
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Push–pull–pull interactions of 2D imide–imine-based covalent organic framework to promote charge separation in photocatalytic hydrogen production†
Islam M. A.
Mekhemer‡
ab,
Mohamed M.
Elsenety‡
ac,
Ahmed M.
Elewa
 a,
Khanh Do Gia
Huynh
a,
Maha Mohamed
Samy
bd,
Mohamed Gamal
Mohamed
bd,
Dalia M.
Dorrah
a,
Dung Chau Kim
Hoang
a,
Ahmed Fouad
Musa
a,
Shiao-Wei
Kuo
d and
Ho-Hsiu
Chou
*aef
a,
Khanh Do Gia
Huynh
a,
Maha Mohamed
Samy
bd,
Mohamed Gamal
Mohamed
bd,
Dalia M.
Dorrah
a,
Dung Chau Kim
Hoang
a,
Ahmed Fouad
Musa
a,
Shiao-Wei
Kuo
d and
Ho-Hsiu
Chou
*aef
aDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: hhchou@mx.nthu.edu.tw
bChemistry Department, Faculty of Science, Assiut University, Assiut, 71515, Egypt
cChemistry Department, Faculty of Science, Al-Azhar University, Cairo, 11884, Egypt
dDepartment of Materials and Optoelectronic Science, Center of Crystal Research, National Sun Yat-Sen University, Kaohsiung 804, Taiwan
eCollege of Semiconductor Research, National Tsing Hua University, Hsinchu 300044, Taiwan
fPhotonics Research Center, National Tsing Hua University, Hsinchu 300044, Taiwan
First published on 27th March 2024
Abstract
Photocatalytic hydrogen production through water splitting provides a promising route towards renewable energy generation. However, constructing photocatalytically active covalent organic frameworks with high charge separation remains challenging. Herein, we demonstrate for the first time the use of 2D imide–imine-based covalent organic frameworks as new photocatalysts for the hydrogen evolution reaction (HER) under visible light irradiation. The main achievement is incorporating donor and dual acceptors, including weak electron-deficient imine and strong electron-deficient imide groups within the 2D COF backbone that create favorable push–pull–pull intramolecular charge transfer to promote charge separation after photoexcitation. DFT and NBO calculations revealed the strong integration of donor and dual acceptors with a synergistic interplay enhancing spatial charge transfer and separation. The synthesized COFs show significantly high thermal stability >400 °C with a high energy barrier for degradation. Moreover, Py-DNII-COF exhibited a 104-fold enhancement in hydrogen evolution compared to TFPB-DNII-COF. Py-DNII-COF demonstrated excellent stability and hydrogen evolution of 625 μmol h−1 g−1 over 48 hours.
Introduction
In the face of escalating energy demands and environmental concerns, photocatalytic hydrogen production has emerged as a promising avenue for clean and renewable energy.1 The utilization of sunlight as a driving force for the generation of hydrogen from water holds tremendous potential, provided efficient and stable photocatalytic materials can be developed.2 Covalent organic frameworks (COFs) are a class of porous crystalline materials with a regular array of covalently-bonded organic units. They have gained attention for various applications, including gas storage,3–5 catalysis,6–9 and electronic devices.10,11 Two-dimensional covalent organic frameworks (2D-COFs) have emerged as promising photocatalysts owing to their unique combination of structural properties, tunable functionalities, and exceptional physiochemical stability. These attributes are challenging to achieve simultaneously in previously reported inorganic and organic amorphous photocatalysts.12–15 In 2014, Lotsch et al. pioneered the use of a hydrazone-linked covalent organic framework (COF) as a photocatalyst for hydrogen evolution (H2 evolution) under visible light irradiation (λ ≥ 420 nm).16 This COF photocatalyst required a Pt co-catalyst and a sacrificial electron donor (SED) for efficient H2 evolution. Building on this approach, researchers continue to design and report a variety of COFs with diverse linkages and topologies for efficient H2 evolution, achieving hydrogen evolution rates as high as 134.2 mmol g−1 h−1.17–20 Moreover, the integration of electron-donating and electron-withdrawing units within the covalent organic framework (COF) backbone to create a periodically-ordered donor (D)–acceptor (A) architecture has emerged as the most common approach for photocatalytic hydrogen evolution.21,22 However, the improvement of robust D–A COFs that can retain their photocatalytic activity over time is critical for real-world applications.23–25 Wu and coworkers reported that the addition of another acceptor to the D–A system often results in broader spectral ranges and enhanced stabilities.26 Recently, our group reported that the incorporation of a donor into dual acceptor systems (D–A–A) offers a promising approach for overcoming the limitations of traditional photocatalysts in H2 evolution.27,28 Consequently, the D–A–A system, which results in the push–pull–pull approach able to enhance light absorption, suppress charge recombination, tune redox potentials, improve stability, and integrate with other systems, makes them a valuable tool for the development of efficient and sustainable solar-driven H2 production technologies, while it is still rarely explored for COFs thus far. In this context, the incorporation of imide and imine moieties into 2D COFs was constructed for the first time while converting 1D ribbons into 2D COFs but without a further study of the catalytic applications.29 Imides, with their strong electron-withdrawing nature, and imines, with their weak electron-deficient capabilities, could create a synergistic interplay within the COF structure.30,31 This synergistic effect is anticipated to facilitate charge separation, promote light absorption, and enhance catalytic activity, ultimately leading to improved hydrogen evolution efficiency.32–34 Nevertheless, the challenge of charge carrier mobility within the COF structure remains. Limited mobility or trapped charges lead to subpar conductivity and thus curtail the system's catalytic proficiency. To conquer this challenge, our focus in this study revolves around the utilization of two-dimensional imide–imine covalent organic frameworks (2D Py-DNII and TFPB-DNII COFs) as novel photocatalysts for the hydrogen evolution reaction (HER) under visible light irradiation. The novelty of our work lies in the pioneering incorporation of donor and dual acceptors of strong electron-deficient imide and weak electron-deficient imine groups within the 2D COF backbone, resulting in a unique push–pull–pull interaction that promotes efficient charge separation and transport upon photoexcitation.Thus, we synthesized 2D COFs of Py-DNII and TFPB-DNII frameworks, leading to the formation of imide–imine linkages. A new approach introduces dual electron-acceptor using push–pull–pull centers into the COF structure to enhance its electrical conductivity and facilitate efficient charge transport. The synthesized COFs were characterized using different spectroscopic techniques. The synthesis of these two-dimensional imide–imine COFs was achieved through a meticulous solvothermal method, ensuring precise control over their structural attributes. Thorough characterization using advanced spectroscopic techniques confirmed the successful implementation of the proposed COF structure. To better understand the electronic structure and the nature of push–pull–pull interactions, we employed Density Functional Theory (DFT) and Natural Bond Orbital (NBO) calculations. These calculations elucidated the strong synergistic interplay between the imide imine linkers, creating a favorable environment for spatial charge separation.
