ways. Uniform KNN imputing assigns equal weights to all neighboring points, whereas distance-based KNN imputing weighs neighbors by the inverse of their distance. Therefore, closer points will have a greater influence than those that are more distant. Iterative imputation, also known as multiple imputation, is an advanced technique that involves an iterative process to fill in missing values. The process is usually based on modeling the relationships between features, and iteratively refining the imputations. Multiple imputations are combined to provide a more robust estimate of the missing values, considering the uncertainty associated with the imputations. The downside of iterative imputing is that it is computationally expensive and does not scale well.
While imputing data can be a valuable technique to enhance the quality and completeness of a dataset, it can also be unproductive (no change in model performance) or even counterproductive (degraded model performance). If the proportion of missing data is large (30–40%), imputing may lead to inaccurate results. It may also be counterproductive in instances where there is a pattern to which data is missing, or where missing data is associated with specific characteristics or factors because the ML model can potentially learn from such correlations. Finally, imputation may be challenging in datasets with intricate dependencies between variables, as is likely the case for OPV materials and devices. Ultimately, imputing data did not change model performance in any measurable way (Fig. S12†). This suggests that the models are limited not only by the quantity of data, but also by their quality.
2.6 Analyzing the dataset
:A combinations that are present in the dataset mostly occur once or twice. Only for pairs of the most common materials (e.g., PTB7-Th and ITIC) are there a significant number of duplicate reports.
Donor material energy levels (HOMO, LUMO) and energies (Eoptg, EHLg) do not fit cleanly within a distribution (Fig. S15†) primarily due to the low number of unique molecular structures (143), while the same features for acceptors form smoother distributions due to the larger number of unique molecular structures (261). In both cases, over-represented materials are clearly visible by the large counts in material property values. Additionally, it should be noted that measuring energy levels of materials is often imprecise, and that there are known issues with the most common technique, cyclic voltammetry (CV).39 Polymer properties are much less frequently reported. We were only able to find 202 values of Đ (36% of the dataset), 185 for Mn (33%), and 106 for Mw (19%). Few papers even mention polymer molecular weight or dispersity, and many that do simply refer to a previous paper where the value was initially reported. The molecular weights and dispersities of commercial materials are barely reported.
:A ratio is incredibly easy to vary, the most common values are 1 and 0.5 (i.e., 1:2 D:A). Similarly, only three solvents (CF, CB, o-DCB) and only two solvent additives (CN, DIO) make up the vast majority of entries. Consequently, relatively little of the solvent property space has been sampled (Fig. S16 and S17†).
Total solids concentration is most often 20 mg mL−1 even though there are reports of concentrations between 6 mg mL−1 and 37.5 mg mL−1 (Fig. S15 and Table S5†). Additionally, data points are concentrated around round numbers such as 10, 15, 20 and 25 mg mL−1. Unfortunately, 115 entries did not have any information about total solids concentration (or individual solution concentration from which we could calculate total solids concentration). This hole in the dataset is particularly egregious since it is one of the easiest numbers to report. The fact that 21% of the entries do not have this data suggests that we as a community of editors and reviewers are not paying close enough attention to the reproducibility of published data. The same can be applied to another parameter that can be recorded with ease: spin coating speed. The two most common values are 2000 rpm and 3000 rpm. However, only 48% of entries (266) reported spin coating speeds even though the entries in this dataset are restricted to those devices fabricated by spin coating. The same biases are also evident in the distributions of annealing temperature and time, although these data are reported more regularly. Finally, although the distribution of active layer thickness values is quite smooth and uniform, the values are centered at 100 nm, with the mode being exactly 100 nm. This is statistically unlikely, and is characteristic of either rounding from human bias, inaccurate measurements, or both.
3 Conclusions
We have curated a first-of-its-kind dataset of OPV device performance, molecular structures, and comprehensive device fabrication parameters, with 558 data points. Assembling the dataset proved to be a significant challenge due to the heterogeneity of data reporting practices in the literature, namely the lack of consistent reporting requirements for device fabrication details and a lack of consistent reporting formats. We then explored a variety of molecular structure encoding methods (from one-hot encoding to graph embeddings) and ML models (from MLR to GNN), and discussed the advantages and disadvantages of each choice.We found the HGB model, a gradient-boosted decision tree-based algorithm, to be best at predicting PCE from molecular structure, and from molecular structure and device fabrication features. Despite modest R2 scores of ca. 0.6, this reflects the state-of-the-art predictive performance for PCE. The same HGB algorithm outperformed models trained on a dataset almost twice the size (ca. 1000 data points) of ours,22 and matched the performance of models when trained on a dataset more than double the size of ours (ca. 1300 data points).29 The performance of the HGB algorithm may be in part attributed to its ability to extract maximal information by incorporating missing values – of which there are many – with its binning strategy.60
There are a number of ways in which model performance could be improved. Firstly, the “big p, small n” problem – where the number of variables or features (p) is much larger than the number of observations or data points (n) – might be overcome by simply adding additional data points. The quality of the dataset could also be improved by including more varied device fabrication procedures for each material combination. The current iteration of the dataset primarily includes only the top performing device from each donor:acceptor combination found in a paper. However, there are often others that may provide additional information despite lower PCE values. Furthermore, most reported PCE values correspond to the best single device, and not an average of multiple replicates, due to current reporting practices.
Our findings underscore the challenges stemming from mining literature data and inherent data quality issues. Although recent advances in literature mining with large language models are promising, the ultimate problem is one of data quality and not quantity.73–75 Standardizing data reporting in OPV-related publications becomes crucial. Establishing minimum reporting standards will streamline literature mining and enable the adoption of ML methodologies within the OPV domain. There has long been discussion in the literature about standards for reporting PCE measurements.76–80 While there are currently no standards or requirements for reporting how OPV devices are made, similar steps forward have recently been proposed for inorganic phosphors81 and perovskite photovoltaics.82
There is much that can be learned from the ongoing discussions within the organic chemistry literature by recognizing the similarities between predicting OPV device performance and predicting reaction yields in organic chemistry sheds light on shared complexities. Both domains face challenges involving two molecular structures as input features, processing variables, sparse data, non-smooth response surfaces, and biases in reported data, notably the underreporting of negative results.70,83–86 Acknowledging these parallels emphasizes the broader challenges in predictive modeling within chemistry-driven domains. Addressing these challenges necessitates enhanced dataset quality and size, and standardized data reporting.85,87,88
This comprehensive understanding highlights the pivotal role of data quality, underscores the importance of standardized reporting practices, and emphasizes the collective effort required to enhance dataset quality and modeling techniques, ultimately unlocking the full potential of ML in driving innovation within the OPV and broader chemistry domains.
4 Methods
All data, code, and figures pertaining to this work are publicly available in a GitHub repository: https://github.com/aspuru-guzik-group/Beyond-Molecular-Structure-ML-for-OPV-Materials-Devices.4.1 Encoding molecular structures
The molecular structures in the original dataset were provided as two large ChemDraw files.23 To automatically extract SMILES strings, the ChemDraw files were first converted to structure-sata files (.sdf), which could then be converted into a pandas dataframe by RDKit's Chem.PandasTools module. However, the labels corresponding to each structure could not be maintained during the conversion. As a result, structures were manually cross-referenced to the ChemDraw files and against the literature. Through this process, we found many identical structures with different labels, and different structures with the same labels.In the file, sidechains were shortened to numbered R groups (e.g. R1, R2), each of which corresponded to a different structure. Automatic R group replacement as implemented in RDKit's ReplaceSubstructs was not possible because SMILES and SMARTS cannot process the presence of different R groups. To automate the sidechain assignment process, we developed a method that uses placeholder metal atoms. Each R group was mapped to a placeholder metal atom and to the corresponding SMILES string. First, the R groups in the SMILES strings were replaced with the corresponding metal atoms. Then, the metal atoms were replaced with the full sidechain in the resulting RDKit Mol object using RDKit's ReplaceSubstructs function. Finally, the cleaned structure was returned as a canonicalized SMILES string.
4.2 Structure verification
When cleaning the data, we found errors in the structural assignments of the original ChemDraw files, as well as in our original conversion from ChemDraw to SMILES. One of the most common errors were misassigned sidechains. Given the dataset's size (558 data points with 143 unique donors and 261 unique acceptors), we implemented a semi-automated cleaning procedure. Structures with multiple unique labels were identified automatically. Tanimoto similarities were calculated for each pair of unique donor or acceptor labels. When the Tanimoto similarity was unity, we reviewed the structures manually. Most often, the same molecular structure was labeled with different names – a common issue in the literature. In those cases, we selected a principal label and replaced all occurrences with that label. Incorrect structures or incorrect sidechain assignments had to be verified manually because no suitable automated solution was identified. For this, we wrote a script to iterate through each unique SMILES, display the structure, check it against the structure in the associated paper (by DOI), and apply corrections if necessary.4.3 Gathering device data
Data are not reported consistently across the literature, whether that be format, completeness or location in the manuscript. When extracting data from papers, certain steps were taken as precautions or to maximize the amount of data extracted. Below we provide as thorough a description as possible of our procedure in order to record possible sources of error, and to guide readers.• Certain features were often simply reported as ranges (e.g., spin speed, thicknesses, etc.). In these instances, we entered the average of the minimum and maximum values.
• When LUMO energies were not directly reported, HOMO + Eoptg was accepted.
• EHLg was calculated from the difference between the fitted HOMO and LUMO energy values.
• HOMO/LUMO: identical donor and acceptor molecules had variable HOMO/LUMO levels from paper to paper. Thus, both previous papers from the same research group or widely accepted universal UPS measurements were utilized in place of the omitted/variable values. Furthermore, when values for Eoptg were not reported, values were extracted by extracting the absorption onset from figures using WebPlotDigitizer (https://automeris.io/WebPlotDigitizer/).89
• Information about polymer molecular weight was very challenging to obtain. Mw, Mn and Đ are not consistently reported. Often, we were required to follow a trail of references through a group's publications to find the original synthesis, and assumed that the molecular weight distribution of the polymer was the same in subsequent reports.
• If two of Mw, Mn and Đ were reported, the third value was calculated.
:A ratios were not always reported, but could often be calculated based on the reported donor and acceptor concentrations. Additionally, when papers reported data from multiple devices with different D:A ratios, they often reported a range of D:A ratios (e.g., 1:1.1 to 1:1.6). In this case, we took the center of the range of weight ratios (e.g., 1:1.35).
• When solvents were presented as a mixture, they were often reported as a percentage by volume (% v/v). From this, a ratio of the two solvents presented was calculated (e.g., 15%, v/v = 6.667:1).
• When recording reported solvent additives, some papers would include the additive, but not provide the additive percentage. In these cases, the solvent additive was included, while the concentration (%) was omitted, resulting in an incomplete data point.
• If a solvent system was reported as a ternary mixture (e.g., o-DCB:CB:DIO 1:1:0.3), the minority solvent was recorded as an additive and its concentration was calculated from the ratio.
• If active layer thickness for a particular device was not reported in a paper, we substituted the thickness of a similar device in the same paper if the total solids concentration and spin coating speed were the same. Otherwise, the data point was omitted.
• Reports of charge extraction by linearly increasing voltage (CELIV)90 measurements for the “faster carrier” were excluded as this did not specify whether hole or electron mobility were recorded.
• From each set of parameters of VOC, JSC, FF, we calculated the PCE with the formula PCE = VOC × JSC × FF. When comparing these with reported values within the dataset, we looked for errors > 1% of the PCE values. In these cases, we compared the values in the dataset and those in the relevant paper. We often found that values had simply been misreported. The most common errors were: (i) inconsistent reporting of FF as either a percentage or a fraction (i.e. a 100-fold discrepancy in FF), (ii) values reported for a different device, or (iii) reported PCE had rounding errors. Because of these discrepancies, we chose to use our calculated PCE instead of the reported PCE values.
4.4 Cleaning fabrication data
The weight ratio of donor to acceptor materials in OPV formulations is most commonly reported as “donor:acceptor”, e.g. 1:1. The ratios were recorded using this convention when gathering the data. To convert this feature into a scalar value, we calculated the ratio of donor to acceptor. For example, 1:1.5 would be encoded as 0.667.
To reduce the number of incomplete data points, we replaced annealing temperatures and times, as well as solvent additives with default values. Devices that were not annealed were assigned default annealing temperatures of 25 °C and times of 0 minutes. Formulations that did not use a solvent additive were assigned a solvent additive label of “_” and an additive concentration of 0%.
Common donor and acceptor materials were assigned many different energy level values in the dataset. However, our models have no way of accounting for that variability, which can arise from a number of possible sources.39 To standardize the reported energy levels, we fit Gaussian distributions to any material with two or more unique energy level values, and replaced them with the fitted mean. However, certain materials were reported to have the same exact energy level in multiple papers. Upon further investigation, we found that this is due to works simply reporting a value from another paper. In instances where the same energy level value occurred more than ten times for a given material, we only counted it once when performing the Gaussian fitting.
4.5 Pre-processing
Molecular encodings (one-hot, SELFIES, BRICS, ECFP) and descriptors (Mordred) were generated from canonical SMILES. One-hot encodings were generated using Scikit-Learn's OneHotEncoder class. SELFIES strings were generated using the SELFIES package's encoder.46 BRICS representations were generated using RDKit's Chem.BRICS module.91 ECFPs were generated separately for the donor and acceptor and concatenated together using RDKit's GetMorganFingerprintAsBitVect method.91 To be able to fully disambiguate molecules with identical conjugated cores only differing by sidechain length, we used ECFP10 fingerprints with 2048 bits (Table S6†). Mordred descriptors were generated using the Mordred Python package.41 All Mordred descriptors with null values or for which the variance in the dataset (donor and acceptor molecules) was zero were removed. Graph embeddings were generated by training a GNN to predict the HOMO, LUMO, and Eoptg for each molecule and then extracting the resulting global feature before the final prediction layer. These embeddings implicitly contain information about the structural and optoelectronic properties of the molecules, which are then appended together for the prediction task.SMILES, SELFIES, and BRICS are alphanumeric representations of variable length, incompatible with ML models. Therefore, they must be tokenized (i.e., converted into numeric representations) and “padded” so that all vectors are of the same size. Tokenizing creates a mapping between the unique symbols in a representation and integer values. The mapping is used as a reference for encoding each individual molecule. In order to achieve a consistent length across the dataset, the tokenized sequences were adjusted to the length of the longest sequence by post-padding with the appropriate amount of zeros.
When training exclusively on molecular structures represented by ECFP, the Tanimoto kernel was used for KRR and GP models.
As described in Fig. 4b and S4,† models trained on molecular structure and processing parameters were supplied with specific subsets of the features, and data points with any missing values were dropped. Data points with missing values were not removed from the training set of the HGB models. When descriptors were used for solvent additives, devices in which solvents additives weren't used were substituted with zeroes, except for the HGB models in which case they were left as missing values.
Certain models such as MLP are sensitive to the scale of feature values. To mitigate this, we applied min–max and/or standard scaling using Scikit-Learn's MinMaxScaler and StandardScaler. In the case of GPs, all features were scaled using the uniform QuantileTransformer. Scaling was also applied to the target values. Typically, target values were subjected to standard scaling, except in the case of MLP model architectures where min–max was also used. Crucially, the features were scaled based on the values in the training set. Scaling training and test sets together introduces data leakage into the model, which results in overestimation of model performance.54,55
4.6 ML model optimization and evaluation
The multioutput ANN was built using PyTorch consisting of 1 input, 1 embedding, 2 ortholinear layers and 1 output layer with 3 output nodes for each target variable. The GNN prediction model was built using Tensorflow and Sonnet, using the GraphNets architecture.48,51,92 For the graph embeddings, two separate networks were used for the donor and acceptor molecules, before the final embedding was appended to give a prediction, similar to work done in Greenman et al.93 The embedder GNNs, based on the ChemProp architecture,50,65 are trained to predict the HOMO, LUMO, and optical gap energies from the molecular graphs, and the embeddings are the global pooled features before the final prediction layer. The multi-output GNN model was trained using the mean squared loss averaged over the properties, until early stopping was reached at the minimal validation loss, with a 85/15 train/validation split. The GP model was built using GPyTorch, with the Tanimoto kernel for the bit-vectors (i.e. fingerprints, and one-hot encoding based features), and the radial basis function kernel for all other features.52,94Model performance was evaluated with 5-fold cross-validation.54 In addition, we used seven different random seeds for 5-fold splitting. In this way, we generated 35 different training and test sets.
Models that were deemed to be more sensitive to hyperparameters (KNN, MLP) were subjected to an inner loop of Bayesian hyperparameter optimization using the BayesSearchCV class of Scikit-Optimize before being trained on the full training set.95 The Bayesian cross-validation search performs 5-fold cross-validation on the training fold and Bayesian optimization to estimate the ideal model hyperparameters efficiently. The hyperparameters from the best model in the inner loop were used for the model trained on the full training set. Ranges for hyperparameter optimization were selected based on common values and expert intuition.
To assess overall model performance, we evaluated average R2, root-mean-square error (RMSE), mean absolute error (MAE), and Pearson correlation coefficient (R) over the 35 test sets. Variation in model performance was assessed based on the standard error over the 35 test sets with a sample size of 7 (the number of independent seeds). Predictions, scores and parity plots for all models can be found in the GitHub repository.
Author contributions
Conceptualization: M. S., N. S., B. L., T.-Q. N., A. A.-G. Data curation: M. S., S. L., D. G. C., M. L. L., K. L., R. S., H.-T. V., H. W., A. Y., Z. Z., N. S., A. P., B. L. Formal analysis: M. S., S. L., G. T. Funding acquisition: T.-Q. N., A. A.-G. Investigation: M. S., S. L., G. T. Methodology: M. S., S. L., G. T. Project administration: M. S. Resources: T.-Q. N., A. A.-G. Software: M. S., S. L., G. T. Supervision: M. S., T.-Q. N., A. A.-G. Validation: M. S., S. L., G. T. Visualization: M. S., S. L., G. T. Writing (original draft): M. S., S. L., D. G. C., G. T. Writing (review & editing): M. S., S. L., G. T., T.-Q. N., A. A.-G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Jie Min for sharing the data from ref. 23 with us. We thank Dr Kjell Jorner, Dr Robert Pollice, Cher-Tian Ser, and Dr Felix Strieth-Kalthoff for helpful discussions. ChatGPT (GPT-3.5) was used to aid in the writing of this manuscript. The authors acknowledge the Defense Advanced Research Projects Agency (DARPA) under the Accelerated Molecular Discovery Program under Cooperative Agreement No. HR00111920027 dated August 1, 2019. The content of the information presented in this work does not necessarily reflect the position or the policy of the Government. G. T. acknowledges support from the Natural Sciences and Engineering Research Council (NSERC) of Canada, and the Vector Institute for Artificial Intelligence. A. A.-G. thanks Anders G. Frøseth for his generous support. A. A.-G. also acknowledges funding by Natural Resources Canada and the Canada 150 Research Chairs program. D. C., K. L., R. S., A. Y., N. S., A. P., B. R. L., and T.-Q. N. thank the US Office of Naval Research (Award No. N00014-21-1-2181) for the support. H. W. acknowledges the support by the National Science Foundation through the Materials Research Science and Engineering Center (MRSEC) at UC Santa Barbara: NSF DMR-2308708 (IRG-1). Z. Z. thanks the support from the Air Force Office of Scientific Research (AFOSR), Grant #FA9550-19-1-0348.References
- P. Heremans, D. Cheyns and B. P. Rand, Strategies for Increasing the Efficiency of Heterojunction Organic Solar Cells: Material Selection and Device Architecture, Acc. Chem. Res., 2009, 42(11), 1740–1747, DOI:10.1021/ar9000923.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Bulk Heterojunction Solar Cells: Morphology and Performance Relationships, Chem. Rev., 2014, 114(14), 7006–7043, DOI:10.1021/cr400353v.
- F. Zhao, C. Wang and X. Zhan, Morphology Control in Organic Solar Cells, Adv. Energy Mater., 2018, 8(28), 1703147, DOI:10.1002/aenm.201703147.
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Solvent Additives: Key Morphology-Directing Agents for Solution-Processed Organic Solar Cells, Adv. Mater., 2018, 30(33), 1707114, DOI:10.1002/adma.201707114.
- L. J. Richter, D. M. DeLongchamp and A. Amassian, Morphology Development in Solution-Processed Functional Organic Blend Films: An In Situ Viewpoint, Chem. Rev., 2017, 117(9), 6332–6366, DOI:10.1021/acs.chemrev.6b00618.
- M. T. Sajjad, A. Ruseckas and I. D. W. Samuel, Enhancing Exciton Diffusion Length Provides New Opportunities for Organic Photovoltaics, Matter, 2020, 3(2), 341–354, DOI:10.1016/j.matt.2020.06.028.
- A. Karki, J. Vollbrecht, A. J. Gillett, S. S. Xiao, Y. Yang, Z. Peng, N. Schopp, A. L. Dixon, S. Yoon, M. Schrock, H. Ade, G. N. M. Reddy, R. H. Friend and T.-Q. Nguyen, The Role of Bulk and Interfacial Morphology in Charge Generation, Recombination, and Extraction in Non-Fullerene Acceptor Organic Solar Cells, Energy Environ. Sci., 2020, 13(10), 3679–3692, 10.1039/D0EE01896A.
- S. Park, T. Kim, S. Yoon, C. W. Koh, H. Y. Woo and H. J. Son, Progress in Materials, Solution Processes, and Long-Term Stability for Large-Area Organic Photovoltaics, Adv. Mater., 2020, 32(51), 2002217, DOI:10.1002/adma.202002217.
- X. Du, L. Lüer, T. Heumueller, J. Wagner, C. Berger, T. Osterrieder, J. Wortmann, S. Langner, U. Vongsaysy, M. Bertrand, N. Li, T. Stubhan, J. Hauch and C. J. Brabec, Elucidating the Full Potential of OPV Materials Utilizing a High-Throughput Robot-Based Platform and Machine Learning, Joule, 2021, 5(2), 495–506, DOI:10.1016/j.joule.2020.12.013.
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R. S. Sánchez-Carrera, A. Gold-Parker, L. Vogt, A. M. Brockway and A. Aspuru-Guzik, The Harvard Clean Energy Project: Large-Scale Computational Screening and Design of Organic Photovoltaics on the World Community Grid, J. Phys. Chem. Lett., 2011, 2(17), 2241–2251, DOI:10.1021/jz200866s.
- S. A. Lopez, B. Sanchez-Lengeling, J. de Goes Soares and A. Aspuru-Guzik, Design Principles and Top Non-Fullerene Acceptor Candidates for Organic Photovoltaics, Joule, 2017, 1(4), 857–870, DOI:10.1016/j.joule.2017.10.006.
- S. A. Lopez, E. O. Pyzer-Knapp, G. N. Simm, T. Lutzow, K. Li, L. R. Seress, J. Hachmann and A. Aspuru-Guzik, The Harvard Organic Photovoltaic Dataset, Sci. Data, 2016, 3(1), 160086, DOI:10.1038/sdata.2016.86.
- Z.-W. Zhao, M. del Cueto, Y. Geng and A. Troisi, Effect of Increasing the Descriptor Set on Machine Learning Prediction of Small Molecule-Based Organic Solar Cells, Chem. Mater., 2020, 32(18), 7777–7787, DOI:10.1021/acs.chemmater.0c02325.
- Z.-W. Zhao, M. d. Cueto and A. Troisi, Limitations of Machine Learning Models When Predicting Compounds with Completely New Chemistries: Possible Improvements Applied to the Discovery of New Non-Fullerene Acceptors, Digital Discovery, 2022, 1, 266, 10.1039/D2DD00004K.
- D. Padula, J. D. Simpson and A. Troisi, Combining Electronic and Structural Features in Machine Learning Models to Predict Organic Solar Cells Properties, Mater. Horiz., 2019, 6(2), 343–349, 10.1039/C8MH01135D.
- H. Sahu, W. Rao, A. Troisi and H. Ma, Toward Predicting Efficiency of Organic Solar Cells via Machine Learning and Improved Descriptors, Adv. Energy Mater., 2018, 8(24), 1801032, DOI:10.1002/aenm.201801032.
- Y. Wen, Y. Liu, B. Yan, T. Gaudin, J. Ma and H. Ma, Simultaneous Optimization of Donor/Acceptor Pairs and Device Specifications for Nonfullerene Organic Solar Cells Using a QSPR Model with Morphological Descriptors, J. Phys. Chem. Lett., 2021, 12(20), 4980–4986, DOI:10.1021/acs.jpclett.1c01099.
- H. Sahu and H. Ma, Unraveling Correlations between Molecular Properties and Device Parameters of Organic Solar Cells Using Machine Learning, J. Phys. Chem. Lett., 2019, 10(22), 7277–7284, DOI:10.1021/acs.jpclett.9b02772.
- H. Sahu, F. Yang, X. Ye, J. Ma, W. Fang and H. Ma, Designing Promising Molecules for Organic Solar Cells via Machine Learning Assisted Virtual Screening, J. Mater. Chem. A, 2019, 7(29), 17480–17488, 10.1039/C9TA04097H.
- B. L. Greenstein, D. C. Hiener and G. R. Hutchison, Computational Evolution of High-Performing Unfused Non-Fullerene Acceptors for Organic Solar Cells, J. Chem. Phys., 2022, 156(17), 174107, DOI:10.1063/5.0087299.
- B. L. Greenstein and G. R. Hutchison, Organic Photovoltaic Efficiency Predictor: Data-Driven Models for Non-Fullerene Acceptor Organic Solar Cells, J. Phys. Chem. Lett., 2022, 4235–4243, DOI:10.1021/acs.jpclett.2c00866.
- B. L. Greenstein and G. R. Hutchison, Screening Efficient Tandem Organic Solar Cells with Machine Learning and Genetic Algorithms, J. Phys. Chem. C, 2023, 127(13), 6179–6191, DOI:10.1021/acs.jpcc.3c00267.
- Y. Wu, J. Guo, R. Sun and J. Min, Machine Learning for Accelerating the Discovery of High-Performance Donor/Acceptor Pairs in Non-Fullerene Organic Solar Cells, npj Comput. Mater., 2020, 6(1), 1–8, DOI:10.1038/s41524-020-00388-2.
- S. Nagasawa, E. Al-Naamani and A. Saeki, Computer-Aided Screening of Conjugated Polymers for Organic Solar Cell: Classification by Random Forest, J. Phys. Chem. Lett., 2018, 9(10), 2639–2646, DOI:10.1021/acs.jpclett.8b00635.
- Y.-C. Lin, Y.-J. Lu, C.-S. Tsao, A. Saeki, J.-X. Li, C.-H. Chen, H.-C. Wang, H.-C. Chen, D. Meng, K.-H. Wu, Y. Yang and K.-H. Wei, Enhancing Photovoltaic Performance by Tuning the Domain Sizes of a Small-Molecule Acceptor by Side-Chain-Engineered Polymer Donors, J. Mater. Chem. A, 2019, 7(7), 3072–3082, 10.1039/C8TA11059J.
- A. Saeki and K. Kranthiraja, A High Throughput Molecular Screening for Organic Electronics via Machine Learning: Present Status and Perspective, Jpn. J. Appl. Phys., 2019, 59(SD), SD0801, DOI:10.7567/1347-4065/ab4f39.
- Y. Huang, J. Zhang, E. S. Jiang, Y. Oya, A. Saeki, G. Kikugawa, T. Okabe and F. S. Ohuchi, Structure–Property Correlation Study for Organic Photovoltaic Polymer Materials Using Data Science Approach, J. Phys. Chem. C, 2020, 124(24), 12871, DOI:10.1021/acs.jpcc.0c00517.
- K. Kranthiraja and A. Saeki, Experiment-Oriented Machine Learning of Polymer:Non-Fullerene Organic Solar Cells, Adv. Funct. Mater., 2021, 2011168, DOI:10.1002/adfm.202011168.
- Y. Miyake and A. Saeki, Machine Learning-Assisted Development of Organic Solar Cell Materials: Issues, Analyses, and Outlooks, J. Phys. Chem. Lett., 2021, 12(51), 12391–12401, DOI:10.1021/acs.jpclett.1c03526.
- Y. Miyake, K. Kranthiraja, F. Ishiwari and A. Saeki, Improved Predictions of Organic Photovoltaic Performance through Machine Learning Models Empowered by Artificially Generated Failure Data, Chem. Mater., 2022, 34(15), 6912, DOI:10.1021/acs.chemmater.2c01294.
- M. Aldeghi and C. W. Coley, A Graph Representation of Molecular Ensembles for Polymer Property Prediction, Chem. Sci., 2022, 13, 10486, 10.1039/D2SC02839E.
- T. B. Martin and D. J. Audus, Emerging Trends in Machine Learning: A Polymer Perspective, ACS Polym. Au, 2023, 3(3), 239–258, DOI:10.1021/acspolymersau.2c00053.
- L. Chen, G. Pilania, R. Batra, T. D. Huan, C. Kim, C. Kuenneth and R. Ramprasad, Polymer Informatics: Current Status and Critical next Steps, Mater. Sci. Eng., R, 2021, 144, 100595, DOI:10.1016/j.mser.2020.100595.
- S. Lo, M. Seifrid, T. Gaudin and A. Aspuru-Guzik, Augmenting Polymer Datasets by Iterative Rearrangement, J. Chem. Inf. Model., 2023, 63(14), 4266–4276, DOI:10.1021/acs.jcim.3c00144.
- J. Lee, S.-J. Ko, M. Seifrid, H. Lee, C. McDowell, B. R. Luginbuhl, A. Karki, K. Cho, T.-Q. Nguyen and G. C. Bazan, Design of Nonfullerene Acceptors with Near-Infrared Light Absorption Capabilities, Adv. Energy Mater., 2018, 8(26), 1801209, DOI:10.1002/aenm.201801209.
- H. Lai, H. Chen, J. Zhou, J. Qu, P. Chao, T. Liu, X. Chang, N. Zheng, Z. Xie and F. He, Isomer-Free: Precise Positioning of Chlorine-Induced Interpenetrating Charge Transfer for Elevated Solar Conversion, iScience, 2019, 17, 302–314, DOI:10.1016/j.isci.2019.06.033.
- J. Qu, D. Li, H. Wang, J. Zhou, N. Zheng, H. Lai, T. Liu, Z. Xie and F. He, Bromination of the Small-Molecule Acceptor with Fixed Position for High-Performance Solar Cells, Chem. Mater., 2019, 31(19), 8044–8051, DOI:10.1021/acs.chemmater.9b02501.
- H. Wang, H. Chen, W. Xie, H. Lai, T. Zhao, Y. Zhu, L. Chen, C. Ke, N. Zheng and F. He, Configurational Isomers Induced Significant Difference in All-Polymer Solar Cells, Adv. Funct. Mater., 2021, 31(26), 2100877, DOI:10.1002/adfm.202100877.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, The Energy Level Conundrum of Organic Semiconductors in Solar Cells, Adv. Mater., 2022, 2202575, DOI:10.1002/adma.202202575.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Renewed Prospects for Organic Photovoltaics, Chem. Rev., 2022, 122(18), 14180, DOI:10.1021/acs.chemrev.1c00955.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, Mordred: A Molecular Descriptor Calculator, J. Cheminf., 2018, 10(1), 4, DOI:10.1186/s13321-018-0258-y.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754, DOI:10.1021/ci100050t.
- J. Degen, C. Wegscheid-Gerlach, A. Zaliani and M. Rarey, On the Art of Compiling and Using “Drug-Like” Chemical Fragment Spaces, ChemMedChem, 2008, 3(10), 1503–1507, DOI:10.1002/cmdc.200800178.
- X. Zhang, G. Wei, Y. Sheng, W. Bai, J. Yang, W. Zhang and C. Ye, Polymer-Unit Fingerprint (PUFp): An Accessible Expression of Polymer Organic Semiconductors for Machine Learning, ACS Appl. Mater. Interfaces, 2023, 15(17), 21537–21548, DOI:10.1021/acsami.3c03298.
- D. Weininger, SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Self-Referencing Embedded Strings (SELFIES): A 100% Robust Molecular String Representation, Mach. Learn.: Sci. Technol., 2020, 1(4), 045024, DOI:10.1088/2632-2153/aba947.
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans, T. Sommer and P. Friederich, Graph Neural Networks for Materials Science and Chemistry, Commun. Mater., 2022, 3(1), 1–18, DOI:10.1038/s43246-022-00315-6.
- B. Sanchez-Lengeling, J. N. Wei, B. K. Lee, R. C. Gerkin, A. Aspuru-Guzik and A. B. Wiltschko, Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules, arXiv, 2019, preprint, arXiv:191010685, DOI:10.48550/arXiv.1910.10685.
- L. Rampášek, M. Galkin, V. P. Dwivedi, A. T. Luu, G. Wolf and D. Beaini, Recipe for a General, Powerful, Scalable Graph Transformer, arXiv, 2023, DOI:10.48550/arXiv.2205.12454.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing Learned Molecular Representations for Property Prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388, DOI:10.1021/acs.jcim.9b00237.
- B. Sanchez-Lengeling, E. Reif, A. Pearce and A. B. Wiltschko, A Gentle Introduction to Graph Neural Networks, Distill, 2021, 6(9), e33, DOI:10.23915/distill.00033.
- G. J. Tom, R. Hickman, A. Zinzuwadia, A. Mohajeri, B. Sanchez-Lengeling and A. Aspuru-Guzik, Calibration and Generalizability of Probabilistic Models on Low-Data Chemical Datasets with DIONYSUS, Digital Discovery, 2023, 2(3), 759–774, 10.1039/D2DD00146B.
- D. Duvenaud, D. Maclaurin, J. Aguilera-Iparraguirre, R. Gomez-Bombarelli, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Convolutional Networks on Graphs for Learning Molecular Fingerprints, arXiv, 2015, DOI:10.48550/arXiv.1509.09292.
- A. Y.-T. Wang, R. J. Murdock, S. K. Kauwe, A. O. Oliynyk, A. Gurlo, J. Brgoch, K. A. Persson and T. D. Sparks, Machine Learning for Materials Scientists: An Introductory Guide toward Best Practices, Chem. Mater., 2020, 32(12), 4954–4965, DOI:10.1021/acs.chemmater.0c01907.
- S. Kapoor and A. Narayanan, Leakage and the Reproducibility Crisis in Machine-Learning-Based Science, Patterns, 2023, 4(9), 100804, DOI:10.1016/j.patter.2023.100804.
- N. Bhatia and Vandana, Survey of Nearest Neighbor Techniques, arXiv, 2010, preprint, arXiv:1007.0085, DOI:10.48550/arXiv.1007.0085.
- R.-R. Griffiths, L. Klarner, H. B. Moss, A. Ravuri, S. Truong, S. Stanton, G. Tom, B. Rankovic, Y. Du, A. Jamasb, A. Deshwal, J. Schwartz, A. Tripp, G. Kell, S. Frieder, A. Bourached, A. Chan, J. Moss, C. Guo, J. Durholt, S. Chaurasia, F. Strieth-Kalthoff, A. A. Lee, B. Cheng, A. Aspuru-Guzik, P. Schwaller and J. Tang, GAUCHE: A Library for Gaussian Processes in Chemistry, arXiv, 2023, preprint, arXiv:2212.04450, DOI:10.48550/arXiv.2212.04450.
- T. Chen and C. Guestrin, XGBoost: A Scalable Tree Boosting System, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794, DOI:10.1145/2939672.2939785.
- T. Duan, A. Avati, D. Y. Ding, K. K. Thai, S. Basu, A. Y. Ng and A. Schuler, NGBoost: Natural Gradient Boosting for Probabilistic Prediction, arXiv, 2020, preprint, arXiv:1910.03225, DOI:10.48550/arXiv.1910.03225.
- G. Ke, Q. Meng, T. Finley, T. Wang, W. Chen, W. Ma, Q. Ye and T.-Y. Liu, LightGBM: A Highly Efficient Gradient Boosting Decision Tree, in Advances in Neural Information Processing Systems, Curran Associates, Inc., 2017, vol. 30 Search PubMed.
- Y. Shi, G. Ke, Z. Chen, S. Zheng and T.-Y. Liu, Quantized Training of Gradient Boosting Decision Trees, in Advances in Neural Information Processing Systems, Curran Associates, Inc., 2022, vol. 35, pp. 18822–18833 Search PubMed.
- L. Grinsztajn, E. Oyallon and G. Varoquaux, Why Do Tree-Based Models Still Outperform Deep Learning on Typical Tabular Data?, arXiv, 2022, preprint, arXiv:2207.08815, DOI:10.48550/arXiv.2207.08815.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Neural Message Passing for Quantum Chemistry, arXiv, 2017, preprint, arXiv:1704.01212, DOI:10.48550/arXiv.1704.01212.
- B. K. Lee, E. J. Mayhew, B. Sanchez-Lengeling, J. N. Wei, W. W. Qian, K. A. Little, M. Andres, B. B. Nguyen, T. Moloy, J. Yasonik, J. K. Parker, R. C. Gerkin, J. D. Mainland and A. B. Wiltschko, A Principal Odor Map Unifies Diverse Tasks in Olfactory Perception, Science, 2023, 381(6661), 999–1006, DOI:10.1126/science.ade4401.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, Chemprop: A Machine Learning Package for Chemical Property Prediction, J. Chem. Inf. Model., 2024, 64(1), 9–17, DOI:10.1021/acs.jcim.3c01250.
- L. Alzubaidi, J. Zhang, A. J. Humaidi, A. Al-Dujaili, Y. Duan, O. Al-Shamma, J. Santamaría, M. A. Fadhel, M. Al-Amidie and L. Farhan, Review of Deep Learning: Concepts, CNN Architectures, Challenges, Applications, Future Directions, J. Big Data, 2021, 8(1), 53, DOI:10.1186/s40537-021-00444-8.
- E. J. Bjerrum, SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules, arXiv, 2017, preprint, arXiv:1703.07076, DOI:10.48550/arXiv.1703.07076.
- N. K. Elumalai and A. Uddin, Open Circuit Voltage of Organic Solar Cells: An in-Depth Review, Energy Environ. Sci., 2016, 9(2), 391–410, 10.1039/C5EE02871J.
- HSPiP Datasets|Hansen Solubility Parameters, https://www.hansen-solubility.com/HSPiP/datasets.php, accessed, 2023-09-12 Search PubMed.
- W. Beker, R. Roszak, A. Wołos, N. H. Angello, V. Rathore, M. D. Burke and B. A. Grzybowski, Machine Learning May Sometimes Simply Capture Literature Popularity Trends: A Case Study of Heterocyclic Suzuki–Miyaura Coupling, J. Am. Chem. Soc., 2022, 144(11), 4819–4827, DOI:10.1021/jacs.1c12005.
- D. Pope and U. Simonsohn, Round Numbers as Goals: Evidence From Baseball, SAT Takers, and the Lab, Psychol. Sci., 2011, 22(1), 71–79, DOI:10.1177/0956797610391098.
- M. Backus, T. Blake and S. Tadelis, Cheap Talk, Round Numbers, and the Economics of Negotiation, National Bureau of Economic Research, 2015, DOI:10.3386/w21285.
- M. P. Polak and D. Morgan, Extracting Accurate Materials Data from Research Papers with Conversational Language Models and Prompt Engineering – Example of ChatGPT, arXiv, 2023, preprint, arXiv:2303.05352, DOI:10.48550/arXiv.2303.05352.
- M. P. Polak, S. Modi, A. Latosinska, J. Zhang, C.-W. Wang, S. Wang, A. D. Hazra and D. Morgan, Flexible, Model-Agnostic Method for Materials Data Extraction from Text Using General Purpose Language Models, arXiv, 2023, preprint, arXiv:2302.04914, DOI:10.48550/arXiv.2302.04914.
- M. Ansari and S. M. Moosavi, Agent-Based Learning of Materials Datasets from Scientific Literature, arXiv, 2023, preprint, arXiv:2312.11690, DOI:10.48550/arXiv.2312.11690.
- V. Shrotriya, G. Li, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Accurate Measurement and Characterization of Organic Solar Cells, Adv. Funct. Mater., 2006, 16(15), 2016–2023, DOI:10.1002/adfm.200600489.
- H. J. Snaith, The Perils of Solar Cell Efficiency Measurements, Nat. Photonics, 2012, 6(6), 337–340, DOI:10.1038/nphoton.2012.119.
- E. J. Luber and J. M. Buriak, Reporting Performance in Organic Photovoltaic Devices, ACS Nano, 2013, 7(6), 4708–4714, DOI:10.1021/nn402883g.
- E. Zimmermann, P. Ehrenreich, T. Pfadler, J. A. Dorman, J. Weickert and L. Schmidt-Mende, Erroneous Efficiency Reports Harm Organic Solar Cell Research, Nat. Photonics, 2014, 8(9), 669–672, DOI:10.1038/nphoton.2014.210.
- A Checklist for Photovoltaic Research, Nat. Mater., 2015, 14(11), 1073, DOI:10.1038/nmat4473.
- R.-S. Liu, Advancing Reporting Guidelines for Optimal Characterization of Inorganic Phosphors, Chem. Mater., 2023, 35(16), 6179–6183, DOI:10.1021/acs.chemmater.3c01743.
- K. P. Goetz and Y. Vaynzof, The Challenge of Making the Same Device Twice in Perovskite Photovoltaics, ACS Energy Lett., 2022, 7(5), 1750–1757, DOI:10.1021/acsenergylett.2c00463.
- F. Strieth-Kalthoff, F. Sandfort, M. Kühnemund, F. R. Schäfer, H. Kuchen and F. Glorius, Machine Learning for Chemical Reactivity: The Importance of Failed Experiments, Angew. Chem., Int. Ed., 2022, 61(29), e202204647, DOI:10.1002/anie.202204647.
- O. Wiest, M. Saebi, B. Nan, J. E. Herr, J. Wahlers, Z. Guo, A. Zuranski, T. Kogej, P.-O. Norrby, A. G. Doyle and N. V. Chawla, On the Use of Real-World Datasets for Reaction Yield Prediction, Chem. Sci., 2023, 14, 4997, 10.1039/D2SC06041H.
- R. Mercado, S. M. Kearnes and C. W. Coley, Data Sharing in Chemistry: Lessons Learned and a Case for Mandating Structured Reaction Data, J. Chem. Inf. Model., 2023, 63(14), 4253–4265, DOI:10.1021/acs.jcim.3c00607.
- P. Raghavan, B. C. Haas, M. E. Ruos, J. Schleinitz, A. G. Doyle, S. E. Reisman, M. S. Sigman and C. W. Coley, Dataset Design for Building Models of Chemical Reactivity, ACS Cent. Sci., 2023, 9(12), 2196–2204, DOI:10.1021/acscentsci.3c01163.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, The Open Reaction Database, J. Am. Chem. Soc., 2021, 143(45), 18820–18826, DOI:10.1021/jacs.1c09820.
- J. R. Kitchin, Examples of Effective Data Sharing in Scientific Publishing, ACS Catal., 2015, 5(6), 3894–3899, DOI:10.1021/acscatal.5b00538.
- A. Rohatgi, Webplotdigitizer: Version 4.6, 2022, https://automeris.io/WebPlotDigitizer Search PubMed.
- O. J. Sandberg and M. Nyman, Charge Extraction by a Linearly Increasing Voltage of Photo-Generated Carriers: The Influence of Two Mobile Carrier Types, Bimolecular Recombination, and Series Resistance, Org. Electron., 2019, 64, 97–103, DOI:10.1016/j.orgel.2018.10.017.
- G. Landrum, P. Tosco, B. Kelley, Ric, Sriniker, Gedeck, R. Vianello, D. Cosgrove, N. Schneider, E. Kawashima, D. N, A. Dalke, G. Jones, B. Cole, M. Swain, S. Turk, A. Savelyev, A. Vaucher, M. Wójcikowski, I. Take, D. Probst, V. F. Scalfani, K. Ujihara, G. Godin, A. Pahl, F. Berenger, J. JLVarjo, Jasondbiggs, strets123 and JP, Rdkit/Rdkit: 2022_09_4 (Q3 2022) Release, 2023, DOI:10.5281/zenodo.7541264.
- P. W. Battaglia, J. B. Hamrick, V. Bapst, A. Sanchez-Gonzalez, V. Zambaldi, M. Malinowski, A. Tacchetti, D. Raposo, A. Santoro, R. Faulkner, C. Gulcehre, F. Song, A. Ballard, J. Gilmer, G. Dahl, A. Vaswani, K. Allen, C. Nash, V. Langston, C. Dyer, N. Heess, D. Wierstra, P. Kohli, M. Botvinick, O. Vinyals, Y. Li and R. Pascanu, Relational Inductive Biases, Deep Learning, and Graph Networks, arXiv, 2018, preprint, arXiv:1806.01261, DOI:10.48550/arXiv.1806.01261.
- K. P. Greenman, W. H. Green and R. Gómez-Bombarelli, Multi-Fidelity Prediction of Molecular Optical Peaks with Deep Learning, Chem. Sci., 2022, 13(4), 1152–1162, 10.1039/D1SC05677H.
- R.-R. Griffiths, L. Klarner, H. B. Moss, A. Ravuri, S. Truong, S. Stanton, G. Tom, B. Rankovic, Y. Du, A. Jamasb, A. Deshwal, J. Schwartz, A. Tripp, G. Kell, S. Frieder, A. Bourached, A. Chan, J. Moss, C. Guo, J. Durholt, S. Chaurasia, F. Strieth-Kalthoff, A. A. Lee, B. Cheng, A. Aspuru-Guzik, P. Schwaller and J. Tang, GAUCHE: A Library for Gaussian Processes in Chemistry, arXiv, 2023, preprint, arXiv:2212.04450, DOI:10.48550/arXiv.2212.04450.
- T. Head, M. Kumar, H. Nahrstaedt, G. Louppe and I. Shcherbatyi, Scikit-Optimize/Scikit-Optimize, 2021, DOI:10.5281/zenodo.5565057.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01942c |
| ‡ Authors contributed equally. |
| § Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |