
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilisation of benzo[c][1,2,5]thiadiazole (BTZ) within polymers of intrinsic microporosity (PIMs) for use in flow photochemistry†
Dominic
Taylor‡
ab,
John M.
Tobin‡
b,
Leonardo
Amicosante
a,
Andrew W.
Prentice
a,
Martin J.
Paterson
 a,
Scott J.
Dalgarno
*a,
Neil B.
McKeown
*b and
Filipe
Vilela
*a
a,
Scott J.
Dalgarno
*a,
Neil B.
McKeown
*b and
Filipe
Vilela
*a
aSchool of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.j.dalgarno@hw.ac.uk; f.vilela@hw.ac.uk
bEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: neil.mckeown@ed.ac.uk
First published on 9th April 2024
Abstract
Polymers of intrinsic microporosity (PIMs) offer a useful combination of processability and inherent microporous features arising from their contorted and rigid structures, with the potential for the immobilisation of light harvesting groups for use in photochemistry. Herein, we report on the chemical incorporation of photosensitisers based on the benzo[c][1,2,5]thiadiazole (BTZ) group into PIM-EA-TB through a copolymerisation strategy, allowing up to 5% BTZ incorporation without compromising processability or microporosity. The BTZ-doped PIMs were then utilised as either homogeneous or heterogeneous visible light photosensitisers of oxygen under both batch and continuous flow conditions. The advantages of the solution processability of these materials was demonstrated by the simple deposition of the photoactive microporous polymer onto glass beads, which were then fixed within a column reactor for continuous flow photochemistry.
Introduction
Photocatalysis has emerged as a sustainable method to achieve clean chemical transformations. In particular, efficient photosensitisation of oxygen to generate the reactive species singlet oxygen (1O2) is an attractive reaction as it allows atmospheric oxygen to be utilised as a powerful reagent.1 Molecular oxygen is unusual in that it exhibits triplet spin multiplicity in its ground state (3O2), which imposes a quantum mechanical barrier on its reaction with most organic substrates.2 However, oxygen possesses a low lying excited electronic singlet state, abbreviated as 1O2, at approximately 95 kJ mol−1 above its ground state. In comparison to 3O2, 1O2 is a much more powerful oxidant with applications in chemical synthesis,3 photodynamic therapy,4 wastewater treatment,5 and the detoxification of hazardous materials.6 One method for generating 1O2 is via photosensitisation, a process which requires only oxygen, light of a suitable wavelength and a photosensitiser possessing an accessible electronically excited triplet state of suitable energy. Upon absorbing light of sufficient energy, a photosensitiser will be excited to its first electronically excited singlet state (S1) and then occupy the triplet state (T1) via intersystem crossing (ISC). From this state, the photosensitiser then engages in triplet–triplet annihilation (TTA) with 3O2, generating 1O2 and regenerating the photosensitiser to its ground state.While organic dyes such as methylene blue and xanthenes remain popular choices for metal-free photosensitisers, recent research has expanded the scope of available photosensitisers to include π-conjugated electron donor–acceptor (D–A) systems.2 This includes D–A systems based on the benzo[c][1,2,5]thiadiazole (BTZ) group (Fig. 1A), which have recently gained attention as flexible photocatalysts and fluorophores.7–9 Recent research has also highlighted the use of photoactive polymer materials as photosensitisers, including photosensitisers attached to Merrifield resins,10–13 cross-linked polymers,14–18 conjugated porous polymers (CPPs),19,20 and covalent organic frameworks (COFs).21 The high surface area that such materials often exhibit provides a high interfacial area for interaction with oxygen while also enhancing interaction with the solvent to aid in the materials dispersibility.22,23 Both of these are factors that have been noted to improve the photosensitisation performance of heterogeneous photosensitisers. However, the heavy degree of cross-linking that these materials exhibit renders them insoluble, consequently categorising them as heterogeneous photosensitisers with inherent nanoscale porosity. While this heterogeneous nature can facilitate the recovery of the photocatalysts following reaction by simple filtration or centrifugation, it also severely limits the processing options that are available for these materials.
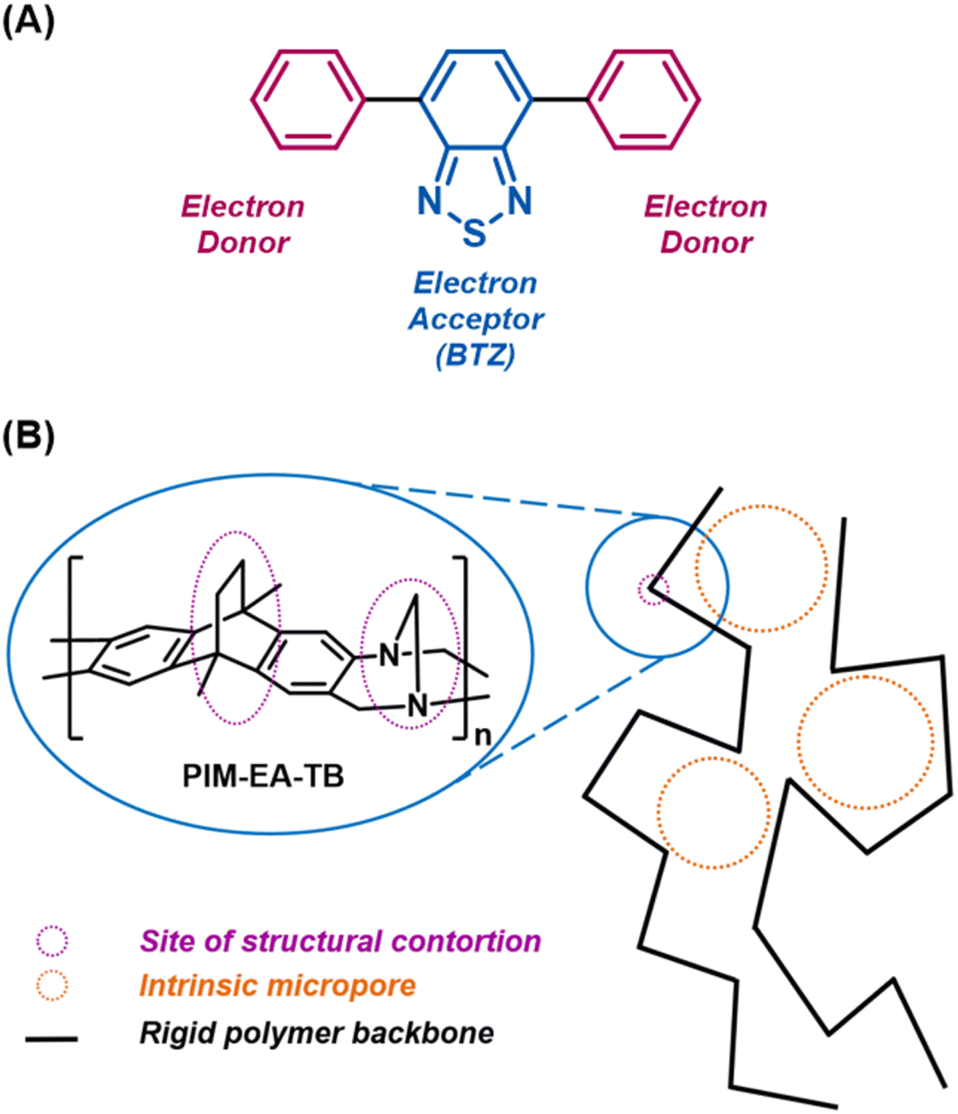 | ||
| Fig. 1 (A) Structure of a BTZ photosensitiser (pH-BTZ) with the electron donor phenyl groups and electron accepting BTZ group highlighted. (B) Illustration of PIM-EA-TB showing the sites of structural contortion. | ||
An alternative to these heavily cross-linked materials are polymers of intrinsic microporosity (PIMs). Due to their highly contorted and rigid backbones, the polymer chains are unable to pack efficiently in the solid state, leading these materials to exhibit intrinsic microporous features (Fig. 1B).24 However, unlike CPPs, COFs etc., some PIMs can be dissolved in common organic solvents,25 allowing amorphous powders or glassy thin films to be formed by precipitation or slow evaporation methods, respectively. This useful combination of intrinsic microporosity with ease of film fabrication has led PIMs to be investigated as gas separation membranes,26–29 in sensing and energy storage,30,31 and as heterogeneous catalysts.32–35 While not so extensively studied, the use of PIMs has also been extended to photocatalytic applications. The prototypical PIM, PIM-1, exhibits intramolecular charge transfer (ICT) between its component terephthalonitrile and spirobisindane units, leading it to fluoresce in both the solid state and in solution.24 Separate research conducted by Smith et al. and Atilgan et al. have explored introducing additional chromophores into PIM-1 as a method to improve the photophysics of the material towards photosensitisation and electron transfer photochemical reactions.34,35
Herein, we demonstrate two distinct strategies for immobilising BTZ photosensitisers within the amine-containing ladder polymer PIM-EA-TB, which is prepared by the formation of Tröger's base (TB) linkages between ethanoanthracene (EA) structural units.27 Firstly, we attempted to trap molecular BTZ photosensitisers within the microporous structure of self-standing thin films of PIM-EA-TB fabricated by slow evaporation from a chloroform solution. However, this approach fundamentally suffered from poor solvent compatibility, with most reaction solvents causing the photosensitiser to leach out of the film. This was then addressed by a copolymerisation strategy that succeeded in the covalent incorporation of BTZ photosensitisers into the backbone of PIM-EA-TB. This latter approach allowed the photosensitising polymer to be used either as part of a homogeneous or heterogeneous system, under both batch and continuous flow conditions. Furthermore, we highlight the ability to employ solution processing techniques to coat the photoactive polymer onto glass beads for use in a continuous flow heterogenous photoreactor.
Results and discussion
To further develop the use of PIMs in photocatalysis, we sought to use PIM-EA-TB either as the matrix for the simple non-covalent incorporation of photosensitisers into its microporous structure or for direct chemical incorporation into the polymer backbone. Crucially, unlike PIM-1, PIM-EA-TB does not exhibit any visible light absorption, which will allow it to adopt the light harvesting properties of the incorporated photosensitiser. While these two approaches could be extended to a wide choice of photocatalysts, we elected to use BTZ-based photosensitisers as they are compatible with the harsh reaction conditions used to prepare the Tröger's base PIMs.Doping PIM-EA-TB films with BTZ photosensitisers
The starting point for testing the viability of PIM-EA-TB as a platform for BTZ photosensitisers was to investigate if photosensitiser immobilisation could be achieved within the micropores of solution cast polymer films. For this purpose, two BTZ photosensitisers were initially tested, pH-BTZ and ThTh-BTZ, which feature absorption maxima in chloroform at 380 nm and 505 nm respectively.7 Thin films of PIM-EA-TB were prepared by dissolving 150 mg of PIM-EA-TB, equivalent to 0.5 mmol of repeat units, in chloroform then slowly removing the solvent under ambient conditions over a period of two days. The initial film, obtained with a solution of PIM-EA-TB containing no photosensitiser, gave a flexible, transparent, and free-standing film (Fig. 2A). The addition of 7.2 mg (25 µmol) of pH-BTZ, gave rise to a transparent film that was yellow in colour (Fig. 2B). Under 4× magnification from an optical microscope, it was observed that this film was homogeneous, with no visible phase separation between pH-BTZ and PIM-EA-TB (Fig. S1†), suggesting pH-BTZ had been successfully incorporated into the polymers microporous structure. Film casting was also attempted with PIM-EA-TB and ThTh-BTZ at the same 25 µmol loading (11.5 mg of ThTh-BTZ per 150 mg of PIM-EA-TB), resulting in the formation of an opaque red film with a visibly rough texture (Fig. 2C). Examination of this film under an optical microscope revealed that crystals of ThTh-BTZ with lengths of over 60 µm had formed, due to the low solubility of this photosensitiser (Fig. S1†).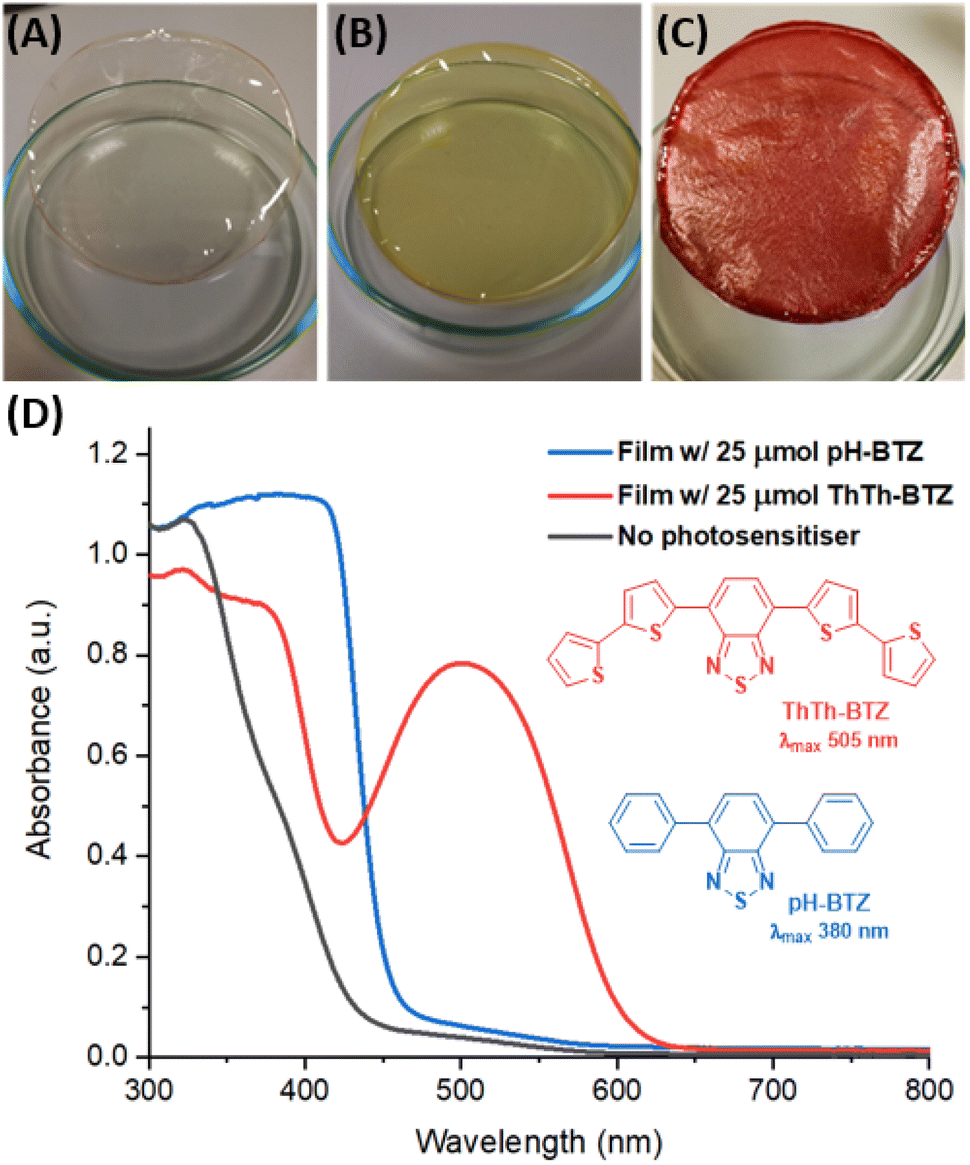 | ||
| Fig. 2 (A–C) BTZ-doped PIM-EA-TB films containing (A) no photosensitiser, (B) 25 µmol pH-BTZ, (C) 25 µmol ThTh-BTZ. (D) Solid state UV-vis absorption spectra of PIM-EA-TB containing no photosensitiser (grey), 25 µmol pH-BTZ (blue) and 25 µmol ThTh-BTZ (red). | ||
Solid state UV-vis absorption spectra of the BTZ doped PIM films revealed that the absorption exhibited by the films broadly reflected the absorption spectra of the molecular photosensitisers they contained (Fig. S2†). The film of PIM-EA-TB containing 25 µmol of pH-BTZ possessed an onset of absorption at approximately 470 nm, rising rapidly to a plateau at 406 nm. This can be compared with the film containing 25 µmol of ThTh-BTZ, which displayed a well-defined wavelength of maximum absorption at 500 nm (Fig. 2D). Due to the lack of any BTZ photosensitiser, the film of PIM-EA-TB by itself did not exhibit any significant absorption of light above 400 nm.
The solvent compatibility of the BTZ doped PIM films was tested to determine which solvents, if any, would be suitable for heterogeneous photosensitisation studies. Exposing the films to various organic solvents either caused the film to dissolve (e.g. CHCl3, DCM) or the photosensitiser to rapidly leach out of the film (e.g. toluene, THF, acetonitrile, methanol). This ultimately confined the solvent for any reaction to water as this solvent was unable to dissolve either the PIM film or the BTZ photosensitiser. This was not an ideal solvent for photosensitisation reactions due to the extremely low lifetime of 1O2 in water (ca. 3.45 µs) in comparison with other solvents.36 Nevertheless, 1O2 production was attempted using the oxidation of 2-furoic acid to 5-hydroxy-2(5H)-furanone using a vial as reaction vessel, the bottom of which was coated with a layer of the pH-BTZ doped PIM-EA-TB.37 However, after 7 days under irradiation from a 420 nm LED, a conversion of only 20% was obtained compared to <2% obtained in the absence of photosensitiser.
Covalent incorporation of BTZ into PIM-EA-TB
To prevent leaching of the photosensitiser and expand the range of usable solvents, we sought to chemically incorporate the pH-BTZ group into the backbone of PIM-EA-TB. Similar approaches have already been explored to generate polymeric BTZ photocatalysts including linear polymers,38,39 cross-linked polymeric gels/networks,14,15 CPPs,22 and COFs.40 However, these approaches often suffer from either a lack of solubility due to covalent cross-linkages or a lack of permanent microporosity in the case of linear polymers, reducing the transport of 3O2 to the photosensitiser and 1O2 to the reagent. While these materials could be fabricated into different formats (such as beads, monoliths, and gels,14 or custom designed 3D printed structures),17,18 these forms were ultimately fixed during the initial polymerisation reaction. In contrast, PIM-EA-TB exhibits substantial microporosity and once synthesised as a powder can be dissolved in various organic solvents and then used for post-synthetic processing techniques.27PIM-EA-TB is typically synthesised by the trifluoroacetic acid (TFA) catalysed reaction of EA and dimethoxymethane (DMM) leading to the in situ formation of the Tröger's base linkage (Fig. 3A).27 We therefore envisaged performing the polymerisation reaction of EA with varying quantities of the p-diamino- analogue of pH-BTZ, pNH2-BTZ, the synthesis of which is provided in the ESI.† This was done with feed ratios containing 1 and 5 mol% of BTZ to yield PIM-BTZ-1% and PIM-BTZ-5%, respectively. These were obtained as yellow powders, compared to the off-white colour of PIM-EA-TB, indicating successful incorporation of the BTZ moiety into the polymer backbone (Fig. 3B). Furthermore, powders of both PIM-BTZ-1% and PIM-BTZ-5% exhibited clearly visible emission under a UV lamp, whereas PIM-EA-TB was non-emissive (Fig. S3†). In order to provide a suitable small molecule analogue to the BTZ doped PIM-EA-TB, the model compound TB-BTZ, was also synthesised by reaction of pNH2-BTZ and p-tert-butylaniline (Fig. 3C). This provided a molecular benchmark for the photosensitisation efficiency and aided in the identification of 1H NMR signals within the polymer (Fig. S4†).
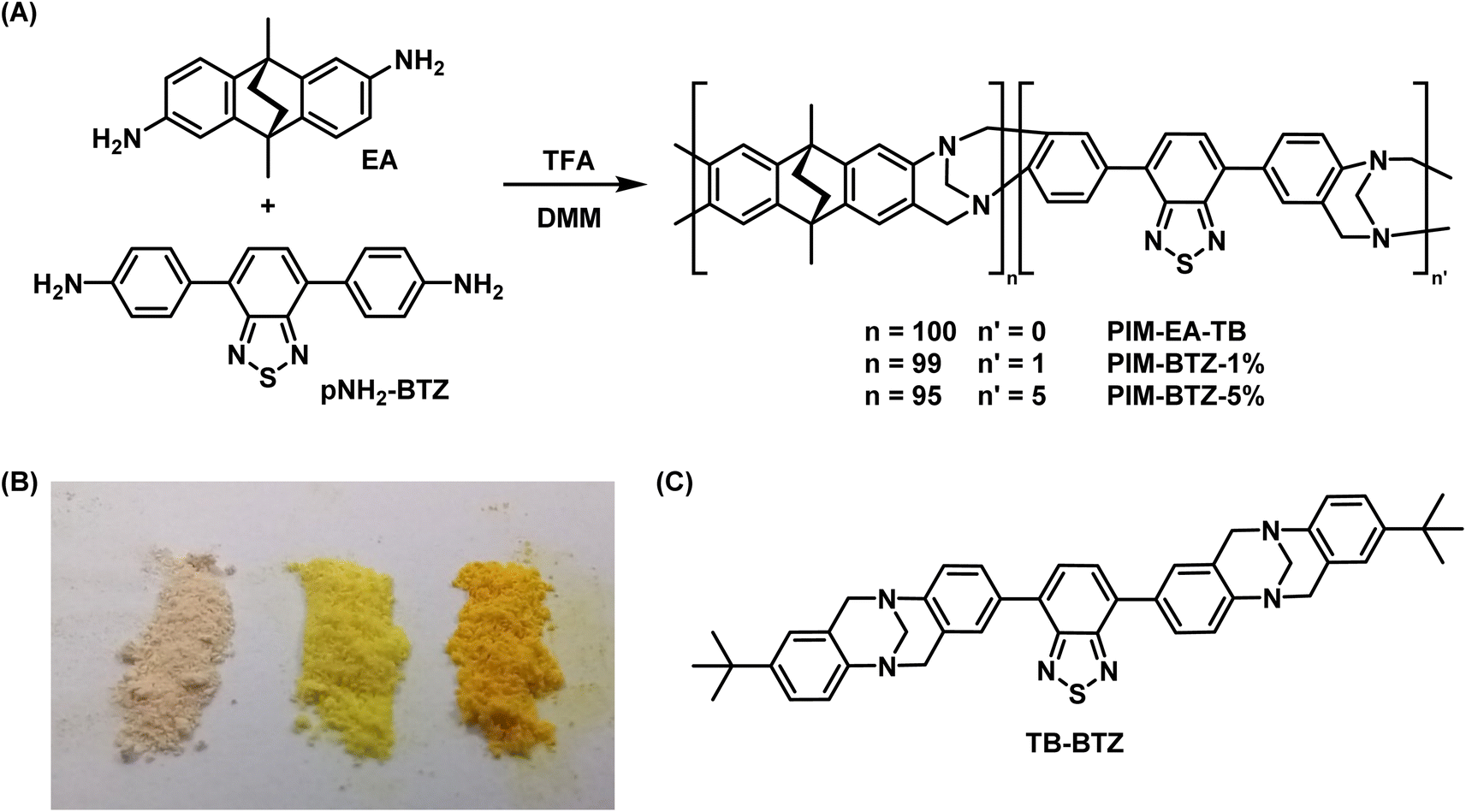 | ||
| Fig. 3 (A) Chemical scheme for the incorporation of pNH2-BTZ into PIM-EA-TB. (B) Powder samples of PIM-EA-TB (left), PIM-BTZ-1% (middle) and PIM-BTZ-5% (right). (C) The structure of model compound TB-BTZ. | ||
The 1H NMR spectra of PIM-BTZ-5% in CDCl3 closely resembled the spectra of PIM-EA-TB with additional peaks in the aromatic region attributable to the BTZ dopant: from this a BTZ content of 4.4% was estimated (Fig. S4†). These additional peaks were also present in the 1H NMR spectra of PIM-BTZ-1%, but were weak and mostly obscured by the neighbouring PIM-EA-TB peaks, making accurate determination of BTZ content difficult. The incorporation of BTZ into the PIM-EA-TB backbone was also confirmed by UV-vis absorption spectroscopy (Fig. 4). The UV-vis absorption spectrum of PIM-EA-TB (in chloroform solution at a concentration of 0.04 mg mL−1) did not feature any significant absorption of light in the visible region with a maximum of absorption below 300 nm. In contrast, PIM-BTZ-5% exhibited a significant absorption maximum located at 412 nm that was attributed to the covalently incorporated BTZ group and closely matched that of model compound TB-BTZ. In the case of PIM-BTZ-1%, the absorption at 412 nm was weaker than PIM-BTZ-5% but still greater than for PIM-EA-TB. Fluorescence spectra of PIM-BTZ-5% and TB-BTZ were also recorded in CHCl3 solution, with maxima identified at 533 nm for both samples (Fig. S5†).
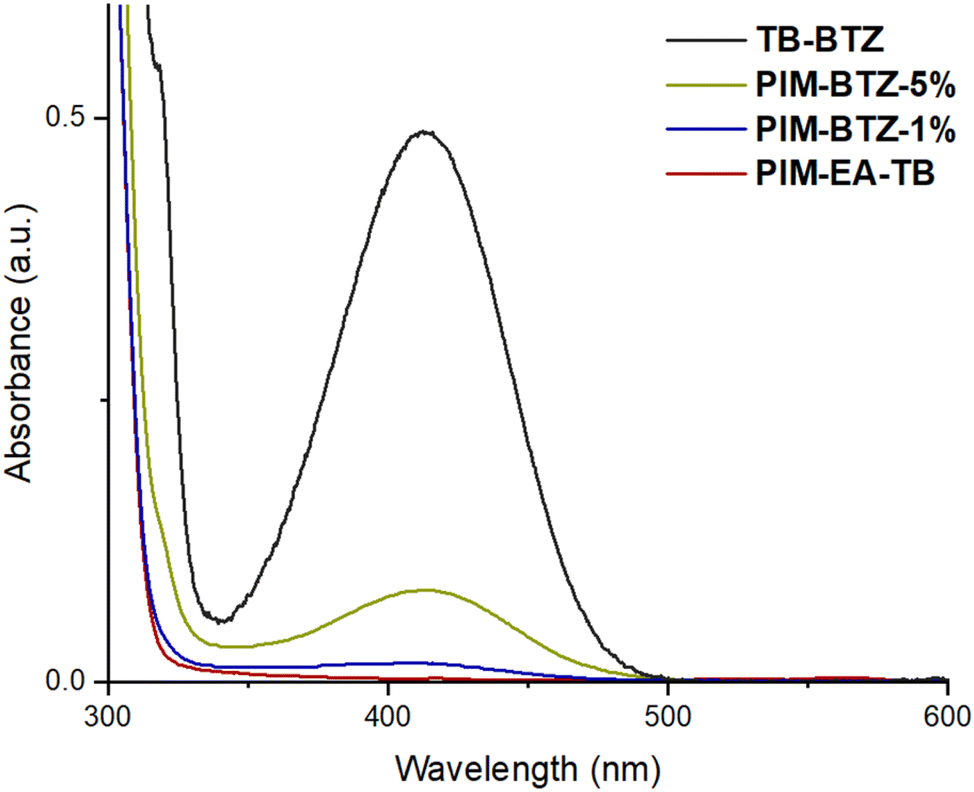 | ||
| Fig. 4 UV-vis absorption spectra for PIM-EA-TB, PIM-BTZ-5%, PIM-BTZ-1% and TB-BTZ. All spectra were obtained in chloroform solution at concentrations of 0.04 mg mL−1. | ||
In order to determine the scope of possible solvents for photocatalytic studies, the solubility of PIM-EA-TB in common solvents was assessed (Table S1†). PIM-EA-TB was mainly soluble in chlorinated solvents (such as chloroform, DCM) as well as in N-methylpyrrolidone and quinoline. The polymer was entirely insoluble in solvents such as acetonitrile or methanol, which would be useful solvents for using PIM-BTZ-1% and PIM-BTZ-5% as heterogeneous suspensions. The solubility of these TB polymers in low boiling point solvents like chloroform allowed free standing thin films of all three polymers to be fabricated by slow evaporation from solution (Fig. S6†).
The N2 gas adsorption and desorption isotherms recorded at 77 K for PIM-BTZ-1% and PIM-BTZ-5% allowed Brunauer–Emmett–Teller (BET) surface areas of 986 and 914 m2 g−1 to be calculated (Fig. S7†). These values are similar to that of PIM-EA-TB (1028 m2 g−1) and indicates that the introduction of the BTZ group results in only a slightly reduction of the BET surface area.27 The decrease in the BET surface area of PIM-EA-TB on increasing percentage of the BTZ unit is presumably due to the introduction of free internal rotation within the polymer chain about the single C–C bonds of the BTZ unit. This is an effect that has previously been observed in PIMs containing a non-rigid or non-contorted comonomer.41 The BET surface areas of PIM-BTZ-1% and PIM-BTZ-5% are larger than the reported surface areas of most other organic polymers (linear or crosslinked) containing BTZ, but is still considerably lower than some crystalline frameworks (see Table S2†). CO2 adsorption isotherms, measured at 273 K, revealed uptakes of 93 and 71 cm3 g−1 at 1 bar of pressure for PIM-BTZ-1% and PIM-BTZ-5% respectively (Fig. S8†). Modelling of the pore size distribution using non-local density functional theory (NLDFT) on the CO2 adsorption isotherms revealed a significant presence of micropores mainly in the region of 0.4–0.7 nm for both materials (Fig. S9 and S10†).
Thermal gravimetric analysis (TGA) revealed that both materials were thermally stable up to 300 °C, with decomposition resulting in a 35% mass loss up to 800 °C (Fig. S11†). Gel permeation chromatography (GPC) was conducted on the synthesised polymers (Fig. S13 and S14†) with weight average molecular weights (Mw) of 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 26000 g mol−1 calculated for PIM-BTZ-1% and PIM-BTZ-5% respectively. These are less than reported Mw for PIM-EA-TB of 155800 g mol−1, which could suggest that an increasing percentage of the BTZ comonomer could hinder polymerisation.27
000 g mol−1 and 26000 g mol−1 calculated for PIM-BTZ-1% and PIM-BTZ-5% respectively. These are less than reported Mw for PIM-EA-TB of 155800 g mol−1, which could suggest that an increasing percentage of the BTZ comonomer could hinder polymerisation.27
Batch photosensitisation studies
In contrast to the BTZ-doped PIM-EA-TB films described above, the chemical incorporation of the photosensitiser into the backbone of the PIM addressed the leaching problem and allowed solvents other than water to be explored. PIM-BTZ-1% and PIM-BTZ-5% were employed as photosensitisers for 1O2 using the oxidation of aryl phosphines as the test reaction. For the initial optimisation studies, triphenylphosphine was selected as the substrate using PIM-BTZ-5% as the photosensitiser. In each reaction, 10 mg of PIM-BTZ-5% was used to give approximately a molar equivalent loading of the photoactive BTZ unit of 5 mol%. Initially, chloroform was selected as the reaction solvent as this was able to fully dissolve PIM-BTZ-5%. After 2 hours of irradiation from a 28 W 410–420 nm LED, a conversion of 45% was measured using crude 31P NMR spectroscopy (Table 1, entry 1). Deuterated chloroform (CDCl3) and dichloromethane (DCM) were both able to fully dissolve PIM-BTZ-5% and resulted in conversions of 57 and 41% respectively (Table 1, entries 2 and 3). This trend in the reaction conversion of CDCl3 > CHCl3 > DCM correlates with the lifetime of 1O2 in each of the solvents (640, 240 and 100 µs respectively).42 In the remaining solvents tested, PIM-BTZ-5% was insoluble although each solvent was able to dissolve the triphenylphosphine substrate. This included acetonitrile (MeCN), methanol (MeOH), toluene and the green solvent dimethyl carbonate (DMC) (Table 1, entries 4–7) with MeCN returning the highest conversion of 27%. While the lower reaction conversion obtained in these solvents can be partially explained by the lower lifetime of 1O2, this cannot be entirely decoupled from factors such as the polymer dispersibility and mass transport of reagents, which are important factors in heterogeneous catalytic systems.|
|
||||
|---|---|---|---|---|
| Entrya | Photosensitiser | Solvent | Time/h | Conversionb/% |
| a Reaction conditions: triphenylphosphine (52 mg, 0.2 mmol), photosensitiser (10 mg of PIM or 1 mol% of pH-BTZ or TB-BTZ), solvent (5 mL), air, 28 W 410–420 nm LED. b Determined by 31P NMR spectroscopy in CDCl3 solution (Fig. S17). c Reaction performed in the dark. d Reaction mixture performed in a Schlenk flask under a nitrogen atmosphere. The reaction was degassed with nitrogen prior to the reaction. e Structures of benchmark photosensitisers are shown in Fig. S18. | ||||
| 1 | PIM-BTZ-5% | CHCl3 | 2 | 45 |
| 2 | PIM-BTZ-5% | CDCl3 | 2 | 57 |
| 3 | PIM-BTZ-5% | DCM | 2 | 41 |
| 4 | PIM-BTZ-5% | MeCN | 2 | 27 |
| 5 | PIM-BTZ-5% | MeOH | 2 | 24 |
| 6 | PIM-BTZ-5% | Toluene | 2 | 21 |
| 7 | PIM-BTZ-5% | DMC | 2 | 4 |
| 8 | — | CDCl3 | 2 | 7 |
| 9c | PIM-BTZ-5% | CDCl3 | 2 | 0 |
| 10d | PIM-BTZ-5% | CDCl3 | 2 | 0 |
| 11 | PIM-EA-TB | CDCl3 | 2 | 5 |
| 12 | PIM-BTZ-5% | CDCl3 | 5 | 97 |
| 13 | PIM-BTZ-5% | CDCl3 | 6 | >99 |
| 14 | PIM-BTZ-1% | CDCl3 | 5 | 85 |
| 15 | TB-BTZ | CDCl3 | 2 | 73 |
| 16e | pH-BTZ | CDCl3 | 2 | >99 |
| 17e | BODIPY | CDCl3 | 2 | 95 |
| 18e | Rose bengal | CDCl3 | 2 | >99 |
| 19e | Methylene blue | CDCl3 | 2 | >99 |
| 20e | TPP | CDCl3 | 2 | >99 |
| 21 | PIM-BTZ-5% | MeCN | 12 | 96 |
| 22 | PIM-BTZ-1% | MeCN | 12 | 64 |
Control experiments were conducted by performing the reaction in the absence of a photosensitiser, performing the reaction in the dark, or by conducting the reaction under a nitrogen atmosphere to preclude oxygen from the reaction mixture (Table 1, entries 8–10). In each case, the reaction conversion was extremely low (0–7%). Having identified CDCl3 as the reaction solvent that afforded the highest conversion, the reaction time was increased from 2 hours to achieve full conversion: after 5 hours a conversion of 97% was obtained while after 6 hours, the conversion was >99% (Table 1, entries 12 and 13). PIM-BTZ-1% was also tested under similar conditions to PIM-BTZ-5%, with a conversion of 85% obtained after 5 hours (Table 1, entry 14).
To provide a molecular comparison for the polymer bound photosensitiser, TB-BTZ was also tested as a photosensitiser. In CDCl3, TB-BTZ at 1 mol% loading was able to achieve 73% after 2 hours of irradiation (Table 1, entry 15). The performance of PIM-BTZ-5% was also benchmarked against the performance of other reported organic photosensitisers, namely pH-BTZ, BODIPY, rose bengal, methylene blue and tetraphenylporphyrin (TPP) (Table 1, entries 16–20). After irradiation for 2 h at 1 mol% loading, these photosensitisers were each able to achieve conversions in excess of 95% under homogeneous conditions. While these results indicate that the performance of the polymer bound photosensitiser was generally slower than small molecule photosensitisers, PIM-BTZ-5% has the advantage of being used either homogeneously in CDCl3 solution or heterogeneously as a suspension in MeCN.
Having identified an optimised set of conditions for the oxidation of triphenylphosphine, the reaction scope was expanded to include various triaryl phosphines (Scheme 1). These oxidations were conducted homogeneously in CDCl3 using PIM-BTZ-5% as the photocatalyst. After each reaction, the conversion was determined by quantitative 31P NMR spectroscopy, then the product isolated by filtering the solution through a small plug of Celite. The Celite effectively trapped PIM-BTZ-5%, leaving the purified product as the filtrate, although the polymer could not be recovered through this method. With each phosphine examined, complete oxidation to form the phosphine oxide was achieved, although both trimesitylphosphine and tri(p-fluorophenyl)phosphine required longer to achieve full conversion (18 h and 10 h respectively). These longer reaction times most likely reflect the steric and deactivating electronic effects of the mesityl and p-fluorophenyl substituents respectively. This observation agrees with previous studies on the reaction mechanism of 1O2 with triarylphosphines, where electron rich phosphines favour σ-electron donation to the π* orbital of 1O2.43
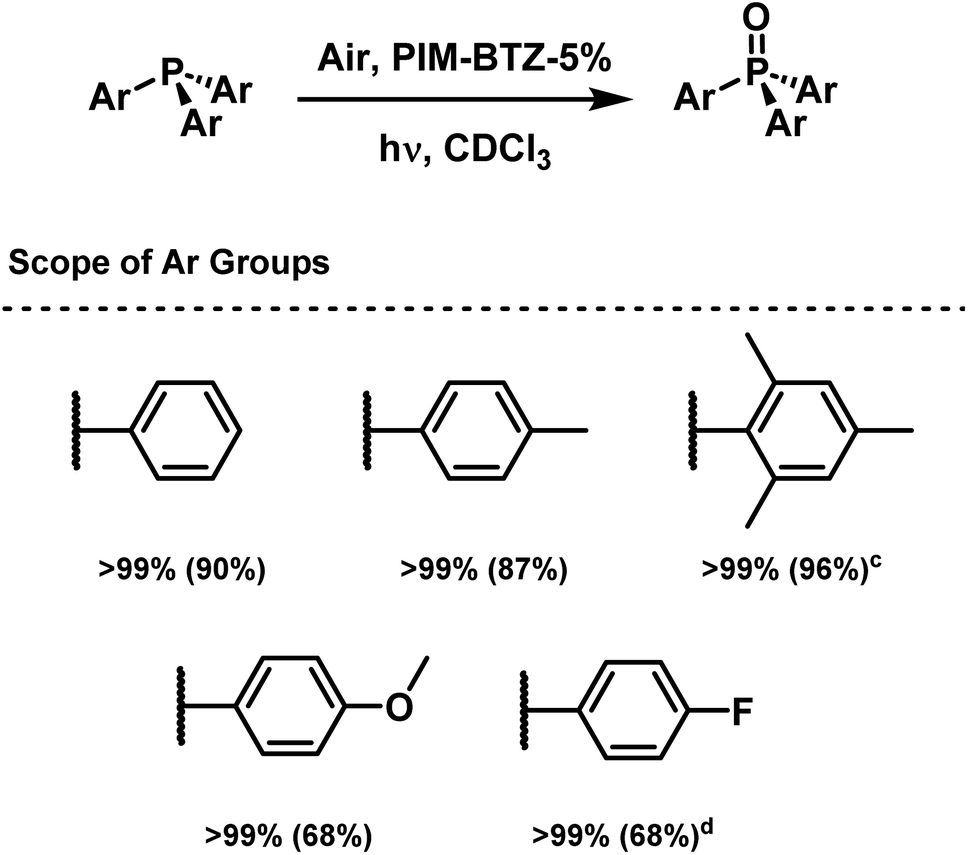 | ||
| Scheme 1 Reaction scope for triarylphosphine oxidation.a,baReaction conditions: triarylphosphine (0.2 mmol), PIM-BTZ-5% (10 mg), CDCl3 (5 mL), air, 28 W 410–420 nm LED. bReaction conversion determined by 31P NMR spectroscopy in CDCl3 solution. Isolated yields following a Celite plug are shown in brackets. cReaction performed for 18 hours. dReaction performed for 10 hours. | ||
While pH-BTZ and TB-BTZ in CDCl3 solution were effective photosensitisers, their isolation would be complicated by the need for chromatographic separation techniques. The use of the PIM as a heterogeneous photosensitiser as a suspension of MeCN was therefore also investigated further. By increasing the irradiation time to 12 h, PIM-BTZ-5% was able to achieve 96% conversion of triphenylphosphine into triphenylphosphine oxide while PIM-BTZ-1% achieved a conversion of 64% (Table 1, entries 21 and 22). This was a feature that was also observed under homogeneous photosensitisation studies, most likely reflecting the lower BTZ content. After performing the reaction with PIM-BTZ-5%, the solid PIM-BTZ-5% was isolated by centrifugation, washed with acetonitrile (7 × 5 mL) then dried under vacuum. The result was clean separation of PIM-BTZ-5% from the phosphine/phosphine oxide mixture, with no visible signs of impurity in the photosensitiser. The reusability of PIM-BTZ-5% under these conditions was also evaluated, demonstrating that the photocatalyst could be used five times without appreciable loss of material or photoactivity (see Fig. S19†).
The heterogeneous photosensitisation using PIM-BTZ-5% in MeCN was further studied by re-examining the scope of phosphine substrates. Near complete oxidation of triphenylphosphine and tri(p-tolyl)phosphine was achieved within 12 h (97 and >99% respectively). However, as was the case for the homogeneous studies, both trimesitylphosphine and tri(p-fluorophenyl)phosphine were slower to oxidise (63 and 82% respectively). Due to the similarity in size between methyl group of tri(p-tolyl)phosphine and the fluoro-group of tri(p-fluorophenyl)phosphine, this difference is most likely not due to size exclusion from the pores of PIM-BTZ-5%, but is due to electronic effects slowing the reaction of 1O2 with the phosphine.43
Continuous flow photosensitisation studies
In order to accelerate the rate of the reaction, the oxidation of triphenylphosphine using PIM-BTZ-5% was also attempted under homogeneous continuous flow conditions. Flow chemistry, in which the reaction mixture is typically passed through narrow diameter transparent polymer tubing, is regarded as a powerful tool for facilitating chemical transformations.44 The use of flow chemistry in photocatalytic reactions has been well documented, with the reactions in particular benefiting from the more efficient and uniform irradiation of the reaction mixture due to the reduction in the pathlength of light.45,46 This can be contrasted with absorption of light in a batch reactor, such as a vial or a flask, where the light intensity varies drastically across the reaction.For the flow experiments, a Vapourtec series E flow reactor and fluorinated ethylene propylene (FEP) tubing with an internal diameter of 1 mm were used. The initial conditions investigated involved pumping a solution of triphenylphosphine and PIM-BTZ-5% in CDCl3 from a vial at a rate of 1 mL min−1 using a peristaltic pump. Concurrently, a second pump was used to circulate air at a rate of 1 mL min−1, with the two phases meeting at a T-junction (Fig. 5A). The resulting system consisting of the CDCl3 solution separated by segments of air was then passed through a 5 mL coiling of FEP tubing under irradiation from the same 28 W 410–420 nm LED employed in the batch photocatalytic experiments. This mixture was cycled continuously for 1 hour before a sample was withdrawn for analysis by 31P NMR spectroscopy. Under these initial conditions, a conversion of 69% was achieved after 1 hour.
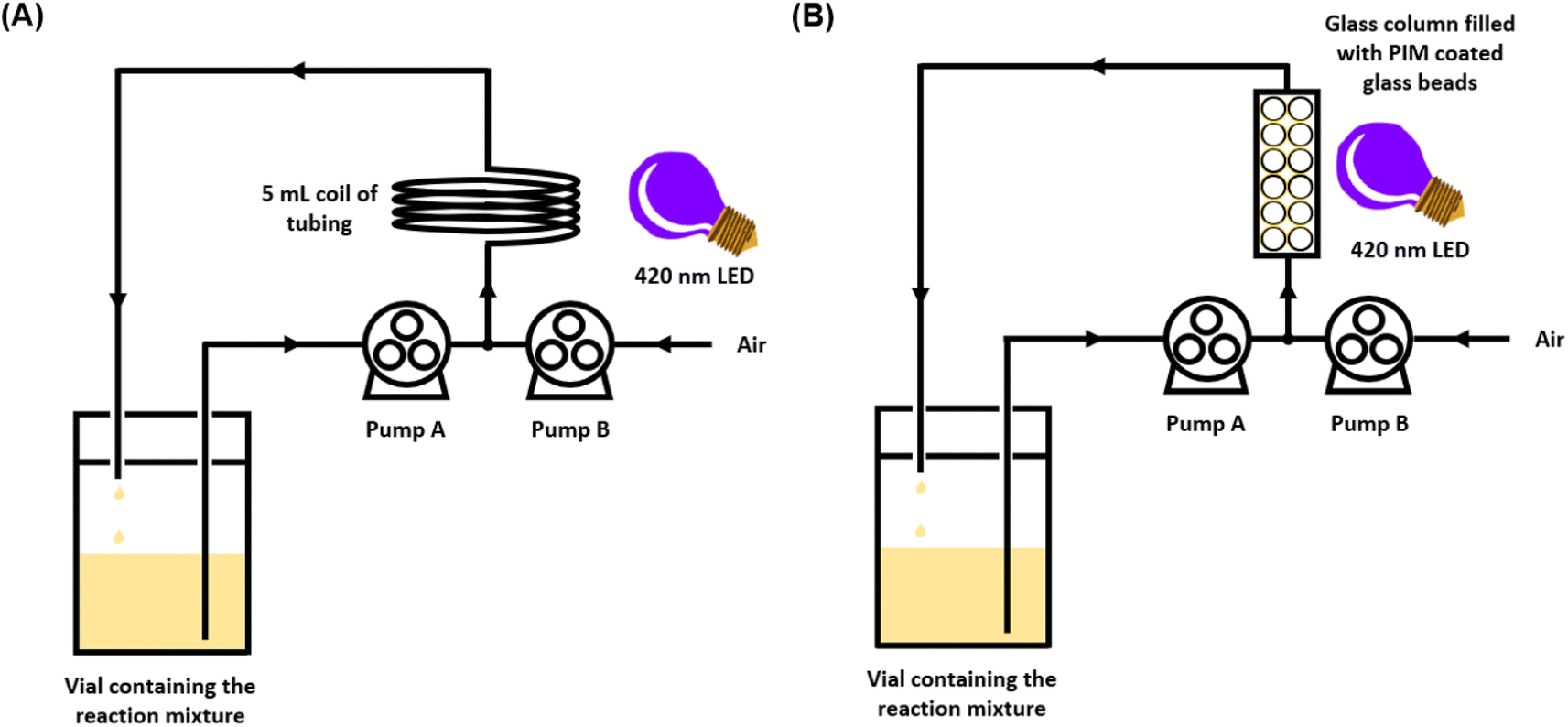 | ||
| Fig. 5 Schematic illustration of the experimental set up for conducting triphenylphosphine oxidation under continuous flow conditions (A) using PIM-BTZ-5% homogeneously within a coil reactor and (B) heterogeneously using PIM-BTZ-5% immobilised on glass beads. | ||
To further increase the rate of the reaction, the flow rates of the solution and of air were varied independently and the conversion after 1 hour measured (see Table S4†). It was observed that reducing the air flow rate relative to the flow rate of the PIM solution increased the reaction conversion. For example, decreasing the flow rate of air to 0.5 mL min−1 increased the conversion of triphenylphosphine to triphenylphosphine oxide to 93% in 1 hour. A possible reason for this is due to the large excess of oxygen available in the reaction. As such, the decrease in the flow rate of air will not affect the reaction rate but will cause an overall drop in the flow rate, increasing the overall residence time of the reaction mixture within the reactor. Overall, a conversion of 93% after 1 hour of irradiation corresponds to an approximate 5-fold acceleration in the rate of the reaction compared to the optimised batch conditions presented in Table 1.
Applying PIM-BTZ-5% as surface coatings
To further develop the applicability of PIM-BTZ-5% for continuous flow photosensitisation, we took advantage of its solution processability to apply the polymer as a surface coating on glass beads. These beads were then packed into a glass column reactor through which the reaction mixture could flow under external irradiation (Fig. 6). This would have the advantage of spatially fixing the polymer photosensitiser in place and greatly simplify catalyst recovery. Glass represents an ideal surface for such applications as it is cheap, inert, and offers a high degree of transparency to allow efficient penetration of light throughout the column reactor. Other methods explored to trap polymer photocatalysts within fixed reactors include the use of polymer monoliths,47,48 Merrifield resins,13 and silica as a support material,49 although such methods offer poor penetration of light due to the opacity of the material. | ||
| Fig. 6 Illustration of the steps involved in the preparation of PIM-BTZ-5% coated glass beads for use in a column reactor under continuous flow. Image on the bottom left shows a single glass bead coated with PIM-BTZ-5% using a fluorescence microscope. | ||
Coating of the glass beads was achieved by slowly concentrating a mixture of 500–750 µm glass beads (5 g) and PIM-BTZ-5% (10 mg) in chloroform under vacuum to a slurry before separating off the glass beads. This yielded glass beads of a clear yellow colour (Fig. S15†), suggesting successful formation of a thin coating. Examination of the coated glass beads under a fluorescence microscope revealed a uniform surface, although some localised defects were observed where beads had stuck together (Fig. 6). This was minimised by spreading the coated glass beads out after coating to prevent clumping. Ultimately, the solvent compatibility of the PIM-BTZ-5% coated glass beads was dictated by the solubility of the PIM coating in the reaction solvent: this precluded the use of solvents such as CDCl3, but allowed the use of non-solvents such as methanol, acetonitrile, etc. Post-coating covalent crosslinking of PIM-BTZ-5% was also investigated by quaternerisation of the Tröger's base linkages with 1,4-bis(bromomethyl)benzene to prevent the coating from dissolving in any solvent (Fig. S16†).50 While crosslinking rendered the polymer coating insoluble in chloroform, it also resulted in a reduction in the mechanical robustness of the polymer coatings, causing them to detach from the beads when tested under flow conditions. Additionally, quaternerisation of TB linkages is known to significantly reduce the surface areas of PIMs due to stronger interactions between the polymer chains and counterions blocking the pores.51 Such a reduction in porosity could also negatively impact the rate of photosensitisation by reducing access of oxygen to the pores.
Testing the 1O2 production ability of PIM-BTZ-5% coated glass beads was carried out using triphenylphosphine oxidation using 5 g of beads contained within a fritted glass column as part of the continuous flow set-up shown in Fig. 5B. In methanol, >90% conversion of triphenylphosphine to triphenylphosphine oxide was achieved within 18 hours, as shown in the kinetic trace in Fig. S20.† To test the consistency of the surface coating method, three batches of separately coated beads were tested, with only minor differences in the rate of conversion observed. Glass beads coated with PIM-BTZ-1% were also tested under continuous flow using methanol as the reaction solvent: after 18 hours, 90% conversion of triphenylphosphine to triphenylphosphine oxide was obtained. The similar performance of PIM-BTZ-5% and PIM-BTZ-1% when employed as surface coatings on glass beads implies that under the continuous flow conditions, an excess of 1O2 is being produced.
The reusability of the PIM-BTZ-5% coated glass beads was also tested across five consecutive runs: washing of the beads between each run was carried out by flowing methanol through the reactor column followed by a stream of air. Across the five runs, the conversion after 18 hours of reaction remained in the region of 86–99%, highlighting both the photostability and robustness of the thin polymer coating (Fig. S21†). 1O2 production was also tested using the oxidation of 2-furoic acid in water, where a conversion of 30% was achieved after 3 days. While still quite slow due to the low lifetime of 1O2 in water, this represents a vast improvement in the reaction conversion in comparison to the 20% conversion obtained after 7 days using BTZ doped PIM-EA-TB films under batch conditions, again emphasising the benefits of continuous flow chemistry.42
DFT modelling of PIM-BTZ-5%
To complement the experimental studies conducted into PIM-BTZ-1% and PIM-BTZ-5%, theoretical calculations were also performed to better understand the electronic structure of the polymer materials (see the ESI† for specific details pertaining to the calculations). To reduce the cost of the computational calculations, TB-BTZ was selected as the theoretical model given the similarity of its absorption/emission spectra with PIM-BTZ-5%. The predicted absorption spectrum, with a maximum located at 393 nm (Fig. S22†), was in good agreement with that observed from experiment, maximum located at 412 nm. The S0 → S1 transition is dominated by an excitation of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (Fig. 7). In keeping with the HOMO–LUMOs of similar BTZ systems, the HOMO orbital featured a significant degree of character on the aryl groups and the benzene ring of the BTZ group.7 This charge is moved from the aryl electron donor groups to the BTZ electron acceptor group in the LUMO, providing evidence of the ICT mechanism commonly associated with BTZ photosensitisers. Other key energy values were calculated for TB-BTZ (Table S5†), including the singlet–triplet energy gap (S0–T1) in chloroform, which was calculated to be 1.38 eV. This is greater than the minimum value of 0.98 eV needed to achieve 1O2 formation, reinforcing the suitability of these material for photosensitisation.2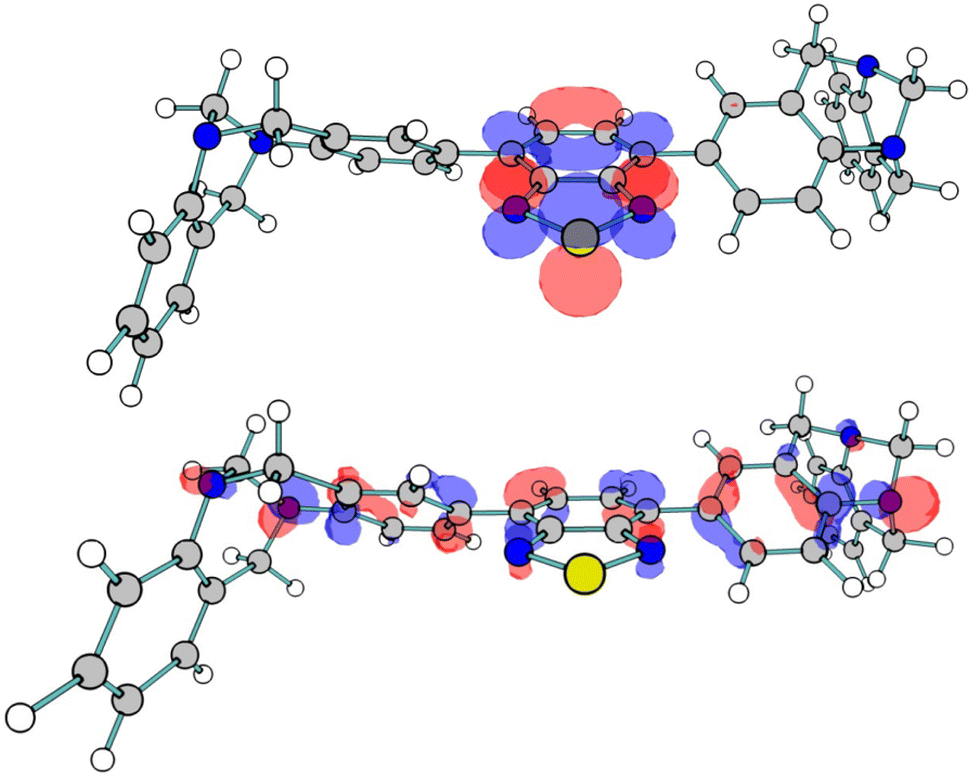 | ||
| Fig. 7 HOMO (bottom) and LUMO (top) for TB-BTZ. | ||
Conclusions
In conclusion, we have investigated the utility of PIM-EA-TB as a solution processible and microporous platform for immobilising visible light photosensitisers based on the BTZ groups. While trapping the BTZ photosensitisers within the pores of the material was fundamentally limited by poor solvent compatibility, covalently incorporating them into the backbone of the material proved effective. Through this strategy, photoactive PIMs with up to 5% BTZ content were prepared without compromising the solution processability or the microporosity of PIM-EA-TB. This allowed PIM-BTZ-5% to be investigated as either a homogeneous or heterogeneous photosensitiser under both batch and continuous flow conditions. Furthermore, the solution processability of PIM-BTZ-5% allowed it to be applied as a coating to glass beads trapped within a column reactor. This augmented the capabilities of the material under continuous flow conditions by simplifying photosensitiser recycling. In future, we envisage that the merger of efficient photocatalysts with solution processible PIMs could facilitate the development of new functional surfaces and expedite research into bespoke continuous flow reactors. The success of this would be underpinned by a fundamental understanding of the structure–property relationship of these materials, including the impact that different contorted building blocks and increasing photocatalyst content had on the structural and optoelectronic behaviour: we aim to report the results of such studies in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FV and SJD thank the Engineering and Physical Sciences Research Council UK (EPSRC) funded (EP/L016419/1) CRITICAT centre for doctoral training. FV thanks Vapourtec for their invaluable technical support. FV and SJD thank the EPRSC for Doctoral Training Partnership funding for LA. MJP gratefully acknowledges funding through the EPSRC, grant No. EP/V006746 and EP/T021675. MJP and AWP also acknowledge support from the Leverhulme Trust (Grant No. RPG-2020-208). NBM thanks the EPSRC for funding through grants EP/V027735/1 and EP/V047078/1. The authors thank Dr Dylan Wilkinson and Prof. Graeme Cooke (University of Glasgow) for their assistance with GPC characterisation of the polymer materials.Notes and references
- I. Pibiri, S. Buscemi, A. Palumbo Piccionello and A. Pace, ChemPhotoChem, 2018, 2, 535–547 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- D. García-Fresnadillo, ChemPhotoChem, 2018, 2, 512–534 CrossRef.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- D. Taylor, T. Malcomson, A. Zhakeyev, S.-X. Cheng, G. M. Rosair, J. Marques-Hueso, Z. Xu, M. J. Paterson, S. J. Dalgarno and F. Vilela, Org. Chem. Front., 2022, 9, 5473–5484 RSC.
- E. Broumidis, C. M. S. Jones, M. Koyioni, A. Kourtellaris, G. O. Lloyd, J. Marques-Hueso, P. A. Koutentis and F. Vilela, RSC Adv., 2021, 11, 29102–29107 RSC.
- D. Taylor, T. Malcomson, A. Zhakeyev, G. M. Rosair, M. J. Paterson, J. Marques-Hueso, S. J. Dalgarno and F. Vilela, RSC Adv., 2023, 13, 5826–5832 RSC.
- E. C. Blossey, D. C. Neckers, A. L. Thayer and A. P. Schaap, J. Am. Chem. Soc., 1973, 95, 5820–5822 CrossRef CAS.
- A. P. Schaap, A. L. Thayer, E. C. Blossey and D. C. Neckers, J. Am. Chem. Soc., 1975, 97, 3741–3745 CrossRef CAS.
- M. I. Burguete, F. Galindo, R. Gavara, S. V. Luis, M. Moreno, P. Thomas and D. A. Russell, Photochem. Photobiol. Sci., 2009, 8, 37–44 CrossRef CAS PubMed.
- C. G. Thomson, C. M. S. Jones, G. Rosair, D. Ellis, J. Marques-Hueso, A.-L. Lee and F. Vilela, J. Flow Chem., 2020, 10, 327–345 CrossRef CAS.
- J. M. Tobin, T. J. D. McCabe, A. W. Prentice, S. Holzer, G. O. Lloyd, M. J. Paterson, V. Arrighi, P. A. G. Cormack and F. Vilela, ACS Catal., 2017, 7, 4602–4612 CrossRef CAS.
- J. Shen, R. Steinbach, J. M. Tobin, M. Mouro Nakata, M. Bower, M. R. S. McCoustra, H. Bridle, V. Arrighi and F. Vilela, Appl. Catal., B, 2016, 193, 226–233 CrossRef CAS.
- C. T. J. Ferguson, N. Huber, T. Kuckhoff, K. A. I. Zhang and K. Landfester, J. Mater. Chem. A, 2020, 8, 1072–1076 RSC.
- A. Zhakeyev, J. Tobin, H. Wang, F. Vilela and J. Xuan, Energy Procedia, 2019, 158, 5608–5614 CrossRef CAS.
- A. Zhakeyev, M. C. Jones, C. G. Thomson, J. M. Tobin, H. Wang, F. Vilela and J. Xuan, Addit. Manuf., 2021, 38, 101828 CAS.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- D. Taylor, S. J. Dalgarno and F. Vilela, in Concepts and Design of Materials Nanoarchitectonics, 2022, pp. 226–246 Search PubMed.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 4, 230–231 RSC.
- N. B. McKeown, Polymer, 2020, 202, 122736 CrossRef CAS.
- N. B. Mc Keown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- Y. Rogan, R. Malpass-Evans, M. Carta, M. Lee, J. C. Jansen, P. Bernardo, G. Clarizia, E. Tocci, K. Friess, M. Lanč and N. B. McKeown, J. Mater. Chem. A, 2014, 2, 4874–4877 RSC.
- M. A. Abdulhamid, H. W. H. Lai, Y. Wang, Z. Jin, Y. C. Teo, X. Ma, I. Pinnau and Y. Xia, Chem. Mater., 2019, 31, 1767–1774 CrossRef CAS.
- B. Riza Putra, M. Carta, R. Malpass-Evans, N. B. McKeown and F. Marken, Electrochim. Acta, 2017, 258, 807–813 CrossRef CAS.
- S. E. Doris, A. L. Ward, A. Baskin, P. D. Frischmann, N. Gavvalapalli, E. Chénard, C. S. Sevov, D. Prendergast, J. S. Moore and B. A. Helms, Angew. Chem., Int. Ed., 2017, 56, 1595–1599 CrossRef CAS PubMed.
- A. Kolodziej, S. D. Ahn, M. Carta, R. Malpass-Evans, N. B. McKeown, R. S. L. Chapman, S. D. Bull and F. Marken, Electrochim. Acta, 2015, 160, 195–201 CrossRef CAS.
- Y. Rong, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard and F. Marken, Electrochem. Commun., 2014, 46, 26–29 CrossRef CAS.
- J. D. Smith, A. M. Jamhawi, J. B. Jasinski, F. Gallou, J. Ge, R. Advincula, J. Liu and S. Handa, Nat. Commun., 2019, 10, 1837 CrossRef PubMed.
- A. Atilgan, Y. Beldjoudi, J. Yu, K. O. Kirlikovali, J. A. Weber, J. Liu, D. Jung, P. Deria, T. Islamoglu, J. F. Stoddart, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2022, 14, 12596–12605 CrossRef CAS PubMed.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- H. Urakami, K. Zhang and F. Vilela, Chem. Commun., 2013, 49, 2353–2355 RSC.
- T. Kuckhoff, J. Heuer, R. Li, K. A. I. Zhang, K. Landfester and C. T. J. Ferguson, RSC Applied Polymers, 2024, 2, 155–162 RSC.
- Y. Gwon, S. Jo, H.-J. Lee, S. Y. Park and T. S. Lee, Polymer, 2021, 229, 124004 CrossRef CAS.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F. S. Xiao, Matter, 2020, 2, 416–427 CrossRef CAS.
- J. Weber, Q. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- J. R. Hurst, J. D. McDonald and G. B. Schuster, J. Am. Chem. Soc., 1982, 104, 2065–2067 CrossRef CAS.
- D. Zhang, B. Ye, D. G. Ho, R. Gao and M. Selke, Tetrahedron, 2006, 62, 10729–10733 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- V. Blanchard, Z. Asbai, K. Cottet, G. Boissonnat, M. Port and Z. Amara, Org. Process Res. Dev., 2020, 24, 822–826 CrossRef CAS.
- Z. Yang, R. Guo, R. Malpass-Evans, M. Carta, N. B. McKeown, M. D. Guiver, L. Wu and T. Xu, Angew. Chem., Int. Ed., 2016, 55, 11499–11502 CrossRef CAS PubMed.
- P. Zheng, W. Xie, Z. Cai, Y. Jiao, Y. Sun, T. Han, X. Ma, N. Li and S. Luo, J. Membr. Sci., 2023, 672, 121425 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation data of monomers and polymers. See DOI: https://doi.org/10.1039/d4ta01009d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |