
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithiophilic interlayer driven ‘bottom-up’ metal infilling in high current density Li-metal anodes†
Syed
Abdul Ahad
a,
Janina
Drews
 bc,
Timo
Danner
bc,
Arnulf
Latz
bcd and
Hugh
Geaney
*a
bc,
Timo
Danner
bc,
Arnulf
Latz
bcd and
Hugh
Geaney
*a
aDepartment of Chemical Sciences, Bernal Institute, University of Limerick, Limerick, V94 T9PX, Ireland. E-mail: hugh.geaney@ul.ie
bInstitute of Engineering Thermodynamics, German Aerospace Center (DLR), Wilhelm-Runge-Straße 10, 89081 Ulm, Germany
cHelmholtz Institute Ulm (HIU), Helmholtzstr. 11, 89081 Ulm, Germany
dInstitute of Electrochemistry, Ulm University (UUlm), Albert-Einstein-Allee 47, 89081 Ulm, Germany
First published on 23rd April 2024
Abstract
Lithium (Li) metal holds great potential for pushing practical energy densities beyond state-of the art Li-ion batteries. However, parasitic problems including Li dendrite formation can result in separator piercing, subsequent short-circuit and ultimately thermal runaway. Here we propose an innovative interlayer strategy that is guided by continuum simulations in 1D and 3D, which shows that materials with low Li nucleation overpotentials and high surface areas can enable spatially controlled plating of Li. This insight inspires an interlayer consisting of highly lithiophilic germanium nanowires (Ge NWs) coated on one side of a carbon cloth (CC). This anode geometry effectively unlocks Li infilling by a “bottom-up” motif during stripping/plating cycles. As a result, dendrite formation is eliminated, with the GeCC interlayer acting as a controlling Li reservoir during stripping/plating cycles. Ultra-stable symmetric cell performance up to 2500 h was achieved, with low overpotentials at high current density (2 mA cm−2) and plating capacity (2 mA h cm−2). Furthermore, aggressive higher current density (4 mA cm−2) and plating capacity (4 mA h cm−2) conditions were enabled by this approach. The high performing GeCC interlayer modified Li metal anodes were tested with LiFePO4 and NMC cathodes, facilitating greatly enhanced cyclic stability compared to control cells.
1. Introduction
Technological advancements and rising consumer demand for portable electric devices and electric vehicles (EVs) have intensified the requirement for high energy density batteries.1–3 Current state-of-the art Lithium (Li)-ion batteries cannot fulfil this demand indefinitely, in part due to the capacity limitation of commercially available graphite anodes (372 mA h g−1).4–6 Li metal as an anode material is an attractive option to enable high energy density batteries; mainly due to its low density (0.534 g cm−3), high theoretical specific capacity (3860 mA h g−1), and lowest redox potential (−3.04 V vs. standard hydrogen electrode (SHE)).7,8 However, rapid development of Li metal anodes is hindered by problems such as inhomogeneous Li plating/stripping, infinite volume expansion, formation of an unstable solid electrolyte interphase (SEI) and eventually dendrite formation during cycling. The formation of dendrites on the Li surface can pierce the cell separator and cause short-circuiting; a common cause of well publicised fires in electronic devices.9–11To overcome the hurdles of Li metal anode development, notable strategies have been examined. (i) The formation of high-modulus artificial solid electrolyte interphase (SEI) layers on the Li surface to avoid dendrite penetration and to avoid excessive electrolyte consumption (which limits cell coulombic efficiencies).12–16 (ii) The development of electrolytes with tailored compositions17–20 and concentrations21–23 along with additives24–26 to avoid dendrite formation during cycling. (iii) The preparation of 3D frameworks to accommodate volumetric expansion of Li metal during stripping/plating cycles.27–30 (iv) The use of functional coating/interlayers of 3D frameworks with carbon derivatives31–33 or elemental coatings such as Al, Si, Sn (and/or their oxides)34–40 to induce lithiophilic effects at the Li electrode/electrolyte interface. For functional coatings on Li metal, lithiophilic–lithiophobic gradient layers on foams (as standalone Li hosts) or via interlayers (coated on Li metal) are an exciting emerging option to guide Li deposition/stripping.35,36,41 Gradient coatings with the lithiophilic side facing towards the metallic Li surface tend to guide Li ions to preferentially lithiate the lithiophilic material, followed by Li deposition on it. In this way, ‘top-down’ deposition of Li that is typically seen for standard Li metal anodes is avoided, with this approach enabling the avoidance of dendrite formation and associated performance failure and/or short circuit.35,42 Despite the identification of various interlayer types to guide Li deposition, further development of simple interlayer architectures which enable higher current density Li stripping/plating and sufficient reservoir space to accommodate high plating capacities of Li is urgently required.
Based on comprehensive continuum simulations, we propose a pioneering strategy to coat highly lithiophilic germanium (Ge) nanowires (NWs) on one side of a flexible carbon cloth (CC), creating a Li-deposition guiding gradient from lithiophilic (Ge) to lithiophobic (carbon). These flexible, mechanically robust and free-standing interlayers can be directly pasted onto Li metal, enabling high performance. To grow Ge NWs, CC was chosen as a base layer since it is known to have good electrical conductivity and porous structure for better electrolyte accommodation.43,44 The GeCC interlayer boasts several advantages compared to lithiophobic CC and bare Li metal anodes such as: (i) the use of Ge NWs enhances the surface area at the Li interface, which reduces the localized current density,45–47 thereby promoting uniform Li deposition on the Li surface. (ii) The formation of a lithiophilic LiGe phase due to the short-circuiting of Ge NWs with the Li metal promotes preferential Li deposition on the lithiophilic side of the interlayer. (iii) Unlike the inherently lithiophobic control CC interlayer, the Ge NWs facing the Li metal in the GeCC interlayer promote a ‘bottom-up’ infilling of Li during Li stripping/plating cycles. Benefitting from the advantages stated above, the LiGeCC anode delivered exceptional long-term symmetric cell performance for 2500 h at a high current density of 2 mA cm−2, with a plating capacity of 2 mA h cm−2. This indicates reversible Li-ion transport during deposition/stripping during long-term cycling even at high current densities (2 mA cm−2, 4 mA cm−2) for the LiGeCC anode. Extensive post-cycling SEM analysis reveals strong interfacial adherence between Li and the lithiated Ge phase. Furthermore, when coupled with a high loading NMC cathode, a LiGeCC anode containing full cell delivered a high average coulombic efficiency of 99.7% with 80% capacity retention after long-term cycling compared to bare Li and LiCC retaining only 5% and 48% capacity.
2. Methodology
2.1 Experiments
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DME, 1:1, vol%) + 0.25 M lithium nitrate along with Celgard separator. The volume of electrolyte used across all the cells was 60 μl. For LiFePO4 (LFP) cathode, the slurry was cast on an aluminum (Al) foil using 80% LFP, 10% carbon black (CB) and 10% PVDF mixed in NMP solvent, followed by drying in a vacuum oven, overnight at 70 °C. The LFP loading obtained was 4.5 mg cm−2 while the NMC811 was procured from NEI Corporation with an active material loading of 10 mg cm−2 (2 mA h cm−2). For high loading NMC811, 1 M LiPF6 (EC:DEC) + 10% FEC electrolyte was used to avoid electrolyte degradation in the tested voltage window. The symmetric cell and half-cell testing along with electrochemical impedance spectroscopy was conducted using either Neware battery cycler or Biologic instrument.
:DME, 1:1, vol%) + 0.25 M lithium nitrate along with Celgard separator. The volume of electrolyte used across all the cells was 60 μl. For LiFePO4 (LFP) cathode, the slurry was cast on an aluminum (Al) foil using 80% LFP, 10% carbon black (CB) and 10% PVDF mixed in NMP solvent, followed by drying in a vacuum oven, overnight at 70 °C. The LFP loading obtained was 4.5 mg cm−2 while the NMC811 was procured from NEI Corporation with an active material loading of 10 mg cm−2 (2 mA h cm−2). For high loading NMC811, 1 M LiPF6 (EC:DEC) + 10% FEC electrolyte was used to avoid electrolyte degradation in the tested voltage window. The symmetric cell and half-cell testing along with electrochemical impedance spectroscopy was conducted using either Neware battery cycler or Biologic instrument.
2.2 Continuum model
The kinetics of Li plating and stripping are described by the common Butler–Volmer equation (eqn (S1) and (S2)†).50–54 Thereby, the reaction is controlled by the overpotential η (eqn (1)), which denotes the deviation of the electrochemical potential (Φs − φe) from the equilibrium (U0).| η = Φs − φe − U0 − (1 − Θ)ηnuc | (1) |
Since plating is taking place on a host structure, there is an additional contribution to the overpotential by nucleation of Li deposits on the electrode surface with the material-specific nucleation overpotential ηnuc. Once the Li nucleated, the continued growth does not require the extra overpotential ηnuc anymore. This is taken into account by the fractional coverage of the electrode structure with Li metal Θ, which is given by eqn (2). Note, that the continuously differentiable power law is chosen to guarantee numerical stability of the simulations.
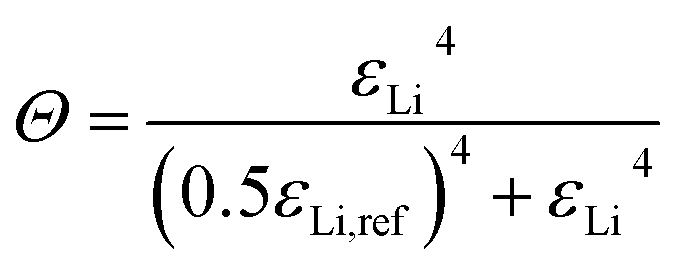 | (2) |
When the volume fraction of plated Li εLi reaches the reference value εLi,ref the electrode surface is fully covered (Θ ≈ 1, eqn (2)) and consequently, the contribution of nucleation to the overall overpotential vanishes (eqn (1)). Thereby, εLi,ref is a model parameter which reflects the microscopic plating morphology and needs to be calibrated thoroughly. The above presented kinetic model for Li plating with nucleation is coupled to a thermodynamic consistent transport theory (eqn (S3)–(S5)†).53 The implementation in the Battery and Electrochemistry Simulation Tool (BEST) enables 3D microstructure resolved simulations.55 Thereby, the previously reported model for Li plating and stripping was extended and for consistency eqn (2) was expressed in terms of the molar quantity of Li metal nLi and nLi,ref, respectively.56 Note, that the reference values εLi,ref and nLi,ref can easily be converted into each other (eqn (S9) and (S10)†). However, the 3D simulation framework does not explicitly resolve changes of electrolyte and Li volume fractions due to the electrochemical reaction and therefore pore clogging and resulting effects can not be depicted. Moreover, due to complexity a propagation of Li growth between voxels is not considered. Therefore, results of 3D simulations are only considered to be reliable in the initial plating phase. To further study the Li filling of the host structure and to analyze the impact of different parameters, the model is additionally implemented in a homogenized 1D framework (Fig. S1†). Thereby, the change of the Li volume fraction is explicitly resolved (eqn (S6)†), whereby the specific surface area available for Li plating is considered by an empirical expression (eqn (S7)†). Finally, the propagation of Li growth between the control volumes of the spatial discretization is considered by introducing an effective value εLi,eff (eqn (S8)†), which replaces εLi in eqn (2) and (S7).† All model extensions in the 1D framework are described in more detail in the ESI (eqn (S6)–(S8)†). Note, that the homogenized 1D approach is thoroughly validated by simulating the Li plating onset in 3D (Fig. S1†).
The model parameters are chosen based on the experimental setup, as well as on literature and manufacturer data. In general, Li deposition on lithiated Ge can happen without a significant nucleation overpotential (ηnuc(Ge) ≈ 0 V), while plating on CC requires to overcome a nucleation barrier of several tens of mV.57 The microstructure for the 3D simulations with a size of 2.5 × 2.5 mm was extracted from micro-CT data of the CC (Fig. S2†) with a resolution of 5 μm and the voxel on the CC surface closest to the current collector was assumed to be covered by Ge NWs, which can not explicitly be resolved. Further details on the parameterization of the model can be found in the ESI (i.e., Tables S1–S3†), including all details on the choice, interpretation, and influence of εLi,ref.
3. Results and discussion
CC was chosen as the starting point for simulations, with the structural characteristics captured by a 3D reconstruction based on micro-CT (Fig. S2†). As reported previously, anodes based on a single active material always favour Li plating on the separator facing side.56,58–62 This phenomenon was also observed in the 3D and 1D simulations of the plain CC anode (Fig. 1a and S3a†) where a clear preference for separator side Li plating was observed. For the longer 1D simulation, even severe limitations caused by pore clogging were found. Such a plating behaviour significantly increases the risk of a detrimental short-circuit by dendrite formation. However, a lithiophilic interlayer with a lower nucleation overpotential than the host framework was found to promote uniform infilling of the CC host structure evolving from the current collector rather than the separator (Fig. 1b, c and S3b†). In the 3D simulations, it can clearly be seen that the initial plating is happening at the Ge interlayer. Videos of the plating process can be found in the ESI (Videos 1–3†). After 100 s at 0.1 mA cm−2, plated Li is only found in the Ge interlayer for a nucleation overpotential at the CC of ηnuc(CC) = 40 mV (Fig. 1c). In contrast, for ηnuc(CC) = 20 mV, plating also occurs at the separator side (Fig. 1b). However, a higher amount of Li deposits are still found on the Ge interlayer side, where all initial nucleation takes place. The subsequent plating on the separator side is very likely a result from not considering propagation of Li growth between the simulation voxels. This is supported by the additional 1D simulations, where a complete bottom-up filling behaviour without any Li deposits on the separator side was observed for ηnuc(CC) = 20 mV during 40 h at 0.1 mA cm−2 (Fig. S3b†).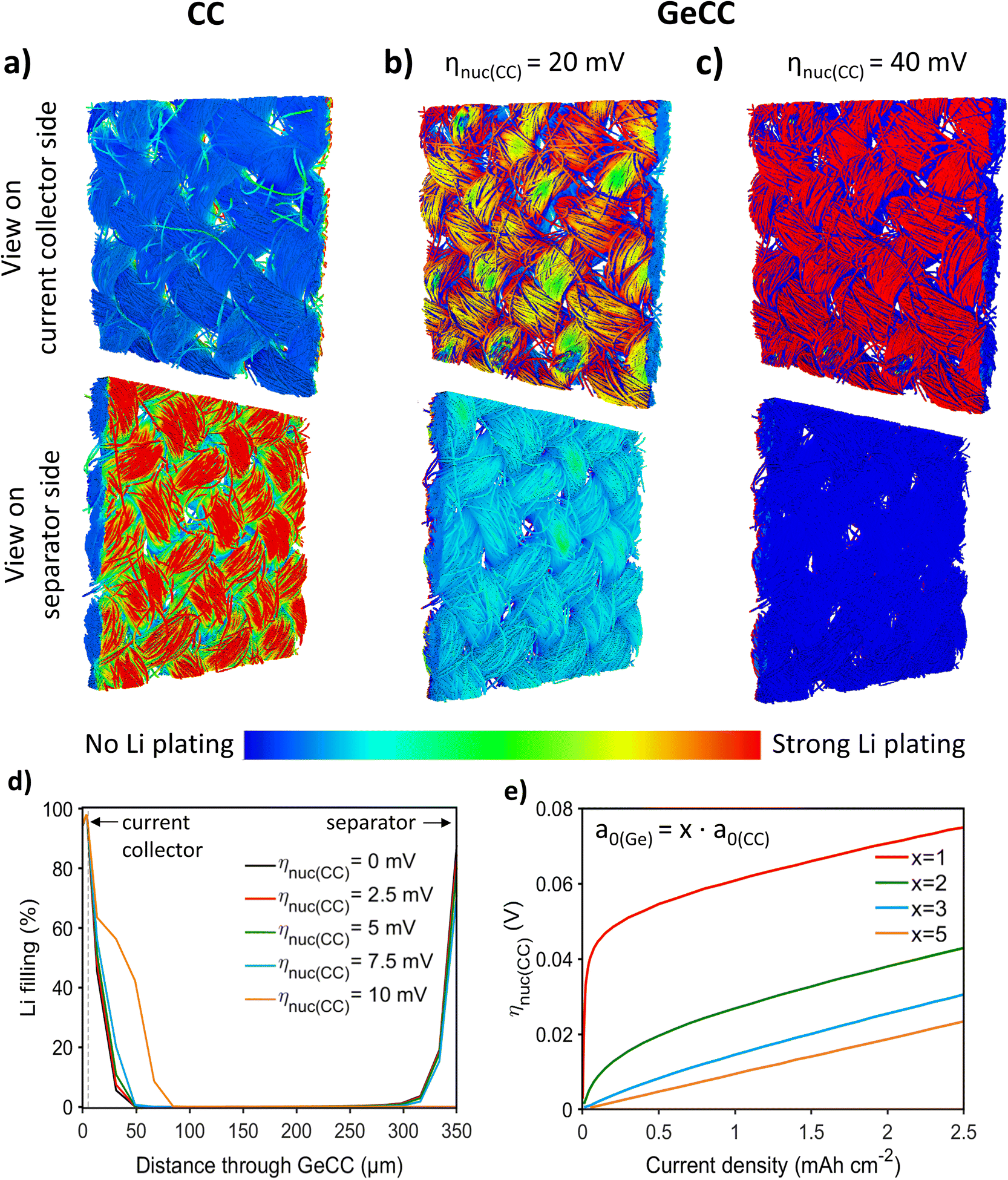 | ||
| Fig. 1 3D and 1D continuum simulations. (a–c) Amount of plated Li on CC and GeCC with ηnuc(CC) = 20/40 mV after 100 s at 0.1 mA cm−2. (d) Influence of ηnuc(CC) on the infilling of GeCC with plated Li after 4 mA h cm−2 at 0.1 mA cm−2. The Ge interlayer is positioned at 0–5 μm and is facing towards the current collector. (e) ηnuc(CC) for which the Li plating mechanism changes from on-top plating to bottom-up infilling with dependence on current density and specific surface area. | ||
Comprehensive simulation studies with the homogenized 1D model show that there is a clear switch between on-top plating and bottom-up filling behaviour when ηnuc(CC) exceeds a certain value (Fig. 1d). For higher current densities this critical nucleation overpotential difference between host structure (CC) and interlayer (Ge) required for a bottom-up infilling increases slightly (Fig. 1e). Consequently, and especially for high current densities, the host structure should be as lithiophobic as possible in terms of a high overpotential for the nucleation of Li deposits, while the interlayer should be very lithiophilic and ideally show no nucleation overpotential for Li plating at all. This finding is in agreement with the 3D simulation results (Fig. 1a–c). Moreover, the 1D simulations indicate that the difference in specific surface area a0 of CC and Ge plays a crucial role (Fig. 1e). Thereby, a gradient with a higher surface area in the lithophilic Ge interlayer compared to the CC main structure is clearly beneficial to initiate the bottom-up infilling. Fig. 1e shows that for such a gradient structure a significantly lower difference in nucleation overpotential can already cause the desired change towards the bottom-up filling behaviour. Thereby, a thin lithophilic Ge layer with only slightly higher (twofold) surface area than the CC is already efficient to prevent on-top plating. Note, that a further increase of the interlayer surface is not very impactful and in general a larger surface area can also enhance undesired side reactions. All in all the simulations strongly imply that a simple coating of the host structure with a lithiophilic material is not as effective as growing NWs and thereby enhancing the surface area of the lithiophilic interlayer. Finally, it is found that the parameter εLi,ref, which represents the Li plating morphology on a microscopic level, can also impact the Li plating behaviour (Fig. S4†). However, more detailed investigations (cf. ESI†) imply that εLi,ref is not crucial for inducing the aspired bottom-up filling. All in all, the choice of material and morphology of the host and interlayer are critical to achieve spatially guided Li plating and thereby avoid pore clogging and Li growth into the separator. Motivated by these simulation results, an interlayer of Ge NWs on CC was developed, since the difference in nucleation overpotential63 and specific surface area of the two materials were expected to favour bottom-up Li growth.
The synthesis route for the lithiophilic GeCC interlayer involves deposition of a tin (Sn) seed catalyst layer on CC using thermal evaporation, followed by growth of Ge NWs using a solvent-vapour growth technique, with diphenylgermane (DPG) as Ge precursor (Fig. S5†).49,64,65 Here we wish to mention that Sn is also a lithiophilic material but Li has lower binding energy with Sn (−2.15 eV) as compared to Ge (−2.98 eV).66 Moreover, the Sn seed is only 0.04 mg cm−2 (∼6%) as compared to a 0.6 mg cm−2 mass loading for the Ge NWs on CC; therefore, we expect that the lithiophilic effect comes mainly from the Ge NWs due to its high mass loading as compared to Sn. To prepare the Ge NW containing interlayer anodes, single side coated GeCC with the lithiophilic Ge NWs pointing towards the Li were pressed on the pristine Li foil (as shown schematically in Fig. 2a). Henceforth, the CC (control samples) and GeCC interlayers pasted on Li anode will be termed as LiCC and LiGeCC respectively. Once the GeCC interlayers were pressed on Li metal, the Li-ion flux is preferentially directed towards the Ge NWs due to its high Li-ion diffusivity, specific surface and lithiophilicity.66 This leads to ‘bottom-up’ Li infilling at the Li-interlayer interface starting from Ge lithiation, followed by Li infilling ‘upwards’ into the CC interlayer during electrochemical process. In contrast, the lithiophobic nature of carbon (C) doesn't help in guiding Li ions towards the Li-interlayer interface, therefore leading to non-uniform Li deposition on the top surface (without Li infilling in the CC).
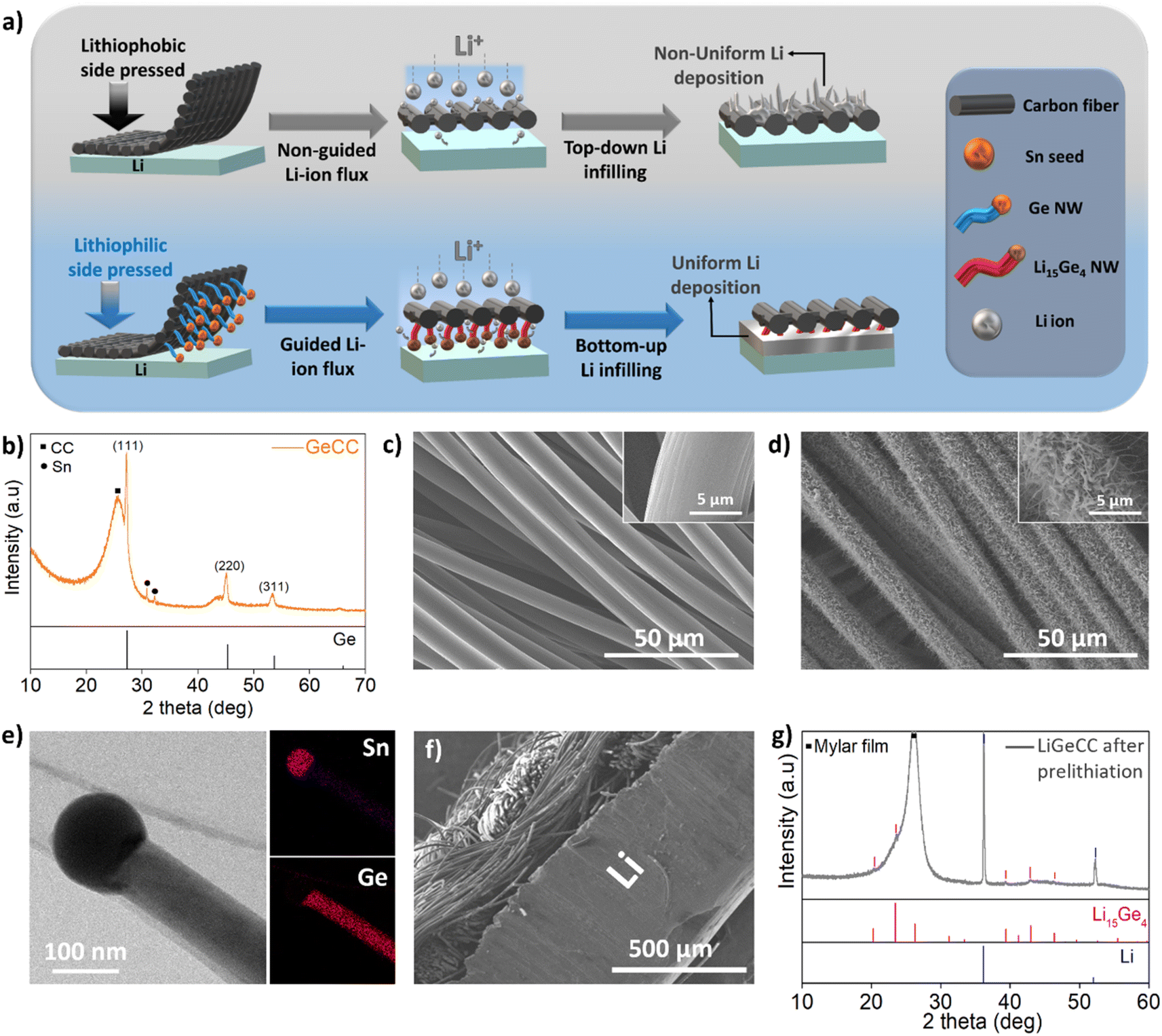 | ||
| Fig. 2 (a) Schematic demonstrating the steps involved in the use of CC (control sample) and GeCC interlayer with Li metal, including its brief mechanism. Synthesis and phase characterization of Ge NWs coated CC interlayer using (b) XRD analysis of GeCC interlayer with corresponding standard pattern of pure Ge (JCPDS no. 04-006-2620). SEM images of (c) CC and (d) GeCC interlayer. (e) TEM image of Ge NWs along with STEM mapping of Sn seed and Ge NW. (f) Cross-sectional SEM image of GeCC pressed on Li foil (LiGeCC). (g) XRD analysis of LiGeCC (after prelithiation) between 10–60° with corresponding standard patterns of pure Li15Ge4 (JCPDS no. 04-004-3527) and Li (JCPDS no. 04-006-5779) phases. | ||
The material characterization of the pristine GeCC interlayer was carried out using X-ray diffraction (XRD) analysis which showed major peaks corresponding to (111), (220) and (311) planes associated with the JCPDS no. 04-006-2620 of crystalline Ge (Fig. 2b). Other than Ge, a major peak around 26° originates from the underlying CC substrate. SEM analysis and micro-CT shows that the bare CC consists of multiple carbon fiber strands woven together, with a single carbon fiber having a smooth surface topology and an average diameter of 9.5 μm (Fig. 2c and S1, S6a†). The 25 nm layer of Sn seed evaporated on one side of CC produced islands of Sn seeds fully covering the CC (Fig. S6b†). After reaction with DPG, the Sn-seeded CC displayed dense growth of several micron-long Sn-seeded Ge NWs (Fig. 2d, inset), with an average diameter of 150.4 nm and an average mass loading of 0.6 mg cm−2 (Fig. S7†). The optical photographs of bare CC (Fig. S8a†), Sn coated CC (Fig. S8b†) and Ge NWs coated CC (GeCC) features a contrast change during each step of the process, while the flexibility of the CC was retained even after the growth of Ge NWs (Fig. S8c†).
Transmission Electron Microscopy (TEM) analysis clearly shows the presence of a Sn seed on top of a Ge NW, with energy dispersive X-ray spectroscopy (EDS) mapping highlighting the uniform distribution of Ge along the NW with Sn signals concentrated in the seed (Fig. 2e). The crystallinity of the Ge NWs and presence of other chemical moieties in GeCC was studied using X-ray photoelectron spectroscopy (XPS), where peak deconvolution confirms the presence of crystalline Ge at 29 eV and Sn at 485.1 eV in their respective core spectra (Fig. S9a–c†).49,67,68 The presence of GeO2 and SnO2 peaks also suggests that surface oxidation of the NWs occurred due to air exposure. Typical carbon moieties such as C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O bonds were also present due to the underlying CC (Fig. S9c†).69 To effectively use GeCC as interlayer in a LMB, the GeCC interlayer was physically pressed on a piece of Li metal with the Ge NW side facing the Li metal (Fig. 2f). This process should immediately start prelithiation of GeCC upon contact with the Li metal since Ge is a highly lithiophilic material.66 To increase the Li+ diffusion and accelerate the pre-lithiation process by self-discharge mechanism, a few drops of 1 M LiTFSI (DOL:DME) + 0.2 M LiNO3 were added on the LiGeCC substrate. After allowing the samples to rest for 10 h, XRD analysis revealed the formation of crystalline Li15Ge4, in addition to the presence of Li metal signals originating from the underlying Li metal in the LiGeCC substrate (Fig. 2g and S10, S11†). The formation of this Li15Ge4 alloy at the Li interface leads to several advantages including: (1) the presence of Li15Ge4 alloy at the LiGeCC interface enhances the Li-ion conductivity, helping Li ion transport and reducing dendrite formation. (2) The NW morphology at the interface minimizes the localized ion current density and promotes uniform Li ion concentration; a property important to decrease dendrite formation.45–47,70 (3) The Li deposition and stripping is directional (as demonstrated below), beginning from the Li15Ge4/Li interface and slowly filling up the rest of the CC.
C, C–O bonds were also present due to the underlying CC (Fig. S9c†).69 To effectively use GeCC as interlayer in a LMB, the GeCC interlayer was physically pressed on a piece of Li metal with the Ge NW side facing the Li metal (Fig. 2f). This process should immediately start prelithiation of GeCC upon contact with the Li metal since Ge is a highly lithiophilic material.66 To increase the Li+ diffusion and accelerate the pre-lithiation process by self-discharge mechanism, a few drops of 1 M LiTFSI (DOL:DME) + 0.2 M LiNO3 were added on the LiGeCC substrate. After allowing the samples to rest for 10 h, XRD analysis revealed the formation of crystalline Li15Ge4, in addition to the presence of Li metal signals originating from the underlying Li metal in the LiGeCC substrate (Fig. 2g and S10, S11†). The formation of this Li15Ge4 alloy at the Li interface leads to several advantages including: (1) the presence of Li15Ge4 alloy at the LiGeCC interface enhances the Li-ion conductivity, helping Li ion transport and reducing dendrite formation. (2) The NW morphology at the interface minimizes the localized ion current density and promotes uniform Li ion concentration; a property important to decrease dendrite formation.45–47,70 (3) The Li deposition and stripping is directional (as demonstrated below), beginning from the Li15Ge4/Li interface and slowly filling up the rest of the CC.
To determine the fundamental Li deposition/stripping performance of CC and GeCC, these interlayers were assembled against Li metal in half-cell configuration to analyse coulombic efficiencies (C.E) at high current (2 mA cm−2) and high areal capacity (2 mA h cm−2). The voltage–areal capacity profiles of CC and GeCC substrates demonstrate initial lithiation of CC and Ge NWs, followed by Li plating below 0 V (Fig. S12a and c†). It is evident that the nucleation overpotentials are low for GeCC (6 mV) as compared to CC (20 mV), due to the incorporation of lithiophilic Ge NWs (Fig. S12b and d†). Once stripped up to 1 V, the GeCC demonstrates much higher C.E, compared to the CC substrate. Long-term C.E testing clearly shows that GeCC delivered stable and high C.E of 98.7% over 250 cycles, while the CC substrate demonstrated massive C.E fluctuations, suggesting poor Li stripping/deposition behaviour (Fig. S12e†). The C.E fluctuations above 100% during Li plating/stripping cycles in CC would likely be due to the formation of soft short-circuits as well as formation and inconsistent reactivation of dead Li during cycling process.71 This again signifies the poor stability of CC compared to the GeCC substrate.
Symmetric cells were assembled using matched Li‖Li, LiCC‖LiCC and LiGeCC‖LiGeCC electrodes with 1 M LITFSI (DOL:DME) + 0.2 M LiNO3 electrolyte. High current density and plating capacities of 2 mA cm−2, 2 mA h cm−2 were used for initial symmetric cell testing. The cycling data clearly shows that LiGeCC outperforms LiCC and bare Li symmetric cells (Fig. 3a). Segmented voltage–time graphs at different time intervals demonstrated that with increasing time, the overpotential of LiCC and Li increased significantly compared to that of LiGeCC cells (Fig. S13a–d†). When the GeCC interlayer was introduced on Li metal with the Ge NWs facing the separator (rather than the Li), the overpotential increased significantly over a short period of time, in direct contrast to LiGeCC (Ge NWs facing Li metal) anode (Fig. S14†). This again signifies the importance of introducing lithiophilic interfacial layer at the Li metal interface. With the GeCC interlayer, a maximum overpotential of 79 mV was noted after 2500 h (Fig. 3b). The constant overpotential obtained in LiGeCC is a testament to the formation of a stable interface between the Li metal–GeCC interlayer, such that the Li plating/stripping is not creating any inactive ‘dead’ Li and excessive SEI formation which would otherwise contribute to high overpotentials.42,72 For the lithiophobic CC (with no Ge NW coating) the overpotential quickly increased to 135 mV after just 400 h. This highlights the dramatic improvement linked to introducing a NW layer of highly lithiophilic material like Ge at the Li interface. The bare Li displayed the highest overpotential of 150 mV in just over 200 h, likely due to excessive electrolyte consumption and dendrite formation.32,73 Electrochemical Impedance Spectroscopy (EIS) was performed to determine the cell resistance of electrodes during cycling using a fitting circuit comprising mainly of ohmic resistance (Rs) of the cell and also the internal resistance (RSEI+ct), comprising of SEI and charge-transfer resistance (Fig. S15 and Table S4†).74,75 The EIS revealed that the internal resistance (RSEI+ct) for LiCC and Li after 1st cycle were 36.2 ohm and 41.7 ohm respectively; almost 2.5 times higher than that of LiGeCC, which was only 15.4 ohm (Fig. 3c). This RSEI+ct decreased to 8.7 ohm after 100 cycles whereas the RSEI+ct of LiCC and Li remained significantly higher compared to LiGeCC symmetric cell (Fig. 3d, inset). The much smaller RSEI+ct value of LiGeCC suggests enhanced interfacial kinetics, due to the formation of the Li15Ge4 phase at the Li electrode–electrolyte interface.36 In contrast, the lithiophobic CC interphase could not induce this effect, which led to 4.5 times higher RSEI+ct compared to the GeCC interlayer after 100 cycles. Overall, the EIS analysis suggests enhanced wettability at the Li electrode–electrolyte interface, with enhanced Li-ion diffusion due to the alloy formation.
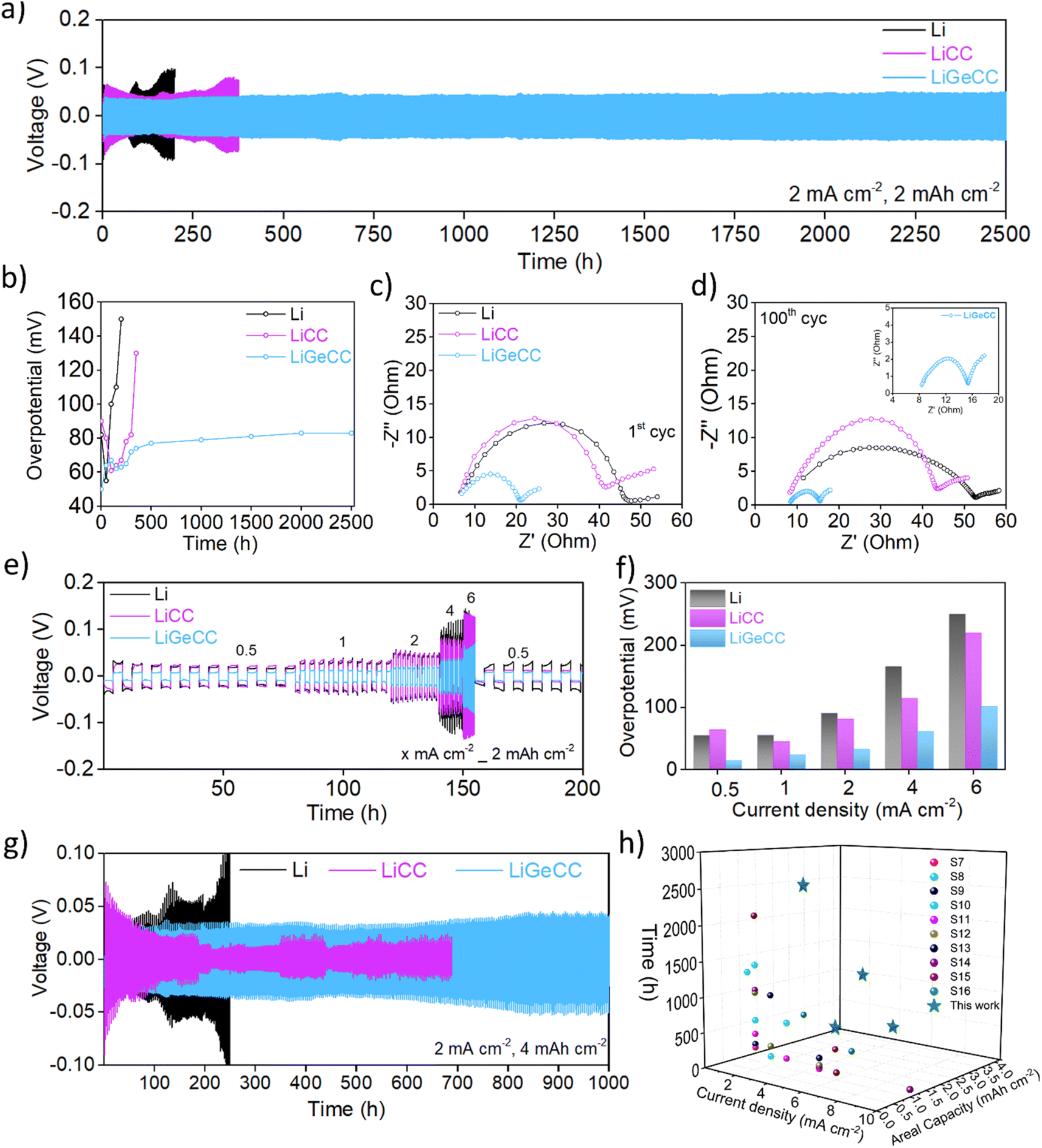 | ||
| Fig. 3 Electrochemical performance of bare Li, LiCC and LiGeCC anodes in symmetric cells. (a) Cyclic performance at 2 mA cm−2, 2 mA h cm−2 current density and plating capacity, respectively. (b) Corresponding overpotential vs. Time plot of Li, LiCC and LiGeCC anode. Electrochemical impedance spectroscopy after (c) 1st cycle and (d) 100th cycle of symmetric cells cycled at current density and plating capacity of 2 mA cm−2, 2 mA h cm−2. (e) Rate capability test at 0.5, 1, 2, 4 and 6 mA cm−2 at a plating capacity of 2 mA h cm−2 of Li, LiCC and LiGeCC. (f) Corresponding overpotential vs. time plot of rate capability test at various current densities. (g) Symmetric cell testing at high plating/stripping capacity of 4 mA h cm−2 at a current density of 2 mA cm−2. (h) Graphical representation of current density vs. areal capacity vs. cycling time of this work in comparison to previously published works. | ||
Rate capability testing was conducted at 0.5, 1, 2, 4 and 6 mA cm−2 with a fixed plating capacity of 2 mA h cm−2 to determine the robustness of the GeCC interlayer. The results showed that the LiGeCC cell maintained the lowest overpotentials, followed by the LiCC and bare Li cell, further suggesting the benefits of introducing the lithiophilic interlayer at the Li interface (Fig. 3e and S16a–c†). Comparatively, at 0.5 mA cm−2, LiGeCC anode had a very low overpotential of 15 mV as compared to 65 mV and 55 mV for LiCC and Li cell respectively (Fig. 3f). This overpotential increased to 100 mV at 6 mA cm−2 for GeCC, while it almost doubled for LiCC (220 mV) and bare Li metal (250 mV). With the current density fixed at 2 mA cm−2 and the plating capacity increased to 4 mA h cm−2, the LiGeCC substrate delivered a stable plating/stripping performance for up to 1000 h, compared to LiCC and Li (Fig. 3g). This suggests that LiGeCC anode can accommodate higher amounts of Li (plating capacity) compared to the LiCC anode. It was also observed that LiCC short-circuited after just 200 h with unstable overpotentials beyond that time.32,33,35 Further testing at high current density and plating capacity of 4 mA cm−2, 2 mA h cm−2 (Fig. S17a†) as well as 4 mA cm−2, 4 mA h cm−2 (Fig. S17b†) showed excellent performance stability of the LiGeCC interlayer for 500 h and 250 h respectively. The overall stability obtained at high current density (2 mA cm−2, 4 mA cm−2) and plating capacity (2 mA h cm−2, 4 mA h cm−2) is superior to previously published reports using interlayers and conventional current density (1 mA cm−2) and/or plating capacity (1 mA h cm−2) for Li metal anodes; again emphasizing the enhanced functionality of the GeCC interlayer for LMBs (Fig. 3h and Table S5†).
To understand the enhanced stability of GeCC compared to the CC interlayer (and unmodified Li), post-mortem SEM analysis was conducted after 100 cycles at current density and plating capacity of 2 mA cm−2, 2 mA h cm−2. The results clearly show non-uniform deposition of Li on the bare Li surface, with apparent cracks and dendrite formation (Fig. 4a(i and ii)). The cross-sectional image further shows the presence of a thick cycled Li layer that is partially detached from the underlying Li; which usually gives rise to the formation of inactive Li as well as poor symmetric cell performance with high interfacial resistance (Fig. 4a(iii)). The LiCC substrate also displayed non-uniform Li deposition, with chunks of Li detaching from the underlying substrate (Fig. 4b(i), inset), causing formation of inactive Li and dendrites (Fig. 4b(ii)). In clear agreement with the continuum simulations (Fig. 1) the cross-sectional image clearly shows build-up of Li on the top surface of CC interlayer rather than infilling Li in CC, starting from the Li-CC interface (Fig. 4b(iii)). Contrary to the Li deposition behaviour on bare Li and on CC, the Li deposition in the presence of the GeCC interlayer is more uniform with no evidence of dendrite formation on the outer surface (separator facing side) of the GeCC (Fig. 4c(i and ii)). Cross-sectional SEM analysis of the LiGeCC reveals a smooth interface between LiGeCC (unlike LiCC), likely due to the lithiophilic LiGe alloy formation at the interface, which helps to uniformly channel Li ions during the stripping/plating process (Fig. 4c(iii)). Once the Li was stripped from the LiCC (Fig. S18a and b†) and LiGeCC (Fig. S18c and d†) electrodes, the CC interlayer (without Ge NW coating) still contains patches of unstripped Li which eventually contributes in the loss of active Li. However, the LiGeCC after Li stripping showed the underling fibrous structure of the parent CC with no major leftover Li, again demonstrating the importance of the lithiophilic interface and its positive effect on promoting complete Li stripping during cycling.
 | ||
| Fig. 4 Morphological evolution of Li, LiCC and LiGeCC substrates in symmetric cells. SEM analysis of (a(i–iii)) Li, (b(i–iii)) LiCC and (c(i–iii)) LiGeCC anode after 100 cycles at current density and plating capacity of 2 mA cm−2, 2 mA h cm−2. SEM analysis of (d(i–iii)) Li, (e(i–iii)) LiCC and (f(i–iii)) LiGeCC anode after 100 cycles at current density and plating capacity of 2 mA cm−2, 4 mA h cm−2. Yellow dotted line represents deposited Li after cycling. All the anodes were examined after the plating step. (g) Schematic illustration explaining the deposition behaviour of Li, LiCC and LiGeCC during cycling. | ||
With the current density kept at 2 mA cm−2, SEM analysis was conducted to determine the Li plating/stripping behaviour once the plating capacity was doubled to 4 mA h cm−2. Here, the dendrite formation on pristine Li post-cycling increased, with a thick layer of cycled Li, detached from the underlying ‘uncycled’ Li surface (Fig. 4d(i and ii)). It is also noticeable that the detached Li layer was thicker (compared to 2 mA h cm−2 plating capacity) further suggesting that the poor performance of Li would also be due to the excessive build-up of detached Li during cycling (Fig. 4d(iii)). Similar analysis of LiCC showed Li plated on the CC surface, with evident cracks and dendrite formation (Fig. 4e(i and ii)). The cracks in the Li surfaces are more evident in the cross-sectional SEM of LiCC, with no evidence of Li plated across the CC interlayer (Fig. 4e(iii)). In contrast, LiGeCC shows uniform deposition of Li metal with nano-sized features on the outer surface of GeCC interlayer once cycled at 4 mA h cm−2 plating capacity (Fig. 4f(i and ii)). As predicted by the simulations (Fig. 1) the cross-sectional SEM analysis reveals a layer of plated Li on the GeCC interlayer–Li interface, clearly signifying that preferential Li deposition starts from the LiGe alloy NWs–Li metal interface and then deposits outward (into the body of the CC) during Li plating cycle (Fig. 4f(iii)). This behaviour of Li deposition inhibits the build-up of Li on the separator facing side of the GeCC, promoting uniform Li infilling in the GeCC interlayer and thereby elimination dendrite formation, unlike what is seen for Li and LiCC anodes (Fig. 4g and S19†). Similar evidence supporting preferential deposition of Li on Ge NW side of GeCC was obtained when 4 mA h per cm2 Li was deposited on CC and GeCC in a half cell configuration (Li vs. CC and Li vs. GeCC). The CC extracted from Li vs. CC half-cell clearly shows dense Li deposition on the CC side facing the separator (Fig. S20a†) while no Li deposition was observed at the CC side facing the Li metal (Fig. S20b†). In comparison, Li metal preferentially deposited on the Ge NW side of GeCC as compared to the CC side of the GeCC facing the separator (Fig. S21c and d†). Further, cross-sectional SEM of these samples suggest no Li infilling in CC (Fig. S21a and b†) as compared to GeCC (Fig. S21c and d†) once 4 mA h per cm2 Li was plated, consolidating our observation that GeCC promotes ‘bottom-up’ Li infilling as compared to CC.
To demonstrate the applicability of the GeCC interlayer with practical Li-ion battery cathodes, Li, LiCC and LiGeCC anodes were assembled against LiFePO4 (LFP) and NMC811 in coin cells. When paired with the LFP cathode, the rate capability test demonstrated the highest capacity values of 170, 162, 146, 121 and 98 mA h g−1 at 0.2, 0.5,1,2 and 3C respectively (Fig. 5a and S22†) for the LiGeCC anode. Comparatively, LiCC–LFP demonstrated lower specific capacity mainly from 1–3C, while Li–LFP overall showed the lowest specific capacity values mainly due to excessive dendrite formation discussed earlier. Further comparison of voltage profiles at 0.5C and 2C showed the highest overpotentials for Li (100 mV–0.5C and 200 mV–2C), followed by LiCC (80 mV–0.5C and 190 mV–2C), while the lowest overpotentials were observed in LiGeCC–LFP cell (75 mV–0.5C and 150 mV–2C) (Fig. 5b and c). A comparison of voltage–specific capacity profiles of Li–LFP, LiCC–LFP and LiGeCC–LFP at 0.2, 1 and 3C is given as well in Fig. S23.† To test long-term cyclability, all anodes were tested against LFP cathodes at 0.5C, with the LiGeCC anode significantly outperforming the LiCC and Li anodes (Fig. 5d). The LiGeCC–LFP cell demonstrated an initial specific capacity of 150 mA h g−1, retaining 110 mA h g−1 after 250 cycles with an average coulombic efficiency of 99.65%. This compared extremely favourably with the Li–LFP and Li/CC–LFP cells which had initial capacities of 144 mA h g−1 and 150 mA h g−1 before failing after 90 and 130 cycles respectively, with much lower average coulombic efficiencies of ∼95%. The corresponding voltage profiles (Fig. S24†) for various cycle number also demonstrated rapid capacity decay in LiCC–LFP (0.45% capacity decay per cycle) and Li–LFP (0.46% capacity decay per cycle) as compared to LiGeCC–LFP (0.10% capacity decay per cycle).
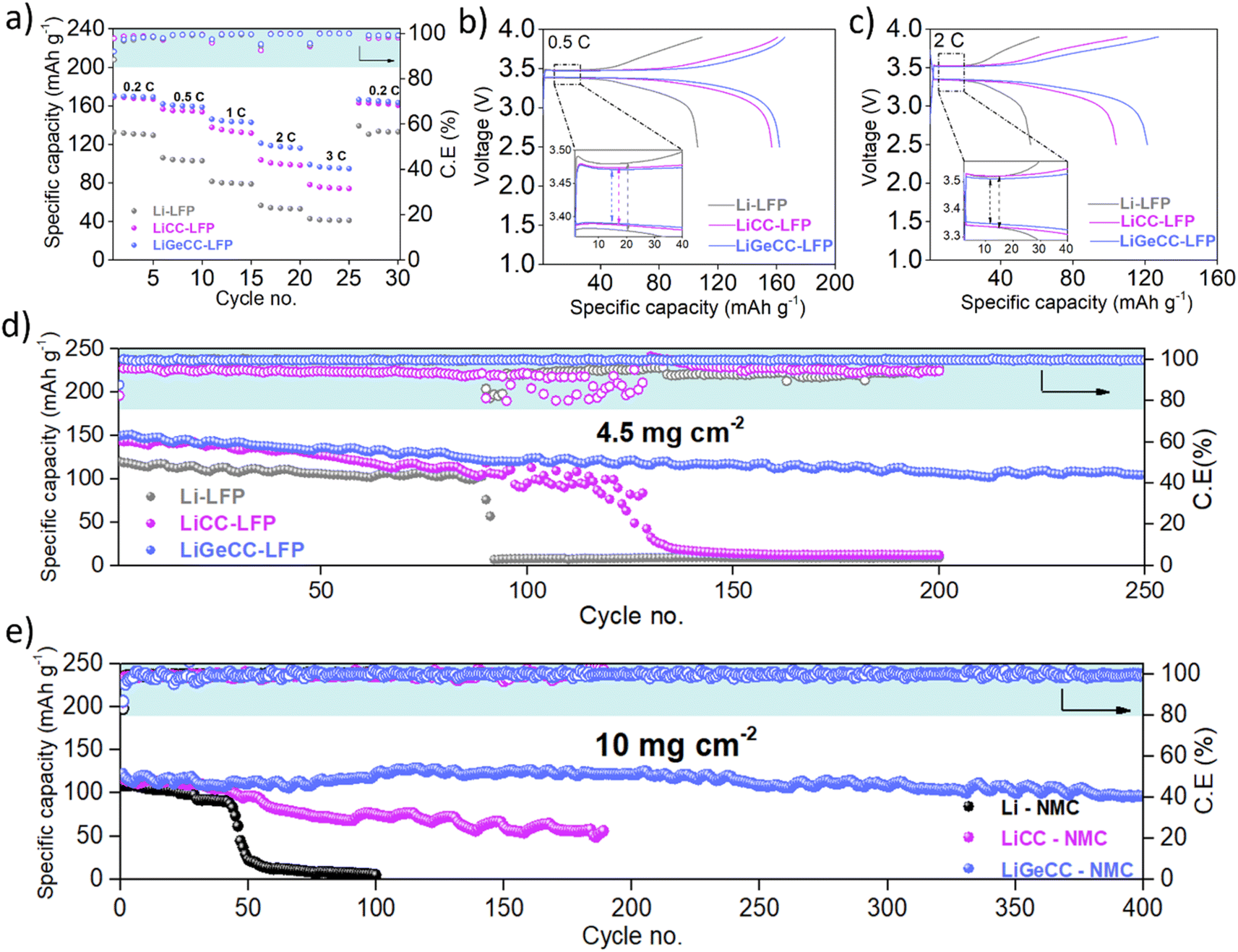 | ||
| Fig. 5 Electrochemical characterization of CC and GeCC interlayer in LFP and NMC cathode systems. (a) Rate capability performance of Li–LFP, LiCC–LFP and LiGeCC–LFP at various C-rates. Corresponding voltage–specific capacity plot at (b) 0.5C and (c) 2C with insets showing overpotential difference between Li–LFP, LiCC–LFP and LiGeCC–LFP. Long-term cycling performance of (d) LFP cathode and (e) NMC cathode with Li, LiCC and LiGeCC anodes. | ||
A high loading NMC811 cathode (10 mg cm−2) was used to determine the performance of the GeCC interlayer in comparison to CC and pristine Li. Long term cycling performance at 0.5C clearly shows that the LiGeCC anode delivered dramatically enhanced performance compared to LiCC and Li anode over the 400 cycles (Fig. 5e). The LiGeCC–NMC delivered an initial specific capacity of 126 mA h g−1 (100 mA h g−1 after 400 cycles) with an average coulombic efficiency of 99.7% and a capacity retention of 80% over 400 cycles. In comparison, Li–NMC and LiCC–NMC demonstrated only 5 mA h g−1 after 100 cycles and 56 mA h g−1 after 190 cycles, showcasing poor capacity retention of 5% and 48%, respectively. The corresponding voltage–capacity graphs show rapid capacity decay in LiCC–NMC and Li–NMC and stable performance for LiGeCC–NMC (Fig. S25†), indicating excellent interfacial stability of the LiGeCC anode. Furthermore, rate capability test of LiGeCC–NMC delivered 180, 158, 135, 122, 84 and 25 mA h g−1 at 0.1, 0.2, 0.5, 1, 2 and 5C respectively (Fig. S26†), whereas the Li–NMC and LiCC–NMC delivered relatively lower specific capacities with the corresponding voltage–capacity graphs given in Fig. S27.† The overall improved performance delivered by LiGeCC with LFP and NMC cathode systems validates the use of highly lithiophilic GeCC interlayer at the Li interface, providing improved Li-interfacial stability with dendrite inhibition which is in agreement with the symmetric cell performance of LiGeCC as well as the results of the continuum simulations.
4. Conclusions
In summary, we have developed an innovative interfacial strategy to inhibit dendrite formation in Li metal anodes. Our simulations show that an interlayer with a low overpotential for Li nucleation and a high specific surface area is key to achieve a bottom-up infilling and avoid Li growth into the separator. This motivated the design of a functional interlayer comprising of Ge NWs coated on the Li facing side of CC. This lithiophilic GeCC interlayer, when in contact with the Li metal anode, induces the formation of a lithiophilic LiGe interlayer, which guides uniform Li deposition (starting from the lithiophilic side towards the lithiophobic side). This promotes Li-interfacial stability for up to 2500 h at a high current density of 2 mA cm−2. In comparison, the lithiophobic control CC cannot help in guiding Li deposition during Li stripping/plating, with Li plating mostly on the outer lithiophobic side of the CC, causing the formation of inactive Li and high overpotentials in comparison to the LiGeCC anode. To verify the findings from symmetric cell studies, high loading NMC811 electrodes, when tested with LiGeCC anode showed exceptional stability over long-term cycling. The design of functional interlayers to induce lithiophilicity at the Li interface, unlocks uniform and guided ‘bottom up’ Li metal deposition/stripping, which is an exciting route towards achieving high current density and long-term cycling in LMBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is supported by Science Foundation Ireland under grant no. 18/SIRG/5484. A. L. gratefully acknowledges financial support through the German Federal Ministry for Education and Research (BMBF) within the Lillint project (FKZ 03XP0225A).References
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- J. Speirs, M. Contestabile, Y. Houari and R. Gross, Renewable Sustainable Energy Rev., 2014, 35, 183–193 CrossRef.
- Q. Wang, B. Liu, Y. Shen, J. Wu, Z. Zhao, C. Zhong and W. Hu, Advanced Science, 2021, 8, 1–25 CAS.
- F. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 RSC.
- F. Wu, O. Borodin and G. Yushin, MRS Energy & Sustainability, 2017, 4, 1–15 Search PubMed.
- M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- K. Zhang, G. H. Lee, M. Park, W. Li and Y. M. Kang, Adv. Energy Mater., 2016, 6, 1–14 Search PubMed.
- J. Wang, J. Yang, Q. Xiao, J. Zhang, T. Li, L. Jia, Z. Wang, S. Cheng, L. Li, M. Liu, H. Liu, H. Lin and Y. Zhang, Adv. Funct. Mater., 2021, 31, 2007434 CrossRef CAS.
- C. Chang, Y. Yao, R. Li, Z. H. Guo, L. Li, C. Pan, W. Hu and X. Pu, Nano Energy, 2022, 93, 106871 CrossRef CAS.
- F. Liu, L. Wang, Z. Zhang, P. Shi, Y. Feng, Y. Yao, S. Ye, H. Wang, X. Wu and Y. Yu, Adv. Funct. Mater., 2020, 30, 2001607 CrossRef CAS.
- P. K. Alaboina, S. Rodrigues, M. Rottmayer and S. J. Cho, ACS Appl. Mater. Interfaces, 2018, 10, 32801–32808 CrossRef CAS PubMed.
- W. Tang, X. Yin, S. Kang, Z. Chen, B. Tian, S. L. Teo, X. Wang, X. Chi, K. P. Loh, H. W. Lee and G. W. Zheng, Adv. Mater., 2018, 30, 1801745 CrossRef PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J. G. Zhang and W. Xu, Nat. Energy, 2017, 2, 17012 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J. G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- H. Kim, F. Wu, J. T. Lee, N. Nitta, H. T. Lin, M. Oschatz, W. Il Cho, S. Kaskel, O. Borodin and G. Yushin, Adv. Energy Mater., 2015, 5, 1–8 CAS.
- P. Xiao, R. Luo, Z. Piao, C. Li, J. Wang, K. Yu, G. Zhou and H. M. Cheng, ACS Energy Lett., 2021, 6, 3170–3179 CrossRef CAS.
- O. B. Chae, V. A. K. Adiraju and B. L. Lucht, ACS Energy Lett., 2021, 6, 3851–3857 CrossRef CAS.
- A. C. Thenuwara, P. P. Shetty, N. Kondekar, S. E. Sandoval, K. Cavallaro, R. May, C. T. Yang, L. E. Marbella, Y. Qi and M. T. McDowell, ACS Energy Lett., 2020, 5, 2411–2420 CrossRef CAS.
- J. Zhao, G. Zhou, K. Yan, J. Xie, Y. Li, L. Liao, Y. Jin, K. Liu, P. C. Hsu, J. Wang, H. M. Cheng and Y. Cui, Nat. Nanotechnol., 2017, 12, 993–999 CrossRef CAS PubMed.
- S. Li, Q. Liu, J. Zhou, T. Pan, L. Gao, W. Zhang, L. Fan and Y. Lu, Adv. Funct. Mater., 2019, 29, 1808847 CrossRef.
- C. Zhang, R. Lyu, W. Lv, H. Li, W. Jiang, J. Li, S. Gu, G. Zhou, Z. Huang, Y. Zhang, J. Wu, Q. H. Yang and F. Kang, Adv. Mater., 2019, 31, 1904991 CrossRef CAS PubMed.
- J. Xie, J. Ye, F. Pan, X. Sun, K. Ni, H. Yuan, X. Wang, N. Shu, C. Chen and Y. Zhu, Adv. Mater., 2019, 31, 1805654 CrossRef PubMed.
- Y. Zhou, Y. Han, H. Zhang, D. Sui, Z. Sun, P. Xiao, X. Wang, Y. Ma and Y. Chen, Energy Storage Mater., 2018, 14, 222–229 CrossRef.
- S. Zhang, J. You, Z. He, J. Zhong, P. Zhang, Z. Yin, F. Pan, M. Ling, B. Zhang and Z. Lin, Adv. Funct. Mater., 2022, 32, 2200967 CrossRef CAS.
- K. Zhang, W. Liu, Y. Gao, X. Wang, Z. Chen, R. Ning, W. Yu, R. Li, L. Li, X. Li, K. Yuan, L. Ma, N. Li, C. Shen, W. Huang, K. Xie and K. P. Loh, Adv. Mater., 2021, 33, 2006323 CrossRef CAS PubMed.
- J. Li, P. Zou, S. W. Chiang, W. Yao, Y. Wang, P. Liu, C. Liang, F. Kang and C. Yang, Energy Storage Mater., 2020, 24, 700–706 CrossRef.
- J. Pu, J. Li, K. Zhang, T. Zhang, C. Li, H. Ma, J. Zhu, P. V. Braun, J. Lu and H. Zhang, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- H. Zhang, X. Liao, Y. Guan, Y. Xiang, M. Li, W. Zhang, X. Zhu, H. Ming, L. Lu, J. Qiu, Y. Huang, G. Cao, Y. Yang, L. Mai, Y. Zhao and H. Zhang, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- L. Wu, G. He and Y. Ding, J. Mater. Chem. A, 2019, 7, 25415–25422 RSC.
- R. He, Y. Wang, C. Zhang, Z. Liu, P. He, X. Hong, R. Yu, Y. Zhao, J. Wu, L. Zhou and L. Mai, Adv. Energy Mater., 2023, 13, 2204075 CrossRef CAS.
- X. Wang, Y. He, S. Tu, L. Fu, Z. Chen, S. Liu, Z. Cai, L. Wang, X. He and Y. Sun, Energy Storage Mater., 2022, 49, 135–143 CrossRef.
- M. Wan, S. Kang, L. Wang, H. W. Lee, G. W. Zheng, Y. Cui and Y. Sun, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- M. L. Meyerson, P. E. Papa, A. Heller and C. B. Mullins, ACS Nano, 2021, 15, 29–46 CrossRef CAS PubMed.
- Y. Sun, C. Zhao, K. R. Adair, Y. Zhao, L. V. Goncharova, J. Liang, C. Wang, J. Li, R. Li, M. Cai, T. K. Sham and X. Sun, Energy Environ. Sci., 2021, 14, 4085–4094 RSC.
- S. Liu, X. Xia, Z. Yao, J. Wu, L. Zhang, S. Deng, C. Zhou, S. Shen, X. Wang and J. Tu, Small Methods, 2018, 2, 1800035 CrossRef.
- Y. Liu, X. Fang, M. Ge, J. Rong, C. Shen, A. Zhang, H. A. Enaya and C. Zhou, Nano Energy, 2015, 16, 399–407 CrossRef CAS.
- L. L. Lu, J. Ge, J. N. Yang, S. M. Chen, H. bin Yao, F. Zhou and S. H. Yu, Nano Lett., 2016, 16, 4431–4437 CrossRef CAS PubMed.
- L. L. Lu, Y. Zhang, Z. Pan, H. Bin Yao, F. Zhou and S. H. Yu, Energy Storage Mater., 2017, 9, 31–38 CrossRef.
- C. P. Yang, Y. X. Yin, S. F. Zhang, N. W. Li and Y. G. Guo, Nat. Commun., 2015, 6, 1–9 CAS.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O'Dwyer and K. M. Ryan, Nano Lett., 2014, 14, 716–723 CrossRef CAS PubMed.
- S. Abdul Ahad, S. Kilian, M. Zubair, V. A. Lebedev, K. McNamara, K. M. Ryan, T. Kennedy and H. Geaney, J. Mater. Chem. A, 2021, 9, 20626–20634 RSC.
- J. Newman and K. E. Thomas-Alyea, Electrochemical Systems, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2004 Search PubMed.
- T. F. Fuller, M. Doyle and J. Newman, J. Electrochem. Soc., 1994, 141, 1–10 CrossRef CAS.
- A. Latz and J. Zausch, Electrochim. Acta, 2013, 110, 358–362 CrossRef CAS.
- A. Latz and J. Zausch, J. Power Sources, 2011, 196, 3296–3302 CrossRef CAS.
- H. R. Shin, S. Kim, J. Park, J. H. Kim, M. S. Park and J. W. Lee, Energy Storage Mater., 2023, 56, 515–523 CrossRef.
- Fraunhofer Institute for Industrial Mathematics (ITWM) and German Aerospace Center (DLR)@Helmholtz Institute Ulm (HIU), BEST – Battery and Electrochemistry Simulation Tool, https://www.itwm.fraunhofer.de/best, accessed 18 October 2023 Search PubMed.
- S. Hein, T. Danner and A. Latz, ACS Appl. Energy Mater., 2020, 3, 8519–8531 CrossRef CAS.
- K. Yan, Z. Lu, H. W. Lee, F. Xiong, P. C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- G. Bucci, T. Swamy, W. Craig Carter and M. Behandish, J. Electrochem. Soc., 2022, 169, 070501 CrossRef CAS.
- R. Zhang, X. Shen, H. T. Ju, J. D. Zhang, Y. T. Zhang and J. Q. Huang, J. Energy Chem., 2022, 73, 285–291 CrossRef CAS.
- J. Yun, B. K. Park, E. S. Won, S. H. Choi, H. C. Kang, J. H. Kim, M. S. Park and J. W. Lee, ACS Energy Lett., 2020, 5, 3108–3114 CrossRef CAS.
- H. R. Shin, S. Kim, J. Park, J. H. Kim, M. S. Park and J. W. Lee, Energy Storage Mater., 2023, 56, 515–523 CrossRef.
- C. Zhang, S. Zhu, D. Wang, C. Lei and Y. Zhao, J. Energy Storage, 2023, 73, 109279 CrossRef.
- K. Yan, Z. Lu, H. W. Lee, F. Xiong, P. C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- H. Geaney, E. Mullane, Q. M. Ramasse and K. M. Ryan, Nano Lett., 2013, 13, 1675 CrossRef CAS PubMed.
- C. A. Barrett, H. Geaney, R. D. Gunning, F. R. Laffir and K. M. Ryan, Chem. Commun., 2011, 47, 3843–3845 RSC.
- J. Zhao, J. Sun, A. Pei, G. Zhou, K. Yan, Y. Liu, D. Lin and Y. Cui, Energy Storage Mater., 2018, 10, 275–281 CrossRef.
- D. McNulty, H. Geaney, D. Buckley and C. O'Dwyer, Nano Energy, 2018, 43, 11–21 CrossRef CAS.
- A. Garcia, S. Biswas, D. McNulty, A. Roy, S. Raha, S. Trabesinger, V. Nicolosi, A. Singha and J. D. Holmes, ACS Appl. Energy Mater., 2022, 5, 1922–1932 CrossRef CAS PubMed.
- K. V. Greco, A. Forner-Cuenca, A. Mularczyk, J. Eller and F. R. Brushett, ACS Appl. Mater. Interfaces, 2018, 10, 44430–44442 CrossRef CAS PubMed.
- A. Zhang, X. Fang, C. Shen, Y. Liu and C. Zhou, Nano Res., 2016, 9, 3428–3436 CrossRef CAS.
- K. L. Jungjohann, R. N. Gannon, S. Goriparti, S. J. Randolph, L. C. Merrill, D. C. Johnson, K. R. Zavadil, S. J. Harris and K. L. Harrison, ACS Energy Lett., 2021, 6, 2138–2144 CrossRef CAS.
- B. Liu, L. Zhang, S. Xu, D. W. McOwen, Y. Gong, C. Yang, G. R. Pastel, H. Xie, K. Fu, J. Dai, C. Chen, E. D. Wachsman and L. Hu, Energy Storage Mater., 2018, 14, 376–382 CrossRef.
- Y. Ma, L. Wei, Y. Gu, L. Zhao, Y. Jing, Q. Mu, Y. Su, X. Yuan, Y. Peng and Z. Deng, Nano Lett., 2021, 21, 7354–7362 CrossRef CAS PubMed.
- L. Tan, S. Feng, X. Li, Z. Wang, W. Peng, T. Liu, G. Yan, L. Li, F. Wu and J. Wang, Chem. Eng. J., 2020, 394, 124848 CrossRef CAS.
- R. Pathak, K. Chen, A. Gurung, K. M. Reza, B. Bahrami, J. Pokharel, A. Baniya, W. He, F. Wu, Y. Zhou, K. Xu and Q. Qiao, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01072h |
| This journal is © The Royal Society of Chemistry 2024 |