Experimental section
Synthesis of COFs-based imide–imine linkages
A 10 mL heavy-walled sealed tube was filled with aromatic aldehyde derivatives (0.022 mmol), naphthalenediimide diamine (DNI-2NH2) (20.00 mg, 0.044 mmol), 1.00 mL of nitrobenzene, and 1.00 mL of mesitylene.29 The reaction mixture was sonicated for 5 minutes, then 10 μL of trifluoroacetic acid (TFA) was added, followed by another 10 minutes of sonication. The tube was plunged into liquid nitrogen, flash-frozen at 77 K, and evacuated to an internal pressure of 100 mTorr. The reaction was then heated to 120 °C for 4 days, resulting in the precipitation of COFs. The solid was isolated by filtration and thoroughly washed with acetone, THF, methanol, and ether. Subsequently, it was subjected to overnight Soxhlet extraction with methanol. Finally, the activated COF-based imide–imine linkages were obtained by oven vacuum activation at 80 °C for 8 h. For more details, see the ESI File (Synthetic procedures of COFs).†Photocatalytic hydrogen production
The photocatalytic reaction was conducted in a 40.00 mL photocatalytic reaction cell. 1 mg of COF powder was combined with 10 mL of a mixed solution containing 80% H2O, 20% N-methyl-2-pyrrolidone (NMP) as co-solvent,35 0.1 M ascorbic acid (AA) as a sacrificial reagent, and 2 wt% of hexachloroplatinic acid as a co-catalyst.36 The mixture was sonicated for 5 minutes to achieve a well-dispersed suspension. To remove any air, the suspension was degassed with argon gas for 10 minutes before being irradiated with a 350 W Xe light source (1000 W m−2; λ = 380–780 nm) under continuous stirring. The reaction was conducted for 5 hours at room temperature. H2 production was confirmed thermally via the back injection of a 0.5 μL sample of the reactor headspace gas onto a semi-capillary column (molecular sieve; diameter: 8 mm; length: 3 m) using a gas chromatograph (GC7920) under isothermal conditions. Additionally, an AQY measurement was performed using a 350 W Xe lamp with a 420 nm band-pass filter. The apparent quantum yield (AQY) was calculated using the following equations.where NA is the Avogadro constant, M is the amount of H2 produced (mol), c is the speed of light h is the Planck constant, S is the irradiation area (cm2), t is the photoreaction time (s), P is the intensity of the irradiating light (W cm−2), and λ is the wavelength of the monochromatic light (m).

Results and discussion
COFs synthesis and characterization
Initially, we conducted theoretical computational investigations to provide preliminary validation of our concepts for both Py-DNII and TFPB-DNII COFs (Fig. 1a). Thus, the monomers of 1,2,4,5-tetrakis-(4-formylphenyl)benzene (TFPB-4CHO) as a weak donor, 1,3,6,8-tetra(4-formylphenyl)pyrene (PyTP-4CHO) as a strong donor and 2,7-bis(4-aminophenyl)-3a,5a,8a,10a-tetrahy-drobenzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)tetraone (DNI-2NH2) as a strong acceptor were prepared according to the reported method with some modifications (Schemes S1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S3†). As discussed in the Synthetic procedures of the monomers section (in ESI†), 1H NMR, 13C NMR, and corresponding data in Fig. S1:S4† provide clear evidence of the successful formation of the expected monomers without residual impurities. Further, the Py-DNII and TFPB-DNII COFs were synthesized using a strategic approach of a Schiff-base condensation reaction of the prepared monomers, wherein DNI-2NH2 was employed as the acceptor while both PyTP-4CHO and TFPB-4CHO served as donor monomers in an equimolar 2:1 stoichiometric ratio (Schemes S3:S4†). This synthesis strategy led to the formation of 2D imide–imine linkages within the COFs' structural backbone. The computational study based on the population analysis and using natural bond orbital (NBO) and electrostatic potential (ESP) distribution (Fig. 1b and c) were used to estimate the distribution of charge and electron density in Py-DNII-COF, respectively. The charge of −0.444 for the nitrogen atom (–N
:S3†). As discussed in the Synthetic procedures of the monomers section (in ESI†), 1H NMR, 13C NMR, and corresponding data in Fig. S1:S4† provide clear evidence of the successful formation of the expected monomers without residual impurities. Further, the Py-DNII and TFPB-DNII COFs were synthesized using a strategic approach of a Schiff-base condensation reaction of the prepared monomers, wherein DNI-2NH2 was employed as the acceptor while both PyTP-4CHO and TFPB-4CHO served as donor monomers in an equimolar 2:1 stoichiometric ratio (Schemes S3:S4†). This synthesis strategy led to the formation of 2D imide–imine linkages within the COFs' structural backbone. The computational study based on the population analysis and using natural bond orbital (NBO) and electrostatic potential (ESP) distribution (Fig. 1b and c) were used to estimate the distribution of charge and electron density in Py-DNII-COF, respectively. The charge of −0.444 for the nitrogen atom (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) of the imine linkage works as the first central position for attracting electrons from the donor moiety. As expected, the significantly higher negative charge of the nitrogen atom of the imide–linkage N–CO– (−0.496) indicates an excess of electrons on the second central position of imide groups.
) of the imine linkage works as the first central position for attracting electrons from the donor moiety. As expected, the significantly higher negative charge of the nitrogen atom of the imide–linkage N–CO– (−0.496) indicates an excess of electrons on the second central position of imide groups.
 | ||
| Fig. 1 Schematic diagram of synthesized COFs (a); Mulliken charge separation of Py-DNII (b); electrostatic potential of Py-DNII (c); molecular orbital visualization of Py-DNII-COF (d) and TFPB-DNII-COF (e). | ||
However, second order perturbation of the Fock matrix in NBO analysis provides a more intuitive picture of chemical bonding and allows for a clearer interpretation of electronic structure transition and charge transfer from donor to acceptor moieties. The results represent a donor–acceptor of most significant translation of the N lone pair of electrons in the imide-linkage to the π* orbital of both adjacent carbonyl groups with interaction energy of 51.42 and 51.26 kcal mol−1. However, the π-orbital of the –CN– of the imine group represents an interaction energy of 10.13 kcal mol−1 to the π* orbital of –C–C in the direction of the amide group and acceptor moiety, in contrast to the lower interaction energy of 8.21 kcal mol−1 for the π* orbital of –C–C in the direction of the pyridine moiety. In addition, there are multiple possible transitions from the bonding orbital to the antibonding orbital in the benzene rings due the conjugated system of the molecule, with interaction energy of 22:14 kcal mol−1. This finding shows that the first central position of imine-linkage might be more prone to withdraw (pull) electrons from the donor pyridine moiety to the second central position of imide (pull), facilitating charge separation on the whole COF. Furthermore, the ESP of the Py-DNII-COF provides insights into the distribution of electron density on the whole COF. The π electrons in the aromatic rings of the tetraphenyl pyrene contribute to the overall electron density of the molecule. However, the lower electron density could imply that the π electron density is more dispersed or delocalized, leading to a less concentrated region of electron density in this part of the molecule, which works as a donor moiety. This is in contrast to the higher electron density distributed on the imide groups of the dinaphthylimide (DNI), which will work as an acceptor moiety in the COF, thus facilitating the withdrawal of electron density from the donor to acceptor through the imide–imine linkage and creating a synergistic interplay within the COF structure. To investigate the hydrogen evolution potential of two synthesized COFs, DFT calculations were employed to determine the energy gaps and energy levels. The Py-DNII and TFPB-DNII COFs exhibited energy gaps of 1.80 eV and 2.69 eV, respectively, as illustrated in Fig. 1d and e. The DFT calculations indicate that the narrow energy gaps observed between the HOMO−1/HOMO and LUMO levels of Py-DNII suggest the likelihood of electronic transitions occurring under visible light, particularly at wavelengths of 457 nm and 688 nm. On the other hand, TFPB-DNII COF exhibits a larger energy gap, resulting in a blue shift in the maximum absorption wavelength, which is detected at 504 nm and 475 nm (Fig. 1e). Furthermore, examining the Py-DNII COF reveals HOMO and LUMO energy levels at −5.16 eV and −3.31 eV, respectively. This finding indicates a strong positive correlation with the potential for proton reduction to hydrogen, emphasizing the significance of these energy levels in the efficacy of the process.
The molecular structures of synthesized COFs were characterized via several spectroscopic techniques. The FTIR spectra of the monomers of DNI-2NH2, TFPB-4CHO, and their corresponding TFPB-DNII-COF are shown in Fig. 2a. The FTIR spectrum of DNI-2NH2 shows the following characteristic bands of ν(–NH2) at 3409 and 3316 cm−1, ν(>CO) at 1702 and 1645 cm−1, ν(C–C) at 1579, σ(C–H) at 980 cm−1 of the aromatic ring, and the stretching vibration of >C–N at 1246 cm−1.28–30 However, the FTIR spectrum of TFPB-4CHO shows ν(>CO) at 1736 and 1697 cm−1, and the ν(C–H) at 2830 cm−1 of aldehyde groups, ν(C–C) at 1582 cm−1, and σ(C–H) at 1002 cm−1 of the aromatic ring. Thus, the disappearance of the –NH2 stretching vibration of DNII-2NH2 indicates that the amine groups were introduced in the formation of TFPB-DNII-COF. In addition, the existence and slight shifting in the peak position of the ν(>CO) at 1705, and 1662 cm−1, ν(C–C) at 1579 cm−1 and ν(>C–N) at 1244 cm−1 confirm that the main monomers (aryl, carbonyl, and imine groups) are still a present in the synthesized DNII-TFPB-COF. As expected, the presence of the >CN stretching vibration at 1602 cm−1 successfully confirms Schiff-base condensation reaction in the formation of TFPB-DNII-COF. Similar findings of vibrational bands were obtained for the formation of Py-DNII-COF, as shown in Fig. 2b.
