
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolarized local excitons assist charge dissociation in Y6-based nonfullerene organic solar cells: a nonadiabatic molecular dynamics study†
Bin
Liu
 *a,
Philip C. Y.
Chow
b,
Junzhi
Liu
c and
Ding
Pan
*ade
*a,
Philip C. Y.
Chow
b,
Junzhi
Liu
c and
Ding
Pan
*ade
aDepartment of Physics, The Hong Kong University of Science and Technology, Hong Kong, China. E-mail: bliuaq@connect.ust.hk; dingpan@ust.hk
bDepartment of Mechanical Engineering, The University of Hong Kong, Hong Kong, China
cState Key Laboratory of Synthetic Chemistry, HKU-CAS Joint Laboratory on New Materials and Department of Chemistry, The University of Hong Kong, Hong Kong, China
dDepartment of Chemistry, The Hong Kong University of Science and Technology, Hong Kong, China
eHKUST Shenzhen-Hong Kong Collaborative Innovation Research Institute, Shenzhen, China
First published on 16th May 2024
Abstract
Y6-based nonfullerene organic solar cells (OSCs) have achieved an outstanding power conversion efficiency (PCE) of over 19% due to the low energy loss and high exciton dissociation efficiency with a small energy offset. However, the exciton dissociation mechanism is still under debate. It is unclear why a small energy offset can lead to efficient exciton dissociation in nonfullerene systems, but causes significant charge recombination in fullerene ones. Most of the previous theoretical studies have focused on the static properties of donor–acceptor heterojunctions, while neglecting excited electron dynamics and nonadiabatic effects. Here, we applied nonadiabatic molecular dynamics simulations to study the charge transfer dynamics in both donor:Y6 and donor:C60 crystalline systems. We found that thermal effects do not significantly influence the exciton dissociation in the NT-4T-FF:Y6 system, which aligns with experimental observations. Based on our simulations, we identified a five-step charge transfer process in nonfullerene systems. While previous studies suggested electrostatic interfacial fields from non-fullerene small molecule acceptors, our research reveals that strong donor–acceptor interactions primarily affect the local exciton states rather than the ground state. Consequently, the polarized local excitons play a key role in reducing the Coulomb attraction between electrons and holes, thus facilitating exciton dissociation with a small energy offset. In contrast, this mechanism is not observed in fullerene OSC systems. Our findings provide a fundamental basis for the further development of novel OSC materials with the potential for achieving even higher PCE.
Introduction
Organic solar cells (OSCs) have become a very promising technology to convert solar energy into electricity due to their unique properties, including unlimited availability, environmentally friendly characteristics, high mechanical flexibility, ease of processing, and low synthesis costs.1–6 Over the past three decades, the power conversion efficiency (PCE) of polymer-small-molecule OSCs, in which the active layer consists of a polymer donor and small molecule acceptor forming an interpenetrating network, has continuously increased from only ∼1% in fullerene solar cells achieved in 1992 (ref. 7) to over 19% in state-of-the-art A-DA’D-A solar cells8,9 (D denotes electron-donating units and A denotes electron-accepting units) through material engineering, such as modification of push–pull strength between D and A units, halogenation, introduction of non-covalent interactions, and side chain engineering.4,10,11 So far, by conducting molecular structure modification, blend morphology optimization, and device engineering, tens of thousands of OSC devices have been reported by the OSC community which mainly rely on the laborious trial-and-error method.12 However, many fundamental questions about the molecular mechanisms still remain unanswered, which hinders the further improvement of OSC performance.The operational mechanism of OSC devices consists of four steps: (1) the generation of Frenkel-type local excitons (LEs) through optical absorption, (2) the diffusion of LEs to the D/A interfaces, (3) the dissociation of LEs into charge-transfer (CT) excitons, and (4) the subsequent separation and collection of charges.11 The LE dissociation efficiency plays a vital role in determining the performance of OSCs, which contributes to larger short-circuit current density (Jsc) and lower energy loss. In conventional fullerene-based OSCs, there exists a notable energy offset greater than 0.3 eV between the LUMO levels of the donor and acceptor materials. This energy offset plays a crucial role as a driving force, allowing for the effective reduction of Coulomb interactions between electrons and holes. As a result, it facilitates the efficient dissociation of LEs into CT excitons.13 Previous studies suggested that the exciton dissociation efficiency exponentially decays on decreasing the energy offset,14,15 resulting in small Jsc and low PCE. Interestingly, nonfullerene OSCs with A-D-A or A-DA’D-A type structures can still maintain high exciton dissociation efficiency when there is a negligible or even zero energy offset between the HOMO or LUMO levels of donors and acceptors. For example, in 2015 Lin et al. introduced an A-D-A type molecule ITIC to the nonfullerene OSC and achieved a PCE of 6.8% with an energy offset of 0.24–0.28 eV.16 In 2019, Yuan et al. made use of an A-DA’D-A type molecule Y6 and achieved a record PCE of 15.7% with an energy offset of 0.09 eV.17 Experimental studies showed that an energy offset of 0–0.1 eV is enough for satisfactory LE dissociation. However, an important question arises regarding why a seemingly insignificant energy offset can lead to efficient exciton dissociation in nonfullerene OSCs, while it fails to achieve the same outcome in fullerene OSCs.
Several exciton dissociation mechanisms have been proposed to elucidate the process of overcoming the Coulomb interaction between electrons and holes in the fullerene based OSCs. These mechanisms include charge delocalization, energy,18,19 entropy,20,21 and disorder effects.22,23 For nonfullerene based OSCs, previous studies suggest that the hybridization between LE and CT states,24,25 the electrostatic potential difference,26 the quadrupole of the Y6 molecule,27 and the interface polarization28 may facilitate charge dissociation. Most previous studies focused on the static properties of donor–acceptor heterojunctions, neglecting excited electron dynamics and nonadiabatic effects. Many of them studied the dissociation of excitons at the CT state into free charges, but ignored the exciton dynamics from the LE state to the CT state.
Here, we conducted Ehrenfest molecular dynamics (EMD) simulations to compare the fullerene based and Y6 based donor–acceptor systems with small driving forces to study photoexcited electron dynamics. EMD combines the time-dependent density-functional theory (TDDFT) with classical equations of motion for nuclei in a mean field manner, and has been recently developed as a powerful tool for simulating time dependent charge separation,29–32 photoisomerization,33 and other processes involving nonadiabatic effects.34,35 We found that the thermal effects are not essential for exciton dissociation in the NT-4T-FF:Y6 system, which is consistent with the current experimental findings. We revealed a five-step exciton dissociation process in the donor polymer-Y6 molecule crystal. Notably, we observed substantial modifications in the dipole moments of various LE states due to the presence of the Y6 molecule. While previous studies suggested electrostatic interfacial fields from non-fullerene small molecule acceptors,26,27 our research reveals that strong donor–acceptor interactions primarily affect LE states rather than the ground state. This phenomenon resulted in charge redistribution and delocalization, thereby reducing the Coulomb attraction between electrons and holes. As a comparison, we did not observe a similar process in the fullerene-based system. Our findings provide a fundamental basis for further advancements in OSC materials. They offer significant potential for achieving enhanced PCE and contribute to ongoing research in this field.
