
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of diverse aluminium concentration gradient profiles in Ni-rich layered cathodes for enhanced electrochemical and thermal performances†
Xinwei
Jiao
a,
Junwei
Yap
a,
Junbin
Choi
a,
Mengyuan
Chen
b,
Devendrasinh
Darbar
c,
Gongshin
Qi
c,
Xiaosong
Huang
*b and
Jung-Hyun
Kim
 *a
*a
aDepartment of Mechanical and Aerospace Engineering, The Ohio State University, Columbus, OH 43210, USA. E-mail: kim.6776@osu.edu
bMaterials & Manufacturing Systems Research, General Motors Global Research and Development, Warren, MI 48092, USA. E-mail: xiaosong.huang@gm.com
cBattery Materials and Systems Research, General Motors Global Research and Development, Warren, MI 48092, USA
First published on 15th April 2024
Abstract
Nickel (Ni)-rich cathodes with elemental concentration gradients within particles have attracted great interest due to their considerably enhanced interfacial stability and electrochemical performance for advanced Li-ion batteries. In this study, we shift our focus from traditional research centered on the optimal chemical compositions in Ni-rich cathode powders with concentration gradients; instead, we decipher the interrelated effects of aluminium (Al) concentration gradient profiles on electrochemical performance and thermal stability. We successfully obtained LiNi0.95Al0.05O2 powders with different Al-gradient profiles by controlling the synthesis parameters, including the co-precipitation schedules of Al/Ni sources and sintering time. Using a combination of focused ion beam (FIB) and energy dispersive spectroscopy (EDS), we analyzed Al dispersion across the particle cross-sections. Remarkably, a greater Al concentration gradient across particles enhanced cathode–electrolyte interphase (CEI) stability during extended cycles, as evidenced by X-ray photoelectron spectroscopy (XPS) and electrochemical impedance spectroscopy (EIS). Consequently, the LiNi0.95Al0.05O2 cathode with greater Al concentration gradients exhibited enhanced capacity retention, C-rate performance, and thermal stability compared to LiNi0.95Al0.05O2 with lower Al gradients or Al-free LiNiO2 cathodes. This result underscores the crucial role of the elemental gradient profile, highlighting it as an equally important aspect in the optimization of cathode chemical compositions.
1. Introduction
The increased environmental awareness has led to the development of hybrid electric vehicles and pure electric vehicles powered by lithium-ion batteries (LIBs). Tremendous advances have been made in developing LIBs, particularly on the cathode side, over the past few decades. The first commercialized cathode material, LiCoO2, has a good energy density and relatively high operating voltage (∼3.9 V vs. Li).1 However, in recent decades, the continued rise of the cobalt price has led to the development of low cobalt content or cobalt-free cathode materials. Spinel LiNi0.5Mn1.5O4 (LNMO), olivine LiFePO4, and Ni-rich layered oxides, including Li[NixCoyMn1−x−y]O2 (x > 0.8) (NCM) and Li[NixCoyAl1−x−y]O2 (x > 0.8) (NCA), have been identified as promising cathode material candidates for next-generation LIBs.Recently, Ni-rich layered oxide cathode materials have gained significant interest due to their higher energy density and reduced raw materials cost. The ultimate goal of this strategy is to develop a pure Ni-layered oxide cathode material, LiNiO2 (LNO), in which all the Co is replaced by Ni, yielding a high specific capacity of 250 mA h g−1.2 However, practical application of the LNO has been hindered by numerous challenges including structural, mechanical, thermal, and electrochemical instability. During charging, LNO experiences three different phase transition stages; one of the most significant phase transitions is from the hexagonal 2 (H2) phase to the hexagonal 3 (H3) phase (4.15 V vs. Li). This phase transition induces a volume change that leads to particle cracking, resulting in a loss of electrical contact between primary particles and reduced electrochemical performance.2–4 In addition, poor thermal stability of LNO negatively impacts the abuse tolerance of LIBs, which is a significant barrier to its commercialization.2 For instance, LNO generates heat at 200 °C, accompanied by O2 generation, when being operated above 4.0 V vs. Li.5 Furthermore, reactive Ni4+ tends to form a rock salt-type phase on the cathode surface upon its contact with the electrolyte, causing low-capacity retention of LNO during long-term cycling.2
Of the many strategies aimed at tackling these difficulties, two particularly promising methods involve the substitution of foreign elements and the application of surface coatings. Substitution with other metal dopants such Mn, Al3+, Ti4+, or Mg2+in lieu of Ni3+. This substitution works to enhance the material's structural and thermal stability by effectively preventing the unwanted mixing of cations, specifically between Ni2+ and Li+ ions.6,7 Meanwhile, these substitution elements can act as “pillars”, enhancing structural stability by suppressing multiple phase transitions and sudden volume changes that occur during charge and discharge processes.2,8,9 In addition, an excessive amount of elemental substitution frequently leads to a compromise between capacity and stability. Hence, the pursuit of an optimal Ni content aims to strike a harmonious balance between capacity and stability in Ni-rich layered cathodes.
While the partial substitution of metal cations can offer an improved performance, it cannot fully protect the cathode particle surface from direct erosion by any harmful species (e.g., HF) in the electrolyte. Therefore, the surface coating of cathode particles has been widely adopted for the Ni-rich cathodes. Coating materials, such as V2O5,10 Al2O3,11 ZnO,12 and ZrP13 can protect the cathode active materials from attack by HF and improve the structural stability and electrochemical performance during repeated cycles. However, the surface coating materials and processes face some challenges that can impair LNO performance. For instance, electrochemical inactivity of a coating material requires its minimal use, making it difficult to control the homogeneity of coated layers.14 Moreover, the Ni-rich NMC's susceptibility to moisture limits the selection of solvents and wet coating processes to avoid harming the particle surface.14 Finally, the coating process requires extra materials and processing costs of cathode active materials.
To overcome the limitations of substitution and surface coating methods for improving the stability of LNO, a concentration-gradient structure within a particle of Ni-rich cathode has been proposed. In this structure the cation content of substituted elements gradually increases from the interior of the cathode particle to the exterior surface of the coating layer, reinforcing resistance to corrosion and suppressing negative impacts from phase transitions and volume changes. Al3+ substitution has been widely used such as in NCA due to its low cost and ability to improve structural stability, making it a viable option for LNO cathodes.15 Recently, Ni-rich cathodes with Al concentration-gradient have been extensively investigated to further improve the stability of LiNiO2 materials, which significantly reduces side reactions between the active Ni4+ and the electrolyte. For instance, Zhang et al.16 reported the performance of a concentration gradient LiNi0.85Co0.12Al0.03O2 cathode by adjusting the calcination temperature to 750 °C, which exhibited good capacity retention during cycling at 1C-rate.
Although many prior reports have highlighted the benefits of concentration gradient cathodes, challenges still exist for the LiNiAlO2 particle with a concentration gradient structure, such as the inclusion of Al complicating the synthesis of hydroxide precursors by disrupting particle growth.15,17 More importantly, there is still a lack of fundamental understanding about how concentration gradient profiles relate to the electrochemical and thermal properties of Ni-rich cathodes. In this study, we synthesized LiNi0.95Al0.05O2 (LNAO) with various Al-concentration profiles from the core to the surface by controlling the synthesis parameters including the co-precipitation schedule and sintering conditions, and the impact of Al-gradient profiles is discussed in conjunction with the electrochemical and thermal behaviors of the LNAO.
2. Experimental section
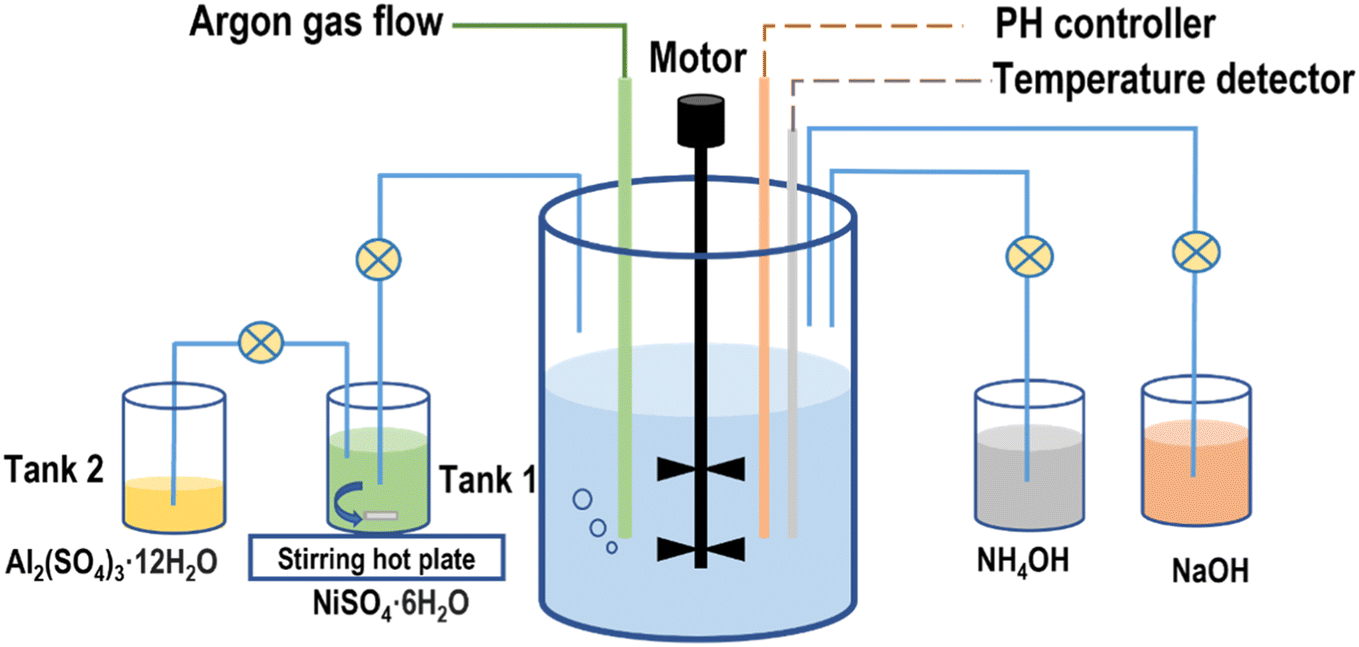
2.1 Material synthesis
The secondary particle precursors, Ni(OH)2 and Ni0.95(OH)1.9–Al0.05(OH)0.15 for LiNiO2 and LiNi0.95Al0.05O2 were synthesized using a co-precipitation reactor (Minifors 2). For the baseline Ni(OH)2 precursors 0.6 M NiSO4·6H2O (Sigma-Aldrich) and 1.21 M NaOH (Sigma-Aldrich) were utilized. An appropriate amount of deionized water and 0.3 M NH4OH (Sigma-Aldrich) were added to the reactor prior to the reaction to maintain the pH value above 11.5. The NiSO4·6H2O solution was slowly pumped into the reactor until the pH value inside the reactor dropped to 11.1. The flow rate of the NaOH aqueous solution was controlled by a PID controller to adjust the pH value, while the appropriate amount of NH4OH solution (0.9 M) were separately fed into the reactor as a chelating agent. The synthesis process for LiNi0.95Al0.05O2 precursor was similar to the Ni(OH)2 reaction. To obtain a concentration gradient structure, a 0.57 M NiSO4·6H2O solution was gradually pumped into the reactor to react with the NaOH solution to form a certain amount of Ni(OH)2 precursors at first. The 0.03 M Al2(SO4)3·12H2O solution was then slowly added into the NiSO4·6H2O solution at 3 h, 5 h, and 6 h since the beginning of the reactions respectively. The two solutions were thoroughly mixed and pumped into the reactor. The injection time of the Al2(SO4)3·12H2O solution added into the NiSO4·6H2O solution is 320 min for the 3 h sample, 200 min for the 5 h sample, and 140 min for the 6 h sample, respectively. Their detailed injection schedules were illustrated in Fig. S1.† The total time required to inject all solution into the reaction vessel was around 10 h. All the co-precipitation reactions were conducted under an argon atmosphere, at 50 °C, with stirring speed maintained at 1000 rpm, and at pH 11.1–11.2 for a total reaction time of 25 h.The precursor powder was obtained, washed with deionized water, and dried in a vacuum oven at 80 °C for 24 h. To synthesize the final products of LiNiO2 and LiNi0.95Al0.05O2, the precursors were thoroughly mixed with a stoichiometric equivalent of LiOH·H2O (Sigma-Aldrich) by hand milling using a mortar and pestle. Samples with pure Ni(OH)2 precursors had a Li/TM molar ratio of 1.05. The mixtures were heated in a tube furnace under an oxygen flow at 710 °C for 15 h. The Ni0.95(OH)1.9Al0.05(OH)0.15 samples with Li/TM ratio of 1.05 were heated in the tube furnace under an oxygen flow at 710 °C for 4, 6, 8 and 15 h, respectively.
2.2 Material and microstructure characterization
The crystal structures of the samples were characterized using X-ray diffractometer (XRD, Bruker D8) with a Cu Kα X-ray source and a diffracted beam monochromator. A scattering angle range (2θ) of 10–80° was used with a step size of 0.02° and a dwell time of 3 s. The resulting XRD patterns were refined by the Rietveld refinement program GSAS-II. The samples for inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer Sciex ELAN 6000) analysis was prepared by dissolving them in 25 mL aqua regia. Scanning electron microscopy (SEM, Thermo Scientific Apreo) was used to characterize the particle morphology and size. The cross-sectional elemental distribution profiles across LiNi0.95Al0.05O2 and precursor particles were analyzed by using focused ion beam (FIB, Helios NanoLab 600) and energy dispersive spectroscopy (EDS). Basic mapping was conducted using low voltage (5 keV) to obtain surface-level information with minimal interaction volume. X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra XPS) analysis was performed for the cycled cathodes. To minimize surface charging, charge neutralization was enabled, and the spectra were calibrated based on C–C peaks at 284.7 eV. The resulting spectra were fitted using CasaXPS software.2.3 Electrochemical and physical characterization
The LNO or LNAO cathode active materials, conductive agent (Super-P), and PVdF binder were mixed in a weight ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7.5:7.5. The electrode materials were mixed with N-methyl-2-pyrrolidone (NMP) solvent and zirconia beads using a planetary mixer (Thinky) to form a slurry at 2000 rpm for 10 minutes. The slurry was then coated onto an Al current collector using the doctor blade technique and vacuum-dried at 80 °C overnight. The final loading mass was 10 mg cm−2 with 35 μm thickness of cathode (excluding an Al-foil). Coin-type half-cells were assembled with a Li metal anode, 1 M LiPF6 in EC:EMC solvents (1:1 weight ratio) as the electrolyte, and a Celgard 2500 polypropylene separator. These cells were tested in a controlled environment at 25 °C, cycling between 3.0 and 4.3 V vs. Li using a battery cycling station (Arbin LBT System). Electrochemical impedance spectrometry (EIS, Gamry E1010E) analyzed cell impedances across frequencies ranging from 106 Hz to 1 mHz at 50% state of charge (SOC). Furthermore, resulting EIS data were characterized using the distribution of relaxation times (DRT) technique to elucidate the physical origin of impedance sources.
:7.5:7.5. The electrode materials were mixed with N-methyl-2-pyrrolidone (NMP) solvent and zirconia beads using a planetary mixer (Thinky) to form a slurry at 2000 rpm for 10 minutes. The slurry was then coated onto an Al current collector using the doctor blade technique and vacuum-dried at 80 °C overnight. The final loading mass was 10 mg cm−2 with 35 μm thickness of cathode (excluding an Al-foil). Coin-type half-cells were assembled with a Li metal anode, 1 M LiPF6 in EC:EMC solvents (1:1 weight ratio) as the electrolyte, and a Celgard 2500 polypropylene separator. These cells were tested in a controlled environment at 25 °C, cycling between 3.0 and 4.3 V vs. Li using a battery cycling station (Arbin LBT System). Electrochemical impedance spectrometry (EIS, Gamry E1010E) analyzed cell impedances across frequencies ranging from 106 Hz to 1 mHz at 50% state of charge (SOC). Furthermore, resulting EIS data were characterized using the distribution of relaxation times (DRT) technique to elucidate the physical origin of impedance sources.
2.4 Thermal stability evaluation
The cycled cathodes were prepared for analysis using differential scanning calorimetry (DSC, TA Instrument Q-2000). The coin cells underwent two initial formation cycles at a rate of C/20, followed by charging to 4.3 V vs. Li at a rate of C/10. Subsequently, the cells were opened in an argon-filled dry box to extract the charged cathode samples. Around 3–5 mg of cathode material containing 25–35 wt% electrolyte was hermetically sealed in a high-pressure-resistant crucible with a gold-plated Cu seal. DSC scanning was performed at a ramping rate of 5 °C min−1 from room temperature to 325 °C.3. Results and discussion
3.1 Effect of synthesis conditions on Al gradient in (Ni, Al)(OH)x precursor
The precursor for the concentration gradient LiNi0.95Al0.05O2 (CG-LNAO) was synthesized through a co-precipitation reaction. The synthesis process of concentration gradient precursors is illustrated in Fig. 1. At the beginning of the reaction, a certain amount of Ni(OH)2 secondary particles was formed by pumping NiSO4·6H2O solution into the reactor. The reaction mechanisms for the production of Ni(OH)2 precursors have been explained as follows:18| Ni2+(aq) + xNH4OH(aq) → [Ni(NH3)n2+](aq) + nH2O + (x − n)NH4OH(aq) | (1) |
| [Ni(NH3)n2+](aq) + yOH− + zH2O → Ni(OH)2(s) + zNH4OH(aq) + (n − z)NH4 | (2) |
 | ||
| Fig. 1 Schematics of co-precipitation synthesis route of concentration-gradient Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors. | ||
In eqn (1) above, ammonia acts as a chelating agent by reacting with Ni2+ to suppress the rapid hydrolysis of Ni2+, preventing the formation of an amorphous and porous structure.19Eqn (2) shows that the Ni(NH3)n2+ complexes react gradually with hydroxide ions to produce dense and spherical precipitates of Ni(OH)2. After a few hours of Ni(OH)2 production, the Al2(SO4)3·12H2O solution in a separated tank (denoted to tank 2) was slowly added to tank 1. The detailed injection schedule is illustrated in Fig. S1.† The well-mixed Ni and Al sulfate solution was then pumped into the reactor to produce the concentration gradient (CG) Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors with a secondary particle morphology. However, at a high pH range, Al does not tend to form a complex with ammonia, leading to the continuous nucleation of Al(OH)3. The numerous Al nucleation at the surface of the existing Ni(OH)2 particles undermined the Ostwald ripening process of the Ni(OH)2 particles, intervening the further growth of CG Ni0.95(OH)1.9–Al0.05(OH)0.15 secondary particle.15 This observation implies that the particle sizes of CG Ni0.95(OH)1.9–Al0.05(OH)0.15 can be controlled by an injection schedule of Al sources during the co-precipitation process. For instance, a longer delay in the Al2(SO4)3·12H2O injection will result in larger secondary particles, and vice versa.
This hypothesis was examined by injecting Al2(SO4)3·12H2O solution into the tank 1 at three different delaying time points: 3 h, 5 h, and 6 h after the initial Ni(OH)2 precipitation. Then, SEM-EDS images obtained from each CG Ni0.95(OH)1.9 Al0.05(OH)0.15 powders were compared in Fig. 2. The results demonstrate that delaying the Al2(SO4)3·12H2O injection from 3 h to 5 h led to an increase in particle sizes from ∼3 μm to ∼10 μm. Both the 3 h and 5 h delayed samples showed even distribution of Ni and Al across various particles, indicating their homogeneous and well-controlled precipitation reaction. For example, the 5 h sample showed a Ni/Al atom ratio of 18.9, which is close to the target ratio of 19.
 | ||
| Fig. 2 SEM-EDS images and cross-sectional EDS line scan analyses of CG Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors made with different Al2(SO4)3·12H2O injection schedules: (a) 3 h, (b) 5 h (c) 6 h after the beginning of simple Ni(OH)2 precipitation. | ||
However, further delay of Al injection to 6 h resulted in the formation of small and non-uniform particles as shown in Fig. 2c. SEM/EDS analysis of multiple particles indicated relatively large variation of Al-concentrations between particles. For example, certain large particles lack Al content as highlighted in Fig. 2c(iii). On the other hand, some small particles contain relatively large amounts of Al (16.56 at%) compared to large particles (9.26 at%) in Fig. S2.† Unlike the samples delayed for 3 or 5 h, the Al(OH)3 nuclei in the 6 h delayed sample were not able to attract enough primary Ni(OH)2 to form on the surface of existing CG Ni0.95(OH)1.9–Al0.05(OH)0.15 particles. This is attributed to the decreasing concentration of NiSO4·6H2O in tank 1 as the delay time increases. As a result, part of Al(OH)3 could rapidly nucleate into Al-enriched secondary particles with lack of morphology control and smaller particle sizes during the later stage of the co-precipitation process, as shown in Fig. 2c(i). The non-uniform distribution of Al compositions and significant particle-size variations in the 6 h delayed sample can negatively impact the performance of the final form of cathode products.
The impact of Al2(SO4)3·12H2O injection schedule on the Al gradient structure in the CG Ni0.95(OH)1.9–Al0.05(OH)0.15 particles were investigated. Fig. 2 also compares Al-concentration gradients from cross-sections of representative particles, which were prepared using FIB and measured using EDS line scans. All the three samples showed a clear Al-concentration gradient across the particle: increasing Al content from particle core to surface. The 3 h sample still has an Al concentration of ∼2.04% at the particle core, suggesting a premature involvement of Al in the precipitation reaction before a sufficient growth of the Ni(OH)2 core particle. By delaying the injection time of Al2(SO4)3·12H2O, the Al-concentration at the particle cores decreased from ∼2.04% (for 3 h) to ∼1.08% (for 5 h) to ∼0.54% (for 6 h). The 6 h sample revealed a gradual increase in Al-concentration from approximately 1.5 μm below the surface, while Al concentration was negligible in most of the particle centers. However, the selected 6 h particle had a Ni/Al atomic ratio of ∼68.93, which is significantly higher than the target ratio of 19. This is because of the Al-deficiency of the large particles as discussed earlier.
The results demonstrate that the 5 h delayed injection schedule of Al2(SO4)3·12H2O offers good Al-gradient profiles and well-developed morphology of secondary particles for the CG Ni0.95(OH)1.9–Al0.05(OH)0.15 precursor. The scheme in Fig. 3 illustrates that an increase in delayed injection time results in a larger particle size but a weakened concentration gradient of Al.
 | ||
| Fig. 3 Scheme of illustration for relationship between particle size and concentration-gradient of Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors with different injection time. | ||
3.2 Effect of sintering time on Al-gradient LiNi0.95Al0.05O2
The CG Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors initially displayed distinct Al concentration gradients, with enrichment at the surface of secondary particles. However, during the sintering process, elemental inter-diffusion between Ni and Al in CG-LNAO can cause degradation of these original profiles. We hypothesized that controlling the sintering time could effectively regulate the concentration gradient behaviors of Al and Ni in the CG-LNAO. To test this hypothesis, CG-LNAO powders were sintered for different lengths of time, ranging from 4 h to 15 h at 710 °C in O2.The dependence of Al-gradient behavior in the CG-LNAO particles on the sintering time could be examined by using cross-sectional FIB/EDS analysis. Fig. S3† shows the SEM images of representative particle cross-sections, which were prepared using FIB, along with the corresponding Al element profiles measured by EDS line scans. At 4 h or 6 h sintering time, relatively high Al concentrations were observed at the surface regions of secondary particles. At 8 h sintering time, Al concentration disparity diminishes between the surface and the bulk, which finally becomes negligible at 15 h. The observed trend can be attributed to the diffusion of Al from the outer layer to the particle core during sintering at 710 °C, which is driven by the concentration gradient of Al across the CG Ni0.95(OH)1.9–Al0.05(OH)0.15 precursor particles (see, Fig. 2).
Further investigation was conducted based on the EDS data to explore the intricate relationship between Al-concentration profiles and sintering time. We normalized the particle radius dimension for a more accurate comparison between the samples and plotted the results in Fig. 4a. The CG Ni0.95(OH)1.9–Al0.05(OH)0.15 precursor was labelled as t = 0 h and plotted together for comparative analysis. As the precursor was sintered with the Li-source, the Al-content in the bulk exhibited a monotonic increase with sintering time (t), ranging from t = 4 h to t = 15 h, as demonstrated in Fig. 4b. The variation in Al-content across the particles was attributed to the diffusion of Al3+ ions from the particle surface towards the bulk, driven by Al-concentration gradient. As a result, the depth of Al-concentration gradient at the surface decreases (see, Fig. 4c) and the difference of Al-gradient (i.e., ΔAl-gradient) between the surface and bulk is being reduced gradually (see, Fig. 4d) with sintering time. Our result unambiguously proves the hypothesis that the dynamics of elemental interdiffusion and concentration gradient structure can be controlled by sintering time. The Al/Ni interdiffusion can also be controlled by sintering temperatures, which is beyond the current scope of this work.
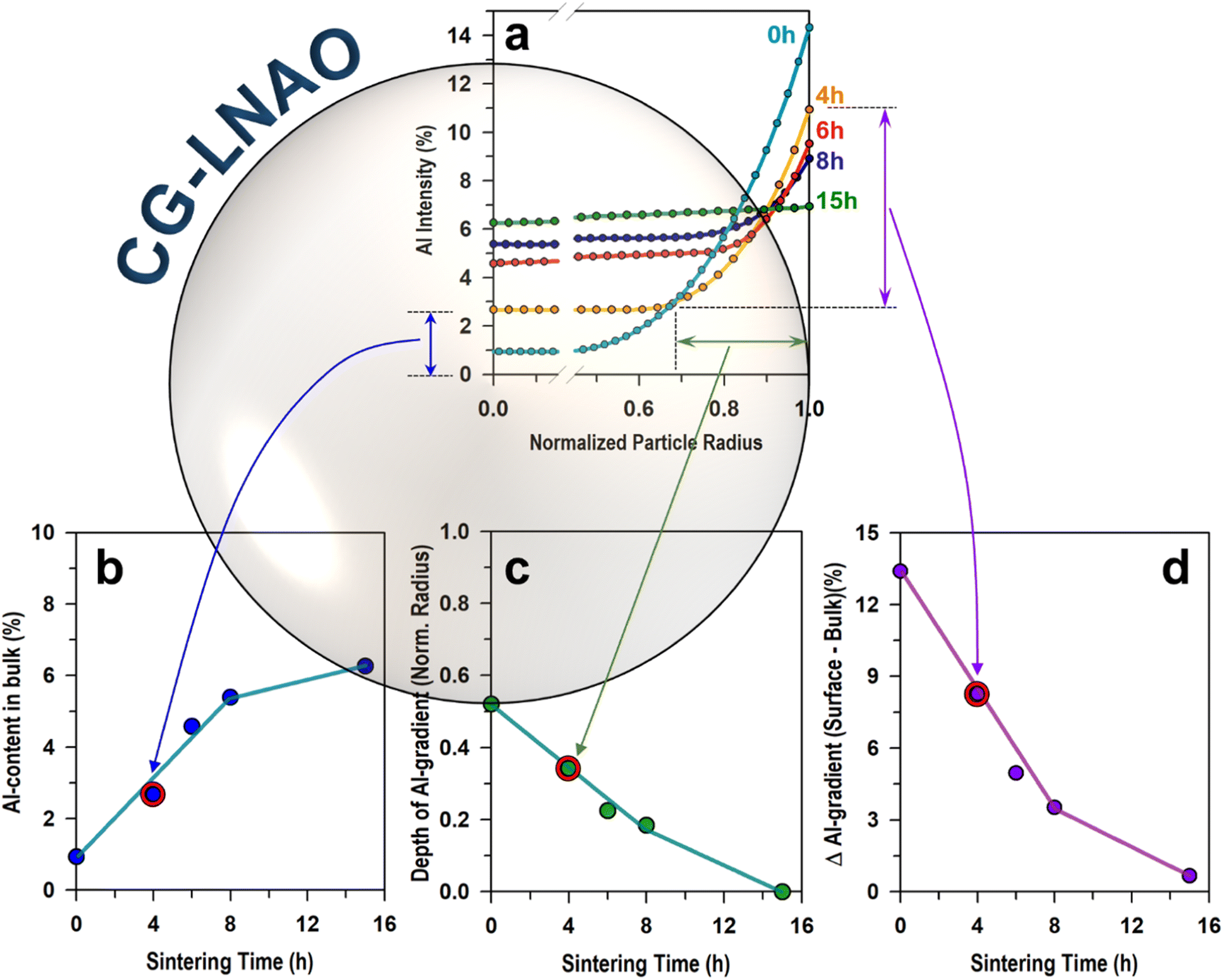 | ||
| Fig. 4 (a) Al-concentration profiles of CG-LNAO particles after sintering at 710 °C for 4 h, 6 h, 8 h, and 15 h in O2. The partial radius was normalized for a comparison. The Al-concentration was obtained from the cross-sectional EDS line scanning. The variation of (b) Al-content in bulk, (c) depth of Al-gradient, (d) ΔAl-gradient (surface vs. bulk) depending on sintering times. The ways to determine each parameter were illustrated for t = 4 h sample. | ||
3.3 Effect of Al-gradient in CG-LNAO on battery performances
Another important prospect of this work is to find the relation between the profiles of Al-concentration gradient and electrochemical properties of the CG-LNAO cathode. Earlier works mainly focused on revealing the benefits of the concentration gradient cathodes over the simply substituted cathodes.20–22 The remaining research question is how the degree of concentration gradient will affect the electrochemical performances. To address this, concentration gradient samples were prepared using different sintering times at 710 °C.Structural analysis of the resulting bare LNO and CG-LNAO powders is shown in Fig. 5 and S4.† All the XRD data were refined based on a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, which has a hexagonal α-NaFeO2 structure. The distinct splits between the (006) and (102) peaks, as well as (108) and (110) peaks, suggest a well-ordered layered structure. The Rietveld refinement analysis revealed the amount of Ni2+ cation mixing in the Li-site, as presented in Table. S1.† The bare LNO sample exhibited 4.95% cation mixing. The CG-LNAO sample showed a varied cation mixing amount depending on sintering time: 2.77% for 4 h (CG-LNAO-4), 2.19% for 6 h (CG-LNAO-6), 2.55% for 8 h (CG-LNAO-8), and 3.12% for 15 h (CG-LNAO-15), respectively. The I(003)/I(104) intensity ratio is also considered as an indicator of the degree of Ni2+ cation mixing in the Li layer.23 The lowest intensity ratio in the bare LNO confirmed more severe cation mixing compared to some CG-LNAO samples, as shown in Table S1.† It is notable that CG-LNAO-4 maintains the highest c values but exhibits more serious cation mixing than CG-LNAO-6. This phenomenon can be attributed to shorter calcination times limiting the diffusion of Li into the material bulk, consequently reducing the Li content in LNAO and leading to Ni2+/Li+ cation mixing at the lithium plane. Besides, the Rietveld refinement of XRD data indicates an increase in cation mixing from 6 h to 15 h of sintering, which can be attributed to an excess amount of Li sublimation during the extended sintering period.
m, which has a hexagonal α-NaFeO2 structure. The distinct splits between the (006) and (102) peaks, as well as (108) and (110) peaks, suggest a well-ordered layered structure. The Rietveld refinement analysis revealed the amount of Ni2+ cation mixing in the Li-site, as presented in Table. S1.† The bare LNO sample exhibited 4.95% cation mixing. The CG-LNAO sample showed a varied cation mixing amount depending on sintering time: 2.77% for 4 h (CG-LNAO-4), 2.19% for 6 h (CG-LNAO-6), 2.55% for 8 h (CG-LNAO-8), and 3.12% for 15 h (CG-LNAO-15), respectively. The I(003)/I(104) intensity ratio is also considered as an indicator of the degree of Ni2+ cation mixing in the Li layer.23 The lowest intensity ratio in the bare LNO confirmed more severe cation mixing compared to some CG-LNAO samples, as shown in Table S1.† It is notable that CG-LNAO-4 maintains the highest c values but exhibits more serious cation mixing than CG-LNAO-6. This phenomenon can be attributed to shorter calcination times limiting the diffusion of Li into the material bulk, consequently reducing the Li content in LNAO and leading to Ni2+/Li+ cation mixing at the lithium plane. Besides, the Rietveld refinement of XRD data indicates an increase in cation mixing from 6 h to 15 h of sintering, which can be attributed to an excess amount of Li sublimation during the extended sintering period.
 | ||
| Fig. 5 XRD data and Rietveld refinement profiles of (a) bare LiNiO2 and (b) CG-LNAO-6. The data for the remaining samples can be found in Fig. S4.† (c) Effect of sintering time on capacity retention of cathodes in half-cells. The CG-LNAO-15 (Li excess) sample was sintered with 7.5% Li excess to compensate the Li sublimation. The first two cycles were conducted at 0.1C, followed by 100 cycles at C/3 rate, in a voltage range of 3.0–4.3 V vs. Li at 25 °C. | ||
The actual chemical compositions of the samples, analyzed via ICP-MS, were summarized in Table S2.† The CG-LNAO-4 has a slight excess Li content of 1.02. It has been reported that such excess Li could be from residue of Li-salts at surface of particles, mainly comprising Li2O, LiOH, and/or Li2CO3.23–25 These residual lithium compounds impede Li+ diffusion and also trigger gelation of the cathode slurry through the cross-linking of PVdF binders.26 With increasing calcination time, a fraction of Li is inevitably lost via its sublimation. After 6 h sintering, the chemical composition of CG-LNAO approaches its theoretical value. However, prolonged sintering (e.g., 15 h) led to unwanted Li loss, resulting in irreversible cation mixing, as shown in Table S2.†
Fig. 5c compares the capacity retentions of CG-LNAO cathodes with different sintering times ranging from 4 h to 15 h. The initial capacities for the 1st and 2nd cycles exhibited similar discharge capacities of 205–215 mA h g−1 at C/10-rate and 25 °C. However, at the 3rd cycle, where C/3-rate cycling began, the discharge capacities of the cathodes dropped rapidly; for example, the bare LiNiO2 (LNO) cathode experienced a capacity drop of ΔQ1–3 = 26 mA h g−1 from the 1st cycle (at C/10-rate) to the 3rd cycle (at C/3-rate). In contrast, the CG-LNAO cathodes sintered for 4–15 h showed fewer capacity drops: ΔQ1–3 = ∼10 mA h g−1 for 4 h and 6 h samples and ∼13 mA h g−1 for 8 h and 15 h samples, respectively.
More importantly, the behavior of the Al concentration gradient significantly impacted the reversibility of CG-LNAO cathodes during extended cycles. Without the Al-concentration gradient, as shown in Fig. 5c, the bare LNO cathode exhibited poor capacity retention in half-cell, delivering only 51.6% of its initial discharge capacity after 100 cycles. In contrast, CG-LNAO cathodes delivered significantly improved capacity retention values of 71.2% for 4 h, 75.9% for 6 h, and 78.4% for 8 h sintering time, respectively, after 100 cycles. The CG-LNAO-15 cathode, however, showed relatively poor capacity retention of 55.5%. To eliminate the possibility of capacity fading caused by the Li deficiency, we increased Li excess amount from 5% to 7.5% during the synthesis of CG-LNAO-15, which is labelled as CG-LNAO-15 (Li excess) in Fig. 5c. However, optimizing the Li content in CG-LNAO-15 failed to improve its capacity retention, confirming that its poor performance is primarily due to the loss of Al concentration gradient across its particles.
The poor capacity retention behaviour of the LNO cathode have been attributed to its undesirable parasitic reactions with electrolyte and significant lattice volume contraction, particularly during the H2 to H3 phase transition.2 These factors could cause active Li+ consumption and accelerate surface structure degradation, leading to the formation of a rock-salt phase at the surface.2,24,27 In contrast, the CG-LNAO with relatively short sintering time (4–8 h) showed a good Al-concentration gradient at particle surfaces, which can passivate the cathode–electrolyte interphase (CEI) and suppress the parasitic reactions in contact with electrolytes. Additionally, the Al content at the surface can inhibit side reactions in contact with moisture in air during storage or electrode/cell fabrication processes.28
Fig. 6a and b provide a comparison of the half-cell voltage profiles and corresponding dQ/dV profiles of bare LNO, CG-LNAO-6, and CG-LNAO-15. The dQ/dV profiles of all cathodes show multiple phase transitions during charging, including hexagonal 1 to monoclinic (H1–M), monoclinic to hexagonal 2 (M–H2), and hexagonal 2 to hexagonal 3 (H2–H3). The CG-LNAO-6 and CG-LNAO-15 cathodes have significantly reduced redox peak intensities at >4.2 V vs. Li, indicating less abrupt H2–H3 phase transition. This result aligns well with an earlier report, which presented that bulk Al-substitution in LNO could suppress the H2–H3 phase transition and reduce volume changes at high voltages.28
 | ||
| Fig. 6 Comparison of (a) voltage profiles (at 2nd cycle), (b) dQ/dV profiles (at 2nd cycle), 2nd cycle Li-ions diffusion coefficient of (c) charge, and (d) discharge processes, (e) fast-charging, and (f) fast-discharging performances of bare LNO, CG-LNAO-6, and CG-LNAO-15 cathodes in half-cells. All the cells were cycled in a voltage range of 3.0–4.3 V vs. Li at 25 °C. | ||
The reduced Ni2+/Li+ cation mixing at Li layer can lower the energy barrier and facilitate Li migration.29 The CG-LNAO-6 exhibits the lowest cation mixing, suggesting its faster Li diffusion compared to bare LNO and CG-LNAO-15. Therefore, to confirm this assumption, the Li-ions diffusion coefficients (DLi+) of the bare LNO, CG-LNAO-6 and CG-LNAO-15 cathodes were assessed using the galvanostatic intermittent titration technique (GITT) after the initial cycle with Li anodes. As shown in Fig. 6c and d, the Li-ions diffusivity of bare LNO undergoes sharp decrease at >4.0 V vs. Li due to its significant volume and internal structure changed during the H2 to H3 phase transition.30 In contrast, both CG-LNAO-6 and CG-LNAO-15 exhibit smoother Li-ion diffusivity during whole charge and discharge processing, attributed to their suppressed phase transition and unit cell expansion along the c-direction. In particular, CG-LNAO-6 exhibits the highest Li diffusion rate among all samples due to its minimal cation mixing and suppressed phase transition at high voltages.
The impact of Al-concentration gradient in CG-LNAO cathodes on fast-charging and fast-discharging capabilities were investigated. Fig. 6e plots the fast-charging performances of half-cells made with various CG-LNAO cathodes. Here, all the cells underwent fast-charging in a range of C/10–5C, followed by discharging at a constant C/10-rate. While all cathodes demonstrated a similar discharge capacity of ∼210 mA h g−1 at C/10, the differences in discharge capacities becomes more pronounced when fast-charging at >1C. Across all C-rates, the CG-LNAO-6 cathode exhibited the highest discharge capacity, followed by the CG-LNAO-15 and the bare LNO. For example, at 5C, the CG-LNAO-6 cathode delivered a discharge capacity of 151.2 mA h g−1, surpassing that of the bare LNO cathode (113.8 mA h g−1). Additionally, Fig. 6f illustrates the fast-discharging performance of all cathodes. It is noteworthy that the CG-LNAO-6 cathode delivered the best rate performance. For example, at 5C discharging, the CG-LNAO-6 delivers 160 mA h g−1, while the CG-LNAO-15 and the bare LNO cathodes, respectively, delivered 147 mA h g−1 and 144 mA h g−1. Despite the same overall amount of Al content in cathodes, the CG-LNAO-15 cathode has inferior fast-charging and fast-discharging capabilities compared to the CG-LNAO-6 cathode. The data from fast charging and discharging are consistent with the trends observed in Li diffusivity, unequivocally validating that the substantial Al concentration gradient in CG-LNAO-6 significantly enhances Li-ion transport across the cathode–electrolyte interface (CEI).
3.4 Effect of Al-gradient in CG-LNAO on CEI layer
Fig. 7 compares the Nyquist plots of bare LNO, CG-LNAO-6 and CG-LNAO-15 cathodes at 50% SOC after 1st, 50th, and 100th cycles. To simplify the analysis, we assume that the contribution of Li SEI on half-cell impedance is identical between cells that underwent the same electrochemical testing history.31 This assumption enables a direct comparison of cathode impedance behaviours. The intercept at the high-frequency region of the Nyquist plot represents the ohmic resistance (Rohmic), which mainly corresponds to the electrolyte resistance. The two semicircles, one in the high-frequency range and one in the low-frequency range, correspond to interface resistances (RInt) from both CEI/SEI and charge transfer resistance (RCT), respectively.32–35 While the Nyquist plots demonstrate the difference in impedance amplitude, they lack resolution in deconvoluting individual impedance sources. Therefore, we used the distribution of relaxation time (DRT) analysis to identify individual impedance sources, as shown in Fig. 7d–f. The DRT plots show four significant sources of electrochemical impedance, each represented by a single peak. The peak around 105 Hz is due to the high frequency noise.35 The P1 (at around 104 to 105 Hz) peak is attributed to the contact impedance such as the cells/Li foil and cathode/Al-foil.35 The P2 (at around 103 to 104 Hz) peak is mainly associated with the CEI layer impedances.36,37 The impedance of Li SEI is observed across wide frequency ranges, including both P2 and P3 (at around 101 to 102 Hz).35 The P4 at around 100 to 101 Hz is attributed to the charge transfer process of cathodes, which experiences a significant decrease upon the introduction of Al concentration gradient.35 | ||
| Fig. 7 Nyquist plots and corresponding DRT analysis data at 1st, 50th, and 100th cycles of half-cell with (a and b) bare LNO, (c and d) CG-LNAO-6, and (e and f) CG-LNAO-15 cathodes at 25 °C. | ||
As demonstrated in Fig. 7, the bare LNO sample has high P2 intensities, which suggests that the bare sample underwent severe parasitic reactions during cycling. In contrast, CG-LNAO-6 exhibits reduced P2 peak intensity, indicating that the optimized concentration gradient structure effectively suppresses the parasitic reactions at CEI. Furthermore, while all cells initially showed negligible RCT peaks after the first formation cycle, extended cycling caused a rapid RCT increase in the bare LNO. The bare LNO exhibits severe parasitic reactions, including the formation of the rock-salt phase and an increase in Ni2+/Li+ disorder,36,38 which can hinder the charge-transfer process. In contrast, the substitution of Al can result in the expansion of a unit cell along the c-direction, which is beneficial for the Li-ion diffusion in the crystal lattice of CG-LNAO.39 Furthermore, Fig. S5† demonstrates the XRD pattern of bare LNO, CG-LNAO-6, and CG-LNAO-15 before and after 100 cycles. For the bare LNO, the intensities of the (101) and (104) peaks were significantly decreased at fully discharged state (3.0 V vs. Li) after 100 cycles. A previous study has reported that the intensities of the (101) and (104) peaks decreased during charging, and the process was found to be reversible during discharging.40 The notable reduction in the peak values of I(101) and I(104) for the bare LNO suggests irreversible changes in the crystal lattice structure, attributed to both Li-loss and the formation of a cation mixing domain, which led to a formation of rock-salt phase.41 In contrast, CG-LNAO-6 and CG-LNAO-15 cathodes remain consistent XRD profiles, suggesting their stable crystalline structure after 100 cycles. This enhancement in crystal structure is closely related to the suppression of H2–H3 phase transformation and higher Li-ion diffusivity. As a result, CG-LNAO-6 were able to sustain low CEI impedance (P2) and charge transfer resistance (P4) values throughout extended battery cycling periods.
To further elucidate the CEI stabilization mechanism of Al-concentration gradient in CG-LNAO cathodes, the surface of cycled cathodes was analysed by using XPS. It has been reported that the surface passivation layer contains both inorganic components and organic species, which are formed due to the complex parasitic reactions between the electrolyte and electrode after extended cycles.42 Continuous parasitic reactions can damage the surface of LiNiO2, promote the regeneration of CEI, and lead to significant capacity fading. XPS analysis could provide useful information about CEI, such as the chemical components, their relative ratio, and the thickness of CEI layers on particles.43–45
Fig. 8 displays the F 1s, C 1s, and O 1s spectra of the LNO, CG-LNAO-6, and CG-LNAO-15 cathodes recovered after 100 cycles. Consistent with our previous report,37,43,44,46 the identified components include electrolyte decomposition products such as lithium alkoxides (ROLi), lithium carbonates (Li2CO3), alkyl carbonate (ROCOOLi), and certain inorganic, including lithium fluorophosphate (LixPOyFz), as well as LiF from salt decomposition. In the F 1s spectra, two distinct peaks at binding energies of ∼685 eV and ∼686.4 eV can be attributed to the M–F (LiF and NiF2) and LixPOyFz, respectively, which are decomposition byproducts of LiPF6.47 It is well-known that moisture in the electrolyte can react with LiPF6 salt, leading to the formation of HF, which can cause transition metal dissolution from an active material and the production of M–F.48,49 The detailed reaction mechanisms of LiPF6 at CEI are listed below.48
| LiPF6 ⇆ PF5 + LiF | (3) |
| PF5 + H2O → POF3 + 2HF | (4) |
| POF3 + Li+ → LixPFyOz + LiF | (5) |
 | ||
| Fig. 8 XPS spectra of surface elements obtained from cycle-aged electrodes recovered from half-cells after 100 cycles in the voltage range of 3.0–4.3 V at 25 °C. (Top row) bare LNO cathode, (2nd row) CG-LNAO-6 cathode, and (3rd row) CG-LNAO-15 cathode. | ||
Compared to the bare LNO and CG-LNAO-15 cathodes, the CG-LNAO-6 cathode shows a lower M–F and LixPFyOz intensity, indicating less severe parasitic reactions occurring at CEI. The F 1s spectrum also revealed the presence of LiPF6 peak from residual salt and the CF2 peak from the PVdF binder. For the C 1s spectra, the C–C (∼284.7 eV) and C–H (∼285.3 eV) peaks are from conductive carbon black and PVdF binder, respectively. In general, the relative intensity of the C–C peak to the C–O peaks reflects the thickness of the CEI layer.44,50,51 The quantitative analysis of the area ratio between C–C and C–O peaks (C–C/C–O) were, respectively, 0.915 for the LNO, 2.14 for the CG-LNAO-6, and 1.68 for the CG-LNAO-15. The relatively high C–C ratio confirms that the CG-LNAO-6 cathode has a thinner CEI layer than the LNO cathode, which explains the lower interfacial impedance of the CG-LNAO-6 cathode. In contrast, the CG-LNAO-15 has a marginal benefit due to its lack of Al-concentration gradient at particle surfaces, which limits its surface passivation function.
The O 1s spectra for the cycled cathodes reveal that the majority of the CEI comprises ROLi, carbonate, ROCOOLi, and LixPOyFz species resulting from electrolyte decomposition or degradation of LiPF6. Compared to the bare LNO sample, CG-LNAO-6 cathode exhibits lower overall peak intensities, particularly reduced coverage of LixPOyFz species, which is consistent with the findings in the F 1s spectra. The M–O peaks were still observable in the CG-LNAO-6 and CG-LNAO-15 cathodes after cycling 100 times. The detection of more bulk O in the cathode indicates a thinner SEI layer on the electrode surface,52 implying fewer side reactions between the electrolyte and the CG-LNAO during cycling.
3.5 Effect of Al-gradient in CG-LNAO on thermal stability
The effect of Al-concentration gradient on thermal stability of CG-LNAO cathode was examined using DSC. For this study, the bare LNO, CG-LNAO-6 and CG-LNAO-15 cathodes were charged to 4.3 V vs. Li after two formation cycles. Fig. 9 shows that the CG-LNAO-6 cathode has a total heat flow of 1.43 W g−1, representing only 25% of the bare LNO (5.72 W g−1). The CG-LNAO-15 cathode has 2.96 W g−1 heat flow, representing 52% of the bare LNO. In addition to the reduced total heat flow, the CG-LNAO-6 cathode demonstrates a broader distribution of heat flow across the temperature range. This result suggests that the CG-LNAO-6 particle can effectively mitigate the thermal runaway phenomenon with its relatively large Al-concentration gradient at the surface. On the other hand, the CG-LNAO-15 particle, with a less significant Al-gradient at the surface, exhibits a less pronounced reduction in thermal runaway. However, its Al-concentration within the bulk particle still enhances its thermal stability compared to the bare LNO. Our results unambiguously prove the benefit of the Al-concentration gradient in terms of electrochemical performances and thermal stability. | ||
| Fig. 9 (a) DSC profiles and (b) amount of heat release from bare LNO, CG-LNAO-6 and CG-LNAO-15 cathodes wetted with liquid electrolyte. All cathodes were retrieved from fully-charged half-cells at 4.3 V vs. Li. | ||
4. Conclusion
We have successfully synthesized CG-LNAO cathode active materials with varying elemental gradients of Al across particles, demonstrating the impact of these gradient profiles on electrochemical performances and thermal stability. Modifying the co-precipitation process by altering the injection schedules of the Al-source allows for control over the particle sizes and Al-gradient profiles of Ni0.95(OH)1.9–Al0.05(OH)0.15 precursors. The subsequent sintering process at 710 °C facilitated elemental inter-diffusion between Ni and Al in CG-LNAO. As a result, the Al content in the bulk, the depth of the Al gradient, and the ΔAl gradient (surface vs. bulk) varied depending on the sintering times. After successfully obtaining various Al-gradient profiles within the particles, their impact on battery performances (e.g., cycle life, fast-charging, fast-discharging), CEI stability, and thermal stability was investigated. The optimized Al-gradient in CG-LNAO-6, obtained by sintering at 710 °C for 6 hours in O2, exhibited high cation ordering, facilitating rapid Li+ transport, and enhanced CEI stability that resulted in improved cycle life, fast-charging/fast-discharging capabilities, and thermal safety. This result underscores the crucial role of the elemental gradient profile, highlighting it as an equally important aspect in the optimization of cathode chemical compositions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the General Motor Global R&D Center. Characterization of this work was supported in part by the Ohio State University Institute for Materials Research.References
- J. Ying, C. Jiang and C. Wan, J. Power Sources, 2004, 129, 264–269 CrossRef CAS.
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef CAS PubMed.
- H. Li, N. Zhang, J. Li and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A2985–A2993 CrossRef CAS.
- Z. Wang, W. Wei, Q. Han, H. Zhu, L. Chen, Y. Hu, H. Jiang and C. Li, ACS Nano, 2023, 17, 17095–17104 CrossRef CAS PubMed.
- D. D. MacNeil, Z. Lu, Z. Chen and J. R. Dahn, J. Power Sources, 2002, 108, 8–14 CrossRef CAS.
- H. Li, M. Cormier, N. Zhang, J. Inglis, J. Li and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A429–A439 CrossRef CAS.
- D. Rathore, C. Geng, N. Zaker, I. Hamam, Y. Liu, P. Xiao, G. A. Botton, J. Dahn and C. Yang, J. Electrochem. Soc., 2021, 168, 120514 CrossRef CAS.
- T. Xu, L. Shen, Y. Wen, Q. Zhou, J. Wang, J. Wu and J. Zheng, J. Electroanal. Chem., 2022, 921, 116675 CrossRef CAS.
- T. He, Y. Lu, Y. Su, L. Bao, J. Tan, L. Chen, Q. Zhang, W. Li, S. Chen and F. Wu, ChemSusChem, 2018, 11, 1639–1648 CrossRef CAS PubMed.
- J. Wang, S. Yao, W. Lin, B. Wu, X. He, J. Li and J. Zhao, J. Power Sources, 2015, 280, 114–124 CrossRef CAS.
- J. Kang and B. Han, ACS Appl. Mater. Interfaces, 2015, 7, 11599–11603 CrossRef CAS PubMed.
- M. Y. Song, H. U. Kim and H. R. Park, Ceram. Int., 2014, 40, 4219–4224 CrossRef CAS.
- H. Hu, Q. Chen, Z. Chen and H. Chen, Appl. Surf. Sci., 2014, 316, 348–354 CrossRef CAS.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef.
- S. Yoon, C. W. Lee, Y. S. Bae, I. Hwang, Y.-K. Park and J. H. Song, Electrochem. Solid-State Lett., 2009, 12, A211 CrossRef CAS.
- Y. Zhang, J. Liu and F. Cheng, Energy Fuels, 2021, 35, 13474–13482 CrossRef CAS.
- A. Liu, N. Zhang, J. E. Stark, P. Arab, H. Li and J. R. Dahn, J. Electrochem. Soc., 2021, 168, 040531 CrossRef CAS.
- K. Heo, J. Lee, Y.-W. Song, M.-Y. Kim, H. Jeong, A. DoCheon, K. Jaekook and J. Lim, J. Electrochem. Soc., 2021, 168, 010521 CrossRef CAS.
- H. Yu, Y. Cao, L. Chen, Y. Hu, X. Duan, S. Dai, C. Li and H. Jiang, Nat. Commun., 2021, 12, 4564 CrossRef CAS PubMed.
- W. Cheng, J. Ding, Z. Liu, J. Zhang, Q. Liu, X. Wang, L. Wang, Z. Sun, Y. Cheng, Z. Xu, Y. Lei, J. Wang and Y. Huang, Chem. Eng. J., 2023, 451, 138678 CrossRef CAS.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- S. Gopukumar, K. Y. Chung and K. B. Kim, Electrochim. Acta, 2004, 49, 803–810 CrossRef CAS.
- L. Zou, W. Zhao, H. Jia, J. Zheng, L. Li, D. P. Abraham, G. Chen, J. R. Croy, J.-G. Zhang and C. Wang, Chem. Mater., 2020, 32, 2884–2892 CrossRef CAS.
- J. Choi, L. Dong, C.-Y. Yu, C. O'Meara, E. Lee and J.-H. Kim, J. Electrochem. Energy Convers. Storage, 2021, 18, 041009 CrossRef CAS.
- W. M. Seong, Y. Kim and A. Manthiram, Chem. Mater., 2020, 32, 9479–9489 CrossRef CAS.
- Y.-K. Sun, D.-J. Lee, Y. J. Lee, Z. Chen and S.-T. Myung, ACS Appl. Mater. Interfaces, 2013, 5, 11434–11440 CrossRef CAS PubMed.
- J. Duan, G. Hu, Y. Cao, C. Tan, C. Wu, K. Du and Z. Peng, J. Power Sources, 2016, 326, 322–330 CrossRef CAS.
- U.-H. Kim, G.-T. Park, P. Conlin, N. Ashburn, K. Cho, Y.-S. Yu, D. A. Shapiro, F. Maglia, S.-J. Kim, P. Lamp, C. S. Yoon and Y.-K. Sun, Energy Environ. Sci., 2021, 14, 1573–1583 RSC.
- H.-H. Ryu, G.-T. Park, C. S. Yoon and Y.-K. Sun, J. Mater. Chem. A, 2019, 7, 18580–18588 RSC.
- W. Liu, Q. Shi, Q. Qu, T. Gao, G. Zhu, J. Shao and H. Zheng, J. Mater. Chem. A, 2017, 5, 145–154 RSC.
- X. Chen, L. Li, M. Liu, T. Huang and A. Yu, J. Power Sources, 2021, 496, 229867 CrossRef CAS.
- N. S. Luu, J.-M. Lim, C. G. Torres-Castanedo, K.-Y. Park, E. Moazzen, K. He, P. E. Meza, W. Li, J. R. Downing, X. Hu, V. P. Dravid, S. A. Barnett, M. J. Bedzyk and M. C. Hersam, ACS Appl. Energy Mater., 2021, 4, 11069–11079 CrossRef CAS.
- B. Manikandan, V. Ramar, C. Yap and P. Balaya, J. Power Sources, 2017, 361, 300–309 CrossRef CAS.
- K. Pan, F. Zou, M. Canova, Y. Zhu and J.-H. Kim, J. Power Sources, 2020, 479, 229083 CrossRef CAS.
- T. Wang, K. Ren, W. Xiao, W. Dong, H. Qiao, A. Duan, H. Pan, Y. Yang and H. Wang, J. Phys. Chem. C, 2020, 124, 5600–5607 CrossRef CAS.
- X. Jiao, L. Rao, J. Yap, C.-Y. Yu and J.-H. Kim, J. Power Sources, 2023, 561, 232748 CrossRef CAS.
- B. Huang, R. Wang, Y. Gong, B. He and H. Wang, Front. Chem., 2019, 7, 107 CrossRef CAS PubMed.
- H. Cao, F. Du, J. Adkins, Q. Zhou, H. Dai, P. Sun, D. Hu and J. Zheng, Ceram. Int., 2020, 46, 20050–20060 CrossRef CAS.
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. Li, E. A. Payzant, D. L. Wood and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef CAS.
- N. Ikeda, I. Konuma, H. B. Rajendra, T. Aida and N. Yabuuchi, J. Mater. Chem. A, 2021, 9, 15963–15967 RSC.
- H. Wang, X. Li, F. Li, X. Liu, S. Yang and J. Ma, Electrochem. Commun., 2021, 122, 106870 CrossRef CAS.
- T. Wang, L. Rao, X. Jiao, J. Choi, J. Yap and J.-H. Kim, ACS Appl. Energy Mater., 2022, 5, 7346–7355 CrossRef CAS.
- L. Rao, X. Jiao, C.-Y. Yu, A. Schmidt, C. O'Meara, J. Seidt, J. R. Sayre, Y. M. Khalifa and J.-H. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 861–872 CrossRef CAS PubMed.
- C.-Y. Yu, X. Jiao, L. Rao, S.-B. Son, E. Lee and J.-H. Kim, Electrochem. Commun., 2022, 138, 107286 CrossRef CAS.
- D. Darbar, E. C. Self, L. Li, C. Wang, H. M. Meyer, C. Lee, J. R. Croy, M. Balasubramanian, N. Muralidharan, I. Bhattacharya, I. Belharouak and J. Nanda, J. Power Sources, 2020, 479, 228591 CrossRef CAS.
- H. Zhu, Z. Wang, L. Chen, Y. Hu, H. Jiang and C. Li, Adv. Mater., 2023, 35, 2209357 CrossRef CAS PubMed.
- M. Liu, J. Vatamanu, X. Chen, L. Xing, K. Xu and W. Li, ACS Energy Lett., 2021, 6, 2096–2102 CrossRef CAS.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CrossRef CAS.
- X. Zheng, W. Liu, Q. Qu, H. Zheng and Y. Huang, J. Materiomics, 2019, 5, 156–163 CrossRef.
- H. Chen, C. Yin and H. Zhou, Ionics, 2021, 27, 4995–5008 CrossRef CAS.
- L. Su, E. Jo and A. Manthiram, ACS Energy Lett., 2022, 7, 2165–2172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00433g |
| This journal is © The Royal Society of Chemistry 2024 |