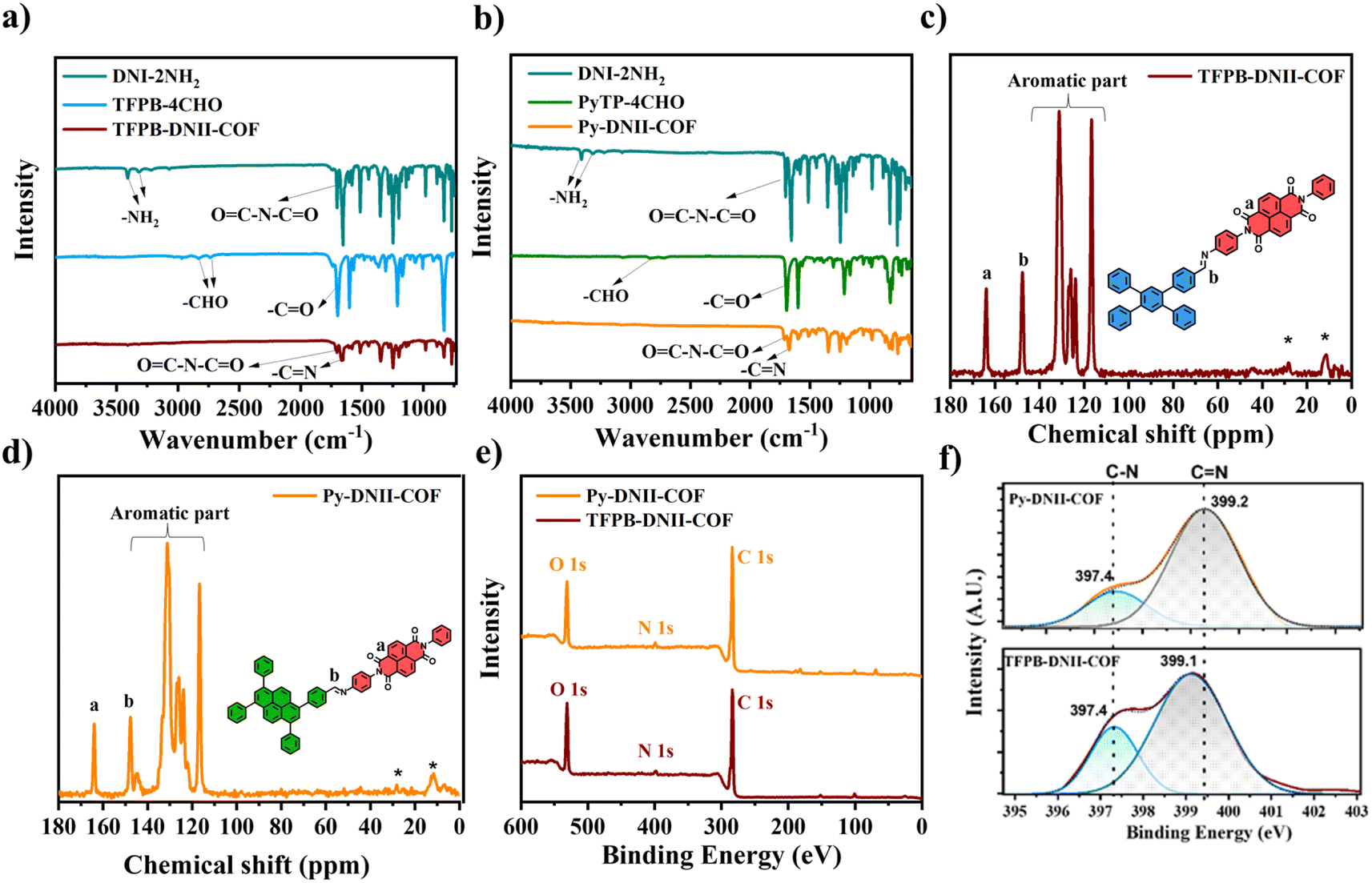 | ||
| Fig. 2 FTIR spectra of TFPB-DNII (a) and Py-DNII-COFs (b); 13C NMR spectra of TFPB-DNII (c) and Py-DNII COFs (d); survey spectra of the X-ray photoelectron spectroscopy (e), core-level deconvolution spectra of N 1s for both Py-DNII and TFPB-DNII COFs (f). | ||
The 13C NMR spectra provide robust structural evidence and support the successful formation of key chemical linkages and functional groups within the TFPB-DNII and Py-DNII COFs, as shown in Fig. 2c and d. These findings are pivotal in characterizing the precise composition and configuration of these COFs, facilitating a comprehensive understanding of their potential applications and properties. In particular, the appearance of the –CN– imine linkage was evident with a characteristic chemical shift at approximately 143 ppm, underscoring the successful Schiff-base formation within the COF structures. The chemical shift at approximately 163 ppm indicates the presence of carbonyl groups within the COFs. Additionally, the spectra exhibited chemical shifts within the range of 150–100 ppm, corresponding to the resonances of aromatic rings. This observation provides further evidence for COF structures, which are consistent with the anticipated structural elements based on the choice of monomers and synthetic strategy.
X-ray photoelectron spectroscopy (XPS) was employed to gain additional insights into the chemical composition of the synthesized COFs. In Fig. 2e, the survey spectra of TFPB-DNII-COF and Py-DNII-COF are presented, highlighting predominant peaks corresponding to the elements C, N, and O. X-ray photoelectron spectroscopy (XPS) further reveals the presence of C 1s and O 1s (Fig. S7†). The deconvolution of the C 1s signal generates peaks at 283.34, 284.53, 287.38, and 288.23 eV, attributable to the C–CC, C–N, CO, and CN bonds for both COFs, respectively (Fig. S7a and c†). The high-resolution O 1s spectrum display peaks at 530.31 and 531.32 eV assignable to the C–O and CO bonds, respectively (Fig. S7b and d†). Furthermore, the high-resolution analysis provides detailed information about the nitrogen-containing functional groups present in the COFs, as shown in Fig. 2f. The N 1s spectra for both COFs reveal distinctive peaks at 397.4 and 399.1 eV, associated with the C–N bond and the imine linkage (CN), respectively, which confirms the successful imide–imine linkage on the synthesized COFs.