Results and discussion
We performed EMD simulations to compare two crystalline donor–acceptor systems, polymer:Y6 and polymer:C60 with three-dimensional periodic boundaries as shown in Fig. 1, to study the exciton dissociation mechanism with a small energy offset. Naphtho[1,2-c:5,6-c′]bis[1,2,5]thiadiazole (NT) based conjugated polymer donors have demonstrated high performance in both fullerene36 and nonfullerene37 OSC devices, so here we chose the polymer NT-4T-FF as the common donor. The fluorination on thiophene rings were used to downshift both HOMO and LUMO levels of the polymer donor to match with those of Y6 and C60 molecules. The LUMO energy offsets of the NT-4T-FF:Y6 and NT-4T-FF:C60 structures calculated with the hybrid B3LYP38–40 exchange–correlation (xc) functional are 0.27 and 0.11 eV, respectively. We simplified the Y6 molecule in our model systems by substituting all alkyl side chains with hydrogen atoms. This modification allows for more efficient theoretical simulations while preserving similar electronic and optical characteristics.31,41 More computation details are given in the Methods section and the ESI.†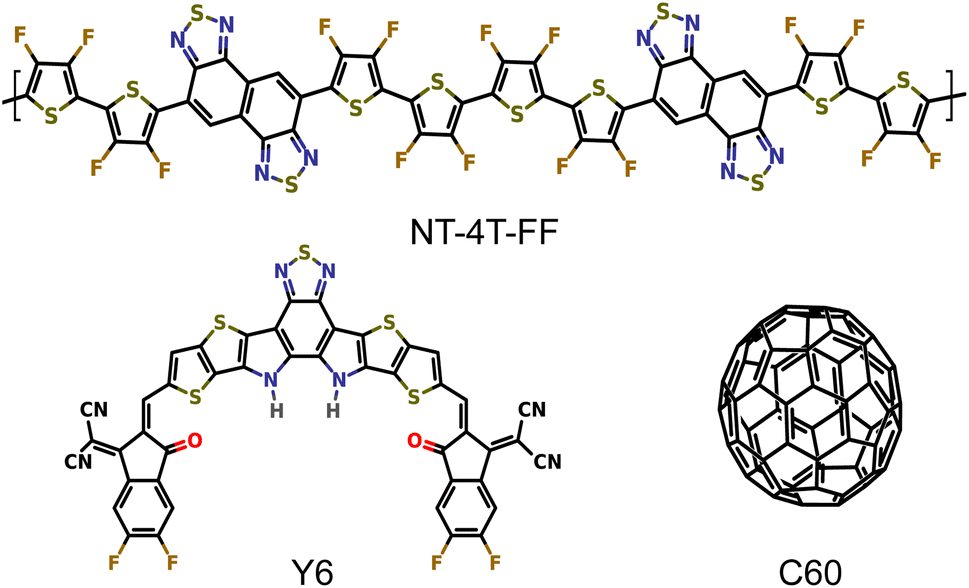 | ||
| Fig. 1 Molecular structures of the donor polymer NT-4T-FF and two acceptors: the Y6 and fullerene (C60) molecules. | ||
We prepared the initial excited state by moving one electron from the HOMO to the LUMO of the polymer donor. Fig. 2 shows the time evolution of the Mulliken charge transfer from the polymer to the Y6 molecule in the EMD and real-time propagation (RTP) TDDFT simulations. In the EMD simulations, we incorporated the non-adiabatic coupling between the electronic and nuclear degrees of freedom, taking into account the thermal effects, whereas in the RTP-TDDFT simulation, the nuclei were fixed at 0 K. As shown in Fig. 2, the electronic and nuclear coupling does not significantly affect the charge transfer process between the polymer and the Y6 molecule, particularly after 150 fs, which suggests that the thermal effects are not essential for the exciton dissociation in the NT-4T-FF:Y6 system. Our finding is consistent with the recent experimental results in nonfullerene OSCs. By using the optical measurement, Ma et al. found that the exciton dissociation into the CT state remained efficient at a temperature as low as 15 K in both P3TEA:SF-PDI2 and PM6:Y6 blends.42 In contrast, phonon assisted exciton dissociation was reported in many fullerene-based OSCs.43,44 For example, Falke et al. found that the electron transfer could be prevented by fixing the nuclear positions in the 4T:C60 crystal system,31 indicating that the thermal effects are more important in fullerene-based OSCs than in nonfullerene ones. The agreement between our simulations and experimental results demonstrates the reliability of our approach in describing the complex dynamics of excited electrons.
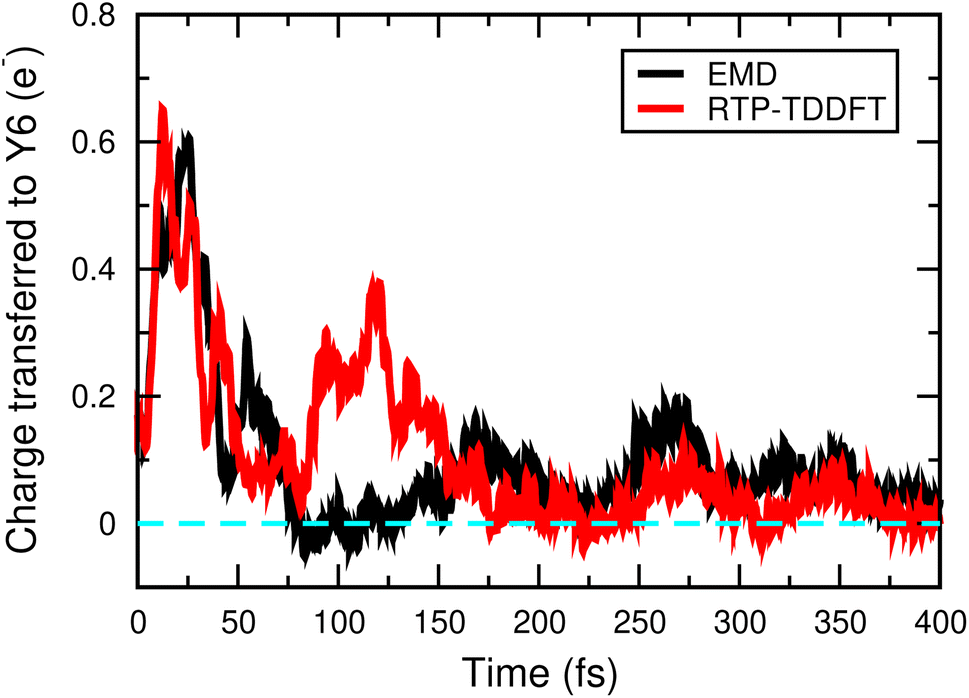 | ||
| Fig. 2 Time evolution of the Mulliken charge transferred from the donor polymer to the Y6 molecule calculated using the Ehrenfest molecular dynamics (EMD, black lines) and the real time propagation approach to time-dependent density functional theory (RTP-TDDFT, red lines). In the EMD simulations, the temperature was 300 K. In the RTP-TDDFT simulations, nuclear positions were all fixed. | ||
Fig. 3 compares the Mulliken charge transfer in the NT-4T-FF:Y6 and NT-4T-FF:C60 systems. We discovered a five-step process of charge transfer, as shown by the charge accumulation on the Y6 molecule in Fig. 3(a). In the first step at t = 0, the lowest local excited state (LE1) was formed by exciting one spin-up electron from the HOMO to the LUMO of the polymer. Fig. 3(b) shows that initially the excited electron density was mainly found around one electron-withdrawing NT unit of the polymer, which is due to the splitting of the degenerated molecular orbitals after the thermal relaxation in the molecular dynamics simulation. In the second step, the localized LE1 charge density was redistributed between two NT units in the polymer in ∼4 fs. To further understand the energy levels of excited states, we took the nuclear coordinates from the EMD simulations and performed the static DFT calculations, in which we excited the electron from the HOMO of the ground state to a certain excited state (see Fig. S5†).33Fig. 4(a) shows that initially the potential energy (VEMD) in the EMD simulations was close to the potential energy of the LE1 state, i.e., the total energy obtained from the static DFT calculation. In 4 fs, VEMD reached the potential energy of the second lowest local excited state (LE2), in which the excited electron was placed in the LUMO+1 of the polymer. The variation of VEMD suggested that the NT-4T-FF:Y6 system changed from the LE1 to LE2 states in the first 4 fs. The excited electron in the LE2 state was more delocalized along the polymer backbone chain, which promotes LE dissociation by increasing the radius of the electron–hole pair and consequently reducing the strength of the Coulomb attraction.19,45 The third step is the exciton dissociation, in which about half of the excited electron was transferred from the polymer to the Y6 molecule between t = 4 and 11 fs as shown in Fig. 3(a). In the third step, VEMD gradually increased to match the potential energy of the third lowest charge-transfer state (CT3), in which the excited electron was in the LUMO+2 of the Y6 molecule, as shown in Fig. 4(b). Fig. 3(b) shows that this charge transfer occurred through the π–π interactions between the NT units of the polymer and the 2-(3-oxo-2,3-dihydroinden-1-ylidene)malononitrile (IC) ending groups in the Y6 molecule, which is consistent with previous theoretical results.46,47 In the fourth step between t = 11 and 26 fs, the transferred electron was redistributed from the two IC ending groups to the whole Y6 molecule. The variation of VEMD shown in Fig. 4(b) suggested that the system changed from the CT3 state to the lowest-lying (CT1) and second lowest-lying charge-transfer (CT2) states, in which the excited electron was in the LUMO and LUMO+1 of the Y6 molecules, respectively. In the final step between t = 26 and 42 fs, as the potential energy of LE1 matched that of the CT1 and CT2 states, electrons were transferred from the Y6 molecule back to the polymer in about 16 fs, resulting in more delocalized charge distribution along the backbone chain of the polymer.
