
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High performance water electrolysis using a poly(fluorene phenylpropylammonium) anion-exchange membrane with 2 M aqueous KOH
Matteo
Rossini
 *a,
Dong
Pan
b,
Burak
Koyutürk
a,
Si
Chen
b,
Amirreza
Khataee
a,
Göran
Lindbergh
a,
Patric
Jannasch
b and
Ann
Cornell
a
*a,
Dong
Pan
b,
Burak
Koyutürk
a,
Si
Chen
b,
Amirreza
Khataee
a,
Göran
Lindbergh
a,
Patric
Jannasch
b and
Ann
Cornell
a
aApplied Electrochemistry, Department of Chemical Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: mrossini@kth.se
bPolymer & Materials Chemistry, Department of Chemistry, Lund University, SE-221 00 Lund, Sweden
First published on 26th April 2024
Abstract
Anion exchange membrane water electrolysis (AEMWE) has great potential to be established as a high-performance and low-capital cost technology for hydrogen production. High current densities can be achieved with a non-platinum group metal (non-PGM) catalyst. However, the harsh operation conditions require stable cell components. Here, we report on the use of a highly stable and ion conductive poly(fluorene alkylene) membrane (PdF–TMA) tethered with trimethylammonium cations via phenylpropyl side chains for AEMWE cells operating with 2 M aqueous KOH. The ether-free PdF–TMA polymer is efficiently prepared by polyhydroxyalkylation to reach a molecular weight of 236 kDa, a high thermal stability, and an ion-exchange capacity of 2.14 mequiv. g−1 (OH− form). Using commercial electrodes of NiFe2O4 (anode) and RANEY® nickel (cathode) and PdF–TMA as an AEM, the output current reached 1 A cm−2 at voltages below 1.9 V at 60 °C. Also, PdF–TMA outperformed AEMION™ in terms of membrane resistance by almost 30% and, after 100 h at 0.5 A cm−2, did not reveal any loss of conductivity, in contrast to AEMION™. Furthermore, both membranes were analysed by 1H NMR spectroscopy after AEMWE tests and the PdF–TMA proved very stable even at 80 °C.
Introduction
Electrochemical production of hydrogen has been carried out at an industrial scale from the 1920s,1 and has attracted great and growing interest in the last few years. Electrical energy, in the best case from renewable sources, is converted into both an efficient fuel and a feedstock for the chemical industry. Nowadays, hydrogen is mainly used in the synthesis of ammonia, methanol and many other bulk chemicals.2 Coal gasification, natural gas reforming and partial oxidation of oil cuts are the most common processes for hydrogen production, which cause an emission of 500 Mt CO2 per year.3 In order to increase hydrogen production by water electrolysis, the cost of electrolysers must be reduced.4 However, when it comes to both alkaline water electrolysis (AWE) and proton exchange membrane water electrolysis (PEMWE), the capital costs are expected to decrease, mainly due to production scale-up, rather than from R&D investments, because of their high readiness level.5 Nevertheless, AWE cannot be operated at high current densities and has a long response time. PEMWE allows a high production rate and dynamic operation, but depends on both scarce and precious iridium catalysts and porous transport layers coated with more platinum group metals (PGMs).6 On the other hand, the inherent limitations of these technologies can be overcome by a hybrid device that brings together the benefits of AWE and PEMWE.7 In anion exchange membrane water electrolysis (AEMWE), an alkaline environment is enabled by the use of an anion exchange membrane (AEM). Hence, a non-PGM catalyst and low-cost nickel or stainless-steel plates can be used, just as in AWE.8 Furthermore, a low ohmic cell resistance and a high current density can be achieved in a zero-gap design with a thin and highly ion conductive membrane, just as in PEMWE.9 In recent years, the conductivity and the stability of AEMs have been significantly enhanced and further developed.10 Additionally, AEMWE has the advantage that AEMs are less permeable to hydrogen than PEMs.10,11 This reduces the risk of forming an explosive atmosphere due to the lower solubility of hydrogen in solutions with high OH− concentration12,13 and the low hydrogen permeability of hydrocarbon-based polymer membranes.Nonetheless, despite significant recent progress in the alkaline stability of AEMs, a high degradation rate still limits the lifetime under real conditions.7 And, according to Du et al., only a limited number of studies have reported stability over more than 100 h and the degradation rate is seldom in the order of 100–200 μV h−1.14 For instance, piperidinium-based AEMs have been shown to be stable for 100 h or more operating in 1 M KOH (or 1 M NaOH)15–17 while commercially available imidazolium (Sustanion™)17–19 and benzimidazolium (AEMION™ or AEMION+™)20–22 based AEMs have also been tested for 100 h or more under similar conditions. Notably, AEMION+™ has been shown to withstand 1 year of operation in a recent study.23 Also, trimethyl-ammonium based membranes are promising candidates for AEMWE, as the commercially available ORION™.24 Jiang et al. have recently reported a membrane that could withstand 3500 h of operation (1 M KOH, 60 °C, 1 A cm−2)25 and Jang et al. assembled a 3-cell-stack which operated for 2000 h under milder conditions.26 AEMWE durability can also reach several hundred hours with carbonate electrolytes,27,28 and more advanced ionomer chemistries have pushed the boundaries of pure water AEMWE to 100–200 h.11,28–30
Also, operating at higher temperature and higher KOH concentration has a positive effect on electrolyte conductivity and catalyst activity, but may further decrease the membrane stability. Along similar lines, some reports have pointed out that the performance of AWE can be enhanced by using a dense polymeric membrane, e.g., polybenzimidazole (PBI), as a separator to absorb electrolyte and conduct the hydroxide anions.13,31 However, in contrast to AEMs, PBI can achieve high conductivities (≈100 mS cm−1) only when placed in contact with 30 wt% KOH solutions,32,33 which decreases the corrosion resistance of plates and other plant components such as pumps, and poses a further challenge to the stability of polymeric materials. The continued development of new membrane polymers over the last decade has greatly improved the stability of AEMs in 1 M aq. KOH solution. Furthermore, some reports have demonstrated the potential application of 2 M KOH.34,35
In the present work a newly synthesized membrane (PdF–TMA) (Scheme 1) and a commercial benchmark membrane (AEMION™) have been studied for application in AEMWE at relatively high concentration of KOH (1–2 M) and temperature (60–80 °C). The high-molecular-weight PdF–TMA membrane polymer was tailored for high conductivity and alkaline stability. Hence, the water uptake was tuned to reach high ion conductivity and alkaline stability. The high molecular weight facilitates mechanical stability, which is generally required at high water uptake. As previously demonstrated, attaching the anion exchange groups to the backbone polymer via flexible spacer units increases the ionic clustering and the conductivity, as well as the alkaline stability.36,37 The membranes were evaluated with commercial electrodes using 1 and 2 M KOH at 60 °C. The PdF–TMA membrane was also evaluated at 80 °C. The resistivity of the membranes was studied in situ by galvanostatic electrochemical impedance spectroscopy (GEIS) before and after a 100 h test. The chemical stability of the AEMs was finally investigated in a post mortem 1H NMR study.
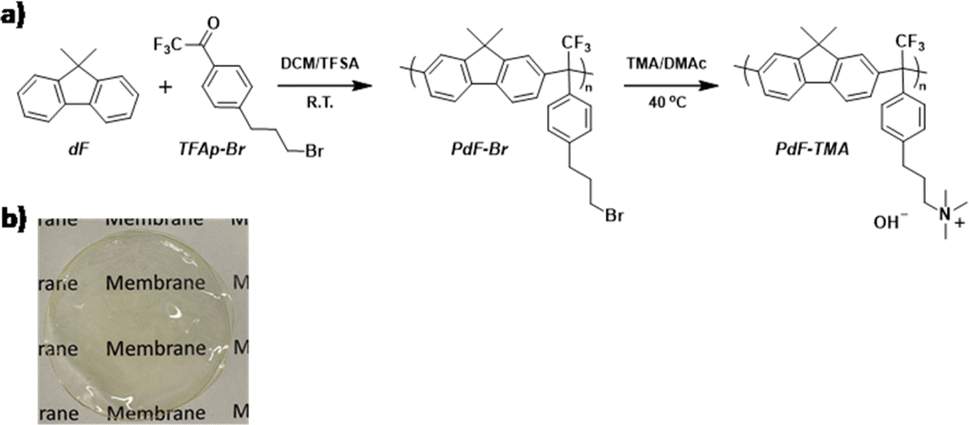 | ||
| Scheme 1 (a) Synthetic route to the poly(fluorene phenylpropylammonium) (PdF–TMA) and (b) photograph of a PdF–TMA membrane. | ||
Experimental
Membrane synthesis
PdF–TMA was synthesized in two steps following a recently reported method.36 First, a precursor polymer (PdF–Br) was prepared via a superacid-mediated Friedel–Crafts polycondensation using 9,9-H,H-dimethylfluorene (dF) and 1-bromo-3-(trifluoroacetylphenyl)-propane (TFAp–Br). In a 25 mL round-bottom flask equipped with an ice/water bath, TFAp–Br (1.68 g) and dF (1 g) were dissolved in dichloromethane (DCM, 6 mL) under stirring. Next, trifluoromethanesulfonic acid (1 mL) was dropwise added to the solution. The reaction proceeded at room temperature for 3 h, before the viscous solution was diluted with chloroform followed by precipitation of the product in methanol (500 mL). A white and fibrous precipitate was collected, dissolved in chloroform and re-precipitated in methanol to obtain PdF–Br in a quantitative yield. Functionalization of PdF–Br using trimethylamine was performed in dimethylacetamide (DMAc) at 40 °C. PdF–Br (0.82 g) was dissolved in 20 mL DMAc, before adding a trimethylamine solution (3 mL, 45 wt% in H2O). The Menshutkin reaction was kept for 7 h, before precipitation of PdF–TMA in diethyl ether. The white powder was washed repeatedly with diethyl ether, before being collected and dried.AEMs were prepared from a 5 wt% PdF–TMA solution in dimethyl sulfoxide (DMSO). The solution was passed through a syringe-driven PTFE filter (Φ = 5 μm) onto Petri dishes (Φ = 5 cm). Membrane casting was performed in an air-ventilated oven at 80 °C for 2 days. The AEMs were then gently peeled off after hydration with water, and stored in deionized water before characterization.
Membrane characterization
The chemical structures of PdF–Br and PdF–TMA were determined by 1H Nuclear Magnetic Resonance (NMR) spectroscopy on a Bruker DRX 400 spectrometer at 400 MHz using DMSO-d6 as solvent. The molecular weight of PdF–Br was characterized by size exclusion chromatography (SEC) using chloroform as eluent, and the glass transition temperature (Tg) of PdF–Br was determined using differential scanning calorimetry (DSC). The ion exchange capacity (IEC) of the PdF–TMA AEM was determined by Mohr titrations. Samples in the Br− form were dried and immersed in aq. NaNO3 (1 M, 25 mL) for 48 h to ensure complete ion exchange. Next, the solution was titrated with 0.1 M AgNO3 using K2CrO4 as colour indicator. The water uptake of the PdF TMA in the OH− form was measured gravimetrically between 20 and 80 °C. Ion-exchange from the Br− to the OH− was done using 1 M aq. NaOH in a N2-flushed desiccator. After ion-exchange, the sample was quickly washed in degassed deionized (DI) water, and equilibrated in DI water for 24 h at 20 °C (or 8 h at higher temperatures), before the weight and dimensions were measured. The KOH solution uptake of PdF TMA was measured similarly. After equilibrations in 1or 2 M aq. KOH solutions, the changes in the weight and dimensions of PdF–TMA were directly recorded. The OH− conductivity of PdF TMA in water was measured using electrochemical impedance spectroscopy (EIS) between 20 and 80 °C.Electrolysis set-up
The as-prepared PdF–TMA membranes and commercial AF1-HNN8-50-X (AEMION™, purchased from Ionomr™) membranes with a thickness of 50 μm were evaluated in an AEMWE setup. Both membrane types were soaked in 1 M KOH for at least 36 h prior to the electrolysis, and the KOH solution was changed twice to fully exchange the I− ions (AEMION™) or Br− ions (PdF–TMA) to OH−. For the electrolysis cell, anodes (NiFe2O4 on stainless-steel felt with Nafion) and cathodes (RANEY® nickel on nickel felt with Nafion) were purchased from Dioxide Material®. Nickel serpentine flow fields with a 5 cm2 area were chosen to withstand the high KOH concentration. The active area was isolated to 1 cm2 with PTFE gaskets ensuring 80% compression of the electrodes. The thickness of the gaskets was 250 μm on the cathode and 500 μm on the anode. A torque of 9 Nm was applied to seal the cell. The flow fields were heated with two heat cartridges, and the temperature controlled by a PID controller with a thermocouple. 1 M or 2 M KOH electrolyte was fed to both the anode and cathode with a peristaltic pump at a flowrate of 3 mL min−1. The electrolyte was pre-heated using two heat exchangers and recirculated after passing through the cell, allowing the gases to disengage in two tanks. An XP4105 potentiostat (Ivium, the Netherlands) with 20 A booster was used for AEMWE trials.Electrochemical characterization
The electrolysis cell was operated at 60 and 80 °C. Before recording a polarization curve, the cell was subjected to 10 cycles between 1.5 V and 1.8 V with a scan rate of 50 mV s−1 in order to stabilize cell performances. Polarization curves were recorded holding the current constant for 120 s at each current level, where the average value of the cell voltage was calculated during the last 30 s. After recording a polarization curve, GEIS was performed at each current density to evaluate the values of high frequency resistance (HFR). Then, the stability of the membranes was evaluated by applying a current density of 500 mA cm−2 for 100 h. A second polarization curve was recorded after the stability test, followed by GEIS measurements.Results and discussion
Ex situ membrane analyses
The molecular weight of the PdF–Br membrane is 236 kDa and was obtained by SEC, while the IEC of the PdF–TMA membrane is 2.14 mequiv. g−1 (OH− form). The commercial benchmark (AEMION™) was chosen to have a similar IEC (2.1–2.5 mequiv. g−1).38Table 1 shows the water uptake, OH− conductivity and swelling ratio (in-plane and through-plane) of the PdF–TMA membrane after being fully anion-exchanged and washed in DI water. The AEM showed a very high conductivity, up to 175 mS cm−1 at 80 °C and a water uptake of 190%. When compared to the selected commercial benchmark AEMION™, the PdF–TMA membrane shows a much lower water uptake and swelling ratio. In fact, the in-plane swelling ratio of AEMION™ is reported to be around 140% at 25 °C,22 thereby indicating a much higher water uptake than that measured for PdF–TMA. In fact, excessive water uptake and dissolution in pure water is reported for non-crosslinked hexamethyl-p-terphenyl–poly(benzimidazolium) (HMT–PBI) membranes such as AEMION™.39 In addition, the hydroxide conductivity is reported to be around 50 mS cm−1 at 40 °C for AEMION™,40 while the conductivity of PdF–TMA was measured to be twice as high. Considering the high conductivity and the anticipated high stability of PdF–TMA, the two membrane types were compared in an electrolysis cell operating with 1 M and 2 M KOH, respectively.
| Temperature (°C) | Water uptake (wt%) | OH− conductivity in water (mS cm−1) | Swelling ratio (%, in-plane) | Swelling ratio (%, through-plane) |
|---|---|---|---|---|
| 20 | 113 | 78.5 ± 2.5 | 25.6 ± 0.2 | 20.1 ± 1.6 |
| 40 | 135 | 104 ± 3 | 30.7 ± 0.3 | 23.4 ± 2.7 |
| 60 | 163 | 136.5 ± 4.5 | 33.3 ± 1.7 | 28.4 ± 2.3 |
| 80 | 190 | 171 ± 4 | 42.1 ± 0.9 | 32.3 ± 1.9 |
Electrolysis using 1 M KOH at 60 °C
Fig. 1a shows the polarization curve of AEMWE obtained before (solid symbols) and after the 100 h stability test (open symbols) at 60 °C using 1 M KOH and two different membranes: PdF–TMA (blue circles) and AEMION™ (AF1-HNN8-50-X, black squares). At low current densities (<0.1 mA cm−2), similar cell voltages were obtained with both AEMs as the electrode kinetics limited the cell performance. However, at higher current densities, lower cell voltages were obtained when the AEMION™ membrane was used. Furthermore, the HFR for the cell with AEMION™ was 30% lower than for the one with PdF–TMA (Table 2), indicating a higher conductivity of AEMION™ in 1 M KOH at 60 °C. These data agree well with the HFR data reported by Pushkareva et al.,41 who used AEMION™ with PGM-free catalysts for AEMWE in 1 M KOH at 60 °C. Also, both AEMs showed almost no variation in HFR at varying current densities, indicating the absence of dehydration of the membrane at higher current densities, since enough water is provided from the KOH electrolyte.42 | ||
| Fig. 1 (a) Polarization curves obtained in 1 M KOH at 60 °C with PdF–TMA (black squares) and AEMION™ (AF1-HNN8-50-X) (blue circles), before (solid symbols) and after the stability test (open symbols). Commercial electrodes with NiFe2O4 (anode) and RANEY® nickel (cathode) were used. (b) Stability evaluations at 500 mA cm−2 in 1 M KOH at 60 °C showing the degradation rate of the PdF–TMA (black) and AEMION™ (AF1-HNN8-50-X, blue) cells. | ||
After the stability evaluation at 500 mA cm−2 for 100 h, both cells showed a significant decrease in performance. Fig. 1b shows the long-term results with degradation rates of 0.19 mV h−1 and 0.26 mV h−1 for the cells with AEMION™ and PdF–TMA, respectively. The HFR values recorded before and after the long-term tests (Fig. 1a) are close to their initial values, suggesting stable membrane operation as the membrane resistance is the main contributor to HFR. The loss of performance at both low and high current densities is likely related to the electrodes rather than to the membranes and is possibly caused by detachment of catalyst particles from the electrodes. Actually, a colour change was observed at the outlet of the cell and the electrode appeared discoloured. We can conclude that the degradation rates are similar for the two AEMs in 1 M KOH at 60 °C as the major cause of loss of performance was due to the electrodes.
Du et al. identified more than 100 h of operation as the benchmark for AEMWE.14 In the present case, we exceeded 100 h of operation with a degradation rate of the AEMION™ membrane below 200 μV h−1, which is quite low when compared with data reported by Du et al. after surveying the performances of AEMWE cells disclosed in the literature.14 This confirms that AEMION™ can be efficiently used for AEMWE due to the high conductivity and stability, as previously reported by Khataee et al.43 Pavel et al.27 have reported 200 μV h−1 after using an A201 membrane (Tokuyama) in a much less aggressive environment (1 wt% K2CO3 at 43 °C) with a higher HFR. With the same materials, Vincent et al.8 have reported 500 μV h−1 (1 wt% K2CO3 at 60 °C). Only Motealleh et al.18 have reported a much lower degradation rate using the SUSTANION™ membrane in 1 M KOH. On the other hand, Park et al. obtained a higher degradation rate with the same membrane.19
Electrolysis using 2 M KOH at 60 °C
Given the high stability of the membranes and to improve the electrode performance, evaluations were also carried out in 2 M KOH. Fig. 2a shows the polarization curves obtained before (open symbols) and after the stability evaluation (solid symbols) in 2 M KOH. Once again, the performance of the two membranes was found to overlap at low current densities. Both cells showed improvements at low current density when 2 M KOH was used instead of 1 M KOH, which agrees with previous reports.41,44,45 This is likely due to the beneficial effects of the higher hydroxide concentration on both the OER and HER kinetics, and on the ion transport. However, the performance of the cell with AEMION™ was limited by an increased membrane resistance at higher current densities. In fact, its HFR increased from 110 mΩ cm2 to 128 mΩ cm2 after shifting to 2 M KOH (Table 2). In contrast, the PdF–TMA membrane showed a remarkable increase in conductivity as the HFR decreased from 162 mΩ cm2 to 86 mΩ cm2 (Table 2). This reduction in HFR by almost 50% allowed the cell with the PdF–TMA membrane in 2 M KOH at 60 °C to reach a lower cell voltage at a higher current density than AEMION™ in 1 M KOH at the same temperature.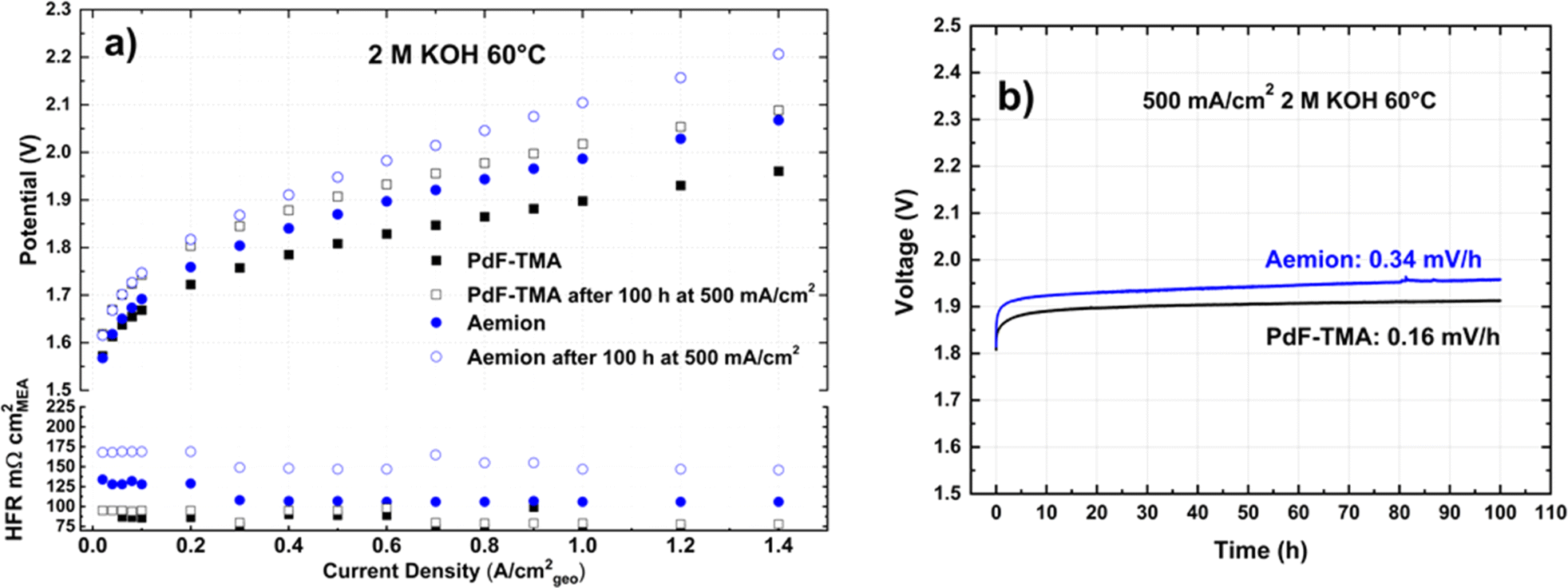 | ||
| Fig. 2 (a) Polarization curves obtained in 2 M KOH and at 60 °C with PdF–TMA (black squares) and AEMION™ (AF1-HNN8-50-X, blue circles) before (solid symbols) and after stability evaluation (open symbols). Commercial electrodes with NiFe2O4 (anode) and RANEY® nickel (cathode) were used. (b) Stability evaluations at 500 mA cm−2 in 2 M KOH at 60 °C showing the degradation rates of the PdF–TMA (black) and AEMION™ (AF1-HNN8-50-X, blue) cells. | ||
The degradation of the cell using PdF–TMA was lower in 2 M KOH (0.16 mV h−1) than in 1 M KOH (0.26 mV h−1). Furthermore, in 2 M KOH the stability evaluations revealed a more precise difference between the two membranes. The cell using PdF–TMA showed a higher stability (0.16 mV h−1) than the one using AEMION™ (0.34 mV h−1). The HFR remained almost constant after the 100 h stability evaluation of PdF–TMA, whereas a significant increase (≈40 mΩ cm2) was observed for AEMION™. This dramatic loss in performance is clear from the polarization curves (Fig. 2a) and the stability results (Fig. 2b). The degradation of the cell with AEMION™ in 2 M KOH may be caused by the combination of electrode and membrane degradation, while the cell with PdF–TMA seemed to only suffer from electrode degradation.
Electrolysis using 2 M KOH at 80 °C
To explore the possibility of further improving the electrolyser performance, both AEMs were evaluated at 80 °C in 2 M KOH (Fig. 3). The increased temperature enhanced the performances at all current densities, favoring electrode kinetics and ion transport. The HFR was found to decrease to 105 and 75 mΩ cm2 for AEMION™ and PdF–TMA, respectively (Table 2). | ||
| Fig. 3 (a) Polarization curves obtained in 2 M KOH and at 80 °C with PdF–TMA (blue circles) and AF1-HNN8-50-X (AEMION™) (black squares) before (solid symbols) and after (open symbols) the stability test. Commercial electrodes with NiFe2O4 (anode) and RANEY® nickel (cathode) were used. (b) Stability evaluations at 500 mA cm−2 in 2 M KOH and at 80 °C indicating the degradation rate of the PdF–TMA (black) and AEMION™ (AF1-HNN8-50-X, blue) cells. | ||
However, the stability evaluation showed a substantial performance loss of the cell with AEMION™ over time. The degradation rate was higher at 80 °C (0.44 mV s−1) than at 60 °C (0.34 mV s−1). Notably, the HFR increased to 180 mΩ cm2. In contrast, the resistance of PdF–TMA did not show any appreciable increase and the HFR of the cell did not exceed 77 mΩ cm2. Also, the performance decay was less evident at 80 °C than at 60 °C (Fig. 3b). In fact, the degradation rate of the cell with PdF–TMA was lower at 80 °C (0.10 mV h−1) than at 60 °C (0.16 mV h−1). This can be explained by a higher activity of the electrodes and a higher conductivity of the electrolyte at 80 °C, which may compensate for any degradation of the electrodes.
Even though the cell using the PdF–TMA membrane showed a similar, or higher, degradation rate in 2 M KOH compared to what Schauer et al. and Hnát et al. achieved with a quaternized qPPO membrane (0.1 mV h−1)34 and a DABCO functionalized PSEBS membrane (0.02 mV h−1),35 respectively, both these AEMs show a lower conductivity than PdF–TMA. In addition, considering that the cell with PdF–TMA in 2 M KOH at 60 °C and 80 °C surpassed the good performance of AEMION™ in 1 M KOH, the PdF–TMA can be considered to be an excellent AEM material for AEMWE applications at high KOH concentration.
NMR spectroscopy of AEMs after stability evaluation
To further investigate the stability of the AEMs, 1H NMR analysis was performed on the membranes before and after the cell evaluation. Fig. 4a shows the spectrum of the pristine AEMION™ membrane (AF1-HNN8-50-X), while Fig. 4b shows the NMR spectrum of the same membrane after evaluation in the electrolysis cell for 100 h at 0.5 mA cm−2 in 1 M KOH at 60 °C. As seen, after these conditions some structural changes were detected (Fig. 4c). This further confirms that the loss of conductivity was due to polymer degradation caused by the high OH− concentration. A similar chemical degradation was also detected after operation at 80 °C (Fig. 4d).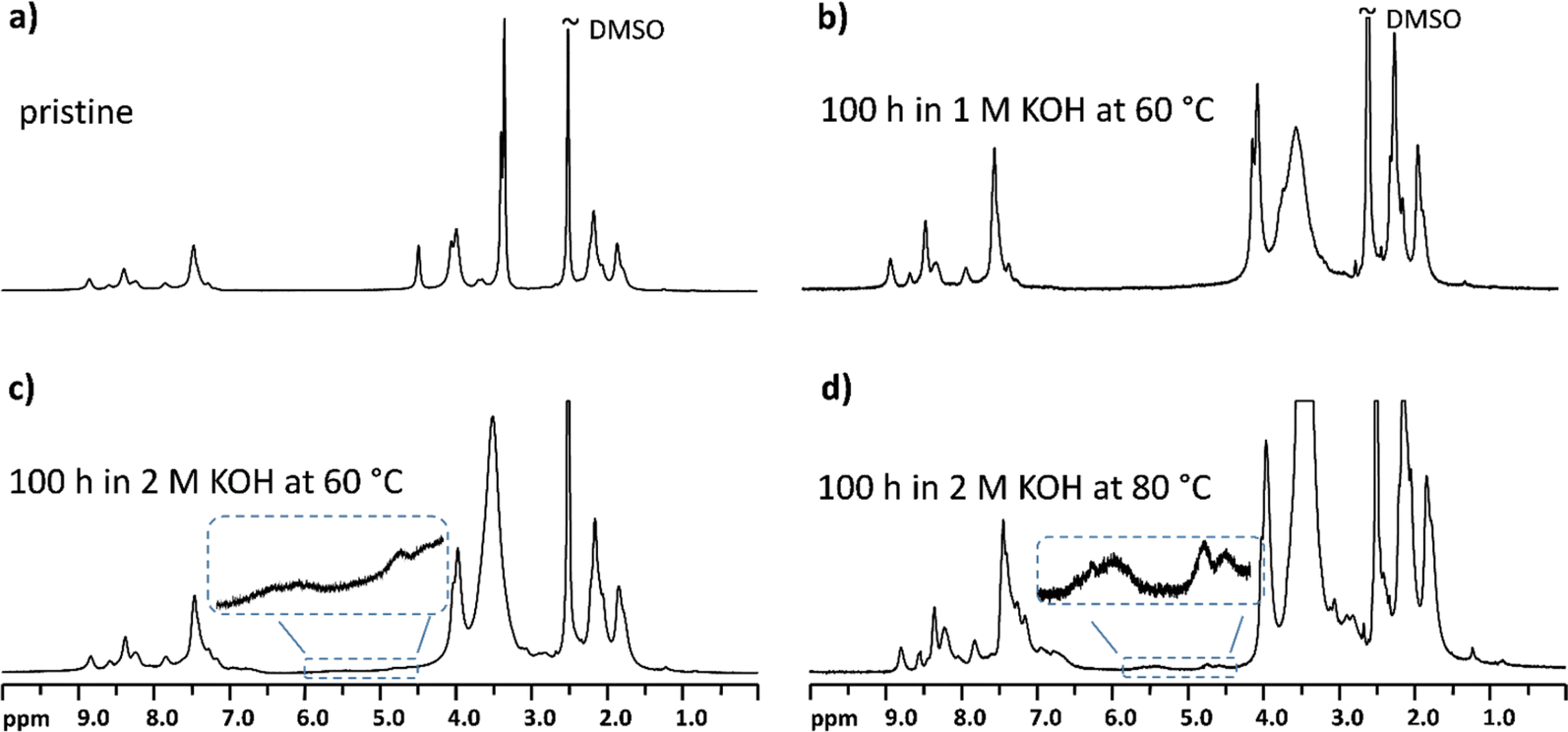 | ||
| Fig. 4 1H NMR spectra of the AEMION™ (AF1-HNN8-50-X) membrane before (a) and after long-term stability evaluation in the electrolysis cell (b–d). | ||
In contrast, the NMR spectra of PdF–TMA remained unchanged (Fig. 5), indicating an intact molecular structure after 100 h of operation in 1 M KOH at 60 °C, and even in 2 M KOH at 80 °C. This supports the finding that the PdF–TMA membrane is highly stable at higher OH− concentrations, as the electrolysis data did not indicate a decrease in conductivity (Table 2).
 | ||
| Fig. 5 1H NMR spectra of the PdF–TMA membrane before (a) and after long-term stability evaluation in the electrolysis cell (b–d). | ||
Electrolyte uptake
As previously reported, the concentration of the KOH electrolyte highly influences the OH− conductivity. A high KOH concentration promotes absorption of hydroxide ions within the membrane and provides extra charge carriers.10 On the other hand, this environment reduces the membrane water content due to a higher osmotic pressure difference, hindering OH− transport through the ionic channels.46 From the results in Table 2, AEMION™ shows a good trade-off between these effects in 1 M KOH, but suffers from a decreased conductivity in 2 M KOH. In contrast, the PdF–TMA membrane shows improved conductivity in 2 M KOH, which may be due to increased KOH absorption. Given these premises, the electrolyte uptake of the two membranes was quantified to understand their different behaviours in the electrolyzer cells at 1 M KOH and 2 M KOH.Table 3 shows the electrolyte uptake and swelling ratio of the two AEMs when immersed in 1 M and 2 M KOH. Considering the electrolyte uptake and through-plane swelling ratio of AEMION™, this membrane tends to absorb more electrolyte solution and swell more at all combinations of temperature and concentration. For this reason, the higher cell resistance recorded for PdF–TMA in 1 M KOH at 60 °C (Table 2) is unlikely due to a higher membrane thickness under the operation conditions. Even though the PdF–TMA membrane has a high conductivity in water (Table 1), AEMION™ becomes more conductive than this membrane in 1 M KOH due to a higher KOH absorption. On the other hand, AEMION™ likely suffers from excessive loss of water in 2 M KOH, and the extra charge carriers cannot improve the conductivity. The PdF–TMA membrane performs better in this harsher environment due to its higher water retention. In fact, even though both membranes show a lower uptake of the electrolyte in 2 M KOH than in 1 M KOH, the decrease in percentage is lower for PdF–TMA than for AEMION™. Not only PdF–TMA can benefit from KOH absorption at this higher concentration, but also the water balance can play a role. As a matter of fact, an excessive water uptake can dilute the charge carriers in the membrane.46
| KOH concentration (M) | AF1-HNN8-50-X (AEMION™) | PdF–TMA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | |||||||||
| Temperature (°C) | 20 | 60 | 80 | 20 | 60 | 80 | 20 | 60 | 80 | 20 | 60 | 80 |
| Electrolyte uptake (%) | 52 | 95 | 108 | 44 | 63 | 67 | 57 | 78 | 84 | 45 | 59 | 62 |
| In-plane swelling (%) | 11.6 ± 4.7 | 18.0 ± 3.7 | 16.7 ± 5.7 | 8.2 ± 4.1 | 9.8 ± 2.5 | 8.9 ± 3.3 | 14.3 ± 2.1 | 20.6 ± 0.9 | 23.5 ± 1.5 | 8.6 ± 1.4 | 0.4 ± 1.6 | 13.9 ± 1.9 |
| Through-plane swelling (%) | 30 | 48 | 50 | 20 | 32 | 36 | 10 | 15 | 18 | 11 | 14 | 18 |
The water uptake directly influences also the degradation of the membrane. When cationic groups are less solvated by water molecules, they become less protected from OH− attack.10 AEMION™ suffered from a high degradation rate when exposed to 2 M KOH, which de-waters the AEM, as previously discussed for the conductivity. PdF–TMA did not reveal any signs of degradation even at 80 °C, while for AEMION™ the degradation accelerated at this temperature. Besides the higher water retention, the improved stability of PdF–TMA is also due to the effect of the flexible phenylpropyl spacer chain (Fig. 4d), which protects the quaternary ammonium cations from hydroxide attack via steric hindrance.10 Notably, PdF–TMA and AEMION™ showed different extents of electrolyte uptake and water retention at similar ion exchange capacities (IEC). Even though the water uptake is highly dependent on this parameter, PdF–TMA may more readily absorb water due to its more flexible structure, which allows the ionic channels to “stretch” and host more water. Also, an improved phase separation may result in water channels with higher internal cation concentrations, which may explain the higher water uptake.
Conclusions
In this work, we have investigated the application of a poly(fluorene alkylene) membrane tethered with trimethylammonium cations via flexible phenylpropyl side chains for AEMWE. Since the membrane displayed a high alkaline stability at 1 M KOH, it was evaluated at higher KOH concentration and higher temperature (2 M KOH, 80 °C) than usual for AEMWE to boost electrolysis performances. Not only did the higher KOH concentration improve the kinetics and ion transport in the electrode, but the PdF–TMA membrane was also shown to be more conductive in 2 M KOH than in 1 M KOH at 60 °C. In comparison, the commercial benchmark AEMION™ membrane showed loss of conductivity and stability in 2 M KOH at 60 °C. Moreover, the conductivity of PdF–TMA increased without compromising its stability when operated at 80 °C in 2 M KOH, thus allowing a high electrolyzer performance. The chemical stability of the membrane was demonstrated by 1H NMR analysis of AEMs after the evaluation. The PdF–TMA membrane shows a lower relative loss of electrolyte when KOH concentration is increased from 1 to 2 M. We speculate that the more flexible structure of PdF–TMA facilitates the retention of the electrolyte solution at high KOH concentrations.Author contributions
Matteo Rossini: conceptualization, methodology, investigation, writing – original draft. Dong Pan: conceptualization, methodology, investigation, writing – review & editing. Burak Koyutürk: methodology, investigation, writing – review & editing. Si Chen: investigation. Amirreza Khataee: supervision, writing – review & editing. Göran Lindbergh: supervision, writing – review & editing. Patric Jannasch: supervision, writing – review & editing. Ann Cornell: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Foundation for Strategic Research (SSF) (project: PUSH, project number: C68943).References
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS
.
-
P. Häussinger, R. Lohmüller and A. M. Watson, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wiley, 1st edn, 2011 Search PubMed
- M. Voldsund, K. Jordal and R. Anantharaman, Int. J. Hydrogen Energy, 2016, 41, 4969–4992 CrossRef CAS
- C. Hank, A. Sternberg, N. Köppel, M. Holst, T. Smolinka, A. Schaadt, C. Hebling and H.-M. Henning, Sustainable Energy Fuels, 2020, 4, 2256–2273 RSC
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed
- I. Vincent, A. Kruger and D. Bessarabov, Int. J. Hydrogen Energy, 2017, 42, 10752–10761 CrossRef CAS
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC
- M. Schalenbach, W. Lueke and D. Stolten, J. Electrochem. Soc., 2016, 163, F1480–F1488 CrossRef CAS
- J. Brauns, J. Schönebeck, M. R. Kraglund, D. Aili, J. Hnát, J. Žitka, W. Mues, J. O. Jensen, K. Bouzek and T. Turek, J. Electrochem. Soc., 2021, 168, 014510 CrossRef CAS
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed
- X. Su, L. Gao, L. Hu, N. A. Qaisrani, X. Yan, W. Zhang, X. Jiang, X. Ruan and G. He, J. Membr. Sci., 2019, 581, 283–292 CrossRef CAS
- M. Liu, X. Hu, B. Hu, L. Liu and N. Li, J. Membr. Sci., 2022, 642, 119966 CrossRef CAS
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, Energy Environ. Sci., 2021, 14, 6338–6348 RSC
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. Richard Ni and L. Meroueh, Int. J. Hydrogen Energy, 2021, 46, 3379–3386 CrossRef CAS
- Y. S. Park, J. Jeong, Y. Noh, M. J. Jang, J. Lee, K. H. Lee, D. C. Lim, M. H. Seo, W. B. Kim, J. Yang and S. M. Choi, Appl. Catal., B, 2021, 292, 120170 CrossRef CAS
- A. G. Wright, J. Fan, B. Britton, T. Weissbach, H.-F. Lee, E. A. Kitching, T. J. Peckham and S. Holdcroft, Energy Environ. Sci., 2016, 9, 2130–2142 RSC
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2, 7903–7912 CrossRef CAS
- B. Chen, P. Mardle and S. Holdcroft, J. Power Sources, 2022, 550, 232134 CrossRef CAS
- M. Moreno-González, P. Mardle, S. Zhu, B. Gholamkhass, S. Jones, N. Chen, B. Britton and S. Holdcroft, J. Power Sources Adv., 2023, 19, 100109 CrossRef
- S. Y. Kang, J. E. Park, G. Y. Jang, O.-H. Kim, O. J. Kwon, Y.-H. Cho and Y.-E. Sung, Int. J. Hydrogen Energy, 2022, 47, 9115–9126 CrossRef CAS
- Z. Jiang, G. Yi, X. Yao, Y. Ma, X. Su, Q. Liu and Q. Zhang, Chem. Eng. J., 2023, 467, 143442 CrossRef CAS
- M. J. Jang, S. H. Yang, M. G. Park, J. Jeong, M. S. Cha, S.-H. Shin, K. H. Lee, Z. Bai, Z. Chen, J. Y. Lee and S. M. Choi, ACS Energy Lett., 2022, 7, 2576–2583 CrossRef CAS
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS PubMed
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS
- J. Xiao, A. M. Oliveira, L. Wang, Y. Zhao, T. Wang, J. Wang, B. P. Setzler and Y. Yan, ACS Catal., 2021, 11, 264–270 CrossRef CAS
- R. Soni, S. Miyanishi, H. Kuroki and T. Yamaguchi, ACS Appl. Energy Mater., 2021, 4, 1053–1058 CrossRef CAS
- D. Aili, M. R. Kraglund, J. Tavacoli, C. Chatzichristodoulou and J. O. Jensen, J. Membr. Sci., 2020, 598, 117674 CrossRef CAS
- M. R. Kraglund, D. Aili, K. Jankova, E. Christensen, Q. Li and J. O. Jensen, J. Electrochem. Soc., 2016, 163, F3125–F3131 CrossRef CAS
- D. Aili, A. G. Wright, M. R. Kraglund, K. Jankova, S. Holdcroft and J. O. Jensen, J. Mater. Chem. A, 2017, 5, 5055–5066 RSC
- J. Schauer, J. Hnát, L. Brožová, J. Žitka and K. Bouzek, J. Membr. Sci., 2015, 473, 267–273 CrossRef CAS
- J. Hnát, M. Plevová, J. Žitka, M. Paidar and K. Bouzek, Electrochim. Acta, 2017, 248, 547–555 CrossRef
- S. Chen, D. Pan, H. Gong and P. Jannasch, Chem. Mater., 2024, 36, 371–381 CrossRef CAS
- D. Pan, P. M. Bakvand, T. H. Pham and P. Jannasch, J. Mater. Chem. A, 2022, 10, 16478–16489 RSC
- AEMION™ technical data sheet.
- T. Weissbach, A. G. Wright, T. J. Peckham, A. Sadeghi Alavijeh, V. Pan, E. Kjeang and S. Holdcroft, Chem. Mater., 2016, 28, 8060–8070 CrossRef CAS
- N. Ziv and D. R. Dekel, Electrochem. Commun., 2018, 88, 109–113 CrossRef CAS
- I. V. Pushkareva, A. S. Pushkarev, S. A. Grigoriev, P. Modisha and D. G. Bessarabov, Int. J. Hydrogen Energy, 2020, 45, 26070–26079 CrossRef CAS
- A. Kiessling, J. C. Fornaciari, G. Anderson, X. Peng, A. Gerstmayr, M. R. Gerhardt, S. McKinney, A. Serov, Y. S. Kim, B. Zulevi, A. Z. Weber and N. Danilovic, J. Electrochem. Soc., 2021, 168, 084512 CrossRef CAS
- A. Khataee, A. Shirole, P. Jannasch, A. Krüger and A. Cornell, J. Mater. Chem. A, 2022, 10, 16061–16070 RSC
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishida and A. Nakano, Int. J. Hydrogen Energy, 2018, 43, 17030–17039 CrossRef CAS
- J. Liu, Z. Kang, D. Li, M. Pak, S. M. Alia, C. Fujimoto, G. Bender, Y. S. Kim and A. Z. Weber, J. Electrochem. Soc., 2021, 168, 054522 CrossRef CAS
- K. F. L. Hagesteijn, S. Jiang and B. P. Ladewig, J. Mater. Sci., 2018, 53, 11131–11150 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2024 |