The XRD pattern of Py-DNII-COF shows intense peaks at 2.55, 4.96, 12.86, 17.31, 22.83, and 24.48° (Fig. 3a), suggesting that the prepared Py-DNII-COF is a crystalline framework. In addition, the Pawley refinement method was used to simulate the optimum structure of Py-DNII-COF. The simulated PXRD pattern matches well with the experimental one, achieving lower residual values of Rwp (6.17%) and Rp (4.18%). The results imply that Py-DNII-COF mainly adopts the eclipsed AA stacking mode (Fig. 3b and c) of the P1 space group with lattice parameters of a (3.89 Å), b (36.74 Å), c (52.33 Å), α (135.07°), β (86.09°), and γ (79.92°). However, the atomic positions are recorded in Table S1.† Scanning electron microscopy (SEM) and high-resolution transmittance electron microscopy (HRTEM) images reveal fluffy aggregation of a needle-like structure for Py-DNII-COF, which could be the result of the strong π–π stacking interactions between adjacent layers with distance of about 4 Å, which is in a good agreement with the simulated XRD result.
 | ||
| Fig. 3 Pawley refinement for Py-DNII-COF with space-filling AA stacking model (a), the top view (b), and side view of AA stacking (c); SEM of Py-DNII-COF (d); HRTEM image [inset of length distribution] of Py-DNII-COF (e). | ||
In addition, the simulated XRD of TFPB-DNII-COF (Fig. S5†) reveals a relatively higher residual value of Rwp (8.89%). The Rp (5.98%) and the simulated model (of a space group P1, with lattice parameters of a (2.84 Å), b (35.11 Å), c (34.66 Å), α (44.29°), β (83.72°), and γ (89.71°)), show a twisted molecular geometry along the COF chains, presenting lower calculated pore width. However, the dihedral angle of 47° was measured from the optimized geometry structure, which shows that the geometry of the compound is not perfectly planar and exhibiting what is known as torsion or steric strain, which could be the reason behind a little higher residual compared to Py-COF. In addition, the SEM and TEM images of TFPB-DNII-COF (Fig. S6†) show the relatively high deformation and irregular shape of the crystals. These observations can impact the compound properties and reactivity, which could be the reason behind the lower activity of TFPB-DNII-COF in the experimental photocatalytic reduction.
Fig. S7† illustrates type (II) isotherms for Py-DNII-COF and TFPB-DNII-COF observed by N2 adsorption–desorption studies, which typically indicate COFs with microporous characteristics. Py-DNII-COF has a significantly higher Brunauer–Emmett–Teller (BET) surface area (183.00 m2 g−1) compared to TFPB-DNII-COF (45.00 m2 g−1). This suggests that Py-DNII-COF has a more extensive internal surface structure, possibly due to its specific structural features. In addition, Py-DNII-COF had an aperture that was spread at an average pore size of 4.1 nm and 7.4 nm (Fig. S8†), which are relatively large pores compared to TFPB-DNII-COF. Furthermore, the low sorption capacity and thus possibly obstructed pores of the TFPB-DNII-COF may either be attributed to their low crystallinity and the ill-defined stacking in the material, or due to residuals present in the pores, or both.37 Thermogravimetric analysis (TGA) conducted in this study was used to measure the weight loss of the material as a function of temperature. Py-DNII-COF and TFPB-DNII-COF were analyzed through TGA/Dr-TGA (differential thermal gravimetric analysis) with a heating ramp 20.00 °C min−1 to 800 °C under N2 gas to show the thermal degradation process of COFs (Fig. S9†) and better understand the mechanism of the degradation process.31 The synthesized COFs show a significantly high thermal stability of 420 and 450 °C for Py-DNII-COF and TFPB-DNII-COF, respectively. The Dr-TGA of Py-DNII-COF shows three steps of degradation. The 1st step is in the range of 350–420 °C, resulting in a weight loss of about 3% due to the removal of the adsorbed solvent molecules trapped inside the micro-pores of COF particles. This was followed by two extra steps at 515 °C and 635 °C with a mass loss of about 12% and 43%, respectively. In addition, TFPB-DNII-COF shows two steps of degradation at about 546 °C and 661 °C with mass loss of 39% and 60%, respectively. Comparing the Td5 and Td10 values (Table S2†) can give us an indication of how these materials react to initial thermal decomposition. TFPB-DNII-COF seems to resist initial decomposition slightly better but undergoes a more complete decomposition at high temperatures, as indicated by the lower char yield. In contrast, Py-DNII-COF starts decomposing at a slightly lower temperature but retains more structure, giving a higher char yield, which could suggest a better performance at high temperatures. Further, the Coats–Redfern model based on TGA data was used to estimate the thermodynamic parameters of a decomposition reaction, and the results are recorded in Table S3.† Regarding Py-DNII-COF, the energy barrier of 27386 J mol−1, 87191 J mol−1, and 55274 J mol−1 at 352 °C, 515 °C, and 635 °C of the three steps is required to overcome the decomposition process. Moreover, the positive enthalpy changes of 22189 J mol−1, 80640 J mol−1, and 47725 J mol−1 indicate the endothermic reaction at all stages. In the case of TFPB-DNII-COF, it shows similar behavior to Py-DNII-COF in the decomposition process. Taken together, there is a decrease in the entropy in each step, while it is the reverse at higher temperatures, which can occur during reactions where gases are produced from COF materials. Furthermore, the change in Gibbs free energy implies that the decomposition reaction is not spontaneous under normal conditions. The increasing magnitude of the change in Gibbs free energy also suggests that any reaction becomes progressively less favorable unless continuously supplied with energy, which indicates a high thermal stability for the synthesized COFs.