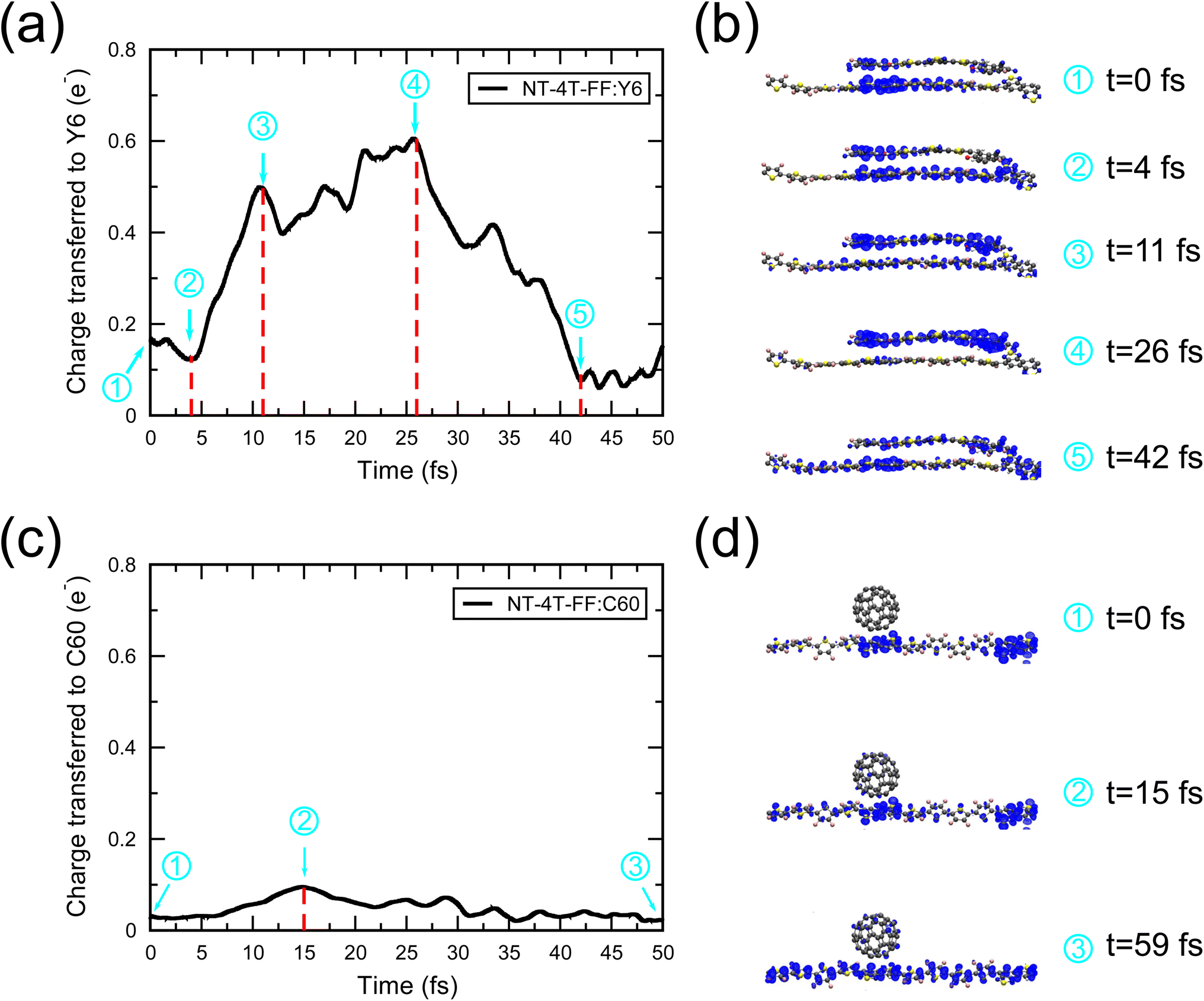 | ||
| Fig. 3 Time evolution of the Mulliken charge accumulation on the Y6 molecule and the C60 molecule transferred from the polymer in the EMD simulations. (a) A five-step charge transfer process is shown in the NT-4T-FF:Y6 system. (c) A three-step charge transfer process is shown in the NT-4T-FF:C60 system. The snapshots of the electron density distribution of the excited electron at each step labelled in (a) and (c) are shown in (b) and (d), respectively. The excited electron density is calculated by subtracting the total electron density at the ground state from the electron density in the EMD simulations, and the positive density difference is shown in blue. | ||
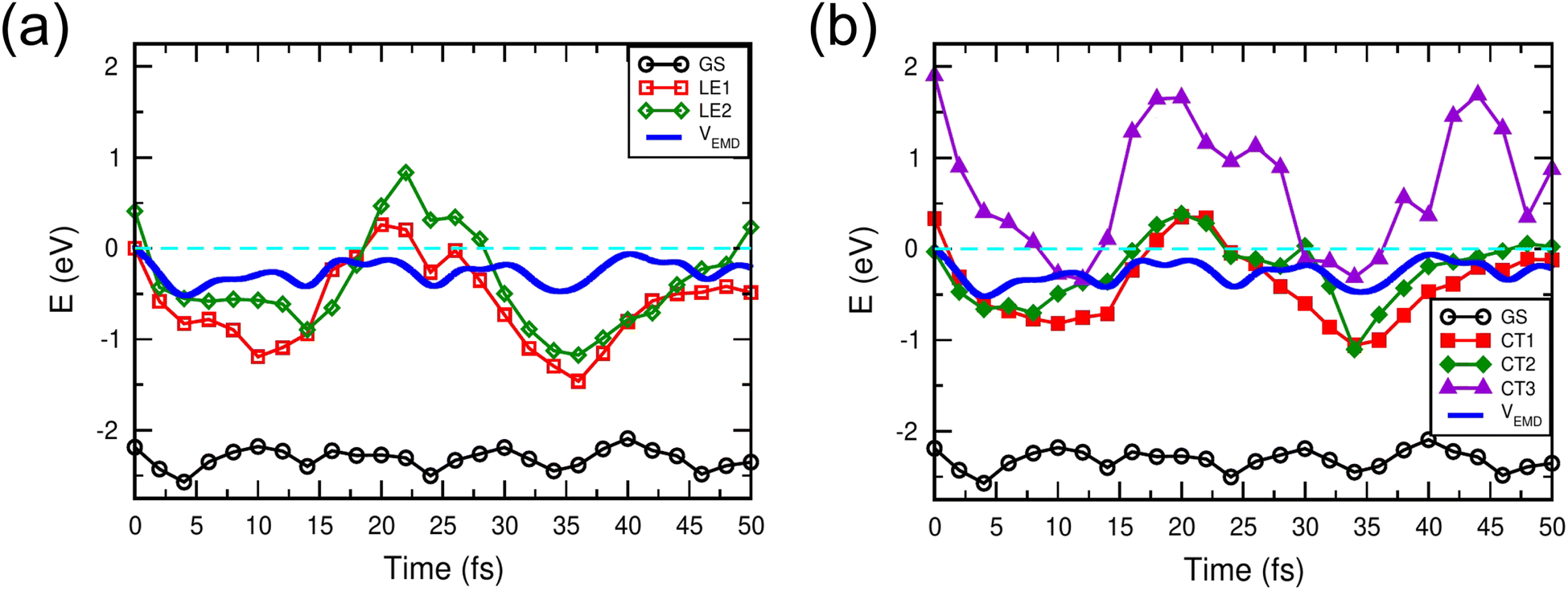 | ||
| Fig. 4 Time evolution of the potential energies in the EMD simulation (VEMD) and static DFT calculations. The LE1 and LE2 states are shown in (a), and the CT1, CT2, and CT3 states are shown in (b). The ground state (GS) and VEMD are shown in both (a) and (b). | ||
Fig. 5 shows a schematic energy diagram to illustrate the exciton dissociation and charge transfer process in the NT-4T-FF:Y6 system. First, the LE1 state was formed upon photoexcitation. In the second step, the excited charge was redistributed along the backbone chain of the polymer, and the system evolved from the LE1 state to the LE2 state. This transition reveals that the lifetime of the LE1 state was approximately 4 fs. In the third step, the excited electron was transferred to the electron-withdrawing IC ending groups in the Y6 molecule, and the system changed from the LE2 state to the CT3 state. The lifetime of LE2 state was about 7 fs. In the fourth step, the system evolved from the CT3 state to the lower-lying CT1 and CT2 states, with a lifetime of 15 fs. During this process, the charge was redistributed along the Y6 backbone chain, resulting in loosely bound electron–hole pairs at the polymer:Y6 interface. In the fifth step, the electron transferred back from the Y6 molecule to the donor polymer, indicating that the CT exciton recombined to become the LE exciton. This process occurred with a lifetime of 16 fs for the CT1–CT2 states. Note that our model system contains only a crystalline donor–acceptor structure, so the charge recombination cannot be avoided. In real OSC devices, the loosely bound CT excitons may further separate into free charges and transfer to neighboring molecules through the π–π intermolecular interactions.41
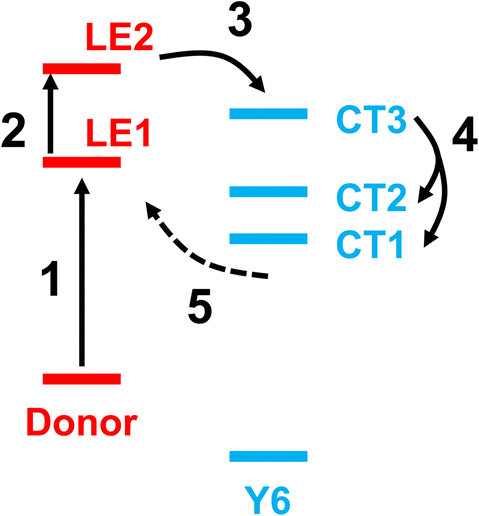 | ||
| Fig. 5 Schematic energy diagram for the five-step charge transfer process in the NT-4T-FF:Y6 system. The LE1 and LE2 states are obtained by exciting the HOMO electron to the LUMO+2 and LUMO+4 orbitals, respectively, where LUMO+x denotes the xth orbital above the LUMO. The HOMO, LUMO+2, and LUMO+4 are all localized on the NT-4T-FF polymer, as shown in Fig. S4.† Similarly, the CT1, CT2, and CT3 states are obtained by exciting the HOMO electron to the LUMO, LUMO+1, and LUMO+3 states, respectively, which are all localized on the Y6 molecule. | ||
Unlike the five-step process in the NF-4T-FF:Y6 system, the charge transfer in the fullerene system consists of three steps as shown in Fig. 3(c) and (d). After the local excitation in the first step, the initially transferred charge from the polymer to the C60 molecule was only about 0.03e, which is negligible as compared to the transferred charge from the polymer to the Y6 molecule (0.17e). Furthermore, during the exciton dissociation process in the second step, the accumulated charge on the C60 molecule was about 0.1e, much smaller than that on the Y6 molecule (0.6e). The lifetime of the LE1 state was about 15 fs. Interestingly, we could not see any charge redistribution process in the C60 molecule. Instead, the CT exciton directly recombined to become the LE in the third step, indicating a strong binding of the electron–hole pair and thus low exciton dissociation efficiency in the NT-4T-FF:C60 system. During the exciton recombination process, the charge transfer between the polymer and C60 was negligibly small, posing challenges in characterizing the lifetime of the CT state. Note that the charge transfer process from the polymer to the C60 molecule was completed within 15 fs, much shorter than that from the polymer to the Y6 molecule (26 fs). Previous experimental studies also showed that the photogenerated excitons in fullerene OSCs have rapid charge separation,31,48,49 whereas the Y6-based systems have relatively slow but still sufficient charge transfer dynamics.41,50,51 Our simulations suggest that the fullerene-based OSC device has limited PCEs due to the poor exciton dissociation efficiency and frequent charge recombination compared to the Y6-based devices, which is consistent with previous experimental studies.7,52,53
The significant charge redistribution occurring on the polymer backbone and the Y6 molecule during the second and fourth steps (Fig. 3(a) and (b)) plays a crucial role in facilitating exciton dissociation. This phenomenon provides a key explanation for the superior performance of Y6 in comparison to fullerene. To conduct a quantitative analysis of the charge redistribution on the polymer backbone chain, we employed maximally localized Wannier functions54 to calculate the change in the dipole moment along the polymer backbone chain in the presence and absence of the Y6 molecule at the ground, LE1, and LE2 states. According to the modern theory of polarization, we cannot uniquely define the total dipole moment of a crystal with periodic boundary conditions, so here we compare the change in dipole moments.55Fig. 6(a) shows that the Y6 molecule affected the dipole moment of the polymer at the LE1 and LE2 states one order of magnitude larger than that at the ground state, indicating that the Y6 molecule substantially polarizes the two LEs. At the LE1 state, the Y6 molecule induced electron accumulation on the left NT unit, whereas at the LE2 state, it caused electron accumulation on the right NT unit (see Fig. S5†). The large difference between the dipole moments at the LE1 and LE2 states reflects the charge redistribution and delocalization along the polymer backbone chain. As a comparison, the C60 molecule affected the dipole moment of the polymer backbone chain little at both ground and excited states, possibly due to the weak interaction between the polymer and the C60 molecule.31
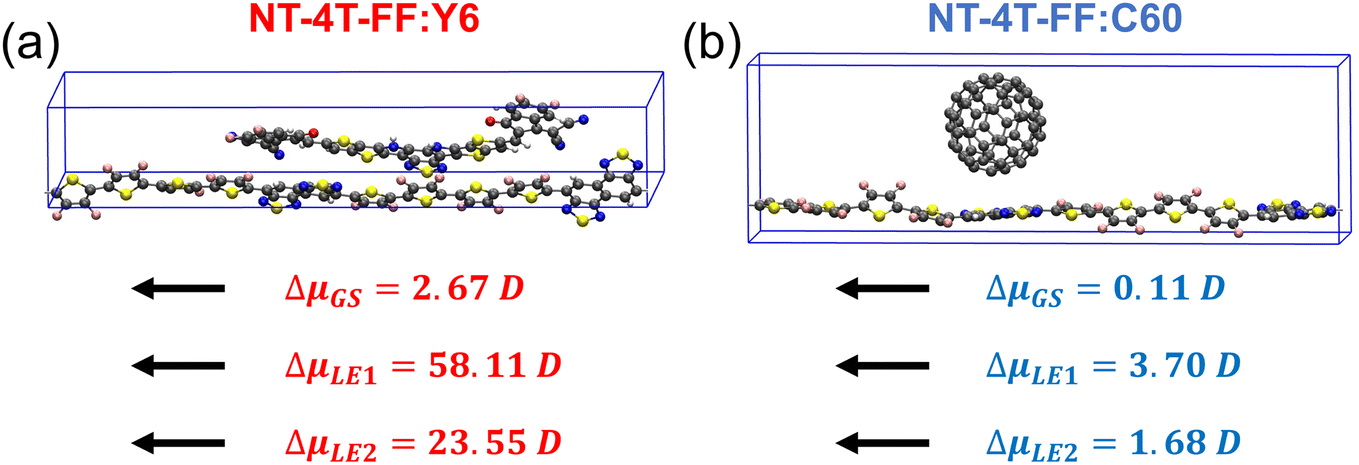 | ||
| Fig. 6 Dipole moment change (Δμ, unit: Debye (D)) along the polymer backbone chain after removing the acceptor molecules at the ground state (GS), and LE1, and LE2 states in (a) the NT-4T-FF:Y6 and (b) the NT-4T-FF:C60 systems calculated at the B3LYP level. Black arrows indicate the direction of the dipole moment change. | ||
The redistribution and delocalization of charges along the Y6 molecule backbone prevent the immediate recombination of separated electrons and holes, and extend the lifetime of CT excitons, which creates more opportunities for enhanced free carrier transport and collection by electrodes.56 In contrast, the charge redistribution observed in the Y6 molecule backbone does not occur in the donor:C60 system. As a result, the exciton dissociation efficiency is lower, and there is a higher possibility for charge recombination in this system, resulting in lower PCE of C60-based OSC devices.
Using the maximally localized Wannier functions, we calculated the exciton binding energy at the CT states:47,57
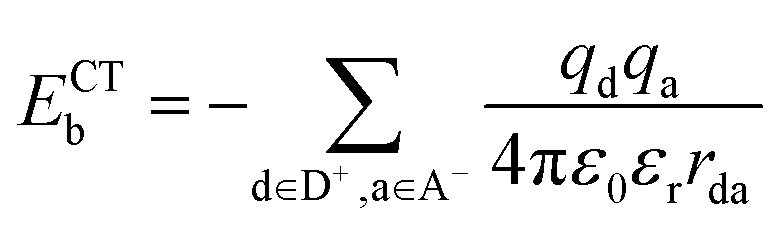 | (1) |
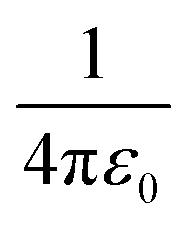 is the Coulomb constant, εr is the relative dielectric constant (εr = 4), qd and qa are the charges of electrons or nuclei in the donor (D+) and acceptor (A−), respectively, and rda is the distance between qd and qa. The position of each electron is given by the center of the corresponding maximally localized Wannier function in the spin polarized calculations. The charges of nuclei are from the pseudopotentials (see the Methods section). In the previous studies, Mulliken charges were used as effective charges, but it is well known that Mulliken charges have a strong basis set dependence, which may give inconsistent charge values.58 Using maximally localized Wannier functions to represent excitons in solids has been recently demonstrated and benchmarked using the ab initio Bethe–Salpeter equation approach.59 We calculated the Coulomb interactions within one unit cell. As shown in Table 1, the exciton binding energies at the CT1, CT2, and CT3 states in the donor-C60 system are 0.26–0.28 eV, which are much larger than 0.07–0.1 eV in the donor:Y6 system.
is the Coulomb constant, εr is the relative dielectric constant (εr = 4), qd and qa are the charges of electrons or nuclei in the donor (D+) and acceptor (A−), respectively, and rda is the distance between qd and qa. The position of each electron is given by the center of the corresponding maximally localized Wannier function in the spin polarized calculations. The charges of nuclei are from the pseudopotentials (see the Methods section). In the previous studies, Mulliken charges were used as effective charges, but it is well known that Mulliken charges have a strong basis set dependence, which may give inconsistent charge values.58 Using maximally localized Wannier functions to represent excitons in solids has been recently demonstrated and benchmarked using the ab initio Bethe–Salpeter equation approach.59 We calculated the Coulomb interactions within one unit cell. As shown in Table 1, the exciton binding energies at the CT1, CT2, and CT3 states in the donor-C60 system are 0.26–0.28 eV, which are much larger than 0.07–0.1 eV in the donor:Y6 system.
| E CTb (eV) | ||
|---|---|---|
| State | NT-4T-FF:Y6 | NT-4T-FF:C60 |
| CT1 | 0.089 | 0.263 |
| CT2 | 0.100 | 0.282 |
| CT3 | 0.069 | 0.277 |
Due to the strong Coulomb attraction, the tightly bound CT excitons cannot be efficiently separated into free charges by thermal energy (∼25 meV) at the typical working temperature. Instead, they have a tendency to recombine into LEs. Thus, previous studies have demonstrated that an energy offset of above 0.3 eV is required in fullerene-based OSCs to overcome the strong Coulomb attraction and facilitate the efficient dissociation of electron–hole pairs.60–62 The charge redistribution and delocalization along the Y6 backbone help to reduce the interaction between electrons and holes, so the CT excitons can be more easily separated into free charges. In addition, previous experimental findings showed that the Y6 molecules have the ability to form polymer-like conjugated backbones with π–π stacking between their end groups, as observed in single crystals. This arrangement facilitates high carrier mobility and ultrafast charge transfer.63 Based on these observations, it is reasonable to anticipate that if the Y6 molecule can establish a conjugated structure with neighboring molecules through π–π stacking, the loosely bound CT excitons will have an increased likelihood of further separating, rather than recombining into LEs.
There is another popular method to obtain nonadiabatic dynamics, i.e., surface hopping. In EMD, nuclei move on an effective potential energy surface obtained by the weighted average of adiabatic states, whereas in surface hopping nuclei evolve on a pure adiabatic potential energy surface, but transitions between different potential energy surfaces are allowed, e.g., by the fewest-switches algorithm.64–66 It may be interesting to also apply the surface hopping method to study the nonadiabatic effects particularly when the system exists in a well localized nonadiabatic coupling region. Note that both the Ehrenfest method and surface hopping may seriously underestimate the dephasing time, due to treating nuclei classically.67
Conclusion
In summary, we employed Ehrenfest molecular dynamics simulations to investigate the dynamics of photoexcited electrons in crystalline NT-4T-FF:Y6 and NT-4T-FF:C60 systems. Our calculations reveal that thermal effects do not significantly influence the exciton dissociation process in the NT-4T-FF:Y6 system, which aligns with experimental observations. Furthermore, we identified a five-step exciton dissociation mechanism in the donor polymer-Y6 molecule crystal. Particularly, the presence of the Y6 molecule induces substantial changes in the dipole moments of both the lowest-lying and higher-lying LE states, one order of magnitude larger than the dipole change at the ground state. Previous studies have suggested that non-fullerene small molecule acceptors may generate significant electrostatic interfacial fields.26,27 However, our current study provides new insights by demonstrating that the strong donor–acceptor interactions primarily influence the excited (LE) states rather than the ground state. Consequently, the polarized LEs lead to charge redistribution and delocalization, which effectively reduce the Coulomb attraction between electrons and holes. Once the electrons are transferred to the Y6 molecule, there is further charge redistribution and delocalization. In contrast, the presence of the fullerene molecule does not significantly alter the dipole moment of various LE states, resulting in less pronounced charge redistribution and delocalization. As a result, a rapid CT recombination process occurs. Our study highlights the crucial role of the charge polarization of the LE states in exciton dissociation. This phenomenon is not observed in the polymer-fullerene system. The insights gained from our investigation provide a fundamental basis for the future development of novel organic solar cell materials, with the potential to achieve even higher power conversion efficiency.Methods
We performed first-principles calculations using the CP2K Quickstep package.68 We used a dual basis set of molecularly optimized Gaussian orbital double-ζ plus polarization quality (DZVP-MOLOPT)69 and plane waves with a density cutoff of 600 Ry. We adopted the Goedecker-Teter-Hutter (GTH) norm-conserving pseudopotentials for the valence electrons.70,71 We used the Perdew–Burke–Ernzerhof (PBE)72 xc functional with Grimme's D3 dispersion correction73 for both adiabatic and non-adiabatic molecular dynamics simulations, and the hybrid B3LYP38–40 xc functional for electronic structure calculations. The force tolerance is 0.01 eV Å−1. Both donor:Y6 and donor:C60 systems have three dimensional periodic boundary conditions. The donor polymer backbone chain is along the x direction. The repeating unit length of the polymer was obtained by optimizing the structure of polymer dimers under open boundary conditions. The lattice constants along the y and z directions were determined by optimizing the total energy of donor–acceptor systems (see the ESI† for more details).We performed Born–Oppenheimer molecular dynamics (BOMD) simulations to initialize the structures of donor–acceptor crystals with thermal relaxation. In the NVT ensemble, we used a time step of 0.5 fs and the temperature was 300 K controlled by canonical sampling through velocity rescaling.74 The trajectory lengths of the donor:Y6 and donor:C60 systems are 15 and 10 ps, respectively. We carried out EMD and RTP-TDDFT simulations after the BOMD simulations. The initial excited state was generated by moving one electron from the HOMO to the LUMO of the polymer donor. In the EMD simulations, the initial velocities of nuclei were adopted from BOMD simulations. We applied a microcanonical NVE ensemble with a time step of 2 as.
In RTP-TDDFT, electrons are propagated according to the time-dependent Kohn–Sham equation:34,75
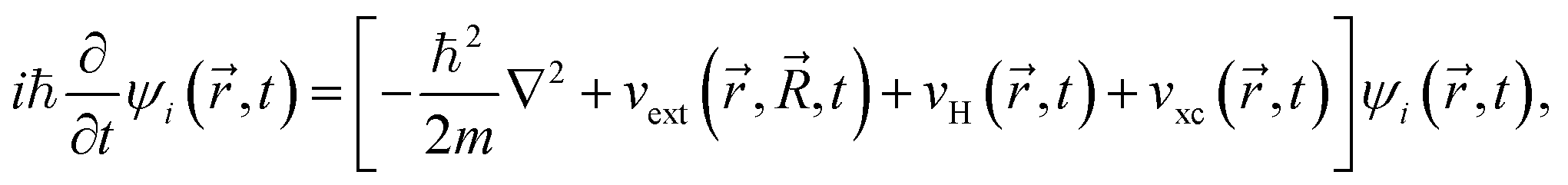 | (2) |
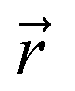 and
and 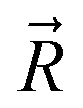 are coordinates of electrons and nuclei, respectively, m is the electron mass,
are coordinates of electrons and nuclei, respectively, m is the electron mass, 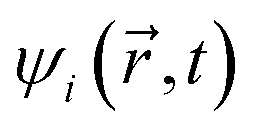 is the ith Kohn–Sham orbital at time t, vext is the external potential generated by nuclei, vH is the Hartree potential, and vxc is the exchange–correlation potential. We applied the adiabatic semi-local density approximation to simplify vxc.76 The Kohn–Sham orbitals are expanded using a linear combination of atomic orbitals:
is the ith Kohn–Sham orbital at time t, vext is the external potential generated by nuclei, vH is the Hartree potential, and vxc is the exchange–correlation potential. We applied the adiabatic semi-local density approximation to simplify vxc.76 The Kohn–Sham orbitals are expanded using a linear combination of atomic orbitals: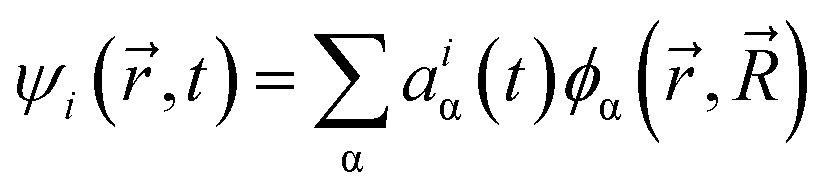 | (3) |
The expansion coefficients aiα(t) are propagated as follows:35
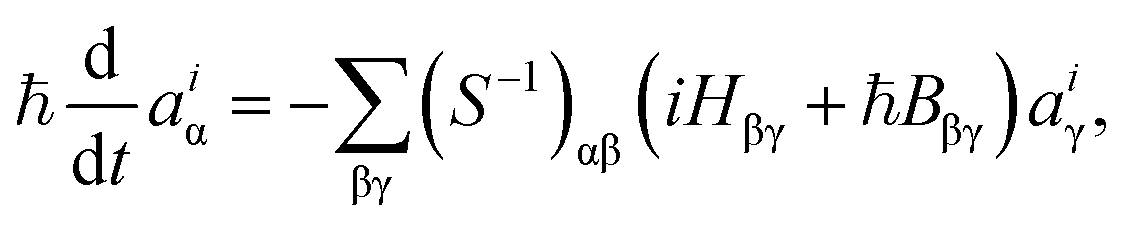 | (4) |
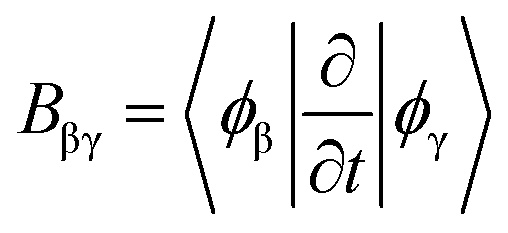 . Note that in some other studies, the nonadiabatic coupling matrix elements were often calculated between Kohn–Sham orbitals.64 We chose the enforced time reversal symmetry propagator.77 In the EMD simulations, nuclei are treated as classical particles moving on an effective potential energy surface:
. Note that in some other studies, the nonadiabatic coupling matrix elements were often calculated between Kohn–Sham orbitals.64 We chose the enforced time reversal symmetry propagator.77 In the EMD simulations, nuclei are treated as classical particles moving on an effective potential energy surface: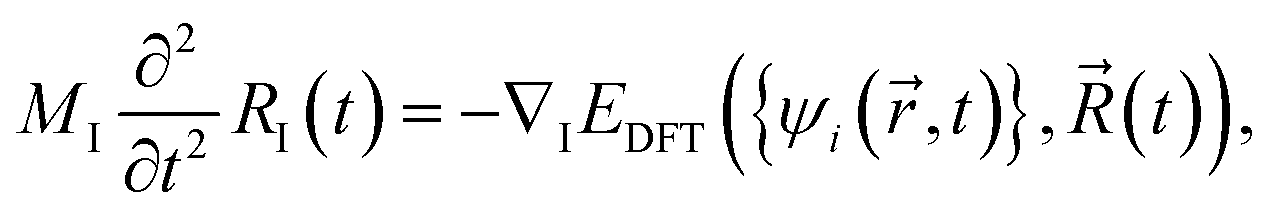 | (5) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. L. and D. P. thank Jianquan Zhang and He Yan for many helpful discussions. D. P. acknowledges support from the Croucher Foundation through the Croucher Innovation Award, the National Natural Science Foundation of China through the Excellent Young Scientists Fund (22022310), the Hetao Shenzhen/Hong Kong Innovation and Technology Cooperation (HZQB-KCZYB-2020083), and the Energy Institute in Hong Kong University of Science and Technology. P. C. Y. C. acknowledges support from the Hong Kong Research Grant Council (16302520) and the National Natural Science Foundation of China through the Excellent Young Scientists Fund (22222905). J. L. acknowledges support from the Hong Kong Research Grants Council (17309023) and the National Natural Science Foundation of China (22122114).References
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated polymer-based organic solar cells, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Recent advances in bulk heterojunction polymer solar cells, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Low bandgap semiconducting polymers for polymeric photovoltaics, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- G. Zhang, J. Zhao, P. C. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Nonfullerene acceptor molecules for bulk heterojunction organic solar cells, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee and T. Wang, et al., A history and perspective of non-fullerene electron acceptors for organic solar cells, Adv. Energy Mater., 2021, 11, 2003570 CrossRef CAS.
- X. Liang, J. Wang, R. Miao, Q. Zhao, L. Huang, S. Wen and J. Tang, The evolution of small molecular acceptors for organic solar cells: advances, challenges and prospects, Dyes Pigm., 2022, 198, 109963 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Photoinduced electron transfer from a conducting polymer to buckminsterfullerene, Science, 1992, 258, 1474–1476 CrossRef CAS PubMed.
- Y. Wei, Z. Chen, G. Lu, N. Yu, C. Li, J. Gao, X. Gu, X. Hao, G. Lu and Z. Tang, et al., Binary organic solar cells breaking 19% via manipulating the vertical component distribution, Adv. Mater., 2022, 34, 2204718 CrossRef CAS PubMed.
- J. Fu, P. W. Fong, H. Liu, C.-S. Huang, X. Lu, S. Lu, M. Abdelsamie, T. Kodalle, C. M. Sutter-Fella and Y. Yang, et al., 19.31% binary organic solar cell and low non-radiative recombination enabled by non-monotonic intermediate state transition, Nat. Commun., 2023, 14, 1760 CrossRef CAS PubMed.
- B. Liu, D. Rocca, H. Yan and D. Pan, Beyond conformational control: effects of noncovalent interactions on molecular electronic properties of conjugated polymers, JACS Au, 2021, 1, 2182–2187 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 1–19 CrossRef.
- Y. Wu, J. Guo, R. Sun and J. Min, Machine learning for accelerating the discovery of high-performance donor/acceptor pairs in non-fullerene organic solar cells, npj Comput. Mater., 2020, 6, 1–8 CrossRef.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Understanding energy loss in organic solar cells: toward a new efficiency regime, Joule, 2018, 2, 25–35 CrossRef CAS.
- S. D. Dimitrov and J. R. Durrant, Materials design considerations for charge generation in organic solar cells, Chem. Mater., 2014, 26, 616–630 CrossRef CAS.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen and J. Zhang, et al., Design rules for minimizing voltage losses in high-efficiency organic solar cells, Nat. Mater., 2018, 17, 703–709 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, An electron acceptor challenging fullerenes for efficient polymer solar cells, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng and P. A. Johnson, et al., Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. Van Der Poll, G. C. Bazan and R. H. Friend, Ultrafast long-range charge separation in organic semiconductor photovoltaic diodes, Science, 2014, 343, 512–516 CrossRef.
- A. C. Jakowetz, M. L. Böhm, A. Sadhanala, S. Huettner, A. Rao and R. H. Friend, Visualizing excitations at buried heterojunctions in organic semiconductor blends, Nat. Mater., 2017, 16, 551–557 CrossRef CAS PubMed.
- B. A. Gregg, Entropy of charge separation in organic photovoltaic cells: the benefit of higher dimensionality, J. Phys. Chem. Lett., 2011, 2, 3013–3015 CrossRef CAS.
- S. N. Hood and I. Kassal, Entropy and disorder enable charge separation in organic solar cells, J. Phys. Chem. Lett., 2016, 7, 4495–4500 CrossRef CAS PubMed.
- H. van Eersel, R. A. Janssen and M. Kemerink, Mechanism for efficient photoinduced charge separation at disordered organic heterointerfaces, Adv. Funct. Mater., 2012, 22, 2700–2708 CrossRef CAS.
- S. Sweetnam, K. R. Graham, G. O. Ngongang Ndjawa, T. Heumüller, J. A. Bartelt, T. M. Burke, W. Li, W. You, A. Amassian and M. D. McGehee, Characterization of the polymer energy landscape in polymer: fullerene bulk heterojunctions with pure and mixed phases, J. Am. Chem. Soc., 2014, 136, 14078–14088 CrossRef CAS PubMed.
- F. D. Eisner, M. Azzouzi, Z. Fei, X. Hou, T. D. Anthopoulos, T. J. S. Dennis, M. Heeney and J. Nelson, Hybridization of local exciton and charge-transfer states reduces nonradiative voltage losses in organic solar cells, J. Am. Chem. Soc., 2019, 141, 6362–6374 CrossRef CAS PubMed.
- G. Han and Y. Yi, Local excitation/charge-transfer hybridization simultaneously promotes charge generation and reduces nonradiative voltage loss in nonfullerene organic solar cells, J. Phys. Chem. Lett., 2019, 10, 2911–2918 CrossRef CAS.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr., A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong and B. Gao, et al., 14.7% efficiency organic photovoltaic cells enabled by active materials with a large electrostatic potential difference, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS.
- L. Perdigón-Toro, H. Zhang, A. Markina, J. Yuan, S. M. Hosseini, C. M. Wolff, G. Zuo, M. Stolterfoht, Y. Zou and F. Gao, et al., Barrierless free charge generation in the high-performance PM6: Y6 bulk heterojunction non-fullerene solar cell, Adv. Mater., 2020, 32, 1906763 CrossRef.
- Z. Tu, G. Han and Y. Yi, Barrier-free charge separation enabled by electronic polarization in high-efficiency non-fullerene organic solar cells, J. Phys. Chem. Lett., 2020, 11, 2585–2591 CrossRef CAS.
- S. Meng, J. Ren and E. Kaxiras, Natural dyes adsorbed on TiO2 nanowire for photovoltaic applications: enhanced light absorption and ultrafast electron injection, Nano Lett., 2008, 8, 3266–3272 CrossRef CAS PubMed.
- S. Meng and E. Kaxiras, Electron and hole dynamics in dye-sensitized solar cells: influencing factors and systematic trends, Nano Lett., 2010, 10, 1238–1247 CrossRef CAS PubMed.
- S. M. Falke, C. A. Rozzi, D. Brida, M. Maiuri, M. Amato, E. Sommer, A. De Sio, A. Rubio, G. Cerullo and E. Molinari, et al., Coherent ultrafast charge transfer in an organic photovoltaic blend, Science, 2014, 344, 1001–1005 CrossRef CAS PubMed.
- E. J. Santos and W. Wang, Ultrafast charge-transfer in organic photovoltaic interfaces: geometrical and functionalization effects, Nanoscale, 2016, 8, 15902–15910 RSC.
- Y. Miyamoto, Y. Tateyama, N. Oyama and T. Ohno, Conservation of the pure adiabatic state in Ehrenfest dynamics of the photoisomerization of molecules, Sci. Rep., 2015, 5, 18220 CrossRef CAS PubMed.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, 2009 Search PubMed.
- S. Andermatt, J. Cha, F. Schiffmann and J. VandeVondele, Combining linear-scaling DFT with subsystem DFT in Born–Oppenheimer and Ehrenfest molecular dynamics Simulations: from molecules to a virus in solution, J. Chem. Theory Comput., 2016, 12, 3214–3227 CrossRef CAS.
- Y. Jin, Z. Chen, M. Xiao, J. Peng, B. Fan, L. Ying, G. Zhang, X.-F. Jiang, Q. Yin and Z. Liang, et al., Thick film polymer solar cells based on naphtho [1, 2-c: 5, 6-c] bis [1, 2, 5] thiadiazole conjugated polymers with efficiency over 11, Adv. Energy Mater., 2017, 7, 1700944 CrossRef.
- L.-H. Chou, T. Mikie, M. Saito, C.-L. Liu and I. Osaka, Naphthobisthiadiazole-Based π-Conjugated Polymers for Nonfullerene Solar Cells: Suppressing Intermolecular Interaction Improves Photovoltaic Performance, ACS Appl. Mater. Interfaces, 2022, 14, 14400–14409 CrossRef CAS PubMed.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber and A. A. Herzing, et al., Crystallography, morphology, electronic structure, and transport in non-fullerene/non-indacenodithienothiophene polymer: Y6 solar cells, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- C. Ma, C. C. Chan, X. Zou, H. Yu, J. Zhang, H. Yan, K. S. Wong and P. C. Chow, Unraveling the Temperature Dependence of Exciton Dissociation and Free Charge Generation in Nonfullerene Organic Solar Cells, Sol. RRL, 2021, 5, 2000789 CrossRef CAS.
- C. Andrea Rozzi, S. Maria Falke, N. Spallanzani, A. Rubio, E. Molinari, D. Brida, M. Maiuri, G. Cerullo, H. Schramm and J. Christoffers, et al., Quantum coherence controls the charge separation in a prototypical artificial light-harvesting system, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- S. Pittalis, A. Delgado, J. Robin, L. Freimuth, J. Christoffers, C. Lienau and C. A. Rozzi, Charge Separation Dynamics and Opto-Electronic Properties of a Diaminoterephthalate-C60 Dyad, Adv. Funct. Mater., 2015, 25, 2047–2053 CrossRef CAS.
- C. Deibel, T. Strobel and V. Dyakonov, Origin of the efficient polaron-pair dissociation in polymer-fullerene blends, Phys. Rev. Lett., 2009, 103, 036402 CrossRef PubMed.
- G. Han, Y. Guo, X. Ma and Y. Yi, Atomistic Insight Into Donor/Acceptor Interfaces in High-Efficiency Nonfullerene Organic Solar Cells, Sol. RRL, 2018, 2, 1800190 CrossRef.
- Z. Cao, S. Yang, B. Wang, X. Shen, G. Han and Y. Yi, Multi-channel exciton dissociation in D18/Y6 complexes for high-efficiency organic photovoltaics, J. Mater. Chem. A, 2020, 8, 20408–20413 RSC.
- C. J. Brabec, G. Zerza, G. Cerullo, S. De Silvestri, S. Luzzati, J. C. Hummelen and S. Sariciftci, Tracing photoinduced electron transfer process in conjugated polymer/fullerene bulk heterojunctions in real time, Chem. Phys. Lett., 2001, 340, 232–236 CrossRef CAS.
- S. Emmerich, S. Hedwig, B. Arnoldi, J. Stockl, F. Haag, R. Hemm, M. Cinchetti, S. Mathias, B. Stadtmüller and M. Aeschlimann, Ultrafast charge-transfer exciton dynamics in C60 thin films, J. Phys. Chem. C, 2020, 124, 23579–23587 CrossRef CAS PubMed.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, Charge separation from an intra-moiety intermediate state in the high-performance PM6: Y6 organic photovoltaic blend, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- C. C. Chan, C. Ma, X. Zou, Z. Xing, G. Zhang, H.-L. Yip, R. A. Taylor, Y. He, K. S. Wong and P. C. Chow, Quantification of Temperature-Dependent Charge Separation and Recombination Dynamics in Non-Fullerene Organic Photovoltaics, Adv. Funct. Mater., 2021, 31, 2107157 CrossRef CAS.
- K. Schulze, C. Uhrich, R. Schueppel, K. Leo, M. Pfeiffer, E. Brier, E. Reinold and P. Bäuerle, Efficient vacuum-deposited organic solar cells based on a new low-bandgap oligothiophene and fullerene C60, Adv. Mater., 2006, 18, 2872–2875 CrossRef CAS.
- X. Xi, W. Li, J. Wu, J. Ji, Z. Shi and G. Li, A comparative study on the performances of small molecule organic solar cells based on CuPc/C60 and CuPc/C70, Sol. Energy Mater. Sol. Cells, 2010, 94, 2435–2441 CrossRef CAS.
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Maximally localized Wannier functions: theory and applications, Rev. Mod. Phys., 2012, 84, 1419 CrossRef CAS.
- N. A. Spaldin, A beginner's guide to the modern theory of polarization, J. Solid State Chem., 2012, 195, 2–10 CrossRef CAS.
- T. F. Hinrichsen, C. Chan, C. Ma, D. Paleček, A. Gillett, S. Chen, X. Zou, G. Zhang, H.-L. Yip and K. S. Wong, et al., Long-lived and disorder-free charge transfer states enable endothermic charge separation in efficient non-fullerene organic solar cells, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- Y. Yi, V. Coropceanu and J.-L. Brédas, A comparative theoretical study of exciton-dissociation and charge-recombination processes in oligothiophene/fullerene and oligothiophene/perylenediimide complexes for organic solar cells, J. Mater. Chem., 2011, 21, 1479–1486 RSC.
- S. C. North, K. R. Jorgensen, J. Pricetolstoy and A. K. Wilson, Population analysis and the effects of Gaussian basis set quality and quantum mechanical approach: main group through heavy element species, Front. Chem., 2023, 11, 1152500 CrossRef CAS PubMed.
- J. B. Haber, D. Y. Qiu, F. H. da Jornada and J. B. Neaton, Maximally localized exciton Wannier functions for solids, Phys. Rev. B, 2023, 108, 125118 CrossRef CAS.
- M. A. Faist, T. Kirchartz, W. Gong, R. S. Ashraf, I. McCulloch, J. C. De Mello, N. J. Ekins-Daukes, D. D. Bradley and J. Nelson, Competition between the charge transfer state and the singlet states of donor or acceptor limiting the efficiency in polymer: fullerene solar cells, J. Am. Chem. Soc., 2012, 134, 685–692 CrossRef CAS PubMed.
- M. Riede, D. Spoltore and K. Leo, Organic solar cells—the path to commercial success, Adv. Energy Mater., 2021, 11, 2002653 CrossRef CAS.
- R. Scholz, R. Luschtinetz, G. Seifert, T. Jägeler-Hoheisel, C. Körner, K. Leo and M. Rapacioli, Quantifying charge transfer energies at donor–acceptor interfaces in small-molecule solar cells with constrained DFTB and spectroscopic methods, J. Phys.: Condens.Matter, 2013, 25, 473201 CrossRef PubMed.
- L. Zhu, M. Zhang, G. Zhou, T. Hao, J. Xu, J. Wang, C. Qiu, N. Prine, J. Ali and W. Feng, et al., Efficient organic solar cell with 16.88% efficiency enabled by refined acceptor crystallization and morphology with improved charge transfer and transport properties, Adv. Energy Mater., 2020, 10, 1904234 CrossRef CAS.
- W. Chu, Q. Zheng, O. V. Prezhdo, J. Zhao and W. A. Saidi, Low-frequency lattice phonons in halide perovskites explain high defect tolerance toward electron-hole recombination, Sci. Adv., 2020, 6, eaaw7453 CrossRef CAS PubMed.
- W. Pei, Z. Wang, W. Xia, Z. Huang, P. Wang, Y. Liu, S. Zhou, Y. Tu and J. Zhao, Rational Design of Full-Color Fluorescent C3N Quantum Dots, J. Phys. Chem. Lett., 2024, 15, 1161–1171 CrossRef CAS PubMed.
- X. Yu, W. Pei, W.-W. Xu, Y. Zhao, Y. Su and J. Zhao, Core-Packing-Related Vibrational Properties of Thiol-Protected Gold Nanoclusters and Their Excited-State Behavior, Inorg. Chem., 2023, 62, 20450–20457 CrossRef CAS PubMed.
- N. L. Doltsinis, Nonadiabatic dynamics: mean-field and surface hopping, Quantum simulations of complex many-body systems: from theory to algorithms, Citeseer, 2002, vol. 10, pp. 377–397 Search PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- M. Krack, Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- L. Genovese, T. Deutsch, A. Neelov, S. Goedecker and G. Beylkin, Efficient solution of Poisson's equation with free boundary conditions, J. Chem. Phys., 2006, 125, 074105 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- E. Runge and E. K. Gross, Density-functional theory for time-dependent systems, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- M. E. Casida, Recent Advances in Density Functional Methods: (Part I), World Scientific, 1995, pp. 155–192 Search PubMed.
- A. Castro, M. A. Marques and A. Rubio, Propagators for the time-dependent Kohn–Sham equations, J. Chem. Phys., 2004, 121, 3425–3433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Five configurations of the NT-4T-FF:Y6 blend, optimized crystalline NT-4T-FF:Y6 structures, one crystalline NT-4T-FF:Y6 blend structure after thermal relaxation, isosurfaces of molecular orbitals of the crystalline NT-4T-FF:Y6 blend at PBE and B3LYP levels, energy diagram for the LE and CT states calculated at PBE and B3LYP levels in a crystalline NT-4T-FF:Y6 blend, optimized structures and molecular orbitals of the Y6 molecule before and after the simplification, and molecular orbitals of two different NT-4T-FF:Y6 configurations obtained from BOMD simulations. See DOI: https://doi.org/10.1039/d4ta01033g |
| This journal is © The Royal Society of Chemistry 2024 |