DOI:
10.1039/D4TA00633J
(Paper)
J. Mater. Chem. A, 2024,
12, 9546-9551
A domino reaction from a sensitive azide: the impact of positional isomerism on chemical reactivity featuring ortho azido/nitro substituted derivatives†
Received
28th January 2024
, Accepted 13th March 2024
First published on 14th March 2024
Abstract
This study highlights for the first time the impact of positional isomerism on chemical reactivity which involves a domino reaction, salt formation, cyclization, and nucleophilic substitution. The unprecedented reactivity occurs upon treating sensitive 5-azido-3,4-dinitropyrazole (1) with nitrogen rich bases. This results in the development of the first examples of fused catenated N5 pyrazolo[1,5-d]tetrazole-based energetic salts. The compound structures of 1, 2, and 3 were obtained by single-crystal analysis. Compounds 2 (Dv: 9202 m s−1; P: 32.08 GPa) and 3 (Dv: 8749 m s−1; P: 31.26 GPa) displayed excellent detonation properties (calculated) and good sensitivities (IS ≥ 20 J; FS ≥ 240 N), which are comparable to RDX. The thermal stability of compound 2 (185 °C) further suggests its significant potential use as a secondary explosive. Compound 1 exhibits high detonation properties (Dv: 9023 m s−1; P: 35.3 GPa) and sensitivity to external stimuli (IS: 8 J; FS: 80 N), indicating it has potential as a metal-free primary explosive.
Introduction
After a decade of effort, high-performance and reduced sensitivity are still the key concerns in the ongoing pursuit of advancement in high-energy materials.1 In the study of energetic materials, positional isomerism has conventionally been associated with its impact on the physical properties, including density, decomposition temperature, sensitivity and so on.2 Notably, there is a lack of documented reports regarding the impact of positional isomerism on the chemical reactivity of energetic materials. Considering the structure–property relationship, fused nitrogen-rich heterocycles are generally considered as potential energetic materials capable of achieving a balance between high performance and stability.3
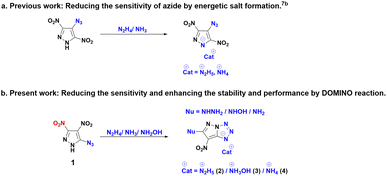
The strategic design of domino reactions, also known as cascade transformations is a powerful tool in synthesizing fused derivatives due to their interconnected molecular structure.4 This unique feature enables sequential reactions within a single synthetic process, reducing the need for intermediate isolation and purification and enhancing overall efficiency. The domino reactions contribute not only to the synthesis atom economy but also promote environmental sustainability and economic viability. Nitrogen-rich explosophores, such as azido (N3), have also emerged as a prominent structural motif in energetic materials, owing to their increased nitrogen content, high heats of formation, and excellent initiating efficiency.5 In comparison to other five-membered nitrogen-rich heterocycles, pyrazoles typically have diverse reactivity and stability.6 Several derivatives of 4-azido-3,5-dinitropyrazole have been reported in the literature, including its energetic salt formation with nitrogen bases.7 Following the progression of the previously documented isomeric derivative, 5-azido-3,4-dinitropyrazole (1) piqued our interest because of its reactivity in the synthesis of several energetic material derivatives.8 Despite its well-documented synthesis in the literature, the energetic properties of compound (1) remain elusive.9
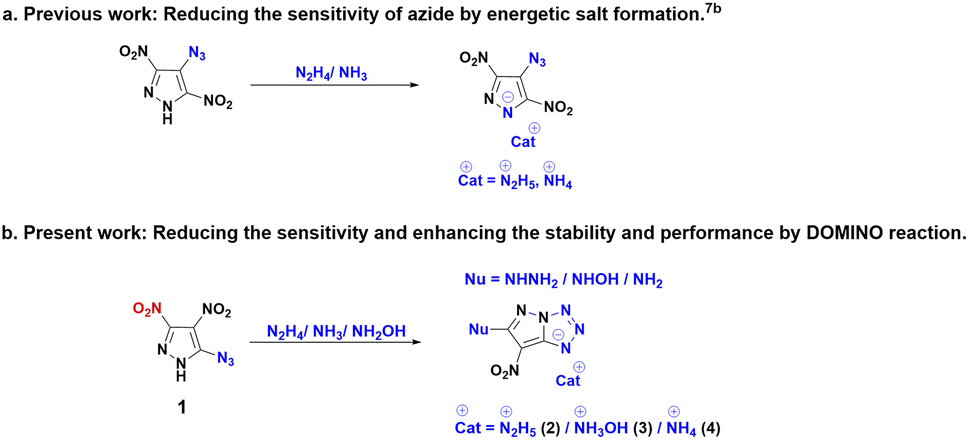
The objective of this study is to examine the energetic properties of 5-azido-3,4-dinitro-1H-pyrazole (1) and its chemical reactivity with nitrogen bases such as hydrazine monohydrate, hydroxylamine, and aqueous ammonia. In contrast to the chemical reactivity of the 4-azido-substituted derivative, which yields energetic salts,7b the isomeric 5-azido substituted derivative undergoes a domino reaction involving salt formation, cyclization, and nucleophilic substitution, due to the presence of an azido group in a position α to an aza atom of the heterocycle (Scheme 1).5c,7c,7d
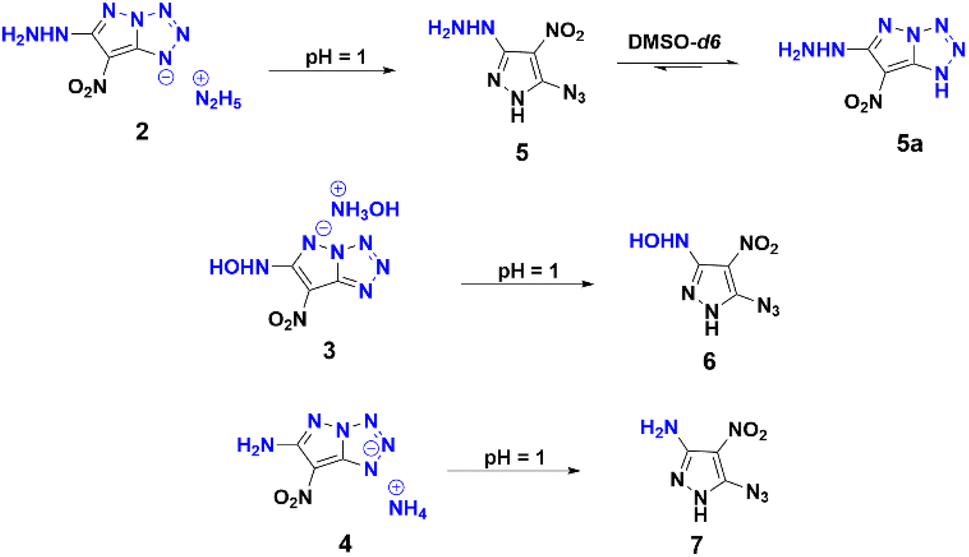
 |
| | Scheme 1 Impact of positional isomerism of azide derivatives with nitrogen bases. | |
Results and discussion
Synthesis
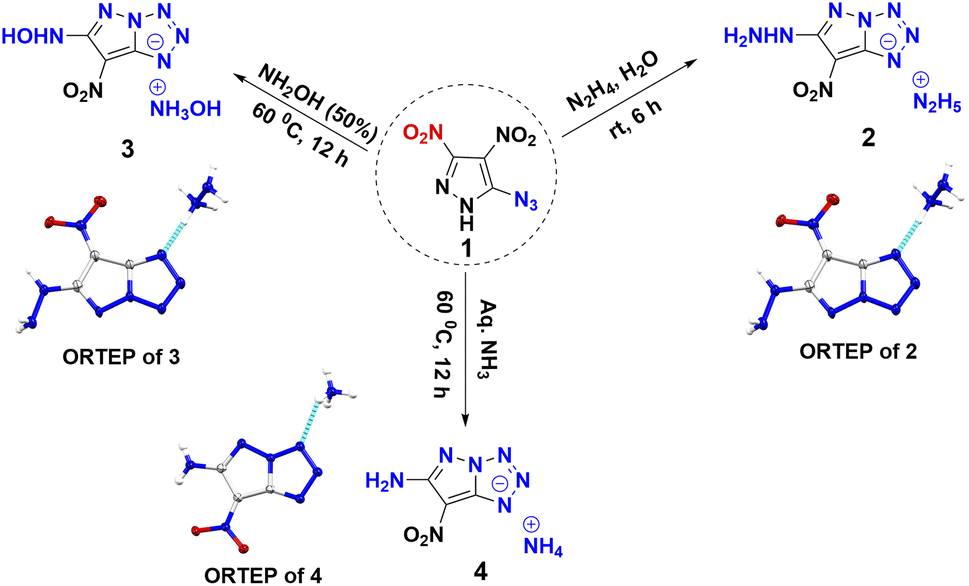
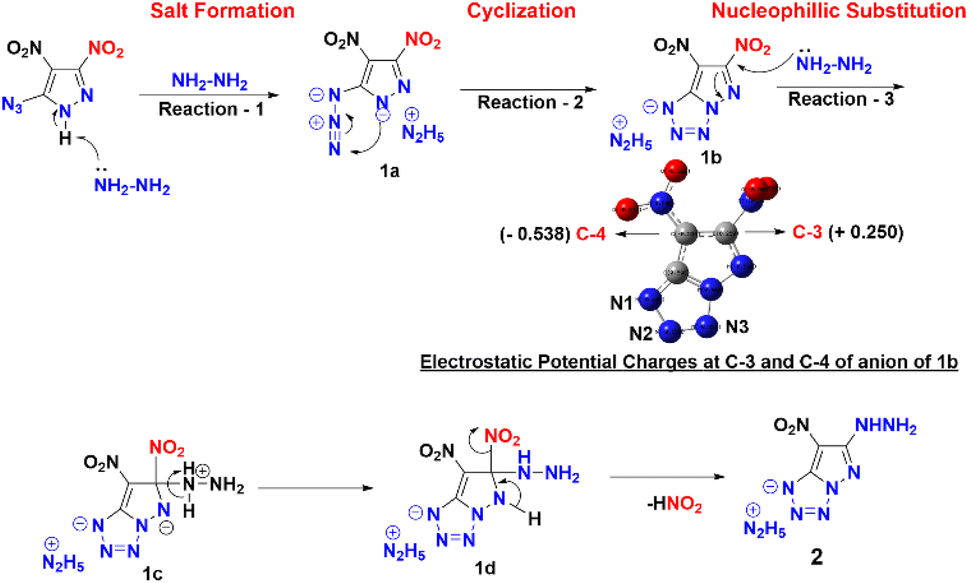
Compound 1 was synthesized in accordance with the modified literature procedure (Scheme S1, ESI†),8 which facilitates safe handling for subsequent reactions. Initially, the reaction of 1 with one equivalent of hydrazine monohydrate in methanol results in precipitation as the temperature is raised from low to room temperature. Notably, the elemental analysis did not agree with the expected hydrazinium salt of 1. Consequently, empirical formula calculations based on percentages of elements suggested a plausible nucleophilic substitution involving one of the nitro groups present in compound 1. Subsequent single X-ray crystal analysis supported the conversion of azido to tetrazole, involving the pyrazole anion (Fig. 1). This investigation culminated in the conclusion that reaction of hydrazine with compound 1 yields compound 2 in a cascade of transformations (Scheme 3) with excess reagent (10 equiv.), as depicted in Scheme 2. Mechanistic insight is provided in Scheme 3. We believe the plausible mechanism which proceeds via the formation of 1a with base (salt formation). In step 2, the direct intramolecular attack of pyrazole anion on the azide leads to the formation of the tetrazole ring of 1b (cyclization). In step 3, the nitrogen positioned α to the C3–NO2 serves as an electron reservoir, facilitating the addition of nucleophile and the subsequent elimination of the C3–NO2 group leading to the final product 2 (nucleophilic substitution). Further, it was confirmed by DFT calculations of electrostatic potential charges at C-3 and C-4 positions of the anion of 1b. Development of substantial positive charge at C-3 in 1b provides additional support for nucleophilic substitution.10
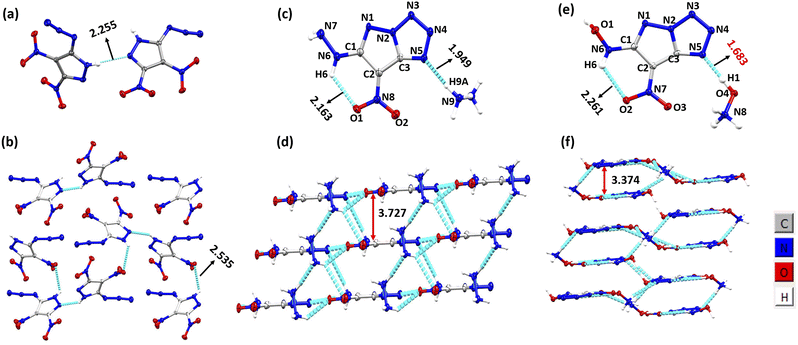 |
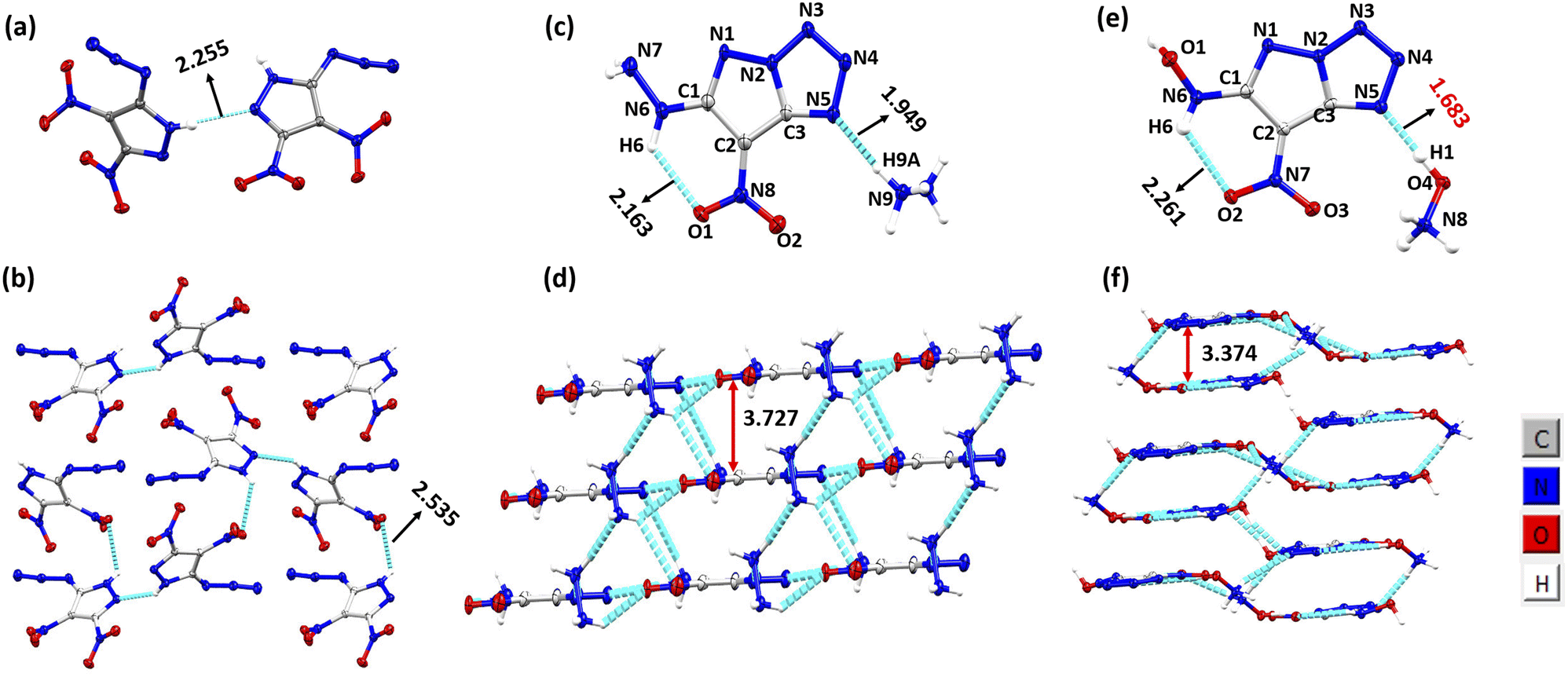
| | Fig. 1 (a) Thermal ellipsoid plot (50%) of 1. (b) Packing and H-bonding in compound 1. (c) Thermal ellipsoid plot (50%) and labelling scheme for 2. (d) Packing and H-bonding in compound 2. (e) Thermal ellipsoid plot (50%) and labelling scheme for 3. (f) Packing and H-bonding in compound 3. | |
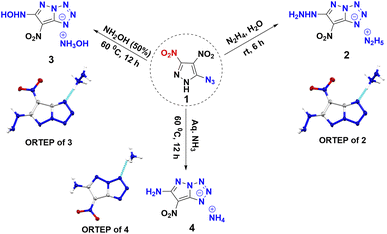 |
| | Scheme 2 Reaction of 5-azido-3,4-dinitropyrazole with nitrogen bases (N2H4/NH2OH/NH3). | |
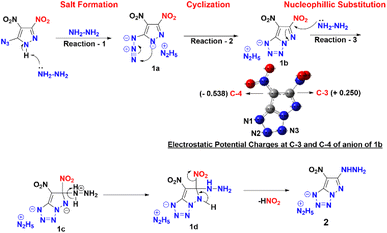 |
| | Scheme 3 Plausible mechanism and electrostatic potential charges at C-3 and C-4. | |
Employing similar reaction conditions with aqueous hydroxyl-amine as a nitrogen base, resulted in a sticky, uncharacterizable compound at room temperature. However, at 60 °C and extended reaction time (12 h), the formation of compound 3 in 71% yield occurs. Given that ammonia is less basic than hydrazine, thermal conditions (60 °C) and extended reaction time (12 h) were required in order to achieve compound 4 in 82% yield. Upon acidification of 2, 3, and 4 with 10% HCl, the azido substituent retained its distinct identity (Scheme 4), a confirmation supported by the characteristic peak range observed at 2150–2123 cm−1 in the IR spectrum. This experimental progression highlights a nuanced and detailed exploration of the reaction dynamics and structural transformation undergone by compound 1.
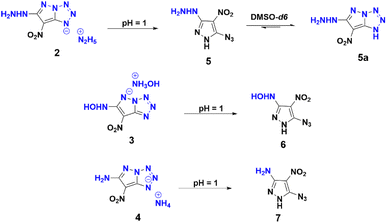 |
| | Scheme 4 Acidification of 2, 3, and 4 with 10% HCl and azido-tetrazole equilibrium of 5 in DMSO-d6. | |
Spectroscopic studies
Full characterization of the compounds was realized by NMR (1H, 13C and 15N), and IR spectroscopy and elemental analysis (ESI†). In particular, the 13C NMR spectrum of compound 5, recorded in DMSO-d6, exhibited distinct signals at 150.2, 141.6, and 107.7 ppm, as shown in Scheme 4. Intriguingly, over a period of time, an additional set of three 13C NMR peaks (154.8, 145.4, and 101.6 ppm) were observed at room temperature, indicating the formation of 5a. This observation indicates that compound 5 exists in equilibrium with 5a in DMSO-d6, which exemplifies the dynamic aspect of its molecular behavior. The 15N NMR spectra of compounds 1, 2, 3, 4, 6, and 7 were recorded in DMSO-d6 and the chemical shifts are reported relative to nitromethane as external standard (ESI†). In the case of compound 4, the 15N peaks corresponding to nitrogens (N1/N2/N3, Scheme 3) involved in the ring formation resonated at −164.89, −23.83, and −45.58 respectively. Conversely, in the free azide state of compound 7, the nitrogen peaks exhibited resonances at −296.44, −146.47, and −130.20 ppm, respectively.
Crystal structures
Single crystals of 1 were grown by the slow evaporation of ethyl acetate. The structure of 1 was solved in the orthorhombic space group P212121 with the cell volume of 689.032(18) Å3. The crystal of 1 has one asymmetric unit in the unit cell, and exhibits an excellent density of 1.919 g cm−3 at 100 K (Fig. 1). Single crystals of 2 were grown by the slow evaporation of a methanol–water system. The structure was solved in the triclinic space group P1 with the cell volume of 201.96(3) Å3. The crystal of 2 has one asymmetric unit in the unit cell, and exhibits a density of 1.778 g cm−3 at 100 K. The torsion angle of N6–C1–C2–C3 is −179.9°, and that of N8–C2–C3–N2 176.7° indicating that the molecule is planar (Fig. 1). Single crystals of 3 were grown by the slow evaporation of methanol. The structure was solved in the triclinic space group P1 with the cell volume of 379.22(3) Å3. The crystal of 3 has one asymmetric unit in the unit cell, and exhibits a remarkable density of 1.911 g cm−3 at 100 K. The torsion angle of N6–C1–C2–C3 is 178.03°, and that of N7–C2–C3–N2-171.97° (Fig. 1). The hydroxylammonium cation has shown strongest hydrogen bonding of distance 1.68 Å (O4–H4⋯N5) in comparison to the hydrazinium cation of hydrogen bond distance 1.949 Å (N9–H9A⋯N5). The other intramolecular hydrogen bonding present in 2 and 3 are N6–H6⋯O1 (2.163 Å) and N6–H6⋯O2 (2.261 Å) respectively. The intermolecular stacking of 3 was closer than that in the corresponding 2. As can be seen that the stacking of 3 was closer than that in the corresponding 2. As can be seen in Fig. 1, the stacking distance of 3 was 3.374 Å, which is remarkably shorter than those of 2 (3.727 Å). Single crystals of 4 were grown by the slow evaporation of methanol–water system. The structure was solved in the space group C2/c with the cell volume of 1430.07(10) Å3. The crystallographic data are provided in ESI.†
Hirshfeld and NCI analysis
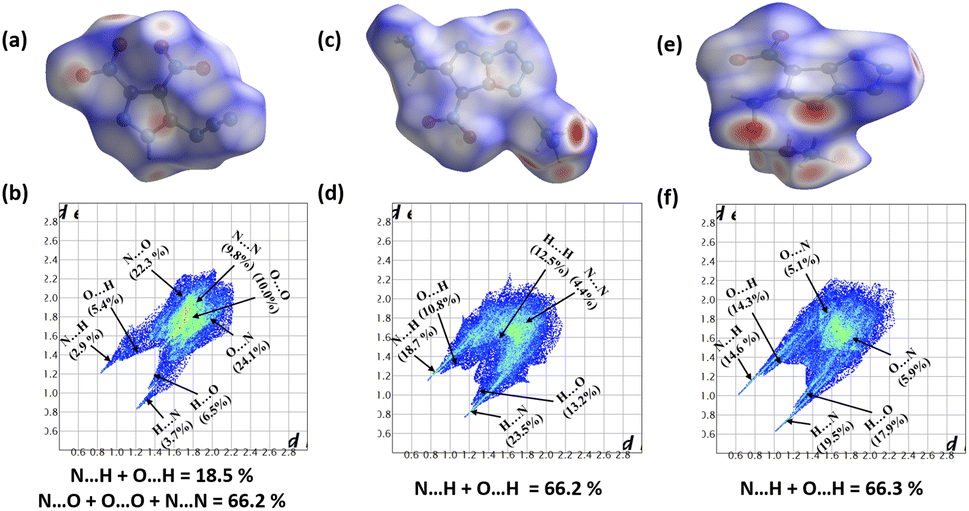
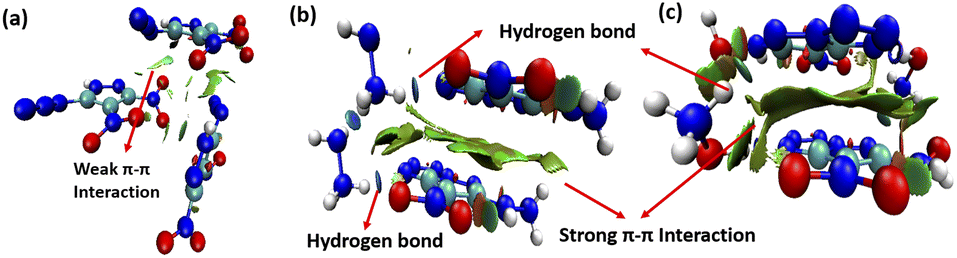
To explore intermolecular interactions in the crystal structures of compounds 1, 2, and 3, we performed an analysis using Hirshfeld surfaces and two-dimensional (2D) fingerprint plots (Fig. 2).11 Strong hydrogen bonds were identified, with N⋯H and O⋯H percentages in 1, 2, and 3 at 6.6%, 66.2%, and 66.3%. Compound 2 showed a significant 42.2% of strong N⋯H interactions, contributing to its higher thermal stability of 185 °C. Compound 1 exhibited a high percentage of N⋯O (46.4%) and O⋯O (10%) interactions, impacting its lower thermal stability and higher sensitivity. Further, a non-covalent interaction (NCI) analysis of compounds 1, 2, and 3 was performed using Multiwfn software, and the resulting plots were analyzed using VMD software (Fig. 3).12 The significant green isosurface indicates π–π interactions, while the blue and green ellipses primarily represent intramolecular hydrogen bonding. Notably, compounds 2 and 3 exhibited strong green isosurface areas due to cationic–anionic interactions. The presence of abundant hydrogen bonds and strong π–π inter-actions contribute to the enhanced physical stability of 2 and 3 compared to 1.
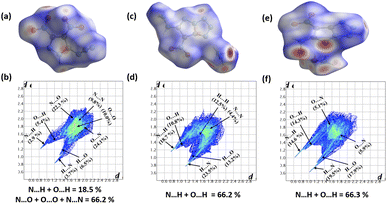 |
| | Fig. 2 Hirshfeld surface graphs and 2D fingerprint plots of 1 (a and b), 2 (c and d), and 3 (e and f). | |
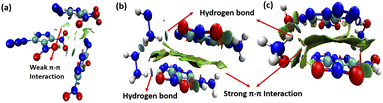 |
| | Fig. 3 Noncovalent interaction plots of gradient isosurfaces 1 (a), 2 (b), and 3 (c). | |
Physicochemical and energetic properties
The physiochemical and energetic properties of all compounds were evaluated and the results tabulated in Table 1. Due to the large number of N–N, and C–N bonds, these compounds have excellent nitrogen percentages (49.25–64.80%), directly contributing to the high heats of formation. The heats of formation (ΔHf) were calculated using the Gaussian 09 suite of programs, employing the isodesmic reaction approach (ESI†).10 The azido group, adding 293 kJ mol−1 to the energy content of the molecule, substantiates the heats of formation values within the range of 442 kJ mol−1 (3) to 662 kJ mol−1 (2), surpassing the heats of formation of lead azide (LA), TNT and RDX. The densities of the compounds were determined using a gas pycnometer at 25 °C. The measured densities were in a range of 1.63 (7)–1.79 (1) g cm−3. Compound 1 and 2 possess the highest densities of 1.79 and 1.75 g cm−3, respectively. Based on the heats of formation and measured densities, the detonation properties (detonation velocity (Dv) and detonation pressure (P)) were computed using the EXPLO5 software (version 7.01.01).13 The evaluated detonation velocities were in the range of 7995 and 9202 m s−1 and detonation pressures between 24.7 and 35.3 GPa. All compounds exhibited a better detonation performance than the traditional explosive TNT (Dv: 7303 m s−1; P: 21.3 GPa) and lead-based primary explosive LA (Dv: 5877 m s−1; P: 33.4). Compounds 1 (Dv: 9023 m s−1; P: 35.3 GPa), and 2 (Dv: 9202 m s−1; P: 32.1 GPa) displayed better detonation properties than RDX (Dv: 8795 m s−1; P: 34.9 GPa) and compound 3 (Dv: 8749 m s−1; P: 31.3 GPa) has detonation properties comparable to RDX. Compared to free azide derivatives (5, 6, and 7), fused derivatives (2, 3, and 4) exhibited better detonation properties.
Table 1 Physiochemical and energetic properties of compounds 1–7
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
LAk |
TNTk |
RDXk |
|
Nitrogen content.
Density determined by gas pycnometer at 25 °C.
Heat of formation.
Thermal decomposition temperature (5 °C min−1).
Impact sensitivity (BAM drophammer).
Friction sensitivity (BAM friction tester).
Detonation velocity.
Detonation pressure.
Crystal density at 100 K in parenthesis.
T
m = Melting Point in parenthesis.
Ref. 7b.
|
|
N
(%) |
49.25 |
64.80 |
51.37 |
60.20 |
60.86 |
52.97 |
57.98 |
28.86 |
18.50 |
37.84 |
|
ρ
(g cm−3) |
1.79 (1.919)i |
1.75 (1.778)i |
1.73 (1.911)i |
1.69 (1.729)i |
1.65 |
1.63 |
1.66 |
4.80 |
1.65 |
1.80 |
| ΔHfc (kJ mol−1) |
559 |
662 |
460 |
442 |
638 |
515 |
526 |
450 |
−59.4 |
92.6 |
|
T
d
(°C) |
141 (Tm = 137)j |
185 |
140 |
173 |
160 |
127 |
156 |
315 |
295 |
204 |
| ISe (J) |
8 |
25 |
20 |
27 |
11 |
9 |
13 |
0.1–1 |
15 |
7.4 |
| FSf (N) |
80 |
240 |
240 |
240 |
120 |
80 |
120 |
0.3–0.6 |
358 |
120 |
|
D
v
(m s−1) |
9023 |
9202 |
8749 |
8360 |
8213 |
8128 |
7995 |
5877 |
7303 |
8795 |
|
P
(GPa) |
35.3 |
32.08 |
31.26 |
26.02 |
26 |
26.37 |
24.74 |
33.4 |
21.3 |
34.9 |
Stability plays a pivotal role in determining the overall performance of energetic compounds, relying on both thermal stability and mechanical sensitivity to external stimuli. Differential scanning calorimetry measurements were conducted to ascertain the thermal behaviour of all compounds at a heating rate of 5 °C min−1. All the compounds except 1 decomposed without melting. The decomposition temperatures of all compounds were in the range from 127 °C (6) to 185 °C (2). Compound 1 decomposed immediately after melting at 137 °C. The thermal stabilities of fused derivatives (2, 3, and 4) are better than that of the neutralized derivatives (5, 6, and 7). This enhanced thermal stability is due to the fact that breaking these bonds or disrupting the extended π-conjugation requires more energy. The impact and friction sensitivities of all the compounds were measured by standard BAM technology (Table 1). The parent compound 1 is less sensitive towards impact (8 J) than expected due to the fact that it is an azole-based compound incorporating an azide besides nitro groups, although it still has to be qualified as sensitive. The friction sensitivity is noticeably higher (80 N) and should therefore only be handled with care. Among all the newly synthesized compounds, the hydroxylamine derivatives (3 (IS: 20 J; FS: 160 N) and 6 (IS: 9 J; FS: 80 N)) have the highest impact values and friction sensitivities due to weaker N⋯H interactions. Compounds 2 (IS: 25 J; FS: 240 N) and 4 (IS: 27 J; FS: 240 N) are less sensitive towards impact and friction. The fused derivatives (2, 3, and 4) are less sensitive (IS: 20–27 J; FS: 160–240 N) than the neutralized derivatives (5, 6, and 7) (IS: 9–13 J; FS: 80–120 N) and better than the commonly used secondary explosive RDX (IS: 7.4 J; FS: 120 N) due to strong π–π interactions.
Conclusions
In conclusion, this innovative study provides insights into the unexplored realm of positional isomerism's impact on chemical reactivity of the isomeric 5-azido-3,4-dinitropyrazole with nitrogen bases, triggering a domino reaction. This resulted in the successful development of a new family of secondary explosives of fused catenated N5 pyrazolo[1,5-d]tetrazole based energetic salts derived from sensitive azide of a primary explosive. Compound 1 displayed outstanding detonation properties (Dv: 9023 m s−1; P: 35.3 GPa) in comparison to RDX and LA. Despite its relatively low decomposition temperature of 141 °C, it qualifies as a primary explosive due to its sensitivity to impact (IS: 8 J) and friction (FS: 80 N). The fused derivatives (2, 3, and 4) demonstrate higher thermal stability and lower sensitivity to impact and friction when contrasted with the neutralized derivatives (5, 6, and 7). Compound 2 has the highest detonation properties (Dv: 9202 m s−1; P: 32.08 GPa) and good thermal stability (Td: 185 °C), positioning it as a promising candidate for application as a secondary explosive.
Author contributions
V. T. and J. S. investigation, methodology, conceptualization and manuscript writing. R. J. S. X-ray data collection and structures solving. V. T. and J. M. S. conceptualization, manuscript writing – review and editing, supervision.
Conflicts of interest
“There are no conflicts to declare”.
Acknowledgements
The Rigaku Synergy S Diffractometer was purchased with support from the National Science Foundation MRI program (1919565). We are grateful for the support of the Fluorine-19 fund.
References
-
(a)
T. M. Klapötke, Chemistry of High-Energy Materials, Walter de Gruyter GmbH & Co KG, Germany, 2017 CrossRef;
(b) H.-H. Licht, Propellants, Explos., Pyrotech., 2000, 25, 126 CrossRef CAS;
(c) T.-Y. Hou, Z. Xu, X.-P. Zhang, Y.-G. Xu and M. Lu, Energ. Mater. Front., 2022, 3, 166 CrossRef CAS;
(d) P. Yin, Q. Zhang and J. M. Shreeve, Acc. Chem. Res., 2016, 49, 4 CrossRef CAS;
(e) D. G. Piercey, D. E. Chavez, B. L. Scott, G. H. Imler and D. A. Parrish, Angew. Chem., Int. Ed., 2016, 55, 15315 CrossRef CAS PubMed;
(f) H. Wie, J. Zhang and J. M. Shreeve, Chem.–Asian J., 2015, 10, 1130 CrossRef PubMed;
(g) Y.-H. Joo and J. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 15081 CrossRef CAS;
(h) V. Thottempudi and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 19982 CrossRef CAS PubMed;
(i) V. Thaltiri, K. Chavva, B. S. Kumar and P. K. Panda, New J. Chem., 2019, 43, 12318 RSC.
-
(a) Q. Sun, W. Chen, N. Ding, C. Zhao, Z. Jiang, S. Li and S. Pang, Chem. Eng. J., 2022, 444, 136539 CrossRef CAS;
(b) J. Xiong, J. Cai, Q. Lai, P. Yin and S. Pang, Chem. Commun., 2022, 58, 10647 RSC;
(c) T. Yan, H. Yang, C. Yang, Z. Yi, S. Zhu and G. Cheng, J. Mater. Chem. A, 2020, 8, 23857 RSC;
(d) Y. Cao, Z. Cai, J. Shi, Q. Zhang, Y. Liu and W. Zhang, Energy Mater. Front., 2022, 3, 26 CrossRef CAS;
(e) F. Chen, Y. Wang, S. Song, K. Wang and Q. Zhang, J. Phys. Chem. C, 2023, 127, 8887 CrossRef CAS;
(f) L. M. Barton, J. T. Edwards, E. C. Johnson, E. J. Bukowski, R. C. Sausa, E. F. C. Byrd, J. A. Orlicki, J. J. Sabatini and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 12531 CrossRef CAS;
(g) D. G. Piercey, D. R. Wozniak, B. Salfer, M. Zeller and E. F. C. Byrd, Org. Lett., 2020, 22, 9114 CrossRef.
-
(a) H. Gao, Q. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 4193 RSC;
(b) K. Mohammad, V. Thaltiri, N. Kommu and A. A. Vargeese, Chem. Commun., 2020, 56, 12945 RSC;
(c) C. Li, T. Zhu, J. Tang, G. Yu, Y. Yang, H. Yang, C. Xiao and G. Cheng, Chem. Eng. J., 2024, 479, 147355 CrossRef CAS.
-
(a) Z. Jin, G. Shen and X. Lv, Chin. J. Chem., 2023, 41, 3751 CrossRef CAS;
(b) D. K. Winter, D. L. Sloman and J. A. Porco Jr, Nat. Prod. Rep., 2013, 30, 382 RSC.
-
(a) D. Chen, H. Yang, Z. Yi, H. Xiong, L. Zhang, S. Zhu and G. Cheng, Angew. Chem., Int. Ed., 2018, 57, 2081 CrossRef CAS PubMed;
(b) M. Benz, T. M. Klapötke, J. Stierstorfer and M. Voggenreiter, J. Am. Chem. Soc., 2022, 144, 6143 CrossRef CAS PubMed;
(c) M. H. V. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilardi, J. Am. Chem. Soc., 2005, 127, 12537 CrossRef CAS PubMed.
-
(a) P. Yin and J. M. Shreeve, Adv. Heterocycl. Chem., 2017, 121, 89 CrossRef CAS;
(b) S. Zhang, Z. Gao, D. Lan, Q. Jia, N. Liu, J. Zhang and K. Kou, Molecules, 2020, 25(1–42), 3475 CrossRef CAS.
-
(a) P. Das, P. Bhatia, K. Pandey and D. Kumar, Mater. Adv., 2024, 5, 171 RSC;
(b) X. Y. Zhang, X. Y. Lin, B. Y. Guo, C. Tan and Y. Han, J. Mol. Struct., 2022, 1267, 133526 CrossRef CAS;
(c) W. Hu, J. Tang, X. Ju, Z. Yi, H. Yang, C. Xiao and G. Cheng, ACS Cent. Sci., 2023, 9, 742 CrossRef CAS;
(d) V. P. Krivopalov, S. G. Baram, A. Yu. Denisov and V. I. Mamatyuk, Izv. Akad. Nauk SSSR, Ser. Khim., 1989, 38, 1839 Search PubMed.
- I. L. Dalinger, T. I. Cherkasova, G. P. Popova, T. K. Shkineva, I. A. Vatsadze, S. A. Shevelev and M. I. Kanishchev, Russ. Chem. Bull., 2009, 58, 410 CrossRef CAS.
- G. Hervé, C. Roussel and H. Graindorge, Angew. Chem., Int. Ed., 2010, 49, 3177 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
-
M. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer 17.5, 2012 Search PubMed.
-
(a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. ContrerasGarcía, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed;
(b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS;
(c) W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
-
M. Suceska, EXPLO5 7.01.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jatinder
Singh
a,
Jatinder
Singh