Photoelectronic properties of imide–imine-based COF
The UV-vis DRS spectra provides valuable insights into the optical absorption properties of the synthesized COFs (Fig. 4a). Notably, the optical absorption responses for both Py-DNII-COF and TFPB-DNII-COF are observed at wavelengths of 640 nm and 670 nm, respectively. Employing Tauc plots (Fig. S11†), the calculated band gaps were found at 2.37 eV and 2.64 eV for Py-DNII-COF and TFPB-DNII-COF, respectively. It is well known that the determination of Eg reveals important information about their electronic structure, which signifies the energy required for an electron to transition from the valence band to the conduction band. A lower band gap of Py-DNII-COF suggests heightened sensitivity to gases characterized by lower ionization energies or those capable of inducing alterations in the electronic structure within this specific energy range, which is particularly suitable for gas evaluation applications. It is worth mentioning that the slightly red shift of Py-DNII-COF in comparison to the TFPB-DNII-COF spectra can be attributed to a higher degree of the planar framework.38 Furthermore, ultraviolet photoemission spectroscopy (UPS) was utilized to study the energy levels of these COFs (Fig. 4b and S12†). The valence band maximum (VBM) and conduction band minimum (CBM) of Py-DNII-COF and TFPB-DNII-COF were −5.07/−2.64, and −5.40/−2.76, respectively (Fig. 4b). Additionally, the correlation between the applied potential and the energy levels of electronic states is intricately governed by the electrochemical system employed in the cyclic voltammetry analysis. VBM was approximated at −5.55 and −5.51 eV for Py-DNII-COF and TFPB-DNII-COF, respectively, as well as the CBM for Py-DNII-COF (−3.18 eV) and TFPB-DNII-COF (−2.87 eV) (Fig. S13†). The UPS and cyclic voltammetry analysis confirmed that both COFs can reduce water to hydrogen (H2). The photoluminescence (PL) characteristics of Py-DNII-COF and TFPB-DNII-COF are presented in Fig. 4c, providing insights into their emission behavior in relation to the recombination mechanism of photoelectrons and holes. In the context of PL spectroscopy, the emission efficiency is intricately linked to the probabilities of radiative and nonradiative transitions.39 Thus, the reduced PL emission, as seen in Py-DNII-COF, may be associated with a higher propensity for nonradiative recombination, whereas the higher emission in TFPB-DNII-COF suggests a more favorable radiative recombination process. The lower emission in Py-DNII-COF prevents the recombination of electron (e−) and hole (h+). These findings are integral for understanding the performance of these Py-DNII-COF in H2 evolution applications. | ||
| Fig. 4 UV-visible absorption spectra (a); band diagram and their thermodynamic equilibrium proton reduction in the vacuum scale (b); steady state PL spectra (c); TRPL spectra (d); transient photocurrents (e); and EIS (f) of both COFs. | ||
Moreover, time-resolved fluorescence decay spectra (TRPL) were recorded to evaluate the excited-state lifetime (Fig. 4d). The weighted average lifetime (τavg) of Py-DNII-COF (9.12 ns) was longer than that of TFPB-DNII-COF (5.93 ns), further reflecting the enhanced separation efficiency and better photocatalytic potential.40 The electrochemical measurements, specifically photocurrent density responses and electro-impedance results, are illustrated in Fig. 4e and f. Additionally, the study of charge separation and migration behaviors involved the examination of transient photocurrents and electrochemical impedance spectroscopy (EIS). The transient photocurrent of Py-DNII-COF (Fig. 4e) exhibited a notable enhancement when compared to TFPB-DNII-COF, suggesting the rapid photoresponse of Py-DNII-COF. EIS measurements of the Nyquist plot (Fig. 4f) showed that Py-DNII-COF exhibits the smallest radius, suggesting its low interface charge-transfer resistance and high conductivity compared to TFPB-DNII-COF.
Photocatalytic hydrogen production
The catalytic efficiency of the COFs was subsequently assessed for visible light-induced hydrogen evolution. Experimental procedures involved suspending the optimized 1 mg of COFs in a solution of NMP and H2O at pH 4 (adjusted with 1 M KOH) while subjecting it to visible light irradiation at 25 °C, as detailed in the experimental section. The introduction of hexachloroplatinic acid facilitated the in situ formation of the platinum (Pt) co-catalyst, effectively mitigating the overpotential for hydrogen evolution. The initial stages of optimization involved the use of triethanolamine (TEOA), triethylamine (TEA), and ascorbic acid (AA) as sacrificial electron donors. Remarkably, Py-DNII-COF demonstrated hydrogen evolution over 5 hours (Fig. 5a), utilizing different sacrificial electron donors (SED). In addition, the optimized photocatalyst quantities of (1, 3, and 5 mg) and co-catalysts were evaluated, as shown in Fig. S14 and S15.†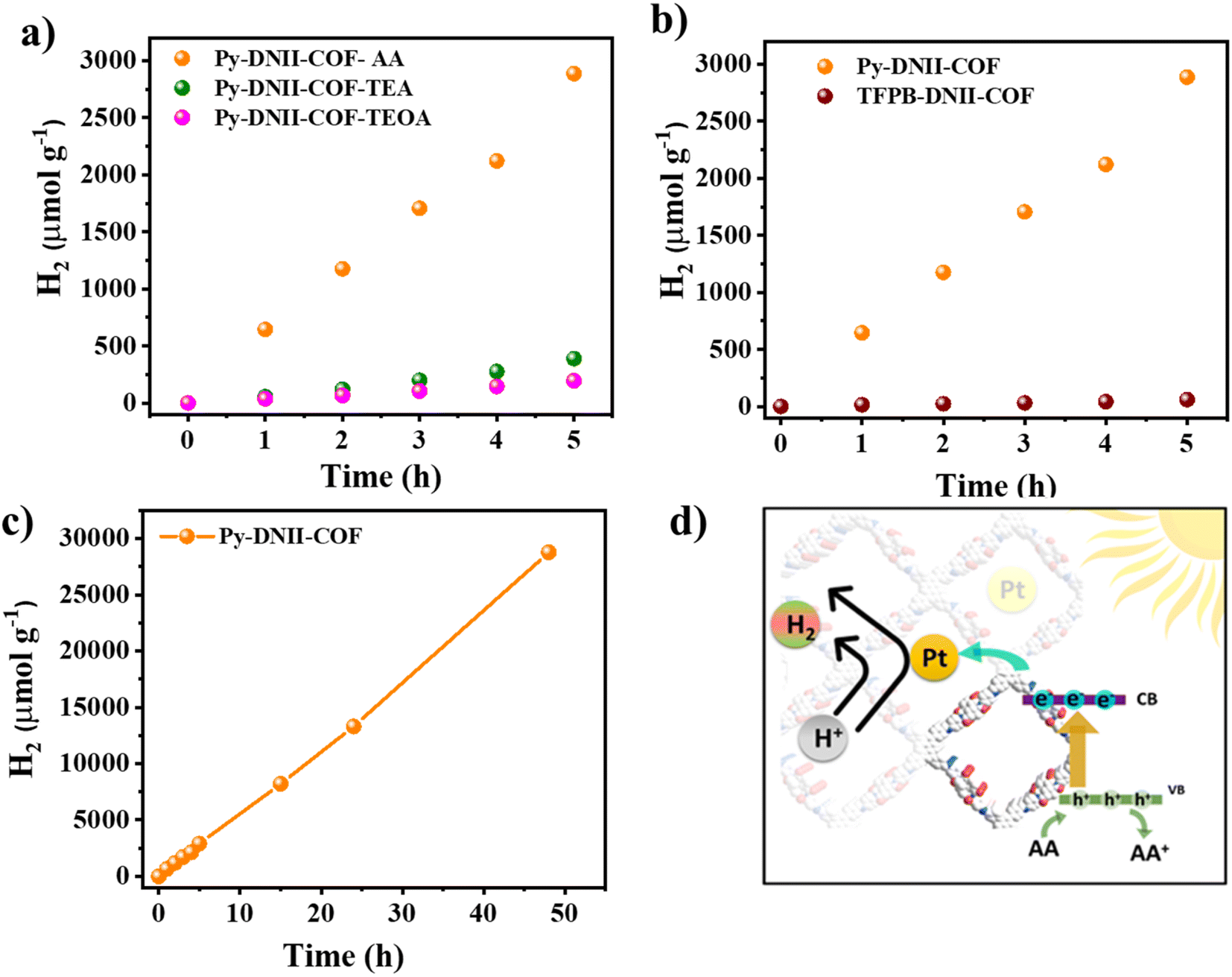 | ||
| Fig. 5 The H2 evolution rate of SEDs (AA, TEA, TEOA) effect on the Py-DNII-COF (a); behavior of Py-DNII-COF in comparison with TFPB-DNII-COF (b); Py-DNII-COF lasting up to 48 h (c); proposed photocatalytic hydrogen production mechanism under visible-light irradiation (d). | ||
Noteworthy is the superior average hydrogen evolution rate of 577 μmol h−1 g−1 for AA, compared to 80 μmol h−1 g−1 and 40 μmol h−1 g−1 for TEA and TEOA, respectively. As anticipated, Py-DNII-COF exhibited a 104-fold enhancement in hydrogen evolution compared to the limited production (6 μmol h−1 g−1) observed in TFPB-DNII-COF (Fig. 5b). Additionally, the calculated AQY for Py-DNII-COF and TFPB-DNII-COF stood at 0.30% and 0.04% at 420 nm, respectively (Fig. S16†). Besides, we investigated the AQY of Py-DNII-COF using band-pass filters (420, 460, and 500 nm) corresponding to its visible light absorption (Fig. S17†). Extended photocatalysis up to 48 hours (Fig. 5c) using Py-DNII-COF revealed remarkable stability with an average hydrogen evolution of about 625 μmol h−1 g−1. Furthermore, Py-DNII-COF achieved good value of 377.5 μmol g−1 h−1 in H2O/AA. Notably, this value was consistent with that achieved in the presence of NMP as a co-solvent (625 μmol g−1 h−1) (Fig. S18†). This underscores water as the primary source of hydrogen, as opposed to the decomposition products of the COF.38 To validate our concept, we compared Py-DNII-COF to other COF-based materials (Table S4†). The phenomenon is illustrated in the proposed photocatalytic reaction mechanism (Fig. 5d). The D–A–A architecture with push–pull–pull phenomenon for Py-DNII-COF creates a directional flow of photogenerated electrons upon light absorption. The excited electron is propelled from the strong donor (pyrene) to the strong acceptor (imide bond) through weak acceptor (imine bond), effectively extending its lifetime and hindering the recombination of the hole remaining on the donor molecule by neutralizing it using ascorbic acid (AA) as a sacrificial reagent. Aligning with Zhen Li et al.‘s findings,41 Pt nanoparticles were found to bond with active sites within the COFs. Consequently, the uniform dispersion of Pt nanoparticles in Py-DNII-COF layers surface, forming Pt–O binding sites, played a pivotal role in enhancing effective charge separation and transfer, thereby elevating the overall efficiency of the photocatalytic system. These outcomes contribute to the broader landscape of materials science and renewable energy research, opening avenues for continued advancements in sustainable technology.
Conclusion
This work reports the successful incorporation of donors, weak electron-deficient imine groups, and strong electron-deficient imide within the 2D COF structure. This deliberate combination creates favorable push–pull–pull interactions, facilitating efficient charge separation and transport post-photoexcitation. Our investigation of 2D imide–imine covalent organic frameworks, specifically Py-DNII and TFPB-DNII COFs, as novel photocatalysts into the catalytic potential for visible light-induced hydrogen evolution provides valuable insights. Through meticulous optimization processes, we determined that Py-DNII-COF, when employed as a photocatalyst, exhibits a significantly enhanced hydrogen evolution rate, particularly when ascorbic acid (AA) serves as the sacrificial electron donor. The 104-fold increase in hydrogen evolution observed over an extended 48 hours period underscores the sustained efficacy of Py-DNII-COF, with water identified as the primary source of hydrogen. Comparative analysis with TFPB-DNII-COF revealed a substantial performance gap, emphasizing the superior catalytic prowess of Py-DNII-COF in harnessing visible light for hydrogen evolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
It is with great gratitude that the authors acknowledge the financial support of the National Science and Technology Council of Taiwan (NSTC 112-2223-E-007-006-MY3; NSTC 111-2628-E-007-009; NSTC 111-2221-E-007-004; NSTC 111-2622-8-007-011; NSTC 112-9017-2, NSTC-112-2221-E-007-081–; NSTC-112-2823-8-007-001–; NSTC-112-2622-E-007-032; NSTC 113-2923-E-007-007-MY3; NSTC 113-2823-8-007-003) as well as the computing time provided by the National Center for High-Performance Computing of Taiwan. The authors thank the Precision Instrument Support Center of National Tsing Hua University for providing measurement and analysis facilities.References
- W. T. Chung, I. M. A. Mekhemer, M. G. Mohamed, A. M. Elewa, A. F. M. EL-Mahdy, H. H. Chou, S. W. Kuo and K. C.-W. Wu, Coord. Chem. Rev., 2023, 483, 215066 CrossRef CAS.
- F. Xu and B. Weng, J. Mater. Chem. A, 2023, 11, 4473–4486 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- Z. Li, Y. Zhi, X. Feng, X. Ding, Y. Zou, X. Liu and Y. Mu, Chem.–Eur. J., 2015, 21, 12079–12084 CrossRef CAS PubMed.
- Z. Li, X. Feng, Y. Zou, Y. Zhang, H. Xia, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825–13828 RSC.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., 2014, 126, 2922–2926 CrossRef.
- J. Liu, N. Wang and L. Ma, Chem.–Asian J., 2020, 15, 338–351 CrossRef CAS PubMed.
- Y. Yusran, H. Li, X. Guan, Q. Fang and S. Qiu, EnergyChem, 2020, 2, 100035 CrossRef.
- X. Wu, G. Chen, J. Wang, J. Li and G. Wang, Acta Phys.-Chim. Sin., 2023, 39, 2212016 Search PubMed.
- W. Wang, W. Zhao, H. Xu, S. Liu, W. Huang and Q. Zhao, Coord. Chem. Rev., 2021, 429, 213616 CrossRef CAS.
- M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120, 8581–8640 CrossRef CAS PubMed.
- T. He, K. Geng and D. Jiang, ACS Mater. Lett., 2019, 1, 203–208 CrossRef CAS.
- F. Li, D. Wang, Q. J. Xing, G. Zhou, S. S. Liu, Y. Li, L.-L. Zheng, P. Ye and J.-P. Zou, Appl. Catal., B, 2019, 243, 621–628 CrossRef CAS.
- C. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef PubMed.
- J. Sheng, H. Dong, X. Meng, H. Tang, Y. Yao, D. Liu, L. Bai, F. Zhang, J. Wei and X. Sun, ChemCatChem, 2019, 11, 2313–2319 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 1–9 CAS.
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, Q. Li, G. Wu, H. Xia, S. Yang and X. Liu, Angew. Chem., 2022, 134, e202208919 CrossRef.
- H. Liu, D. Wang, Z. Yu, Y. Chen, X. Li, R. Zhang, X. Chen, L. Wu, N. Ding and Y. Wang, Sci. China Mater., 2023, 66, 2283–2289 CrossRef CAS.
- Z. Li, T. Li, J. Miao, C. Zhao, Y. Jing, F. Han, K. Zhang and X. Yang, Sci. China Mater., 2023, 66, 2290–2298 CrossRef CAS.
- G. B. Wang, H.-P. Xu, K.-H. Xie, J.-L. Kan, J. Fan, Y. J. Wang, Y. Geng and Y. B. Dong, J. Mater. Chem. A, 2023, 11, 4007–4012 RSC.
- Z. Xie, X. Yang, P. Zhang, X. Ke, X. Yuan, L. Zhai, W. Wang, N. Qin, C. X. Cui and L. Qu, Chin. J. Catal., 2023, 47, 171–180 CrossRef CAS.
- J. You, Y. Zhao, L. Wang and W. Bao, J. Cleaner Prod., 2021, 291, 125822 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou and X. Li, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- X. Li, S. Cui, D. Wang, Y. Zhou, H. Zhou, Y. Hu, J. Liu, Y. Long, W. Wu and J. Hua, ChemSusChem, 2014, 7, 2879–2888 CrossRef CAS PubMed.
- I. M. A. Mekhemer, Y. Wu, A. M. Elewa, W. Chen, C. Chueh and H. Chou, Sol. RRL, 2024, 2300994 CrossRef CAS.
- M. H. Elsayed, M. Abdellah, A. Z. Alhakemy, I. M. A. Mekhemer, A. E. A. Aboubakr, B. H. Chen, A. Sabbah, K. H. Lin, W. S. Chiu and S. J. Lin, Nat. Commun., 2024, 15, 707 CrossRef CAS PubMed.
- H. L. Nguyen, C. Gropp and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 2771–2776 CrossRef CAS PubMed.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng and J. Liu, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed.
- X. Song, Y. Wu, X. Zhang, X. Li, Z. Zhu, C. Ma, Y. Yan, P. Huo and G. Yang, Chem. Eng. J., 2021, 408, 127292 CrossRef CAS.
- A. Bafaqeer, M. Tahir and N. A. S. Amin, Appl. Catal., B, 2019, 242, 312–326 CrossRef CAS.
- W. Jiang, W. R. Cui, R. P. Liang and J. D. Qiu, Chem. Eng. J., 2021, 417, 128034 CrossRef CAS.
- J. Z. Cheng, L. L. Liu, G. Liao, Z. Q. Shen, Z. R. Tan, Y. Q. Xing, X. X. Li, K. Yang, L. Chen and S. Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- M. M. Samy, I. M. A. Mekhemer, M. G. Mohamed, M. H. Elsayed, K. H. Lin, Y. K. Chen, T. L. Wu, H.-H. Chou and S. W. Kuo, Chem. Eng. J., 2022, 464, 137158 CrossRef.
- A. M. Elewa, A. F. M. EL-Mahdy, A. E. Hassan, Z. Wen, J. Jayakumar, T. L. Lee, L. Y. Ting, I. M. A. Mekhemer, T. F. Huang and M. H. Elsayed, J. Mater. Chem. A, 2022, 10, 12378–12390 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- C. Wang, F. Zheng, L. Zhang, J. Yang and P. Dong, Appl. Surf. Sci., 2023, 640, 158383 CrossRef CAS.
- F. Ma, Q. Tang, S. Xi, G. Li, T. Chen, X. Ling, Y. Lyu, Y. Liu, X. Zhao and Y. Zhou, Chin. J. Catal., 2023, 48, 137–149 CrossRef CAS.
- M. Wang, Z. Wang, M. Shan, J. Wang, Z. Qiu, J. Song and Z. Li, Chem. Mater., 2023, 35, 5368–5377 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01108b |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |