
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid nanoreactors formed by interpolyelectrolyte complex formation: a colloidal platform for light-driven catalysis†
Afshin
Nabiyan
*abc,
Christof
Neumann
 d,
Andrey
Turchanin
bcd and
Felix H.
Schacher
*abce
d,
Andrey
Turchanin
bcd and
Felix H.
Schacher
*abce
aInstitute of Organic Chemistry and Macromolecular Chemistry (IOMC), Friedrich-Schiller University Jena, Lessingstraße 8, D-07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, D-07743 Jena, Germany
cCenter for Energy and Environmental Chemistry Jena (CEEC Jena), Philosophenweg 7a, 07743 Jena, Germany
dInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Lessingstraße 10, 07743 Jena, Germany
eHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstraße 12-14, 07743 Jena, Germany
First published on 1st May 2024
Abstract
Multicompartment micelles can combine several chemical functionalities in close proximity and are therefore promising candidates for a new class of nanomaterials. In this study, we report a photocatalytically active multi-compartment micellar nanoreactor for visible light driven hydrogen evolution. The micellar nanoreactors are formed based on interpolyelectrolyte complexes (IPECs) of polydehydroalanine-based (PDha) graft copolymers with varying charge. Effects of the 3D confinement and the ratio of positive to negative charges, Z(±) of the graft copolymer micellar IPECs were also investigated in view of the resulting hydrogen evolution activity of our model system using Eosin Y and platinum nanoparticles as catalytically active components. We show that within a certain Z(±) regime stable micellar IPECs are formed and the hydrogen evolution activity can be tuned upon mixing the interpolyelectrolyte complexes with certain Z ratios (Z(±)).
Introduction
Over the last few years, energy and environmental concerns regarding our planet have prompted many scientists to develop new routes to green and efficient energy storage and conversion systems.1 In this way, self-assembly processes of macromolecules and nanostructures provide new opportunities for designing novel and complex materials and soft matter matrices which can serve for energy transformation.2–4 Micelles as one type of supramolecules typically form by self-assembly processes of macromolecules in selective solvents, often in aqueous media. It has been shown that confinement within a micellar nano-environment can have drastic effects on a given chemical reaction, including inhibition or acceleration.5–9Even more, polymer and block copolymer self-assembly has paved the way for the development of functional materials with wide-ranging use such as in energy storage/conversion,10–12 membranes,13 bio-applications,14 and lubrication.15 Amongst the properties that determine the outcome of polymer self-assembly, the architecture of individual building blocks plays a critical role and may be used to direct aggregation pathways. Here, one particularly interesting example are graft copolymers, macromolecules featuring a polymeric backbone and grafts of varying density and chemical functionality.16–18 Such materials feature unique rheological characteristics, partially due to high entanglement molecular weights and extended backbone conformations due to the steric repulsion of the side chains upon reaching high grafting densities.19,20 Graft copolymers are also interesting building blocks in self-assembly due to the intrinsic immiscibility of backbone and side chain grafts.21,22 Understanding and controlling such self-assembly processes allows for the preparation of functional materials for various kinds of applications.17,22
Many efforts have been made in the past few years to obtain well-defined micellar aggregates and, alongside, polymeric nanoparticles from graft copolymer systems.17,21–23 Typical morphologies for micelles from graft copolymers in solution are cylinders,17,24 spheres,17,25 and vesicles,17,26,27 but also more complex structures like multicompartment micelles have been reported.17,28,29 Complex compartmentalized nanostructures are promising candidates for the generation of inorganic–organic hybrid structures.30–32 Multicompartment micelles have also shown promising results in catalysis and nanotechnology because of the discrete subdomains within either their core or corona.33
In general, there are different approaches for the preparation of multicompartment micelles in solution.29,33–35 The most common way is to use a selective solvent to induce self-assembly,29 where the driving force for structural evolution originates from the mutual incompatibility of the unlike segments.29,34,36,37 Another facile and straightforward method to induce co-assembly of block copolymers is the formation of interpolyelectrolyte complexes (IPECs) through electrostatic interactions between two oppositely charged segments, either inter- or intramolecular.31,32,37,38 Furthermore, these interpolyelectrolyte complexes are capable of undergoing dynamic polyion exchange reactions, rendering dynamic colloidal objects which are responsive to changes in pH or salinity.31,32,37–39
IPEC formation represents a facile and straightforward route to direct co-assembly processes between oppositely charged macromolecules. A large variety of parameters such as the charge-to-charge stoichiometry (Z±), the solution conditions, or the molecular weight of the involved building blocks can control these co-assembly processes.32 Moreover, interpolyelectrolyte complexation can provide us unique opportunities to precisely regulate both the properties and the morphology of the resulting colloidal structures.32 In addition, during the last few years, this area for IPECs has been further extended.33,40,41 Many research groups developed either double hydrophilic or amphiphilic diblock copolymers comprising ionic and non-ionic segments, polyampholytic triblock terpolymers, branched polyelectrolytes (polyelectrolyte stars and cylindrical polyelectrolyte brushes), or ionic dendrimers to achieve IPEC micelles of varying architecture.22,42–44 Variations of pH or ionic strength of the surrounding microenvironment within IPECs provide further means for structure regulation of the introduction of dynamics.28,29,42,44–47 Such materials become interesting for applications related to sensing, structuring, or the controlled uptake and release of guest substances, especially in the case of biologically active examples.41–43
Here, we introduce a method for the direct co-assembly of graft copolymers in aqueous solutions to create photo-catalytically active micellar IPECs. For this, we synthesized polyampholytic polydehydroalanine-based (PDha) graft copolymers with different charged grafts. This allows the formation of micellar IPECs in aqueous solution and, additionally, can be used to in situ form catalytically active Pt nanoparticles. Subsequently, the resulting micellar IPECs were loaded with a suitable dye as photosensitizer (PS) and the resulting micellar hybrid system was assessed for visible-light-driven hydrogen production. Our findings reveal that the ratio of positive to negative charges, Z(±), in micellar IPECs, can modulate hydrogen evolution activity. Furthermore, an optimized Z(±) ratio and spatial confinement within micellar IPECs further seem to contribute to enhanced photocatalytic activity.
Results and discussion
Polyampholytic polydehydroalanine (PDha) is a polymer carrying a high density of functional (charged) groups, specifically –NH2 and –COOH, in every repeat unit (see Fig. 1). We have recently demonstrated that PDha is a versatile polyampholyte accessible through either free or controlled radical polymerization.11,48,49 We also employed the PDha backbone, specifically the –NH2 functional groups, for post-polymerization modification reactions, achieved through either aza-Michael addition reactions or ring opening of the epoxy functional groups.11,50 We also described PDha and its corresponding graft copolymers as interesting materials for coatings of magnetic iron oxide and TiO2 nanoparticles,11,51 dispersion agents for carbon nanotubes and hydrophobic organic dye photosensitizers,12,49 smart templates for the formation of alloy nanoparticles,23 and as a platform for constructing smart sensors.50 Herein, the aim was to utilize PDha-based graft copolymers as fundamental units for constructing micellar IPECs through electrostatic complexation. In this approach, two graft copolymers are illustrated in Fig. 1, polydehydroalanine-graft-2-acrylamido-2-methylpropane sulfonic acid (PDha-g-AMPS) (serving as polyanion) and polydehydroalanine-graft-((3-acrylamidopropyl) trimethyl ammonium chloride-co-viologen) modified by 2-(ethoxy methyl)oxirane (PDha-g-(APTMA-co-VEMO)) (acting as polycation). Both materials were employed to create micellar IPECs for the subsequent immobilization of catalyst/photosensitizer combinations. We performed the modification of PDha under basic aqueous conditions to attach AMPS and APTMA as well as the ethyl viologen functionalized by 2-(ethoxy methyl) oxirane (VEMO). Thereby, PDha with an apparent molecular weight of Mn around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was synthesized according to our previously reported method and the successful synthesis was proven using 1H-spectroscopy and size exclusion chromatography (SEC) measurements (ESI, Fig. S1–S4†).23 However, based on our last reports, we could control the kinetics and accordingly the degree of functionalization of the PDha backbone with varying time of the corresponding grafting reaction.50 Consequently, the quantities of grafted AMPS, VEMO, or APTMA side chains could be tuned to 35%, 11%, and 40%, respectively, ensuring the production of graft copolymers with adequate solubility in both acidic and neutral aqueous solutions. In case of VEMO, the grafting reaction leads to the formation of lightly crosslinked and branched PDha graft copolymers.
000 g mol−1 was synthesized according to our previously reported method and the successful synthesis was proven using 1H-spectroscopy and size exclusion chromatography (SEC) measurements (ESI, Fig. S1–S4†).23 However, based on our last reports, we could control the kinetics and accordingly the degree of functionalization of the PDha backbone with varying time of the corresponding grafting reaction.50 Consequently, the quantities of grafted AMPS, VEMO, or APTMA side chains could be tuned to 35%, 11%, and 40%, respectively, ensuring the production of graft copolymers with adequate solubility in both acidic and neutral aqueous solutions. In case of VEMO, the grafting reaction leads to the formation of lightly crosslinked and branched PDha graft copolymers.
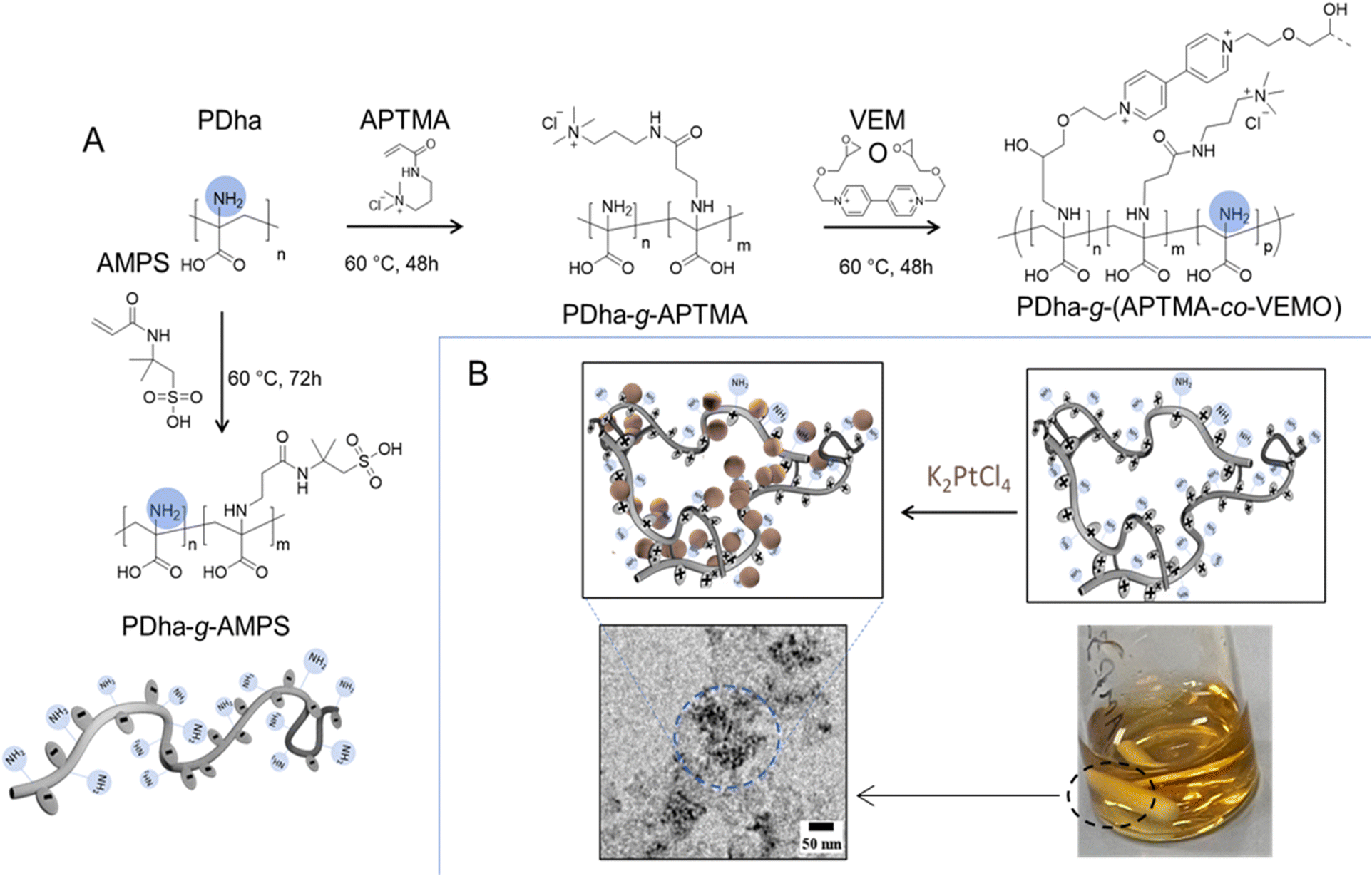 | ||
| Fig. 1 (A) Synthesis route of PDha-g-AMPS (polyanion) and PDha-g-(APTMA-co-VEMO) (polycation), graft copolymers and (B) schematic illustration of the Pt nanoparticle synthesis by PDha-g-(APTMA-co-VEMO) and TEM image of Pt@PDha-g-(APTMA-co-VEMO). | ||
The viologen modifier among the introduced grafts in the side chain was synthesized with modifications following the protocol outlined by Tsarevsky et al.52 The degree of functionalization was determined using 1H NMR spectroscopy, consistent with our previous work.11,50 As expected, the resulting graft copolymer exhibited solubility across the entire pH range. Analysis by NMR (1H and 13C) spectroscopy and Fourier transform infrared spectroscopy (FT-IR, refer to Fig. S1–S4†) confirmed the presence of the PDha backbone, viologen, and charged side chains. Additionally, SEC traces revealed an elution profile displaying an overall monomodal distribution (Fig. S4†).
Formation and solution behavior of micellar IPECs
The construction of micellar IPECs through interpolyelectrolyte complexation offers a facile and straightforward method for the direct co-assembly of macromolecules. Typically, the spontaneous formation of IPECs structures occurs when two oppositely charged polyelectrolytes are mixed, and is driven by attractive electrostatic interactions.35 In our case here, solutions of oppositely charged graft copolyelectrolytes from PDha-g-AMPS and PDha-g-(APTMA-co-VEMO) were mixed in deionized water at total concentrations of 1.0 mg mL−1 (Fig. 2A).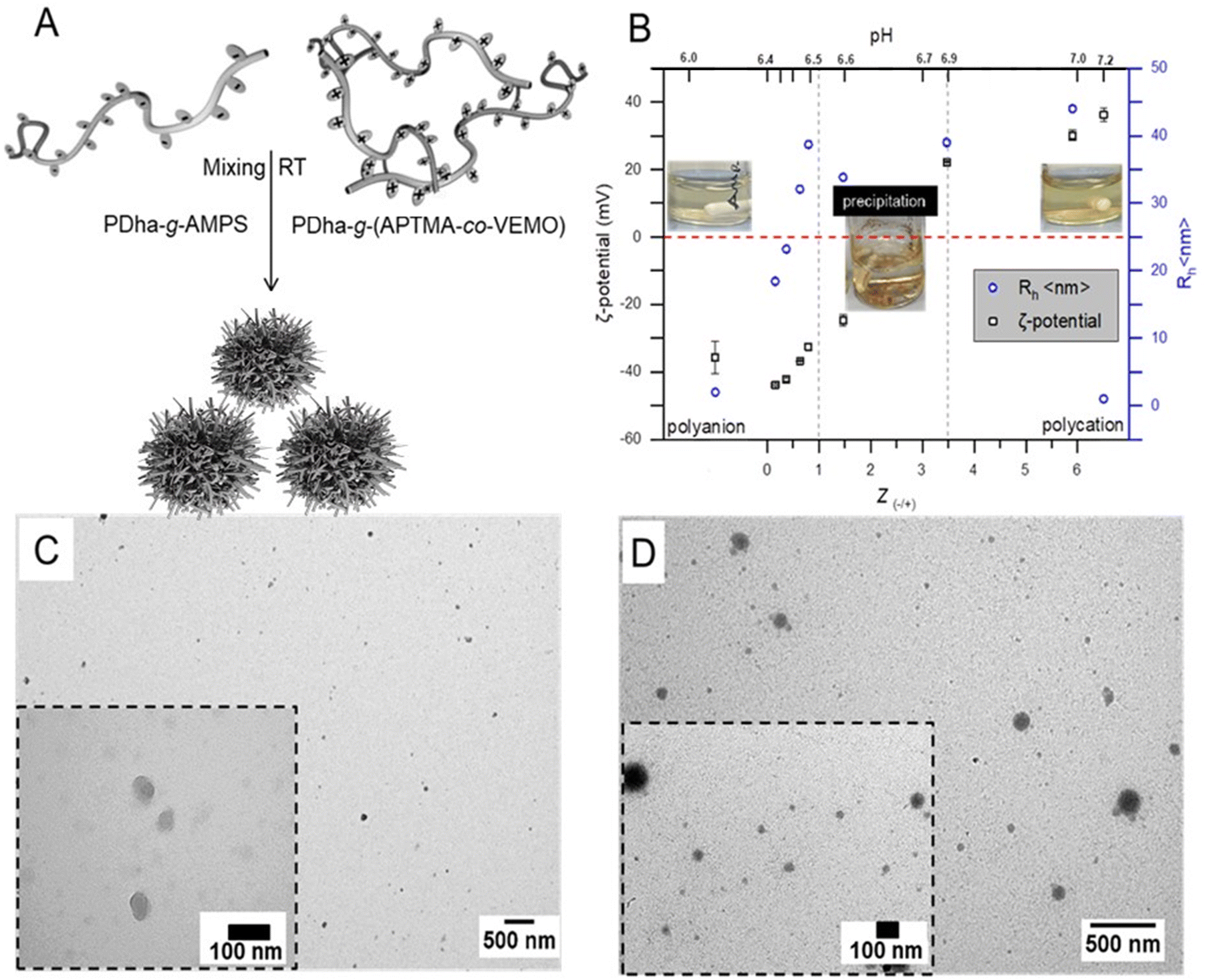 | ||
| Fig. 2 (A) Schematic depiction of the complex formation between PDha-g-AMPS (polyanion) and PDha-g-(APTMA-co-VEMO) (polycation), graft copolymers, (B) hydrodynamic radii (blue) and ζ-potential (black) over different charge ratio, Z of IPECs micelles formed in mixtures with a total constant concentration of 1.0 mg mL−1, (C) and (D) are TEM micrographs of the IPECs micelles and Pt@IPECs micelles with Z = 0.6 respectively. | ||
Hereby, PDha-g-AMPS represents a strong polyanion, which is charged (sulfonate) over the entire pH range from 1–14.53,54 It was also chosen due to its high hydrophilicity and good water solubility. Along the same lines, PDha-g-(APTMA-co-VEMO) resembles a strong polycation due to the ammonium groups of APTMA and also VEMO. In addition, VEMO is supposed to enhance the charge transfer between the catalytically active sites (i.e., Pt nanoparticles) and photosensitizers during photocatalytic reactions.55,56 Additionally, VEMO acts as a crosslinker for PDha, creating a network for the formation of Pt nanoparticles (Fig. 1, PDha-g-(APTMA-co-VEMO)).
To induce IPEC formation, water was chosen as solvent at a graft copolymer concentration of about 1.0 mg mL−1 for both graft copolymers and the resulting aggregates were investigated using dynamic light scattering (DLS) and transmission electron microscopy (TEM).
PDha-g-AMPS and PDha-g-(APTMA-co-VEMO) were initially dissolved separately in water, resulting in clear solutions, but with a slight brownish colour in case of PDha-g-(APTMA-co-VEMO). For these initial solutions, DLS revealed the formation of soluble unimers and possibly small aggregates, with radii ranging from 2 to 6 nm (Fig. 2 and S5†). Especially in the case of PDha-g-(APTMA-co-VEMO), we attribute the formation of small aggregates to the presence of the viologen moieties (Fig. S5†).
Nevertheless, we hypothesized that variations in pH would not only affect the net charge and charge density along the backbone of both graft copolymers but also induce changes in their conformation and size. Under basic conditions, we observed Rh within the range of 2.0–4.0 nm, which we attribute to fully stretched backbone chains due to electrostatic repulsion from the negatively charged carboxylate and sulfonate groups. In contrast, at acidic and neutral pH levels, the Rh increased to values above 4 nm. One plausible explanation for this phenomenon is that under these conditions, intramolecular interactions between AMPS or APTMA and the carboxylic moieties of PDha become more pronounced, counteracting electrostatic repulsion (Fig. S5†).
IPEC formation was induced by dropwise mixing of both solutions under continuous stirring. When samples are mixed at room temperature, solution mixtures immediately turned turbid, indicating the formation of aggregates (Fig. 2B). The self-assembly of the corresponding micellar IPECs was followed by DLS, zeta-potential measurements, and TEM.
DLS radii for mixtures of PDha-g-AMPS and PDha-g-(APTMA-co-VEMO) at different ratios in water at a total concentration of 1 mg mL−1 is shown in Fig. 2B (blue). Our data indicates IPECs with a nearly narrow size distribution are formed. Moreover, the hydrodynamic radii of complexes with different ratios of Z(−/+) are also investigated and they are depicted in Fig. 2B. The adjustment of the charge-to-charge ratio Z = [−]/[+] = [SO3−]/[N+] was mainly based on the grafted charged groups in both cases (as calculated by NMR data). At Z = 0.16, Rh increases from 2 nm to 18 nm, indicating the formation of micellar IPECs. Further increasing the Z ratio results in a steady increase in the radius of the micellar IPECs measured by DLS. However, the formed IPECs precipitate when a critical Z(±) value is reached above 1. At Z = 1.0, the formed IPECs start to show decreasing size. Macroscopic precipitation occurred at 0.8 < Z ≤ 3.47 after 12 hours under constant stirring (Fig. 2). We attribute this to decreased electrostatic stabilization and internal hydrogen bonding occurring between PDha chains.32
To gain further insight into the structure of the IPEC, transmission electron microscopy was performed. Typically, samples for TEM measurements were prepared by drop-coating micellar solutions, approximately 0.1 g mL−1, onto carbon-coated copper grids. TEM imaging revealed mostly spherical objects with an average diameter of ∼15–60 nm. As a representative example, TEM micrographs of IPEC nanoparticles with Z = 0.6 are depicted in Fig. 2C and S8.† Our investigation confirms the spherical nature of the micelles and reveals average radii of approximately ∼20–30 nm. This value is lower than that we observed by DLS (45 nm), which is likely due to dehydration during sample preparation.
Immobilization of catalyst and photosensitizer within micellar IPECs
Micellar IPECs have demonstrated significant potential for incorporating various payloads and have been utilized as carriers for DNA, gene delivery and encapsulation.35,57–59 Hereby, we are now particularly interested in loading the formed IPECs with a photosensitizer (PS) and a catalyst (CAT) for subsequent light-driven catalysis.Eosin Y (EY) is an organic PS that has been successfully utilized for photocatalytic hydrogen generation. It possesses a large quantum yield of triplet formation, a long excited-state lifetime, and suitable redox properties of its excited states.60–62 We aimed to incorporate the EY in our polyanion (e.g., PDha-g-AMPS), as such materials have been previously employed in catch-and-release studies of charged dyes.63 Therefore, we studied the solution behavior of PDha-g-AMPS in the presence of EY by DLS, which showed an increase in size (EY@PDha-g-AMPS) compared to the pure graft copolymer (Rh 3 to 8 nm), confirming likely attractive interactions (Fig. S5B†).
Pt nanoparticles have demonstrated excellent electrocatalytic and photocatalytic performance.64 Our previous study indicated that PDha graft copolymers could serve as a platform for the preparation of nanoparticles and alloys.23 Here, we utilized PDha-g-(APTMA-co-VEMO) for the template-assisted synthesis of Pt nanoparticles. This also renders a hybrid material which is colloidally stable in water. Initially, PDha-g-(APTMA-co-VEMO) was dissolved in water, followed by the addition of a fresh solution of potassium tetrachloroplatinate. Subsequently, Pt nanoparticles were synthesized under UV light irradiation (Fig. 1, 3 and S6†).23 The formation and morphology of Pt nanoparticles were investigated using X-ray photoelectron spectroscopy (XPS) and TEM (Fig. S7–S9†).
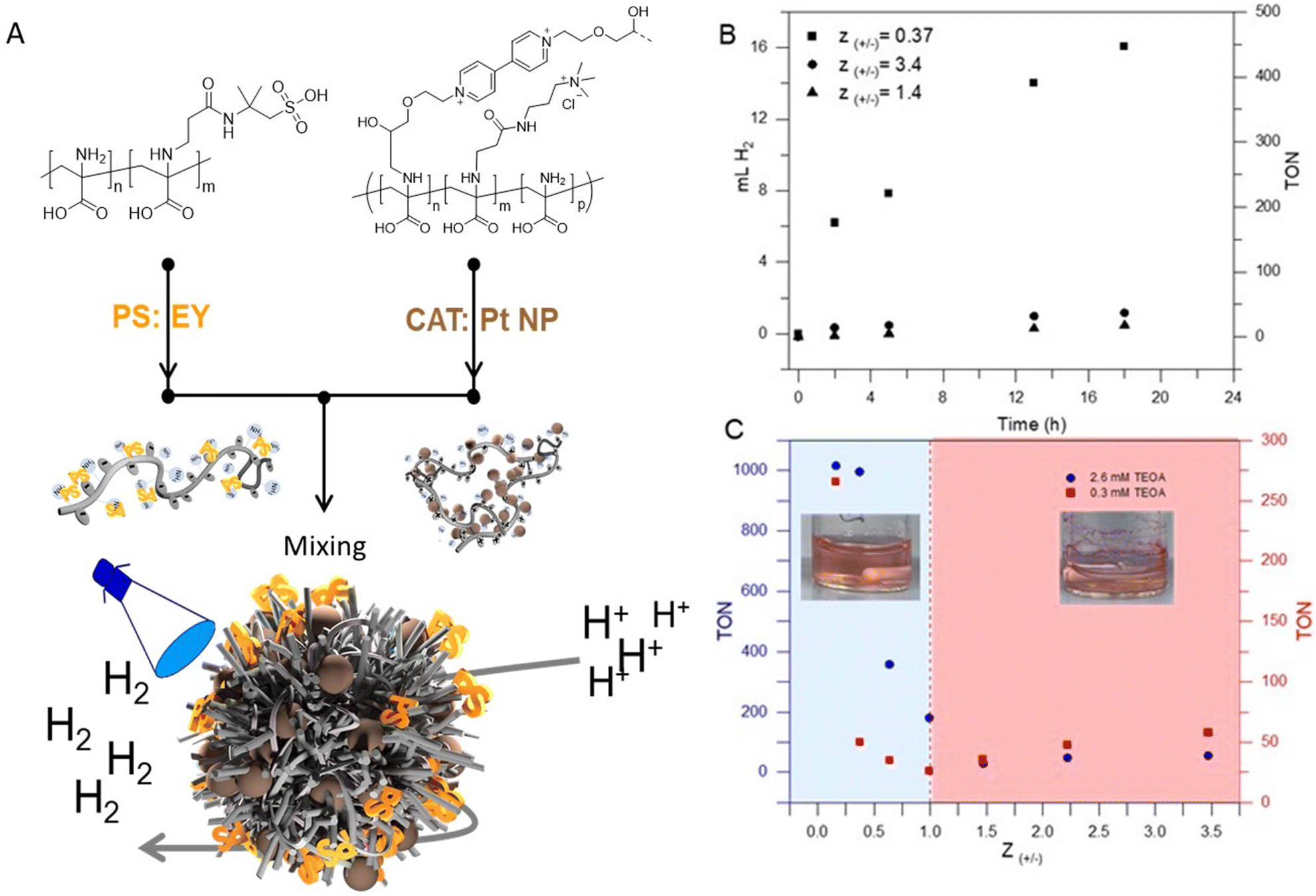 | ||
| Fig. 3 (A) Schematic depiction of the formation of photocatalytically active micellar IPECs from EY@PDha-g-AMPS (polyanion) and Pt@PDha-g-(APTMA-co-VEMO), (B) time-dependent hydrogen evolution of different IPECs complexes (TEOA 2.6 M) and (C) depict the net charge-dependent hydrogen evolution of IPECs with different TEOA concentrations that were irradiated for 24 hours. (B and C measured at λ > 450, total micellar concentration was 1 mg mL−1, and for EY 0.05 mM, and for K2PtCl4 0.002 mM were used. The turnover number (TON) is defined as the ratio of the moles of hydrogen produced to the moles of the Pt precursor initially introduced during the micellar formation from a fresh aqueous solution of K2PtCl4). | ||
From TEM investigations (Fig. S7†), we extracted also information on both the shape and size of the nanoparticles, which mostly appeared as spherical dots with an average diameter of <3 nm. XPS (Fig. S9†) revealed the Pt 4f7/2 and 4f5/2 doublet with binding energies of 71.4 and 74.8 eV, respectively. These are typical values for Pt0, supporting the formation of Pt nanoparticles. However, XPS also showed signals associated with the graft copolymer backbone, such as C 1s, N 1s and S 2p (Fig. S9†).
With the embedded PS and CAT in our designed polycation and polyanion respectively, at hand, the possibility of forming catalytically active micellar IPECs was investigated. Hereby, IPECs were prepared by mixing different ratios of positive to negative charges, Z(±), of individual graft copolymer electrolytes, and the resulting nanostructures were analyzed in detail with TEM, ξ-potential, and DLS (Fig. 2D, S6 and S8†).
As illustrated in Fig. S6,† there was a distinct increase in particle size upon adding Pt@PDha-g-(APTMA-co-VEMO) solution (from 8 nm to 25 nm at Z = 0.16–0.37), supporting the formation of micellar IPECs. Upon addition of further polycation (i.e., Pt@ PDha-g-(APTMA-co-VEMO)), sizes further increased and hydrodynamic radii were within the range of 20–150 nm (Fig. S6†). However, upon exceeding Z-ratios of 1, also here macroscopic precipitation could be observed. We attribute this to rather fast secondary aggregation.
We also monitored the ζ-potential of the formed micellar IPECs (Fig. S6A†), and found increasing values during the addition of Pt@PDha-g-(APTMA-co-VEMO) (−20 to +1 mV), suggesting the existence of overall neutral aggregates. TEM observations again revealed a spherical morphology and as a representative sample, TEM micrographs of micellar IPECs with Z = + 0.6 are shown (Fig. 2D and S8†). Both structures with about 15–20 nm in radius, as well as larger nanoobjects (>100 nm), could be observed. Finally, the obtained IPECs are analyzed by XPS, and here also the corresponding signals for Pt nanoparticles can be found (Fig. S10†).
A comparison between micellar IPECs with and without load suggests that micelles with a slightly larger size are formed after loading (Fig. S8†). Considering this observation, it is crucial to ascertain whether the Z value of the copolymer alters due to the presence of the load (i.e., CAT or PS). In other words, depending on the Z ratio, the quantity of CAT or PS may fluctuate, also depending on the proportion of each graft copolymer (i.e., PDha-g-(APTMA-co-VEMO) or PDha-g-AMPS). This fluctuation could also impact the catalytic activity of micelles. In our case, we aimed to minimize the impact of CAT/PS loading on the Z ratio of IPECs micelles by carefully controlling the loading amounts of CAT and PS.
Hereby, the loading process can be understood via the role of the –NH2 groups of the PDha backbone and the interaction between PS/CAT and the respective graft copolymers. We assume that in both the cationic and anionic segments, the –NH2 group of the PDha backbone plays an active role in anchoring and loading either PS or CAT nanoparticles. These –NH2 groups along the PDha backbone also aid in stabilizing the final complexes. The employed PS carries a negative charge and could indirectly influence the amount of –NH2 groups and the necessary cationic segment for neutralization (e.g., APTMA in PDha-g-(APTMA-co-VEMO) graft copolymers). CAT nanoparticles on the other hand are hydrophobic, and might block the –NH2 groups, reducing the overall solution stability. To balance all these effects, the overall amount of catalyst was kept rather low. On the other hand, the amount of PS was kept constant and dialysis against water was used to remove free PS. This altogether allowed us to ensure that the overall Z ratio for the herein used micellar IPECs is rather constant but, of course, is difficult to determine exactly.
Light-driven HER catalysis
In general, photocatalytic systems for H2 production comprise a photosensitizer (PS), an electron relay, a sacrificial electron donor, and a heterogeneous or homogeneous catalyst (CAT).62,65 In many systems, the photon-to-energy conversion efficiency is typically low due to the poor electron transport and connection between PS and CAT. In most cases, PS and CAT are not chemically bonded and the PS/CAT interaction is limited by diffusion.65 Colocalization using soft matter matrices is a promising strategy to enhance efficient charge separation and transport in such systems.9,12,66,67 In our case, the aim is to exploit the 3D confinement of both PS and CAT within micellar IPECs for efficient light-driven catalysis.Photocatalytic hydrogen evolution experiments were carried out using micellar IPECs loaded with Eosin Y (EY) as PS and Pt nanoparticles as catalyst (Fig. 3A). Triethanolamine (TEOA) was used as a sacrificial electron donor under blue LED light irradiation (λ > 450 nm). Control photocatalytic experiments confirmed that no measurable H2 evolution was detected in the absence of either irradiation or PS.
Additionally, we studied the effect of TEOA concentration by maintaining a constant catalyst amount and irradiation time, revealing that the activity increased with increasing TEOA concentration up to 2.6 mM (Fig. S11†). Beyond this concentration, the activity slightly decreased, which we attribute to increasing sample viscosity.
To observe and compare the performance of micellar IPECs, hydrogen evolution over time upon irradiation at Z-ratios of 0.37, 1.4, and 3.4 in H2O was plotted and is shown in Fig. 3B. It's evident from Fig. 3B that IPECs with Z values > 1.0 exhibit lower HER activity (TON < 100) compared to IPECs with Z values < 1.0 under the excitation of visible light. This is primarily due to the likely low solution stability and precipitation. To compare our system with other examples in the literature, Table S1† lists the photocatalytic H2 activity of various catalytic systems using EY as the photosensitizer. As can be seen, our system seems to be competitive, suggesting that incorporating PS/CAT within micellar IPECs could establish an efficient pathway for light-driven catalysis. An interesting observation is comparing IPECs with a Z value of around 3.4 to those with Z values of 1.4, where slightly higher activity for hydrogen evolution can be found (i.e., TON around 40 in 18 hours). We attribute this to the potentially positive net charge of the micellar IPECs, which may lower the attraction towards protons, leading to lower HER activity. On the other hand, IPECs with Z around 0.37 show TONs of 468 after 18 h of irradiation. Presumably, such IPECs provide good solution stability and probably close contact between both CAT and PS. Presumably, such IPECs provide good solution stability and likely close contact between both CAT and PS, facilitating electron transport and charge transport.
Next, we further focused on the effects of the net charge of micellar IPECs on hydrogen evolution. As illustrated in Fig. 3C, structures with different net charge exhibit varying activity for hydrogen production at comparable TEOA concentrations. Generally, micellar IPECs with Z < 1 display higher activity if compared to IPECs with Z > 1. However, HER activity was almost negligible at 1.0 ≤ Z ≤ 2.0, presumably due to relatively low dispersion stability. This was also observed as rapid and macroscopic precipitation during preparation (Fig. 3C).
As the above results indicated, the Z value has a significant effect on both the formation of the micellar IPECs and their activity in hydrogen production. To understand the effect of the Z ratio on the solution behavior, we compare the size from DLS with the hydrogen production activity for each specific Z value.
DLS (Fig. 2B and S6†) reveals three distinct stages of aggregate formation. Initially, micelles with a negative net charge form when Z > 0.37. Subsequently, aggregates with very low stability and low dispersion stability are found in the range of 3.4 > Z > 0.8. Finally, nanoaggregates with positive net charge are observed when Z > 3.4.
It is well-known that micelles formed by polyions are favored thermodynamically, when there is a concentration range of positive and negative charges of the polyelectrolytes close to 1:1.68,69 In our case, as we approach a 1:1 ratio, larger micelles form, likely due to an increase in the number of aggregates until we reach the composition range where precipitation occurs. We assume that this is due to a low amount of hydrophilic groups and the presence of fewer excess (negative or positive) charges. In this process, we expect that the hydrophilicity of the PDha backbone is also affected. PDha exhibits characteristics of a potential polyzwitterion, with an isoelectric point around pH 5. When the polycations and polyanions are mixed, the negatively charged units (such as AMPS units) initially neutralize and bind to the (APTMA-co-VEMO) units. Within the pH range where the COOH groups of the PDha backbone remain deprotonated, it is likely that the (APTMA-co-VEMO) units also partially neutralize the COO− groups of the PDha backbone. Consequently, only the amine groups remain, but nevertheless this does not seem to provide sufficient dispersion stability.
Our catalytic results (Fig. 3B) indicate that micellar IPECs with an excess net charge exhibit improved catalytic activity. In particular, micellar IPECs with excess negative charge show the most significant improvement in hydrogen production activity compared to those with a presumably denser core (e.g., micelles with a range of positive and negative charges of the polyelectrolytes close to 1:1). This observation suggests that it is important to form micelles that are stable but are also quite strongly swollen with water due to an excess of either negative or positive charge. This observation indicates that the Z value is not the sole factor controlling micellar stability and catalytic performance. The superior performance of negatively charged micellar IPECs may be attributed to their ability to control parameters such as proton attraction and diffusion, which are crucial for hydrogen production.
Conclusions
In this work, we show the formation of micellar IPECs between negatively charged PDha-g-AMPS and PDha-g-(APTMA-co-VEMO) graft copolymers and the use of such nanostructures for embedding different catalytically active building blocks. In particular, we used Eosin Y as PS and Pt nanoparticles as CAT. The resulting 3D confinement within the micellar IPECs presumably leads to close proximity between PS and CAT and – depending on loading and overall charge-to-charge ratio – varying activity for light-driven HER.The herein demonstrated activity of micellar IPECs with varying polycation/polyanion ratios allows the formulation of design guidelines for tailormade soft matter matrices for light-driven catalysis. In particular, IPECs with an overall negative net charge show significantly higher activity, with a maximum turnover number of ∼1000 (216 μmol mg−1 h−1) relative to the used CAT under optimal conditions over 24 h light irradiation. Such IPEC aggregates could serve as model systems for the confinement and control over light-driven reactions. Further photocatalytic investigations with, for example, other PS and CAT such as acridinium or BODIPY will be the subject of further studies.
Author contributions
A. N.: conceptualization, methodology, investigation, validation, formal analysis, writing – original draft. C. N.: methodology, investigation, XPS analysis. A. T.: review & editing. F. H. S.: conceptualization, methodology, resources, formal analysis, writing – review & editing, supervision, project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the Deutsche Forschungsgemeinschaft (DFG NA 1866/1-1) and within the framework of TRR234 “CataLight” (Project No. 364549901, projects B03, B05, and Z02). C. N. and A. T. acknowledge the financial support of the DFG through research infrastructure grant INST 275/357-1 FUGG-Projektnummer 313713174. We are thankful for the cryo-TEM/TEM facilities of the Jena Center for Soft Matter (JCSM), which were established with a grant from the German Research Council (DFG) and the European Fonds for Regional Development (EFRE). We further acknowledge Jonas Eichhorn for TEM measurements.Notes and references
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557 CrossRef.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 151683 Search PubMed.
- Y. Zhong, S. Liu, J. Wang, W. Zhang, T. Tian, J. Sun and F. Bai, APL Mater., 2020, 8, 120706 CrossRef CAS.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS PubMed.
- J. Fendler, Catalysis in Micellar and Macromoleular Systems, Elsevier, 2012 Search PubMed.
- G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- P. Qu, M. Kuepfert, E. Ahmed, F. Liu and M. Weck, Eur. J. Inorg. Chem., 2021, 2021, 1420–1427 CrossRef CAS.
- A. Nabiyan, M. Schulz, C. Neumann, B. Dietzek, A. Turchanin and F. H. Schacher, Eur. Polym. J., 2020, 140, 110037 CrossRef CAS.
- T. Niitani, M. Amaike, H. Nakano, K. Dokko and K. Kanamura, J. Electrochem. Soc., 2009, 156, A577 CrossRef CAS.
- A. Nabiyan, J. B. Max, C. Neumann, M. Heiland, A. Turchanin, C. Streb and F. H. Schacher, Chem.–Eur. J., 2021, 27, 16924–16929 CrossRef CAS PubMed.
- D. Costabel, A. Skabeev, A. Nabiyan, Y. Luo, J. B. Max, A. Rajagopal, D. Kowalczyk, B. Dietzek, M. Wächtler and H. Görls, Chem.–Eur. J., 2021, 27, 4081–4088 CrossRef CAS PubMed.
- D. J. Goldfeld, E. S. Silver, M. R. Radlauer and M. A. Hillmyer, ACS Appl. Polym. Mater., 2020, 2, 817–825 CrossRef CAS.
- J. Brubert, S. Krajewski, H. P. Wendel, S. Nair, J. Stasiak and G. D. Moggridge, J. Mater. Sci.: Mater. Med., 2016, 27, 1–12 CrossRef CAS.
- M. J. Derry, T. Smith, P. S. O’hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS.
- L. Gu, Z. Shen, C. Feng, Y. Li, G. Lu, X. Huang, G. Wang and J. Huang, J. Mater. Chem., 2008, 18, 4332–4340 RSC.
- C. Feng, Y. Li, D. Yang, J. Hu, X. Zhang and X. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- C. Hagiopol, Copolymerization: toward a Systematic Approach, Springer Science & Business Media, 1999 Search PubMed.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- A. N. Le, R. Liang and M. Zhong, Chem.–Eur. J., 2019, 25, 8177–8189 CrossRef CAS PubMed.
- Y. Cui, X. Jiang, C. Feng, G. Gu, J. Xu and X. Huang, Polym. Chem., 2016, 7, 3156–3164 RSC.
- A. Nabiyan, J. B. Max and F. H. Schacher, Chem. Soc. Rev., 2022, 51, 995–1044 RSC.
- J. B. Max, K. Kowalczuk, M. Köhler, C. Neumann, F. Pielenz, L. V. Sigolaeva, D. V. Pergushov, A. Turchanin, F. Langenhorst and F. H. Schacher, Macromolecules, 2020, 53, 4511–4523 CrossRef CAS.
- X. Zhang, Z. Shen, C. Feng, D. Yang, Y. Li, J. Hu, G. Lu and X. Huang, Macromolecules, 2009, 42, 4249–4256 CrossRef CAS.
- C. Cai, J. Lin, T. Chen and X. Tian, Langmuir, 2010, 26, 2791–2797 CrossRef CAS PubMed.
- C. Wang, G. Li, S. Tao, R. Guo and Z. Yan, Carbohydr. Polym., 2006, 64, 466–472 CrossRef CAS.
- K.-J. Gao, G. Li, X. Lu, Y. G. Wu, B.-Q. Xu and J.-H. Fuhrhop, Chem. Commun., 2008, 1449–1451 RSC.
- D. Ikkene, A. A. Arteni, C. Boulogne, J. L. Six and K. Ferji, Macromolecules, 2022, 55, 4268–4275 CrossRef CAS.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2–19 CrossRef CAS.
- F. H. Schacher, T. Rudolph, M. Drechsler and A. H. E. Müller, Nanoscale, 2011, 3, 288–297 RSC.
- F. Schacher, E. Betthausen, A. Walther, H. Schmalz, D. V. Pergushov and A. H. E. Müller, ACS Nano, 2009, 3, 2095–2102 CrossRef CAS PubMed.
- D. V. Pergushov, E. V. Remizova, J. Feldthusen, A. B. Zezin, A. H. E. Müller and V. A. Kabanov, J. Phys. Chem. B, 2003, 107, 8093–8096 CrossRef CAS.
- J. Huang, Y. Guo, S. Gu, G. Han, W. Duan, C. Gao and W. Zhang, Polym. Chem., 2019, 10, 3426–3435 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- D. V. Pergushov, A. H. E. Müller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- T. P. Lodge, A. Rasdal, Z. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS PubMed.
- A. Laschewsky, Curr. Opin. Colloid Interface Sci., 2003, 8, 274–281 CrossRef CAS.
- F. Schacher, A. Walther and A. H. E. Müller, Langmuir, 2009, 25, 10962–10969 CrossRef CAS PubMed.
- E. Betthausen, M. Drechsler, M. Förtsch, F. H. Schacher and A. H. E. Müller, Soft Matter, 2011, 7, 8880–8891 RSC.
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- A. C. Rinkenauer, A. Schallon, U. Günther, M. Wagner, E. Betthausen, U. S. Schubert and F. H. Schacher, ACS Nano, 2013, 7, 9621–9631 CrossRef CAS PubMed.
- T. Nomoto, S. Fukushima, M. Kumagai, K. Machitani, Y. Matsumoto, M. Oba, K. Miyata, K. Osada, N. Nishiyama and K. Kataoka, Nat. Commun., 2014, 5, 1–10 Search PubMed.
- M. Wu, Y. Zhu and W. Jiang, Angew. Chem., Int. Ed., 2018, 57, 3578–3582 CrossRef CAS PubMed.
- C. V. Synatschke, T. I. Löbling, M. Förtsch, A. Hanisch, F. H. Schacher and A. H. E. Müller, Macromolecules, 2013, 46, 6466–6474 CrossRef CAS.
- J. Sun and Z. Li, Macromolecules, 2020, 53, 8737–8740 CrossRef CAS.
- E. K. Penott-Chang, D. V. Pergushov, A. B. Zezin and A. H. E. Muller, Langmuir, 2010, 26, 7813–7818 CrossRef CAS PubMed.
- C. V. Synatschke, F. H. Schacher, M. Förtsch, M. Drechsler and A. H. E. Müller, Soft Matter, 2011, 7, 1714–1725 RSC.
- M. Billing and F. H. Schacher, Macromolecules, 2016, 49, 3696–3705 CrossRef CAS.
- J. B. Max, D. V. Pergushov, L. V. Sigolaeva and F. H. Schacher, Polym. Chem., 2019, 10, 3006–3019 RSC.
- J. B. Max, A. Nabiyan, J. Eichhorn and F. H. Schacher, Macromol. Rapid Commun., 2021, 42, 2000671 CrossRef CAS PubMed.
- M. von der Lühe, A. Weidner, S. Dutz and F. H. Schacher, ACS Appl. Nano Mater., 2017, 1, 232–244 CrossRef.
- Z. Wang and N. V. Tsarevsky, Polym. Chem., 2016, 7, 4402–4410 RSC.
- M. G. Kellum, A. E. Smith, S. K. York and C. L. McCormick, Macromolecules, 2010, 43, 7033–7040 CrossRef CAS.
- L. A. McCullough, B. Dufour and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5386–5396 CrossRef CAS.
- H. Kotani, K. Ohkubo, Y. Takai and S. Fukuzumi, J. Phys. Chem. B, 2006, 110, 24047–24053 CrossRef CAS PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- K. H. Parsons, M. H. Mondal, C. L. McCormick and A. S. Flynt, Biomacromolecules, 2018, 19, 1111–1117 CrossRef CAS PubMed.
- M. Ruano, A. Mateos-Maroto, F. Ortega, H. Ritacco, J. E. F. Rubio, E. Guzmán and R. G. Rubio, Langmuir, 2021, 37, 6189–6200 CrossRef CAS PubMed.
- Z. Sui, J. A. Jaber and J. B. Schlenoff, Macromolecules, 2006, 39, 8145–8152 CrossRef CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35–41 CrossRef CAS.
- X. Liu, Y. Li, S. Peng, G. Lu and S. Li, Int. J. Hydrogen Energy, 2013, 38, 11709–11719 CrossRef CAS.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- P. Biehl, P. Wiemuth, J. G. Lopez, M.-C. Barth, A. Weidner, S. Dutz, K. Peneva and F. H. Schacher, Langmuir, 2020, 36, 6095–6105 CrossRef CAS PubMed.
- S. Min and G. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CrossRef CAS.
- A. Lewandowska-Andrałojć, D. Larowska, E. Gacka, T. Pedzinski and B. Marciniak, J. Phys. Chem. C, 2020, 124, 2747–2755 CrossRef.
- H. Sai, A. Erbas, A. Dannenhoffer, D. Huang, A. Weingarten, E. Siismets, K. Jang, K. Qu, L. C. Palmer and M. O. De La Cruz, J. Mater. Chem. A, 2020, 8, 158–168 RSC.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- B. Hofs, A. De Keizer and M. A. Cohen Stuart, J. Phys. Chem. B, 2007, 111, 5621–5627 CrossRef CAS PubMed.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, Langmuir, 2024, 40, 2015–2027 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01824a |
| This journal is © The Royal Society of Chemistry 2024 |