
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper shape-templated N-doped carbons: exercising selective surface area control for lithium-ion batteries & beyond†
Samantha N.
Lauro
 a,
James N.
Burrow
b,
Benjamin G.
Broekhuis
b,
Philippe E.
Papa
ab and
C. Buddie
Mullins
*abc
a,
James N.
Burrow
b,
Benjamin G.
Broekhuis
b,
Philippe E.
Papa
ab and
C. Buddie
Mullins
*abc
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712-1224, USA. E-mail: mullins@che.utexas.edu
bMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712-1589, USA
cTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712-1591, USA
First published on 26th February 2024
Abstract
Doped-carbons are attractive alternatives to traditional graphite anodes in lithium-ion batteries because they maintain the low cost and operating voltage window of carbon while providing higher capacities. Key challenges lie in creating useful doped-carbons with controllable properties and understanding which properties are most beneficial for these materials. Here, N-doped carbons are synthesized via a metal-templating platform, which provides selective control of both the chemical composition and surface area, allowing the two properties to be decoupled from one another. Copper metal particles in dendritic, spheroidal, and flake shapes are utilized as metal templates with graphitic carbon nitride precursor to create N-doped carbons. The resulting carbons are chemically similar, defined by XPS, pXRD and Raman spectroscopy. However, the surface area and pore distributions are distinctly different, as quantified by gas adsorption analysis and observed via SEM, which results in capacity differences of 100 mA h g−1 when applied as active materials in lithium-ion batteries.
1. Introduction
Lithium-ion batteries (LIBs) have become a pivotal technology in enabling the global transition to renewable energy sources.1 Technological advancements demanding batteries with performance tailored towards each application have led to extensive research into novel chemistries for both electrolytes2–4 and active materials,5–7 with one such component being the anode. The traditional anode active material in LIBs is graphite, in which lithium ions are stored between the carbon sheets by intercalation, resulting in a specific capacity of ∼372 mA h g−1.8 Graphite is low in cost and abundant compared to other active materials, but higher energy and power densities are needed for future technologies. Doped-carbons are an alternative anode material to graphite, with synthetic platforms that can employ virtually any organic or bio-derived precursor, leading to a variety of materials that can be produced with different dopants, surface areas, pore distributions, and crystallinities.9,10Nitrogen (N)-doped carbons have been of specific interest because N-doped carbons are simple to synthesize, and can have increased specific capacities. N can be doped into sp2 hybridized carbon structures in one of four typical bonding motifs – pyrrolic, pyridinic, graphitic, and oxidized.11 Pyrrolic and pyridinic N groups are sp2 hybridized, substituted into five and six member carbon rings, respectfully, while graphitic and oxidized N groups are sp3 hybridized, substituted into six member carbon rings.9 Pyridinic and pyrrolic N functionalities are generally preferred for LIB anodes as the lone pair provides an additional reversible lithiation site, boosting capacity, while graphitic N functionalities are less preferred due to their tendency to bind irreversibly with lithium.12 The concentration of N dopant is also critical, as excessive dopant levels can decrease the electrical conductivity of the material.13 Therefore, it is important that both the type and quantity of N dopants are balanced.
There any many synthetic methods to produce N-doped carbons, usually consisting of the carbonization of a nitrogen-containing precursor.14 The precursor usually falls into one of three categories-bio-derived precursors, such as an organic waste product from the food industry, lab-synthesized nitrogen containing polymers, or graphitic carbon nitride. Bio-derived precursors are readily available, such as food and animal waste,15,16 and is an attractive platform as a solution for reducing waste products. However, these materials often have low purity, since many dopants besides nitrogen are present, making chemical analysis of the source of LIB performance benefits challenging to isolate. Polymeric compounds on the other hand, such as polyacrylonitrile and polyaniline, can be synthetically controlled to include only desired dopants, highly purified, and produce unique nanostructures, but the synthetic process may involve toxic chemicals and be time intensive, and the resulting N-dopant levels after carbonization are moderate (usually less than 20 atomic%).17,18 In contrast, graphitic carbon nitride (g-C3N4) can be produced from inexpensive precursors such as urea and melamine,19 consists of only carbon and nitrogen, and can result in N-doped carbons with nitrogen contents upwards of 30 atomic%, making it an ideal material to use for fundamental studies on N-doped carbon anodes. Carbon nitride can be converted to an N-doped carbon through pyrolysis, sometimes in the presence of metals, metal-salts, and salt mixtures.20,21
One synthetic technique, metal-templating, has shown promise in controlling the final properties of N-doped carbons.22–24 While metals such as zinc and copper have been readily used as nanostructured hosts for LIB anodes,25–28 in the context of carbon synthesis, metal-templating refers to the process in which a carbon precursor is pyrolyzed in the presence of a metal, usually as a mixture of powders. The metal is removed after the carbonization, often by acid-washing, to yield resulting carbon materials with unique properties. Fahlman et al. mixed magnesium particles with graphitic carbon nitride (g-C3N4) before pyrolysis to create N-doped carbon anodes with highly preferred pyrrolic N functionalities.20 Mao et al. created hollow N-doped carbon spheres by using a zinc template, and altered the N concentration by performing the pyrolysis over a range of temperatures (650–850 °C).29 Previous work from our group investigated the use of iron and copper metal templates, and revealed that the choice of metal allowed for control over the type of N dopants present.24 Iron templates resulted in N-doped carbons with preferred graphitic N functionalities, while copper promoted pyridinic and pyrrolic functionalities, with the latter being more preferential for anodes of LIBs. Copper is a unique metal-template option, because it remains a solid at the temperatures of carbon pyrolysis (unlike zinc and magnesium, with melting points at ∼420 °C and 650 °C, respectively), and does not form a carbide intermediate like other templates (such as zinc and iron). Copper is low in cost, abundant, and can be produced in different nanostructured-shapes at the commercial level and in research labs, providing an array of particle size and shape options to choose from.30–32 As a metal template, changing the contact surface area between the carbon and copper resulted in different physical morphologies of the N-doped carbons. Physical morphology encompasses particle shape, size, texture, surface area, and porosity, which in an LIB anode influences ionic transport through both the electrode film and through the carbon particles, solid electrolyte interphase growth (SEI), and the contribution of capacitive and pseudo-capacitive surface storage mechanisms to overall observed specific capacity and rate capability.33–36
Importantly, the tunability of this synthetic platform enables fundamental studies on the role of different properties of N-doped carbons for a given application. A majority of previous studies have shown that the choice of metal template dictates the surface chemical bonding motifs of these N-doped carbons, and has focused on changing the chemical identity of the metal (i.e., magnesium, zinc, iron, copper).20,22–24 In contrast, the present study investigates and demonstrates that altering the physical shape of the metal template controls the resulting physical morphology (and porosity) of the carbons, without significantly impacting surface chemistry.
2. Experimental
2.1 Materials
All materials were used as received without further purification. Urea (synthetic grade), copper spheroidal powder (99%), copper dendritic powder (99.7%), nitric acid (70%, ACS reagent grade), and dimethyl carbonate (DMC, >99%) were purchased from Millipore Sigma. Copper flake powder (99.9%) was acquired from Beantown Chemical, lithium foil (750 μm thick, 99.9%) from ThermoFisher Scientific, and lithium bis(fluorosulfonyl)imide salt (LiFSI, 99%) from Oakwood Chemical. 1-Methyl-2-pyrrolidone (NMP, 99%), ethylene carbonate (EC, >99.0%), and fluoroethylene carbonate (FEC, >98.0%) were acquired from Tokyo Chemical Industry. Copper foil, polyvinylidene fluoride (PVDF, >99.5%) and conductive carbon black (SuperP) were obtained from MTI Corporation.2.2 Carbon synthetic methods
Urea derived graphitic carbon nitride (g-C3N4) was synthesized by a thermal condensation method.37 Briefly, 75 g urea was placed in an alumina crucible covered with aluminum foil. The crucible was heated in a box furnace under air atmosphere using a ramp rate of 2.3 °C min−1 and held at 550 °C for four hours. The crucible cooled naturally to room temperature and the carbon nitride was collected. To create nitrogen doped carbons, 1.2 g of g-C3N4 was mixed with 1.2 g of copper metal particles with a mortar and pestle. Three different samples were prepared, where the copper particles were either dendritic, spheroidal, or flake shaped. These shapes were according to the manufacturer label. The mixture was then transferred to a covered alumina crucible and carbonized under flowing argon (120 cc min−1) in a quartz tube furnace. The temperature was ramped at a rate of 5 °C min−1 and held at 40 °C for one hour to purge residual air from the tube, then held at 100 °C for one hour to remove residual water, and carbonized at a final temperature of 750 °C for two hours before cooling naturally back to room temperature. The resulting carbons were washed with concentrated nitric acid (HNO3) three times to remove the copper and residual salt by-products and then washed with excess deionized water until pH neutral. The N-doped carbons were dried in a box furnace at 120 °C overnight and collected. The N-doped carbons are named by the shape of copper template used (Dendritic, Spheroidal, Flake) and therefore correspond to dendritic-shape copper, spheroidal-shape copper, and flake-shaped copper templated carbons, respectively.2.3 Material characterization
Crystallite size was determined by obtaining powder X-ray Diffraction (pXRD) patterns by using a Rigaku MiniFlex 600II (Cu Kα radiation, λ = 1.5418 Å) in continuous scan mode from 2θ = 10° to 80° with a step width of 0.02 at 2.5° min−1 with a single crystal zero background silicon sample holder. The crystallite sizes and d-spacings were determined by using a Lorentz fit for peaks at approximately 24° and 44° to account for diffraction of 002 and 101 planes, respectively. The degree of graphitization of each sample was determined by Raman spectroscopy. Spectra were obtained with a Renishaw inVia Raman Microscope equipped with a 100 mW green laser which was employed to excite the samples at 514 nm with a spectral shift centered at 1500 cm−1. The ID/IG ratio was determined by fitting the spectra with four Voigt peaks representing the D*, D, D**, and G bands at approximately 1230, 1366, 1521, and 1595 cm−1, respectively, and using the ratio between the D and G band intensity. X-ray photoelectron spectroscopy (XPS) spectra were acquired on a Kratos Axis Ultra X-ray photoelectron spectrometer. CasaXPS software was used to fit survey scans and the high-resolution spectra of each elemental region (C 1s, N 1s, O 1s), and all spectra were shifted to account for the adventitious carbon peak at 284.7 eV.21 Scanning electron micrographs (SEM) were collected on a FEI Quanta 650 ESEM at a scanning voltage of 30.0 kV. Textural properties of the resulting carbons were studied by acquiring isotherms of N2 at 77 K and CO2 at 273 K using a Quantachrome Autosorb iQ-MP (Anton Paar). Before gas adsorption analysis, around 100 mg of powdered sample was loaded into glass sample tubes and degassed under dynamic vacuum at 200 °C overnight. With ASiQ WinTM software, surface areas were quantified with multi-point BET analysis with optimized point selection to account for the Rouquerol criteria and the microporous nature of these materials. These multi-point BET analyses are provided in the ESI (Fig. S7†). Pore size distributions were derived from the adsorption branch of the isotherm with quenched-solid density functional theory (QSDFT) assuming a convolution of spherical, cylindrical, and slit-shaped pores for N2 adsorption, while non-local density functional theory (NLDFT) was employed for analysis of ultra-microporosity from CO2 isotherms at 273 K on carbon. The total pore size distribution was acquired by stitching together results from CO2 (<1.5 nm) and N2 (1.5–50 nm). Microporosity was defined as the cumulative pore volume at a pore diameter of 2 nm. Total pore volume (up to 200 nm) was quantified using total quantity of adsorbed N2 at P/P0 = 0.99.2.4 Electrochemical measurements
Electrode films were fabricated by mixing the active carbon materials with a conductive carbon black additive (SuperP) and polyvinylidene fluoride (PVDF) binder in an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 mass ratio in 1-methyl-2-pyrrolidone (NMP) solvent. The slurries were coated onto 8 μm thick copper foil using a doctor blade set at 150 μm, and films were transferred to an oven to dry for 3 hours at 120 °C before pulling vacuum for 12 hours to remove residual solvent. Resulting electrodes had a mass loading between 1.5–1.8 mg cm−2 and film thicknesses ∼80 μm.
:10:10 mass ratio in 1-methyl-2-pyrrolidone (NMP) solvent. The slurries were coated onto 8 μm thick copper foil using a doctor blade set at 150 μm, and films were transferred to an oven to dry for 3 hours at 120 °C before pulling vacuum for 12 hours to remove residual solvent. Resulting electrodes had a mass loading between 1.5–1.8 mg cm−2 and film thicknesses ∼80 μm.
Materials were tested electrochemically in stainless steel 2032 coin cells assembled in an argon atmosphere glovebox using three 500 μm thick stainless steel spacers, one circular spring, and one polypropylene separator (Celgard 2400) flooded with 150 μL of electrolyte. The slurry-cast materials were cut into 13 mm diameter electrodes and tested against a 750 μm thick lithium metal counter electrode using a 1.2 M lithium bis(fluorosulfonyl)imide (LiFSI) in 3:7 (v/v) ethylene carbonate (EC):dimethyl carbonate (DMC) with 5% v/v fluoroethylene carbonate (FEC) electrolyte system.
Long-term and variable rate cycling tests were performed using a multi-channel battery test system (Neware BTS4000-5V10 mA) using a 10 mV–3 V potential window. Long-term cycling consisted of three conditioning cycles at a rate of 0.02 A g−1 and 200 subsequent cycles at a rate of 0.2 A g−1. Variable rate cycling consisted of three cycles at 0.02 A g−1 followed by five cycles of each faster rate (0.05 A g−1, 0.1 A g−1, 0.2 A g−1, 0.5 A g−1, 1.0 A g−1, and 2.0 A g−1) and recovered at a rate of 0.2 A g−1. Cyclic voltammograms were obtained over the potential window of 10 mV–3 V vs. Li/Li+ at a scan rate of 0.1 mV s−1 on a multi-channel battery test system (Arbin BT 2043). All reported electrochemical results are from duplicate cells (Fig. S7 and S8†).
3. Results & discussion
Fig. 1a outlines the synthetic procedure. Commercially available copper particles of three different morphologies – dendritic, spheroidal, and flake shaped (as labelled by the manufacturers) – were used as templates. N-doped carbons were produced by carbonizing a mixture of the copper templates with urea-derived g-C3N4 at 750 °C for two hours. The resulting product was washed with concentrated nitric acid (HNO3) to remove the metal templates and any salt by-products.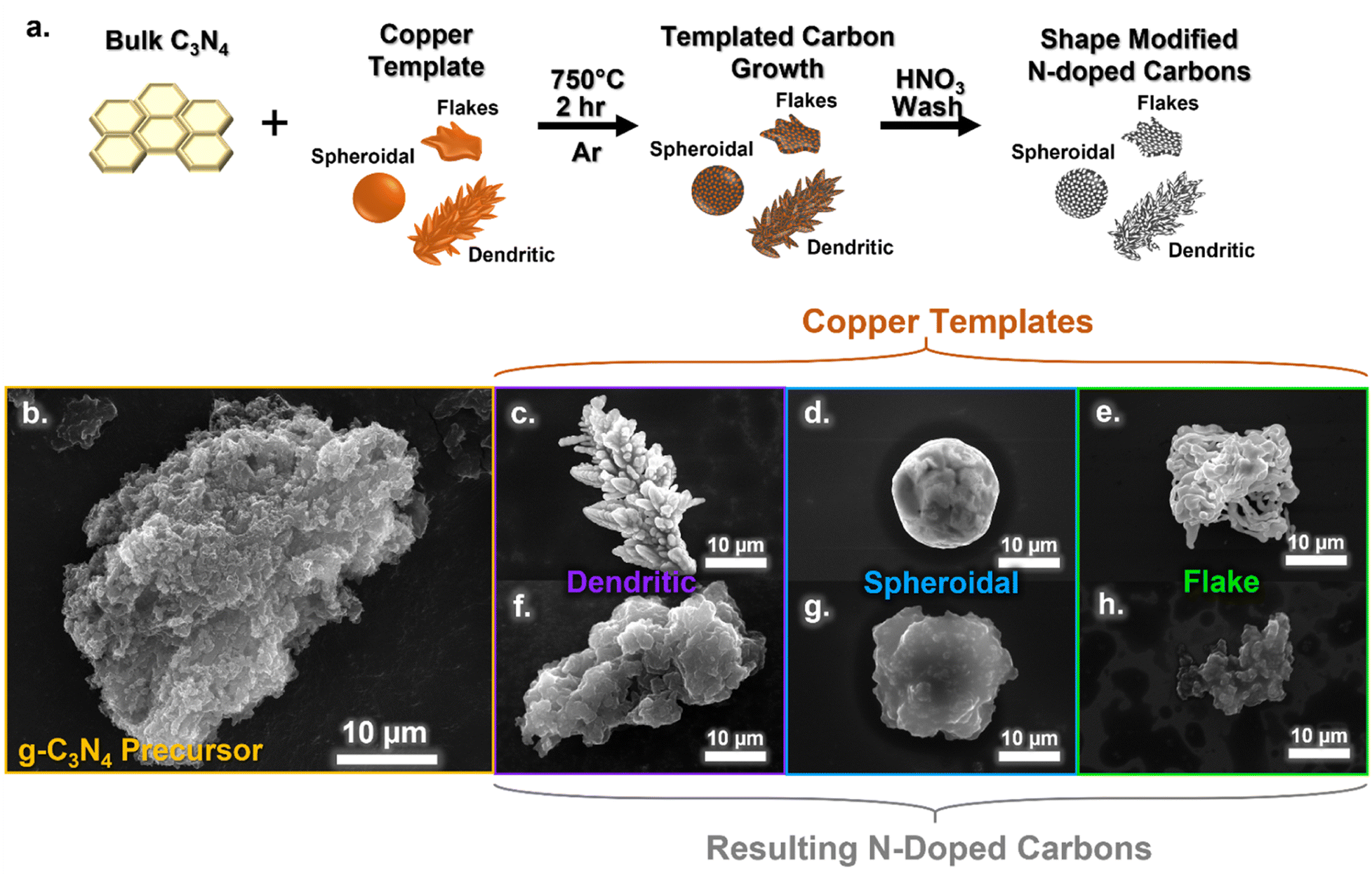 | ||
| Fig. 1 Schematic illustration of N-doped carbon synthesis procedure for using different shaped copper metal templates (a). SEM images of g-C3N4 precursor (b), dendritic copper particle (c), spheroidal copper particle (d), flake copper particle (e),and the N-doped carbons produced using dendritic (f), spheroidal (g), and flake (h) shaped copper templates. | ||
Scanning electron microscopy (SEM) revealed the morphology of the resulting carbon materials. The g-C3N4 precursor exhibited a highly textured, amorphous particle morphology as seen in Fig. 1b, which was not retained in the final N-doped carbons (Fig. 1f–h). The copper metal templates have distinct morphological differences: the dendritic copper has a branched morphology and asymmetrical shape (Fig. 1c); the spheroidal copper is round, with a smooth surface (Fig. 1d); and the flake copper appears to be an agglomeration of secondary string-like particles (Fig. 1e). The resulting N-doped carbons had morphologies reminiscent of their respective templates and visually distinctive from each other. The dendritic templated carbon (Fig. 1f) exhibited a textured surface with grooves and wrinkles, with approximately 30-micron sized particles, and asymmetrical particles shapes. The spheroidal templated carbons (Fig. 1g) were smaller in size (∼5–10 μm) and had a smoother observed texture, while the flake templated carbons (Fig. 1h) were irregular in shape and size (around 10 μm). Additional SEM images of the carbon materials are shown in Fig. S1,† and population images of the copper templates and carbons are shown in Fig. S2.† The carbon secondary particle size appears to be related to the size of the copper templates used, with large dendritic templates resulting in larger sized N-doped carbon particles, and smaller spheroidal templates resulting in smaller carbon particle sizes. The metal template remains in a solid phase during synthesis (as opposed to salt-melts), so the surface area of the copper templates available to the g-C3N4 is likely responsible for particle shape and morphology of the final N-doped carbons. Going forward, the carbon nomenclature will be indicated by their copper template shape (i.e. dendritic, sphere, and flake carbon).
To better understand the nanostructure of the resulting carbon particles, powdered X-ray diffraction (XRD) was performed (Fig. 2a). All of the carbon samples produced a broad diffraction pattern with a large peak centered at approximately 2θ = 27.5°, representing the 002 reflection. The broadening of this peak as compared to the sharper peak from the g-C3N4 precursor represents a turbostratic carbon structure, which is indicative of crystallites with short-range but a lack of long range order.38 Scherrer analysis gives an estimation of the crystallite parameters, shown in Table S1.† The d-spacing in the 002 plane is ∼3.4 Å for all of the carbons. However, there is a slight difference in the lateral crystallite coherence length (La), with the dendritic carbon having a larger La of 8.71 Å compared to the spheroidal and flake carbon, which had La values of 6.98 and 6.45 Å, respectively. The La represents the in-plane order of the crystallite and is directly related to the defect density.39 Therefore, as a larger La value correlates to longer range order, the dendritic carbon likely exhibited slightly fewer structural defects than the spheroidal and flake carbons. Nonetheless, all three samples show similar crystal structures overall. The graphitic order of the carbons was further compared via Raman spectroscopy (Fig. 2b). The ID/IG ratios calculated from the D and G bands peak intensities (example fitting shown in Fig. S3 and values in Table S2†) were found to be nearly identical for the three carbons (ID/IG values of 1.20, 1.19, and 1.19 for flake, spheroidal, and dendritic carbons, respectively).
 | ||
| Fig. 2 Characterization of dendritic, spheroidal, and flake templated carbons. (a) XRD patterns of carbons and g-C3N4 precursor. (b) Raman spectra of carbons. (c) Summary of elemental composition of carbons from XPS analysis of survey spectra. (d) Summary of nitrogen bonding motifs in carbons from XPS analysis of the N 1s spectra. | ||
Only very small differences in the surface chemistry of the carbons were observed with X-ray photoelectron spectroscopy (XPS). The total elemental composition summarized in Fig. 2c is in line with previous studies of copper templating,24 comprised of ∼63% carbon, 30% nitrogen, 5% oxygen, and less than 2% of residual copper. The corresponding XPS survey spectra can be found in Fig. S4.† The N bonding motifs were deconvoluted by fitting the N 1s region in Fig. S5† with four peaks at binding energies of ∼398.1, 398.7, 399.8, and 400.8 eV to represent the pyridinic, pyrrolic, graphitic, and oxidized N functionalities, respectively.40 The difference in chemical motifs is near identical, with only 1% or less difference between the carbons, and preferential formation of pyridinic N from the use of copper (Fig. 2d).
As demonstrated above, the carbons resulting from the different copper templates were nearly identical in chemical structure and nanocrystalline (dis)order. In contrast, the physical morphology differed significantly depending on the morphology of the templates employed. While SEM provided a qualitative view of the carbon particle morphology at the micron-scale, gas sorption isotherms of nitrogen (N2) and carbon dioxide (CO2) were used to generate a more quantitative comparison of the carbons' surface areas and pore size distributions,41–44 as summarized in Fig. 3a. The dendritic carbon exhibited the smallest BET surface area,44 measured at 308 m2 g−1, while the spheroidal carbon had the largest at 406 m2 g−1. Meanwhile, the flake carbon had an intermediate surface area of at 380 m2 g−1. This difference in exposed surface area is primarily attributed to the volume of mesopores. Although the cumulative volume of micropores is similar between the templated carbons, each carbon exhibits a unique pore size distribution (Fig. 3b). While all of the carbons displayed a similar volume of micropores (<2 nm), the volume of meso- (2–50 nm) and macropores (50–200 nm) in the dendritic carbon was less than half of that in the spheroidal and flake carbon, with the spheroidal carbon having the largest volume of these types of pores. This difference in the volume of pores with larger characteristic length scales (meso/microporosity) is influenced by the amount of contact area with the metal template. However, increased exposed surface area accompanies increases in porosity, and a larger electrochemically active surface area requires the formation of more solid electrolyte interphase (SEI) for passivation. Since the SEI consumes the Li inventory and is associated with irreversible capacity losses during initial cycling, increased porosity does not always correlate with improved electrochemical performance of carbon anodes in LIBs. In this way, templating the nanostructure, while keeping surface chemistry and crystalline/defect structure consistent, can enable improved performance by exerting a finer control on porosity and surface area, which influence SEI formation and rate performance.
 | ||
| Fig. 3 Surface area characterization of dendritic, spheroidal, and flake templated carbons. (a) BET derived surface area and pore volume of carbons. (b) Pore size distributions derived via combination of quenched solid and nonlocal density functional theory (QSDFT and NLDFT) analysis of N2 and CO2 isotherms, respectively. | ||
As mentioned previously, pyridinic and pyrrolic nitrogen defects have been shown to provide reversible lithium-ion storage sites due to the availability of lone pairs of electrons, while graphitic defects have often resulted in irreversible capacity loss.45 Pender et al. demonstrated that carbons derived from Cu-templated g-C3N4 exhibited pyridinic and pyrrolic defects and obtained high capacities for Li storage.24 The chemical composition of the anode largely influences the mechanisms of lithium-storage, especially for faradaic processes; however, physical morphology also influences the final LIB performance. For example, for capacitive and/or pseudocapacitive storage mechanisms, large (electrochemically-active) surface area is critical.46 Moreover, highly porous structures often result in increased capacity at fast rates of charge/discharge due to a mixture of increased contribution from capacitive storage processes (which occur over smaller time scales than faradaic storage processes)47 and more electrolyte penetration, increasing the ease of mass transport.48 Additionally, particle size and shape also influence how the anode material is packed into electrode films, which can alter the porosity and electronic conductivity of the film, further altering electrochemical performance.49,50 As a result, even with identical chemical makeup, careful tuning of the physical particle morphology is imperative for the development of high quality battery anodes.
The electrochemical performance at variable cycling rates for each of the templated carbons is shown in Fig. 4a. Lithium half-cells of the dendritic, spheroidal, and flake carbons were cycled for five consecutive cycles at current densities of 0.05, 0.12, 0.2, 0.5, 1.0, and 2.0 A g−1 to evaluate the difference in rate capability between the materials. All reported cycling is shown as an average between duplicate cells. Individual cell cycling is shown in Fig. S7† and capacities reported with standard deviation in Table S5.† At the slower cycling rate of 0.05 A g−1, the dendritic carbon delivers the largest capacity of 640 mA h g−1, compared to the flake and spheroidal carbons which delivered 610 and 544 mA h g−1, respectively, which are near double the capacity of traditional graphite at the same cycling rate, ∼330 mA h g−1.51 In contrast, at rates of 0.2 A g−1 and above, the dendritic carbon achieved the smallest specific capacity of the three carbons. For example, at a fast charge/discharge rate of 2.0 A g−1 (∼3.7 mA cm−2), the dendritic carbon exhibited only 17 mA h g−1 of capacity compared to 54 and 92 mA h g−1 for spheroidal and flake carbon, respectively. The difference in performance between the templated carbons can be further understood by examining the differential capacity curves (dQ/dV, mA h g−1 V−1) for insight into the lithium storage processes. The normalized differential capacities at 0.05 A g−1 for the dendritic, spheroidal, and flake copper are overlayed in Fig. 4b (derived from the charge discharge profiles in Fig. 4c–e). The extraction of lithium from the N-doped carbons can be attributed to a mixture of three processes: faradaic storage in the carbon layers (<∼0.8 V), (pseudo)capacitive storage at defects and in pores (∼0.8–2.2 V), and reversible binding of lithium-ions to nitrogen sites (>∼2.2 V).24,52–54 Although there is significant overlap in the specific potentials at which these lithium storage mechanisms occur, our choice of representative potential ranges is in accordance with previous literature on copper templated N-doped carbons.24 As clearly visible in Fig. 4b, the dendritic carbon exhibited proportionally less capacity contribution from (pseudo)capacitance in pores and defects than did the flake or spheroidal carbons. This result explains the reduced specific capacity of the dendritic carbon at faster rates, where capacitive mechanisms of lithium-ion storage become dominant, and can be directly related to the smaller BET surface area and porosity of the dendritic carbon.55 The significance of this difference is further exemplified in the long-term cycling at a rate of 0.2 A g−1, shown in Fig. 5. Over the course of 200 cycles, the dendritic, spheroidal, and flake carbons demonstrated average capacities of 416, 483, and 541 mA h g−1, respectively. Despite identical chemical composition, more than 100 mA h g−1 of capacity difference between the dendritic and flake carbon material seemingly resulted from the differences in physical morphology and nanostructure alone. However, while the spheroidal carbon exhibited the largest measurable BET surface area of the three templated carbons, the flake carbon achieved the largest capacities at faster cycling rates. This difference in performance between the flake and spheroidal carbon is likely due to the difference in the pore-size distribution, especially in the ultra-micropore (<0.7 nm) region, as shown in Fig. 3b. The flake carbon exhibits three populations of ultra-micropores at approximately 0.35, 0.48, and 0.6 nm in size, while the spheroidal and dendritic carbons only exhibit a single population of approximately 0.52 nm. The full mechanistic role of ultra-micropores in lithium storage is currently a topic of heavy debate in the literature. Kang et al. has reported that ultra-micropores <0.4 nm increase carbon rate performance by a suggested size-sieving mechanism, where the pores are accessible to lithium-ions but inaccessible to electrolyte molecules, reducing decomposition and irreversible capacity loss.56 Lee et al. utilizes a first principles approach to demonstrate that lithium-ions can occupy the pore cavity, surfaces, and walls in micro and ultra-micropores.57 Ultra-microporosity is suggested to provide active sites for lithium-ions, with thin walls which shorten diffusion time and increase capacitive-like storage.58,59 The flake carbon exhibits this increased capacitive contribution from the middle region (approximately 0.8–2.6 V) of the dQ/dV lithium extraction curve at 0.2 A g−1 compared to the other two carbons (Fig. S9†). Other morphological differences, which cannot be captured by BET analysis alone, may also play a role in influencing the lithium-accessible surface area, which is defined by the ability of lithium to reach electrochemically active surfaces sites, spanning the nano and micro-scale of the active material particle surface area to the macro organization of pore channels within the electrode composite film itself. The shape and size of the active material particles can change how the electrode film packs, which can alter the size, length, and continuity of pores in the film, and subsequently change the ionic diffusion pathway through the electrolyte, though is difficult to quantify and visualize in amorphous carbon systems due to the other carbonaceous components used in the electrode film.34,49,60
 | ||
| Fig. 4 Variable rate cycling of the dendritic, spheroidal, and flake copper shape templated carbons at current densities of 0.05, 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1 with recovery at 0.2 A g−1. (a) Normalized differential capacity curves (dQ/dV vs. V) of lithium extraction at 0.05 A g−1 cycling rate for dendritic, spheroidal, and flake carbons. (b) Charge–discharge profiles from variable rate cycling of dendritic (c), spheroidal (d), and flake (e) templated carbons. | ||
 | ||
| Fig. 5 Long-term cycling of dendritic, spheroidal, and flake carbons at a cycling rate of 0.2 A g−1 over 200 cycles. | ||
The surface area of carbon anodes is also well-known to influence the first-cycle irreversible capacity loss due to the formation of the solid electrolyte interphase (SEI).61 After cycling, post-mortem analysis was conducted on all carbon electrodes, via SEM and energy dispersive X-ray (EDX) analysis shown in Fig. S10.† SEI is observed to have grown for all carbons, though no film fracture or cracking is observed indicating minimal volumetric expansion and structural stability. EDX reveals the presence of carbon, nitrogen, oxygen, sulfur and fluorine in the SEI, an expected result from the breakage of the carbonate solvents and LiFSI salt used in the electrolyte. Cyclic voltammetry (CV) (Fig. S11†) performed at a scan rate of 0.1 mV s−1 and analysis of the peak area decrease between the first and second CV cycles reveal that 38, 43, and 42% of the charge passed in the initial cycle resulted in irreversible capacity loss for the dendritic, spheroidal, and flake carbons. Although only a maximum of a 5% difference in first cycle irreversibility was observed between samples despite significant morphological differences, the dendritic carbon (with the smallest measured surface area) did show the smallest absolute capacity loss compared to the spheroidal and flake carbons. Both the peak corresponding to SEI formation and the peaks related to faradaic reactions occurred at the same potentials for all carbon samples (1.2 and 0.1 V vs. Li/Li+, respectively), indicative of their similar chemical composition. The CV curves also have a non-negligible box shape, further indicating (pseudo)capacitive contributions towards the overall Li-ion storage capacities.47
Overall, our results suggest that the flake carbon achieves the largest capacities in the present study for both the long-term and variable rate cycling, and maintains higher and competitive capacities to graphite under the same cycling rate conditions.51 However, additional considerations, such as initial capacity loss, capacity fade, and lithiation potential/overall cell voltage, are important factors to consider when optimizing anode active materials for LIBs for industrial scalability. The long-term cycling in Fig. 5, for example, has both capacity growth and decay present in the profile. The flake, spheroidal, and dendritic carbons start with initial capacities of 473, 442, and 405 mA h g−1, respectfully, and increase over the first 125 cycles to values of 578, 503, and 450 mA h g−1, representing a capacity increase of 18, 12, and 10%, respectfully. This phenomena is commonly observed for N-doped carbon anodes in LIBs, and is ascribed to increased electrolyte penetration with the activation of defects and pores.24,62 After the 125th cycle, capacity fade starts to occur, with final capacities at the 200th cycle reaching values of 535, 458, and 402 mA h g−1 for the flake, spheroidal, and dendritic carbons, respectfully, representing a capacity fade of approximately 7.4, 9, and 10.6% from the highest values reached at the 125th cycle. This flux in capacity is informative from a research perspective, but problematic for real-world batteries, which need to provide consistent, predictable performance per application needs. The flake carbon has the highest percent of capacity increase at the 125th cycle and smallest amount of capacity fade at the 200th cycle, while the dendritic carbon demonstrates the opposite trend, suggesting that purposeful structuring of carbon materials may aid in alleviating capacity fade in N-doped carbon anodes. Additional scalability concerns are the coulombic efficiency for these materials, tabulated in Table S6,† at approximately 99% for all the carbons, which is high for research materials, but falls shy of the 99.9% required for commercial applications. As has been well detailed by Gomez-Martin et al.,54 emphasis should also be placed on the origin of capacity gains, as capacitive contributions achieved at higher voltages are not necessarily practical in real-world applications. Further future work is required to optimize these materials for commercial use, but for this study battery performance testing is simply utilized to exemplify the importance of isolating physical vs. chemical contributions for evaluation of a material.
4. Conclusions
Copper shape-templating created an experimental platform to selectively create pyridinic-preferred N-doped carbons with different physical morphologies. The chemical composition and crystalline structure were confirmed to be near identical for all carbons through XPS, pXRD, and Raman spectroscopy, while the physical morphology differed as qualitatively observed via SEM of differing secondary particle shapes. Surface area and porosity differences quantified through gas physisorption analysis revealed that surface area differences up to 100 m2 per gram could be obtained with differing micropore network distributions, despite the use of the same chemical precursors. These morphological differences influence the electrochemical evaluation of doped carbon materials, resulting in specific capacity differences of up to ∼100 mA h g−1 for chemically identical materials. The dendritic, spheroidal, and flake copper shapes are commercially available templates that are easily obtained at scale. Future work investigating the use of more custom copper shapes, such as rods and nanoparticles, as well as investigating other metals templates in this manner, such as zinc and iron, will aid in creating an expansive platform of tunable heterogenous carbons. The insight gleaned from studies that isolate the individual influence of each chemical and physical material property on LIB performance inform how carbon materials should be evaluated and purposefully designed for each specific technological application, from batteries to carbon capture to water splitting and beyond.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Welch Foundation for support of this work (grant F-1436 (CBM)) as well as support (for SNL) from the U.S. Department of Defense (National Defense Science and Engineering Graduate Fellowship Program).References
- B. Diouf and R. Pode, Renewable Energy, 2015, 76, 375–380 CrossRef.
- S. Choi and G. Wang, Adv. Mater. Technol., 2018, 3, 1700376 CrossRef.
- E. Quartarone and P. Mustarelli, J. Electrochem. Soc., 2020, 167, 050508 CrossRef CAS.
- J. Xing, S. Bliznakov, L. Bonville, M. Oljaca and R. Maric, Electrochem. Energy Rev., 2022, 5, 14 CrossRef CAS.
- H. Cheng, J. G. Shapter, Y. Li and G. Gao, J. Energy Chem., 2021, 57, 451–468 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- J. Jyoti, B. P. Singh and S. K. Tripathi, J. Energy Storage, 2021, 43, 103112 CrossRef.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef CAS PubMed.
- Z. Wu, K. Sun and Z. Wang, Batteries, 2022, 8, 246 CrossRef CAS.
- Y. Yuan, Z. Chen, H. Yu, X. Zhang, T. Liu, M. Xia, R. Zheng, M. Shui and J. Shu, Energy Storage Mater., 2020, 32, 65–90 CrossRef.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS.
- G. M. Veith, L. Baggetto, L. A. Adamczyk, B. Guo, S. S. Brown, X.-G. Sun, A. A. Albert, J. R. Humble, C. E. Barnes, M. J. Bojdys, S. Dai and N. J. Dudney, Chem. Mater., 2013, 25, 503–508 CrossRef CAS.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2009, 113, 19082–19090 CrossRef CAS.
- M.-M. Titirici, R. J. White and L. Zhao, Green, 2012, 2, 25–40 CrossRef CAS.
- V. Selvamani, R. Ravikumar, V. Suryanarayanan, D. Velayutham and S. Gopukumar, Electrochim. Acta, 2016, 190, 337–345 CrossRef CAS.
- J. Ou, H. Deng, H. Zhang and H. Wang, Diamond Relat. Mater., 2022, 124, 108931 CrossRef CAS.
- J. Zhu, C. Chen, Y. Lu, Y. Ge, H. Jiang, K. Fu and X. Zhang, Carbon, 2015, 94, 189–195 CrossRef CAS.
- T. Zhu, J. Zhou, Z. Li, S. Li, W. Si and S. Zhuo, J. Mater. Chem. A, 2014, 2, 12545–12551 RSC.
- Y. Zheng, Z. Zhang and C. Li, J. Photochem. Photobiol., A, 2017, 332, 32–44 CrossRef.
- J. Chen, Z. Mao, L. Zhang, D. Wang, R. Xu, L. Bie and B. D. Fahlman, ACS Nano, 2017, 11, 12650–12657 CrossRef CAS PubMed.
- J. N. Burrow, J. P. Pender, J. V. Guerrera, B. R. Wygant, J. E. Eichler, D. C. Calabro and C. B. Mullins, ACS Appl. Nano Mater., 2020, 3, 5965–5977 CrossRef CAS.
- M. Du, X. Tian, R. Ran, W. Zhou, K. Liao and Z. Shao, Energy Fuels, 2020, 34, 11557–11564 CrossRef CAS.
- Y. Tang, X. Wang, J. Chen, X. Wang, D. Wang and Z. Mao, J. Alloys Compd., 2021, 858, 157720 CrossRef CAS.
- J. P. Pender, J. V. Guerrera, B. R. Wygant, J. A. Weeks, R. A. Ciufo, J. N. Burrow, M. F. Walk, M. Z. Rahman, A. Heller and C. B. Mullins, ACS Nano, 2019, 13, 9279–9291 CrossRef CAS PubMed.
- H. Lin, Z. Zhang, Y. Wang, X. L. Zhang, Z. Tie and Z. Jin, Adv. Funct. Mater., 2021, 31, 2102735 CrossRef CAS.
- J. H. Kwak, S. Park, S. Shin, S. Park, C. Kang, S.-H. Yu, J. Moon and H.-D. Lim, Energy Storage Mater., 2023, 59, 102762 CrossRef.
- H. Qiu, T. Tang, M. Asif, X. Huang and Y. Hou, Adv. Funct. Mater., 2019, 29, 1808468 CrossRef.
- G. Wang, X. Xiong, P. Zou, X. Fu, Z. Lin, Y. Li, Y. Liu, C. Yang and M. Liu, Chem. Eng. J., 2019, 378, 122243 CrossRef CAS.
- Y. Tang, X. Wang, J. Chen, X. Wang, D. Wang and Z. Mao, Carbon, 2020, 168, 458–467 CrossRef CAS.
- L. Avramović, V. M. Maksimović, Z. Baščarević, N. Ignjatović, M. Bugarin, R. Marković and N. D. Nikolić, Metals, 2019, 9, 56 CrossRef.
- D. Mott, J. Galkowski, L. Wang, J. Luo and C.-J. Zhong, Langmuir, 2007, 23, 5740–5745 CrossRef CAS PubMed.
- I. Lisiecki, F. Billoudet and M. P. Pileni, J. Phys. Chem., 1996, 100, 4160–4166 CrossRef CAS.
- M. Shaker, A. A. S. Ghazvini, F. R. Qureshi and R. Riahifar, Carbon Lett., 2021, 31, 985–990 CrossRef.
- L. Bläubaum, F. Röder, C. Nowak, H. S. Chan, A. Kwade and U. Krewer, ChemElectroChem, 2020, 7, 4755–4766 CrossRef.
- S. Müller, J. Eller, M. Ebner, C. Burns, J. Dahn and V. Wood, J. Electrochem. Soc., 2018, 165, A339 CrossRef.
- M. Jana, A. Sil and S. Ray, J. Phys. Chem. Solids, 2014, 75, 60–67 CrossRef CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- G. A. Zickler, B. Smarsly, N. Gierlinger, H. Peterlik and O. Paris, Carbon, 2006, 44, 3239–3246 CrossRef CAS.
- N. Hellgren, R. T. Haasch, S. Schmidt, L. Hultman and I. Petrov, Carbon, 2016, 108, 242–252 CrossRef CAS.
- Intergovernmental Panel on Climate Change (IPCC), in Climate Change 2013 – the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2014, pp. 1–30 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- A. V. Neimark, Y. Lin, P. I. Ravikovitch and M. Thommes, Carbon, 2009, 47, 1617–1628 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- C. Ma, X. Shao and D. Cao, J. Mater. Chem., 2012, 22, 8911–8915 RSC.
- S. Li, J. Qiu, C. Lai, M. Ling, H. Zhao and S. Zhang, Nano Energy, 2015, 12, 224–230 CrossRef CAS.
- Y. Gogotsi and R. M. Penner, ACS Nano, 2018, 12, 2081–2083 CrossRef CAS PubMed.
- F. Zhang, K.-X. Wang, G.-D. Li and J.-S. Chen, Electrochem. Commun., 2009, 11, 130–133 CrossRef CAS.
- M. Nikpour, B. A. Mazzeo and D. R. Wheeler, J. Electrochem. Soc., 2021, 168, 120518 CrossRef CAS.
- J. Zhang, J. Qiao, K. Sun and Z. Wang, Particuology, 2022, 61, 18–29 CrossRef CAS.
- D. Jang, S. Suh, H. Yoon, J. Kim, H. Kim, J. Baek and H.-J. Kim, Appl. Surf. Sci. Adv., 2021, 6, 100168 CrossRef.
- Y. Mao, H. Duan, B. Xu, L. Zhang, Y. Hu, C. Zhao, Z. Wang, L. Chen and Y. Yang, Energy Environ. Sci., 2012, 5, 7950–7955 RSC.
- Y. Tang, J. Chen, Z. Mao, C. Roth and D. Wang, Carbon Energy, 2023, 5, e257 CrossRef CAS.
- A. Gomez-Martin, J. Martinez-Fernandez, M. Ruttert, M. Winter, T. Placke and J. Ramirez-Rico, Carbon, 2020, 164, 261–271 CrossRef CAS.
- L. Zhang, X. Yang, F. Zhang, G. Long, T. Zhang, K. Leng, Y. Zhang, Y. Huang, Y. Ma, M. Zhang and Y. Chen, J. Am. Chem. Soc., 2013, 135, 5921–5929 CrossRef CAS PubMed.
- D. Kang, H.-K. Kim, H.-J. Kim and Y. Han, J. Alloys Compd., 2022, 900, 163420 CrossRef CAS.
- Y. C. Lee and S. C. Jung, Nanoscale Adv., 2022, 4, 5378–5391 RSC.
- D. Yan, D.-Q. Wang, X.-L. Dong and W.-C. Li, ACS Appl. Energy Mater., 2022, 5, 13511–13519 CrossRef CAS.
- X. Yang, C. Li, G. Zhang and C. Yang, J. Mater. Sci., 2015, 50, 6649–6655 CrossRef CAS.
- J. Ye, A. C. Baumgaertel, Y. M. Wang, J. Biener and M. M. Biener, ACS Nano, 2015, 9, 2194–2202 CrossRef CAS PubMed.
- X. Zhao, Y. Chen, H. Sun, T. Yuan, Y. Gong, X. Liu and T. Chen, Batteries, 2023, 9, 226 CrossRef CAS.
- J. Dong, Y. Xue, C. Zhang, Q. Weng, P. Dai, Y. Yang, M. Zhou, C. Li, Q. Cui, X. Kang, C. Tang, Y. Bando, D. Golberg and X. Wang, Adv. Mater., 2017, 29, 1603692 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00427b |
| This journal is © The Royal Society of Chemistry 2024 |