
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic design of active and stable bimetallic PtTi nanoparticle electrocatalysts for efficient oxygen reduction at fuel cell cathodes†
Antonia
Herzog‡
 a,
Stefanie
Kühl§
a,
Jiasheng
Lu
a,
Raffaele
Amitrano
a,
Sören
Selve
b,
Johannes
Schmidt
c,
Thomas
Merzdorf
a and
Peter
Strasser
*a
a,
Stefanie
Kühl§
a,
Jiasheng
Lu
a,
Raffaele
Amitrano
a,
Sören
Selve
b,
Johannes
Schmidt
c,
Thomas
Merzdorf
a and
Peter
Strasser
*a
aDepartment of Chemistry, Chemical Engineering Division, Technical University Berlin, 10623 Berlin, Germany. E-mail: pstrasser@tu-berlin.de
bCenter for Electron Microscopy (ZELMI), Technical University Berlin, 10623 Berlin, Germany
cDepartment of Chemistry, Functional Materials, Technical University Berlin, 10623 Berlin, Germany
First published on 14th August 2024
Abstract
We explore and utilize correlations between wet-chemical synthesis parameters and the resulting atomic geometry and crystal phase structure of carbon-supported bimetallic PtTi/C alloy nanoparticles with excellent electrocatalytic reactivity and chemical stability during the oxygen reduction reaction in acidic electrolyte environments. We systematically vary wet-chemical synthesis parameters such as reductive annealing temperatures and precursor ratios and study their effect on the characteristics of atomic-scale materials, such as phase structure, crystallite particle size distribution, alloying degree, and ordering degree, using X-ray scattering, spectroscopy and high-resolution electron microscopy coupled to structure and crystal phase modeling and deconvolution using Rietveld techniques. While annealing parameters controlled the ratio of ordered and disordered PtTi alloy phases, precursor ratio adjustment revealed a previously elusive critical Ti threshold ratio, where the formation of undesired TiO2 phases remained suppressed, while the PtTi alloying degree and phase structure remained preserved. The resulting ca. 3 nm sized bimetallic PtTi nanoparticle electrocatalyst showed excellent Pt mass-based oxygen reduction reaction activity at 0.9 VRHE in acidic environments as well as favorable performance and compositional stability during accelerated stress tests relative to a Pt reference catalyst. This behavior could be linked to the stable PtTi and intermetallic Pt3Ti phases. The synthetic conditions uncovered herein offer wet-chemical access to TiO2-free high performance PtTi nanoparticle alloy ORR catalysts for fuel cell cathodes.
Introduction
Proton-exchange membrane fuel cells (PEMFCs) hold great promise as an environmentally friendly power source for transportation, stationary and portable applications by converting hydrogen into electricity. Today's PEMFCs operating under acidic conditions have relatively low operating temperatures, a high power density and quick-start and on–off cycling characteristics.1 Despite these advantages, current PEMFCs face challenges in catalyst performance. Specifically, improving platinum (Pt) utilization and durability necessitates the development of more reactive, cost-effective, and durable cathode electrocatalysts. It is essential to address the substantial overpotential losses associated with the oxygen reduction reaction (ORR), which is known for its sluggish kinetics.2–5 While Pt-based catalysts are favored for their high activity and stability, their high cost and scarcity drive the exploration of alternatives like Pt–metal alloys, including PtNi, PtCo, PtFe, and PtCu, as well as non-precious metal catalysts like transition metal–nitrogen–carbon (M–N–C) complexes.6–11 Current research focuses on enhancing the electrocatalytic activity of Pt alloys by weakening the binding energies of chemisorbed Oad and OHad intermediates to the nanoparticle surface, which has been attributed to both ligand and strain effects on the basis of the metal d-band model.12Under acidic conditions, the performance of ORR catalysts is especially critical due to the environment causing catalyst degradation. A major challenge with using Pt alloys that include significant amounts of platinum group metal (PGM)-free 3d metals is the corrosive surface leaching of these 3d metals from the alloy. This leaching, which also affects Pt to a lesser extent, occurs during the preparation of the catalyst ink, the coating of the catalyst layer, and the operation of the PEMFC. Leached metal ions can then poison ionomer sulfonate groups, which no longer contribute to proton transport. Leached metal ions may also be reduced inside the membrane or even on the anode, depending on their nobility and the detailed operation conditions of the PEMFC. As a result, the catalytic performance of alloy nanoparticle cathodes and the overall performance of the entire single PEMFC tend to decay over time,13 with the cathode losing its initial reactivity benefits due to ligand and strain effects.14 Therefore, mitigating metal leaching from PEMFC cathodes during the entire PEMFC life cycle is a scientific and technical priority.
To overcome the cathode leaching challenge, Pt alloys with highly corrosion-stable alloy components, such as valve metals, in particular PtTi alloys, have been suggested as promising stable cathode electrocatalysts.15,16 Pt and Ti are known for their good corrosion resistance over a wide range of chemical environments. Also, the strong Lewis acid/base interaction between Pt and Ti creates a large negative enthalpy of formation, resulting in strong chemical bonds between Pt and Ti in PtTi alloys.17 In this way, PtTi alloys could concomitantly benefit from reduced 3d-metal leaching and Pt dissolution, especially at high electrode potentials.13 He et al.18 conducted a high-throughput combinatorial screening of Pt alloy ORR cathodes to study the relationship between activity and stability in various Pt alloy films. Their findings indicated that the PtTi alloy exhibited modest improvements in catalytic activity compared to pure Pt, while maintaining good chemical stability. Disordered Pt75Ti25 alloy nanoparticles supported on carbon were reported with a twofold catalytic ORR reactivity improvement over a commercial Pt/C catalyst.19 Additionally, ordered intermetallic PtxTiy structures could lead to a further stability benefit over randomly disordered alloy structures due to their enthalpy of alloy formation.16,20 Moreover, theoretical investigations using density functional theory (DFT) have been performed on core–shell nanoparticles with pure Pt as the outermost monolayer and a Pt3Ti bulk crystal structure in the layers beneath to study clusters up to sizes of 1.7 nm.21 It was found that a downshift in the d-center occurs, leading to more occupied orbitals, which result in weaker binding linked to improved ORR kinetics in PEMFCs. However, despite all these activity and stability benefits, an important caveat regarding Ti-containing fuel cell cathode catalysts consists of the critically required absence of TiO2 phases that were shown to become Fenton active reagents upon Ti ion leaching.22 Hence, PtTi bimetallic ORR catalysts must be designed and synthesized in the absence of separate TiO2 phases, which is a synthetic challenge as far as wet chemical routes are concerned. Generally, the extreme oxophilicity and low electronegativity of the early 3d transition metals make it rather challenging to synthesize PtTi alloy nanoparticles with small particle sizes using scalable wet-chemical techniques. Apart from methods based on melting, gas-phase deposition and reduction,20,23,24 or mechanical alloying of raw metals,20,25–28 only a few research groups succeeded in the preparation of PtTi alloy nanoparticles via wet chemical reduction of metal salt precursors under an inert gas atmosphere.19,24,29 However, PtTi nanoparticle agglomeration during the thermal annealing process at higher temperatures limits the usefulness of many of the previous synthesis techniques. In order to address the aggregation of nanoparticles, Pt3Ti alloy nanoparticles were synthesized at 700 °C in a surfactant-free-KCl matrix30 or sulphur anchoring was used to limit particle aggregation.31 The present work addresses the insufficient deconvolution of the resulting Ti phase structure of PtTi alloys, in particular with respect to the formation of a separate nanocrystalline Ti2O phase that could result in Ti ion leaching under electrochemical ORR test conditions.
In this study, we explore the synthesis–structure–reactivity relationships of bimetallic PtTi alloy ORR electrocatalysts in acidic environments for their use and relevance as cathode catalysts for PEMFCs. We investigate the preparation and formation processes of PtTi alloys at varying synthesis parameters during wet impregnation and post-synthetic thermal annealing. First, the effect of the catalyst reduction temperature during thermal annealing on the phase structure, particle morphology and particle reactivity of the resulting PtTi alloys was analyzed. We demonstrate how the reduction temperature controls the formation of distinct PtTi face centered cubic (fcc) and intermetallic ordered Pt3Ti phases in different ratios. We also explored the effect of the initial Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ti molar precursor ratio on the resulting phase composition. We succeeded in suppressing the formation of TiO2 phases and determined the maximum possible Ti ratio that can be incorporated into Pt to form PtTi alloy phases. These PtTi/C catalysts showed unprecedented electrocatalytic ORR performances compared to previous PtTi particles, combined with good performance stability.
:Ti molar precursor ratio on the resulting phase composition. We succeeded in suppressing the formation of TiO2 phases and determined the maximum possible Ti ratio that can be incorporated into Pt to form PtTi alloy phases. These PtTi/C catalysts showed unprecedented electrocatalytic ORR performances compared to previous PtTi particles, combined with good performance stability.
Results and discussion
PtTi/C catalysts with different crystal structures and Ti contents
The fabricated catalysts were characterized by inductively coupled plasma-optical emission spectroscopy (ICP-OES), transmission electron microscopy (TEM), scanning TEM (STEM), high-resolution TEM (HRTEM), energy dispersive X-ray spectroscopy (EDX), and X-ray photoelectron spectroscopy (XPS) analysis. Additionally, X-ray diffraction (XRD) combined with Rietveld quantitative phase analysis was applied for the investigation of the phase composition as well as structural parameters like the crystallite size and lattice constant.
Fig. 1 displays the TPR profiles of the PtTiOx/C composite and the as-received catalyst Pt/C obtained between 210 °C and 850 °C under a flow of 4 vol% H2 in Ar. The TPR profile of the PtTiOx/C composite shows two H2-consumption peaks with maxima at 265 °C and 432 °C. By contrast, the TPR profile of the as-received Pt/C catalyst features only a broad peak with a maximum at around 550 °C and weaker intensity. In accordance with prior literature, the first H2-consumption peak of PtTiOx/C is likely related to the reduction of PtOx crystallites to metallic Pt.34,35 Accordingly, the second H2-consumption peak at 432 °C is assigned to the reduction of TiOx/TiO2 to metallic Ti. While the reduction of bulk TiO2 occurs above 600 °C,35 the present results suggest that the presence of Pt may facilitate the reduction of TiOx nano-crystallites, in agreement with reports on Pt/TiO2 particles.34 Furthermore, the reduction profile shows a significant shoulder in the second peak at around 350 °C, indicating a two-step reduction process of TiO2, which is explainable by the reduction of Ti4+ to Ti2+via intermediate Ti3+, resulting in a TiOx compound (x < 2).35 On the other hand, the shoulder might also be associated with the reduction of Ti species interacting with the Pt–TiOx interface site.36 However, the TiOx/TiO2 reduction starts at around 300 °C and reaches a maximum rate of reduction at 432 °C before it terminates at around 800 °C. We note that in the present material system, a previously suggested reduction temperature of 700 °C24 did not correspond to any characteristic threshold in the TPR profiles. It is feasible that larger H2 flow rates may favor faster reduction and consequently lower temperatures of complete reduction.
 | ||
| Fig. 1 Temperature-programmed reduction profiles of PtTiOx/C and Pt/C recorded at a heating rate of 10 K min−1 in 4 vol% H2/Ar. TCD = thermal conductivity detector. | ||
The results of the presented TPR measurements guided the temperature selection for the subsequent reductive annealing steps: 430 °C (peak maximum of the reduction of TiOx), 700 °C (close to the end of reduction of TiOx) and 800 °C (end of reduction of TiOx). At the lowest reduction temperature (430 °C), the reductive annealing time was extended to 16 h (four times longer than at 700 and 800 °C) to prevent incomplete reduction. The resulting three distinct catalysts are henceforth labeled “PtTi/C-T”, where T represents their respective reductive annealing temperature. For the three obtained catalyst samples, ICP-OES analysis revealed a similar actual atomic Pt:Ti composition of about 40 at% Pt and 60 at% Ti (Pt60Ti40) with a Pt weight loading of ∼24 wt% on the carbon support (see Table S1†). An explanation for the slight change in the metal loading of the different PtTi/C catalysts compared to Pt/C is given on page 2 of the ESI.†
The crystalline phase structure of the catalysts after reductive annealing was analyzed by XRD and compared to that of the as-received Pt/C catalyst, as illustrated in Fig. 2. The XRD pattern of the as-received Pt/C catalyst closely matches that of the pure Pt face centered cubic phase (fcc, PDF card #4-0802, space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Additionally, the XRD pattern of the impregnated PtTiOx/C composite before thermal annealing in Fig. S1† is very similar to that of the as-received catalyst, since the TiOx layer is only coated on the surface of the Pt particles. In the case of the annealed PtTi/C catalysts, the reflections of the fcc phase are slightly shifted towards higher angles compared to Pt/C as indicated for (111) in Fig. 2. Furthermore, the (222) reflex of the fcc plane becomes visible at ∼86° 2θ due to a more defined crystalline structure after annealing. The shift in the peak position can be taken as a clear indication of the formation of a PtTi alloy, as the incorporation of smaller Ti atoms into the Pt fcc lattice causes a contraction of the Pt fcc lattice. The patterns of the reduction temperatures of 430 and 700 °C predominately display an fcc phase. At the highest reduction temperature of 800 °C, an ordered intermetallic Pt3Ti phase (PDF card #03-065-3259, space group Pmm) could be identified by the superlattice diffraction peaks at 22.9° (100), 32.5° (110), 52.5° (210), 57.9° (211), 72.7° (300) and 77.6° 2θ (310). In fact, the pattern of PtTi/C-700 °C suggests the presence of intermetallic Pt3Ti phases, showing a slight bump at the (110) peak position at 33.3° 2θ. The more pronounced formation of the Pt3Ti phase at higher reduction temperatures is in accordance with the study of Ding et al.19 Besides, the XRD patterns of the alloys treated at higher reduction temperatures demonstrate a significant decline of the peak width, which indicates significant particle growth. Such particle growth can be attributed on one hand to the incorporation of Ti atoms in the Pt lattice, and on the other hand to a thermal effect since the thermally annealed Pt/C-700 °C catalyst also shows significant particle growth; see the XRD pattern of Pt/C-700 °C in Fig. S2† and the corresponding crystallite sizes in Table S2† for details. Additionally, all PtTi/C catalysts showed hints of a coexisting anatase TiO2 phase (PDF card #01-089-4921) with small reflexes at 25.4°, 54.5° and 62.9° 2θ. Furthermore, the reflection at 25.5° 2θ in all patterns corresponds to the (002) lattice plane of graphitic carbon from the support (carbon black) – most visible in the pattern of Pt/C.
m). Additionally, the XRD pattern of the impregnated PtTiOx/C composite before thermal annealing in Fig. S1† is very similar to that of the as-received catalyst, since the TiOx layer is only coated on the surface of the Pt particles. In the case of the annealed PtTi/C catalysts, the reflections of the fcc phase are slightly shifted towards higher angles compared to Pt/C as indicated for (111) in Fig. 2. Furthermore, the (222) reflex of the fcc plane becomes visible at ∼86° 2θ due to a more defined crystalline structure after annealing. The shift in the peak position can be taken as a clear indication of the formation of a PtTi alloy, as the incorporation of smaller Ti atoms into the Pt fcc lattice causes a contraction of the Pt fcc lattice. The patterns of the reduction temperatures of 430 and 700 °C predominately display an fcc phase. At the highest reduction temperature of 800 °C, an ordered intermetallic Pt3Ti phase (PDF card #03-065-3259, space group Pmm) could be identified by the superlattice diffraction peaks at 22.9° (100), 32.5° (110), 52.5° (210), 57.9° (211), 72.7° (300) and 77.6° 2θ (310). In fact, the pattern of PtTi/C-700 °C suggests the presence of intermetallic Pt3Ti phases, showing a slight bump at the (110) peak position at 33.3° 2θ. The more pronounced formation of the Pt3Ti phase at higher reduction temperatures is in accordance with the study of Ding et al.19 Besides, the XRD patterns of the alloys treated at higher reduction temperatures demonstrate a significant decline of the peak width, which indicates significant particle growth. Such particle growth can be attributed on one hand to the incorporation of Ti atoms in the Pt lattice, and on the other hand to a thermal effect since the thermally annealed Pt/C-700 °C catalyst also shows significant particle growth; see the XRD pattern of Pt/C-700 °C in Fig. S2† and the corresponding crystallite sizes in Table S2† for details. Additionally, all PtTi/C catalysts showed hints of a coexisting anatase TiO2 phase (PDF card #01-089-4921) with small reflexes at 25.4°, 54.5° and 62.9° 2θ. Furthermore, the reflection at 25.5° 2θ in all patterns corresponds to the (002) lattice plane of graphitic carbon from the support (carbon black) – most visible in the pattern of Pt/C.
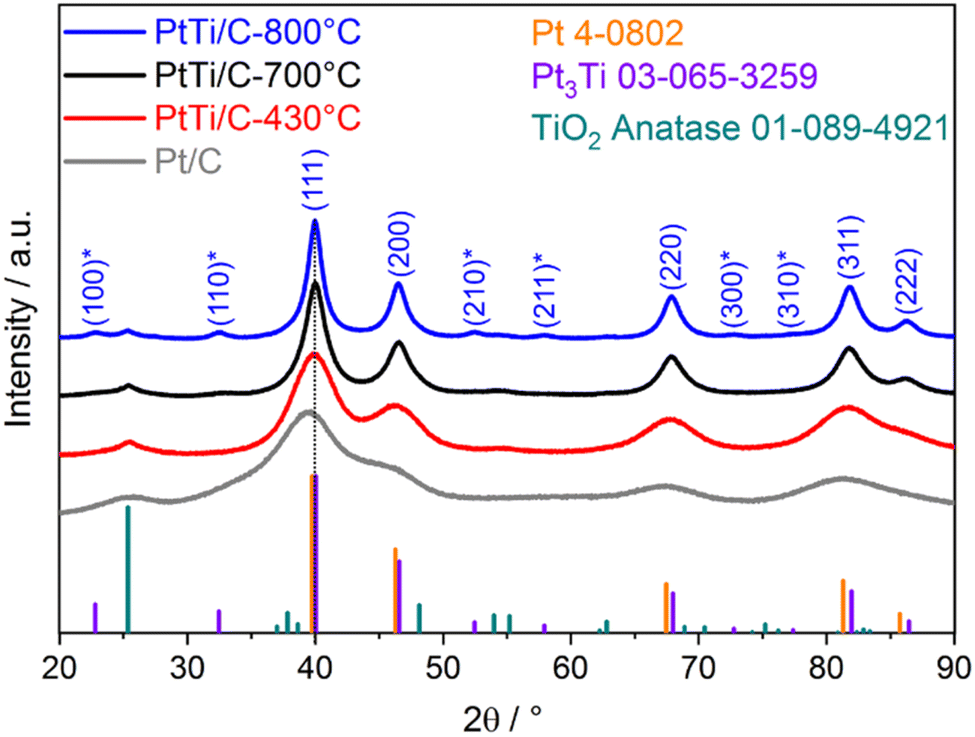 | ||
| Fig. 2 XRD patterns of the three PtTi/C catalysts as a function of reductive annealing temperature. The reference diffraction patterns of the different crystal structures are displayed as colored bars at the bottom. The indices of the crystal planes correspond to the intermetallic Pt3Ti phase, where * denotes the superlattice planes. The dotted line at the (111) reflection illustrates the shift between the patterns. | ||
Rietveld refinement of each pattern revealed more intricate details of the structural changes upon reductive annealing. The pattern deconvolution and refinement are exemplarily shown for PtTi/C-700 °C in Fig. S3.†
First, Rietveld analysis confirmed the presence of the corresponding phases described above, with PtTi fcc, TiO2 anatase and graphite present in all three PtTi/C samples. As already suspected, the intermetallic Pt3Ti is present after reduction at 700 and 800 °C. The resulting Rietveld-derived experimental phase composition (with the graphite phase subtracted) is presented in Fig. 3a. The analysis revealed a decreasing proportion of PtTi fcc at higher reduction temperatures, while the proportion of the intermetallic Pt3Ti phase increased from 700 to 800 °C. This indicates that Pt3Ti might be the thermodynamically more stable phase compared to PtTi fcc. The binary Ti–Pt phase diagram in Fig. S4† also indicates the formation of PtTi phases at lower temperatures, starting from 600 °C, whereas the Pt3Ti phase forms only at higher temperatures, as reported in the literature for Pt3Ti nanoparticles.37 Besides, all PtTi/C catalysts possess a considerable proportion of a TiO2 phase in the form of anatase, which only slightly increases at higher reduction temperatures. The formation of TiO2 is likely due to an excess of the Ti precursor in all catalysts. The prevalence of anatase over rutile TiO2 aligns with the composite phase diagram for TiO2,38 and the anatase phase has been observed to remain stable over a wide temperature range up to 1100 °C for TiO2 nanoparticles.39
 | ||
| Fig. 3 Structural impact of annealing temperature as derived from Rietveld refinement: (a) phase composition with the graphite phase subtracted, (b) lattice constant and (c) crystallite size (LVol-B) of Pt(Ti) fcc (orange) and Pt3Ti (violet). Dashed lines are a guide to the eye. | ||
All PtTi/C samples have a smaller lattice constant of the fcc phase compared to Pt/C (Fig. 3b), indicating a lattice contraction due to alloying of Ti with Pt, as already observed from the peak shift in the XRD patterns. The lattice constant of the fcc phase decreased from 3.93 Å in Pt/C to 3.91 Å in PtTi-430 °C, while it further decreased to 3.895 Å at the higher reduction temperatures of 700 °C and 800 °C. Consequently, Ti was only partially alloyed with Pt at 430 °C, while a maximum amount of alloyed PtTi was obtained at 700 °C, since the lattice constant and, thus, the alloying grade at the higher reduction temperature of 800 °C remained constant. Moreover, the lattice constant of Pt3Ti at 700 and 800 °C is in the same range as that of the fcc phase and agrees well with the experimental value of 3.897 Å of Pt3Ti in the literature.30 The detailed values of the structural parameters are given in Table S3.†
In Fig. 3c the mean crystallite sizes of the two PtTi phases are plotted, where PtTi/C-430 °C shows only a slightly bigger size than Pt/C. The size of the fcc phase increased significantly from 430 to 700 °C, while the sizes of PtTi/C-700 °C and PtTi/C-800 °C are similar. In contrast, the crystallite sizes of the intermetallic Pt3Ti phase substantially increased from 700 to 800 °C. This implies that the formation of bigger Pt3Ti crystallites was favored at a reduction temperature of 800 °C, corresponding to its increased proportion.
All annealed catalysts were investigated by TEM to study the morphology and changes in the mean nanoparticle size upon annealing. Representative TEM micrographs of the electrocatalysts (Fig. 4a–c) demonstrated a homogeneous distribution of the PtTi nanoparticles on the carbon support, with nearly spherical shapes at each annealing temperature. No agglomerated particles could be found before annealing and at 430 °C, while only a few agglomerates appeared at higher annealing temperatures of 700 and 800 °C. However, the TEM-based particle size increased upon annealing (Fig. 4d); the particle size distributions are depicted in Fig. S5.† The particles at the lowest reduction temperature of 430 °C show an average particle size of 2.1 ± 0.2 nm, which is comparable with 2.2 ± 0.2 nm of the as-received Pt/C catalyst (see the TEM image and size distribution in Fig. S6†), in accordance with the XRD results. At 700 °C, the particle size increased to 2.7 ± 0.3 nm, which is attributed to particle growth/agglomeration during thermal annealing as well as of the higher introduction of Ti into the Pt particles. At the highest reduction temperature of 800 °C, the particle size increased further to 3.3 ± 0.4 nm, which can be considered as a pure thermal effect, since Ti was not further alloyed with Pt at 800 °C, as revealed by Rietveld refinement (Fig. 3b). Interestingly, the comparison of the TEM average sizes to the crystallite sizes of PtTi and Pt3Ti obtained by Rietveld refinement shows that the TEM-derived particle sizes represent the average size of the crystallites. Consequently, the nanoparticles appear to exist largely as separate crystallites of each phase.
 | ||
| Fig. 4 (a–c) TEM micrographs of PtTi/C samples annealed at different temperatures; (d) particle sizes as a function of annealing temperature – error bars represent the calculated error of each particle size. | ||
STEM and EDX mapping in Fig. S7a and b† illustrate the spatial distribution and morphology of the Pt(Ti) and TiO2 phases within the PtTi/C-700 °C sample on the carbon support in greater detail. Adjacent to the Pt(Ti) particles, the carbon support is substantially covered with large flakes and particles of TiO2. Additionally, TiO2 partially covers some of the Pt(Ti) particles. High-resolution TEM (HRTEM) images in Fig. S7c and d† further highlight the distinct lattice planes of the various particles, revealing large TiO2 particles of around 4 nm in size, consistent with the crystallite size of 4.1 nm obtained from XRD analysis (Table S3†). The significant presence and formation of TiO2 may hinder the accessibility of the PtTi particles and obstruct the carbon support for the electrochemical measurements. Fast Fourier Transform (FFT) analysis of selected areas of the HRTEM image in Fig. S8† further reveals the characteristic lattice planes of fcc Pt(Ti) and TiO2 anatase. This aligns with the corresponding EDX analysis of a large particle area, which shows a high Ti to Pt ratio of 70:30 at% due to the presence of TiO2 particle (Fig. S7e†). In contrast, the EDX quantification of individual Pt particles reveals varying amounts of Ti, ranging from 7 to 20 at% within the Pt(Ti) particles (Fig. S9†).
We can conclude from this detailed structural study that a reduction temperature of 700 °C can be used to fully alloy Ti with Pt without exhibiting considerable particle growth. Thus, it will be further utilized as the reduction temperature.
:Ti composition ratio constant. These catalysts contained, in addition to two distinct PtTi phases, an additional TiO2 phase in the form of anatase, which was probably formed due to an excess of the initial Ti precursor. In order to adjust the Ti composition towards a maximum amount of incorporated Ti in the PtTi lattice without forming a prominent TiO2 phase, catalysts with different Pt:Ti compositions were prepared by reducing the initial amount of Ti precursor during the wet impregnation, followed by constant annealing conditions at 700 °C. The resulting catalysts are henceforth labeled “PtTi/C-x”, with x being the respective factor of the amount of initial Ti precursor compared to PtTi/C-700 °C. Therefore, the PtTi/C-700 °C reference catalyst is hereafter referred to as PtTi/C-1.0. Further details of the initial amounts of Pt and Ti are listed in Table S4†.
ICP-OES analysis (Fig. 5a) revealed a clear correlation between the resulting Pt:Ti atomic composition and the initial Ti amount during wet impregnation; the actual atomic Pt:Ti composition varied stepwise from 59 at% Pt and 41 at% Ti in PtTi/C-0.5 to 42 at% Pt and 58 at% Ti in PtTi/C-1.0.
 | ||
| Fig. 5 Compositional and structural properties as a function of the initial Ti amount: (a) PtTi composition; (b) XRD patterns with the reference powder diffraction patterns of the different crystal structures displayed as colored bars in the bottom; (c) phase distribution as derived from Rietveld refinement with the graphite phase subtracted; (d) crystallite size (LVol-B) of PtTi fcc (orange), Pt3Ti (violet) and TiO2 (blue) – dashed lines are a guide to the eye. | ||
The crystalline structure of the resulting catalysts was analyzed by XRD, and the patterns are depicted in Fig. 5b. All patterns are clearly described by an PtTi fcc structure, an intermetallic Pt3Ti phase at the (110) peak position at 33.3° 2θ and a little bump of graphite (002) at around 25° 2θ. The TiO2 anatase phase becomes evident for PtTi/C-1.0.
Using Rietveld refinement, the structural changes upon variation of the Ti amount were analyzed. The refinement deconvolution of PtTi/C-0.65 is presented in Fig. S10† and the detailed values of the structural parameters are given in Table S5.† In Fig. 5c, the Rietveld-derived experimental phase composition (graphite phase subtracted) is presented. The results show that the patterns of PtTi/C-0.5 and −0.65 are fully accounted for by a PtTi fcc and a Pt3Ti phase, whereas the formation of an anatase phase was still observed in PtTi/C-0.75 with a phase content of 13 wt%, which increased to 31 wt% in the reference PtTi/C-1.0. If the Ti amount was even further increased to a factor of 1.5, the formation of a rutile phase alongside to an anatase phase could be observed in the XRD pattern of PtTi/C-1.5 (Fig. S11†). Thus, a higher amount of a TiOx coating on Pt/C favored the conversion to rutile during thermal annealing at 700 °C, which usually takes place at temperatures between 550 °C and about 1000 °C and strongly depends on the impurities or dopants and on the morphology of the sample.40
The Rietveld results further suggested that the ratio between PtTi fcc and the intermetallic Pt3Ti phase (∼70:30 wt%) was not significantly affected by the employed Ti amount. Furthermore, the lattice constants of all samples remained almost the same (Table S5†), showing no clear indication of a higher alloying grade with increasing Ti amount.
Also, the crystallite sizes of the PtTi and Pt3Ti phases, as illustrated in Fig. 5d, evidenced only slight variations with increasing Ti amount, with PtTi fcc slightly trending toward smaller sizes and Pt3Ti towards larger sizes. In all cases, the PtTi fcc phase exhibited a larger crystallite size in the range of 3.3 to 3.9 nm than the intermetallic Pt3Ti phase in the range of 1.6 to 2.2 nm. Additionally, the crystallite size of TiO2 remains constant at around 4.0 nm.
From the structural analysis, we can conclude that excess Ti amounts in the PtTi/C-0.75 and −1.0 catalysts were to a large extent not alloyed with Pt. This is supported by the relative proportion between PtTi fcc and Pt3Ti phases and their consistent lattice constants, as well as by the increasing proportion of TiO2 phases upon increasing Ti amount and the observation of large TiO2 particles in PtTi/C-1.0 in the STEM and EDX mapping (Fig. S7a and b†). Excess Ti leading to the formation of the undesired anatase TiO2 phase was observed starting with the PtTi/C-0.75 catalyst and was further increased for the PtTi/C-1.0 catalyst. Even though the experimental lattice constants cannot ensure a higher alloying degree of Ti in the PtTi/C-0.65 catalyst compared to the PtTi/C-0.5 catalyst, PtTi/C-0.65 can be considered as the catalyst with the maximum alloyed Ti, since it contains the largest Ti content with no crystalline TiO2 phase.
The XPS spectra of PtTi/C with varying initial Ti amounts, as shown in Fig. S12,† offer deeper insights into the electronic structure of the catalysts. The Pt 4f7/2 peak for all PtTi/C catalysts is centered at 71.4 eV, compared to 71.1 eV for pure Pt/C,41 indicating potential Pt–Ti alloying.37,42 For Ti, the Ti 2p3/2 peak, typically around 459 eV for Ti4+, shifts to lower binding energies in the 0.5–0.75 Ti samples, confirming Pt–Ti alloying aligning with the XRD results.43 In contrast, higher initial Ti loadings shift the Ti 2p3/2 peak to higher binding energies, indicating increased TiO2 formation and decreased alloying. Additionally, a small peak at 455.2 eV indicates metallic Ti from Pt3Ti and is observed in the 0.5–0.75 Ti samples.37,43
Furthermore, the structurally most favorable catalyst PtTi/C-0.65 with its adjusted initial Ti content was investigated in more detail using TEM to study the morphology and changes in the mean nanoparticle size. A representative TEM micrograph with the corresponding size distribution can be found in Fig. S13,† demonstrating homogeneously distributed PtTi nanoparticles with a mean diameter of 2.8 ± 0.3 nm and nearly spherical shapes. Surprisingly, these morphological results of PtTi/C-0.65 revealed almost no difference to PtTi/C-1.0 (see PtTi/C-700 °C in the annealing study), suggesting that the varying Ti content did not have any influence on the morphology and sizes, since the particles should have the same amount of alloyed Ti as revealed by XRD.
STEM and EDX mapping of the PtTi/C-0.65 sample in Fig. S14† reveal the distribution of Pt(Ti) particles on the carbon support, with only a minor presence of small amorphous TiO2 particles (∼2 nm) detected. These small TiO2 particles are less likely to significantly block the active sites of the PtTi catalyst and to decrease the electrochemical surface area of the support compared to the large crystalline TiO2 particles in PtTi/C-1.0 (Fig. S7a and b†). Additionally, HRTEM images in Fig. S15 and S16† confirm the fcc structure of PtTi and the intermetallic structure of Pt3Ti, as evidenced by the reflections from FFT analysis.
We conclude that by adjusting the initial Ti amount at a reduction temperature of 700 °C, it is possible to prepare a structurally favorable electrocatalyst, PtTi/C-0.65, with a maximum amount of Ti being alloyed in the PtTi and Pt3Ti phases without the major formation of undesired TiO2 crystallites.
Electrocatalytic ORR activity and stability
 | ||
| Fig. 6 (a) ECSA and (b) Pt mass activity of PtTi/C-0.5, −0.65 and −1.0 after 25 (black) for 1000 (green) and 10000 (red) voltage cycles – values calculated from relative stabilities. Pt/C mass activity after 25 voltage cycles is given for comparison. | ||
The electrocatalytic activity for the reduction of molecular oxygen to water (ORR) was measured by anodic linear sweep voltammetry (LSV). The detailed experimental LSV curve of the PtTi/C-1.0 catalyst compared to the as-received and annealed Pt/C reference catalysts is plotted in Fig. S17c.† An evaluation of the reproducibility and error associated with the RDE tests of the catalyst ink is provided in section 20 of the ESI.† All activated PtTi/C catalysts showed a significantly enhanced Pt mass-based activity compared to the as-received Pt/C with a four-fold higher activity (Fig. 6b and S17d†). Furthermore, the measured activity of the PtTi/C catalysts exceeded the values of comparable PtTi/C catalysts reported in the literature.19,24 When adjusting the Ti amount from PtTi/C-0.5 and from PtTi/C-1.0 to PtTi/C-0.65, the resulting Pt mass activity at 0.9 VRHE increased from 0.63 and from 0.59 to 0.68 A mgPt−1, respectively (Fig. 6b). Overall, the difference in activity is most probably related to the increased amount of TiO2 species on the particles, which is also reflected by the lower ECSA of PtTi/C-1.0 as well as the increased amount of TiO2 on the carbon support and the particles (Fig. S7†).
Additionally, the ORR performance at annealing temperatures of 430, 700, and 800 °C was assessed and correlated with TEM particle sizes (Fig. S18†). As the annealing temperature increased, the ECSA decreased due to particle growth. The mass activity significantly increased for the catalysts treated at 700 °C and 800 °C compared to the 430 °C treatment with only 0.41 A mgPt−1, indicating enhanced activity from Pt–Ti alloying. The 800 °C-treated alloy showed slightly lower activity than the 700 °C-treated one, likely due to increased particle size as also observed for bare Pt particles44 and additional effects from the presence of Pt3Ti and TiOx/TiO2. Therefore, the subsequent stability measurements will focus on the best performing 700 °C-treated PtTi/C catalyst using different initial Ti amounts.
000 (10k) voltage cycles in N2-saturated 0.1 M HClO4 solution. In doing so, the potential range was set from nearly the maximum power, approaching full load conditions (+0.60 VRHE) of fuel cell applications, to nearly open circuit voltage (+0.95 VRHE), approaching no-load conditions. This potential range is suggested by the Department of Energy (DOE) to investigate catalyst stability.45 The corresponding CVs and LSVs of PtTi/C-0.65 are presented in Fig. S20a–d.†
The electrochemical stability of the catalysts is illustrated in Fig. 6. These values were calculated from the relative change of the absolute experimental values, which tend to vary in terms of their initial activities at the onset of the electrochemical stability measurements.
The ECSA values of the catalysts changed only slightly during voltage cycling (Fig. 6a). Furthermore, all PtTi/C catalysts showed a similar loss of Pt mass activity during voltage cycling. After 1000 cycles of AST, the Pt mass activity decreased by ∼20%, while after 10000 cycles of AST, it decreased by ∼35%. However, the electrochemical stability of the present PtTi/C catalysts was substantially superior to that previously reported in the literature, where the activity decreased by more than 50% after applying 10000 voltage cycles.24 Furthermore, the Pt mass activity of the PtTi/C catalysts after 10000 cycles was more than twofold that of the as-received Pt/C catalyst after only 25 cycles.
To correlate the high stability of the PtTi/C catalysts, we further characterized PtTi/C-0.65 after 10000 voltage cycles of AST. Grazing incidence X-ray diffraction (GIXRD) performed on the catalyst loaded on a RDE (Fig. S21†) revealed high stability of the crystalline phases of PtTi/C-0.65 after the electrochemical stability test, showing even slightly sharper Pt3Ti reflections and a shift of PtTi fcc from 39.9° to 40°, as shown in Fig. S22 and S23.† This behavior of higher Pt–Ti alloying is further confirmed by XPS analysis of PtTi/C-0.65 after the AST (Fig. S24†), showing an increase in the binding energy of Pt 4f7/2 and a decrease in the binding energy of Ti 2p3/2.
For further investigation, analysis of the morphology of PtTi/C-0.65 after 10000 voltage cycles of AST was performed by TEM. A representative TEM image together with the corresponding particle size distribution histogram is presented in Fig. S25.† The average particle size slightly shifted from 2.8 ± 0.3 nm before performing the AST to 3.0 ± 0.4 nm after performing 10000 voltage cycles, probably due to particle growth indicated by the size distribution (Fig. S25†). The observed variations in the particle size were consistent with the changes in the ECSA during voltage cycling, which can lead to a decrease in catalyst performance. Additionally, EDX measurements before and after applying 10000 voltage cycles revealed a decrease in the Ti content of about 20 at%, as shown by the Pt:Ti compositions in Table S7.†
Thus, the high stability of the PtTi/C catalyst can be attributed to the stable intermetallic PtTi and Pt3Ti phases.
Conclusions
We conducted an in-depth study on the synthesis, structure, and catalytic reactivity of disordered and ordered PtTi/C alloy electrocatalysts for the ORR in acidic environments. By meticulously adjusting the reduction temperature and initial Ti precursor amount, we successfully prepared homogeneously distributed, TiO2-free PtTi/C alloy nanoparticles, approximately 3 nm in size, achieving excellent structural, morphological, and catalytic performance characteristics.Using H2-TPR measurements, we selected reduction temperatures for thermal annealing at 700 and 800 °C that revealed PtTi alloying. XRD, Rietveld, XPS, HRTEM, and EDX analysis confirmed the formation of an intermetallic Pt3Ti phase alongside a PtTi fcc phase. Systematic variation of the initial Ti amount did not influence the molar ratios of the resulting PtTi fcc and intermetallic Pt3Ti phases but did affect the formation of an undesired anatase TiO2 phase. This indicates a maximum limit for Ti incorporation into the Pt particles.
The PtTi/C alloys exhibited outstanding catalytic ORR reactivity, achieving up to 0.68 A mgPt−1 at 0.9 VRHE under acidic electrolyte conditions, outperforming typical Pt/C catalysts by a factor of five. Their excellent electrochemical stability, due to the stable fcc PtTi and Pt3Ti phases, makes them highly attractive for high-durability applications. Despite concerns about the Fenton activity of Ti cations leached from TiO2 supports, our fully alloyed PtTi/C nanoparticles, free from TiO2 phases, present a promising new family of PEM fuel cell cathode ORR electrocatalysts. Future validation studies in membrane electrode assemblies (MEAs) are necessary to evaluate the behavior of PtTi/C alloys in realistic environments.
Experimental
Synthesis
Furthermore, the wet impregnation of the commercial Pt/C catalyst was modified with an initial Ti amount of 0.88 mmol using the 0.5- (0.44 mmol), 0.65- (0.57 mmol), 0.75- (0.66 mmol) and 1.5-fold (1.32 mmol) amounts of titanium(IV) butoxide, followed by constant conditions of reductive annealing at 700 °C.
Physical and chemical characterization
:H2SO4:HCl in a 1:1:3 ratio) and heated by microwave irradiation to 180 °C for 20 min, with a ramping step of 10 min. The samples were diluted with Milli-Q water (>18 MΩ cm) to reach appropriate emission intensity. Standards with known concentrations were co-analyzed with the samples.
Electrochemical characterization
Electrochemical measurements were performed in a three-compartment glass cell with a rotating disk electrode (5 mm diameter, glassy carbon, Pine Instrument) and a potentiostat (Biologic) at room temperature. A Pt mesh and a Hg/Hg2SO4 (MMS) electrode (in saturated K2SO4)46,47 were used as the counter electrode and reference electrode, respectively, and inserted into the electrolyte (0.1 M HClO4). All potentials reported in this paper were normalized with respect to the reversible hydrogen electrode (RHE). To accurately calibrate the potentials from E vs. Hg/Hg2SO4 to E vs. RHE, the potentials were experimentally calibrated by flowing H2 over a rotating Pt disk electrode in 0.1 M HClO4, as detailed in the literature.48For the ink preparation, a mixture of 79.6 vol% of ultrapure water, 0.4 vol% of a Nafion solution (5 wt% in isopropyl alcohol) and 20.0 vol% of isopropyl alcohol was added to an exactly determined amount of 2 to 6 mg of the catalyst powder, depending on the Pt loading of the catalyst sample and the desired Pt loading on the GC. After sonification for 30 min, 10 μL of the fabricated ink was drop-cast on the GC electrode surface to obtain a Pt loading of 10, 15 or 20 μgPt cm−2. The electrochemical activation of each catalyst was performed via potential cycling for 25 cycles (+0.05 − +1.0 VRHE with 100 mV s−1) in N2 saturated 0.1 M HClO4. The electrochemically active surface area (ECSA) was determined by averaging the integrated areas of the hydrogen adsorption and desorption peaks of the last cycle using 210 μC cmPt−2.The catalytic activity was measured by linear sweep voltammetry in an oxygen saturated electrolyte (+0.05 − +1.0 VRHE with 5 mV s−1 at 1600 rpm). All presented current densities are iR corrected, where the uncompensated ohmic resistance (R) was determined by potential electrochemical impedance spectroscopy (PEIS) at +0.4 VRHE.
Stability measurements were performed via potential cycling for 1000 or 10000 cycles (+0.6 − +0.95 VRHE with 100 mV s−1) in N2 saturated 0.1 M HClO4. Before and after stability testing, three cyclic voltammograms (+0.05 − +1.0 VRHE with 100 mV s−1) were recorded.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. H., S. K., and P. S. contributed to writing the original manuscript. S. K. and P. S. supervised the study. A. H. designed and performed the sample synthesis in consultation with S. K. and T. M. A. H. also conducted and analyzed the electrocatalytic measurements and performed the XRD measurements, including data analysis. J. L. and R. A. carried out the GIXRD measurements and analysis. S. K. performed the XRD Rietveld analysis. S. K. conducted the TEM measurements, while S. S. performed the STEM, EDX, and HRTEM measurements. J. S. conducted the XPS measurements, and A. H. analyzed the XPS data. P. S. secured the funding. All authors contributed to the discussion and interpretation of the data and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Central Facility for Electron Microscopy, Technical University Berlin (ZELMI) for their support during the TEM, STEM, and HRTEM measurements, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 403371556 – GZ: INST 131/789-1 FUGG. We thank Astrid Müller-Klaucke for the ICP-OES measurement. Financial support was given by the German Ministry of Education and Research (BMBF) via the project “LoPlaKats” (number 03SF0527A). This project has received partial funding from the Clean Hydrogen Partnership under grant agreement No. 101101346 (HIGHLANDER) and grant Agreement No. 10110149 (BRAVA). This joint undertaking received support from the European Union's Horizon 2020 Research and Innovation program, Hydrogen Europe and Hydrogen Europe Research. The views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union. Neither the European Union nor the Clean Hydrogen Joint Undertaking can be held responsible for them.References
- F. Barbir and S. Yazici, Int. J. Energy Res., 2008, 32(5), 369–378 CrossRef CAS.
- R. P. O'Hayre, F. B. Prinz, S.-W. Cha and W. G. Colella, Fuel Cell Fundamentals, Wiley, Hoboken, 3rd edn, 2016, p. 1 Search PubMed.
- P. Strasser, Chem. Ing. Tech., 2009, 81(5), 573–580 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53(1), 102–121 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
- H. Yano, M. Kataoka, H. Yamashita, H. Uchida and M. Watanabe, Langmuir, 2007, 23(11), 6438–6445 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1(7), 552–556 CrossRef CAS PubMed.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129(42), 12624–12625 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasche and P. Strasser, J. Electrochem. Soc., 2012, 159(4), B394–B405 CrossRef CAS.
- M. Oezaslan, F. Hasche and P. Strasser, J. Electrochem. Soc., 2012, 159(4), B444–B454 CrossRef CAS.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, X. Ren, J. Zhang and M. An, J. Mater. Chem. A, 2020, 8(44), 23187–23201 RSC.
- Y.-W. Zhang, Bimetallic Nanostructures: Shape-Controlled Synthesis for Catalysis, Plasmonics, and Sensing Applications, John Wiley & Sons Incorporated, Newark, 2018, p. 1524 Search PubMed.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46(3–4), 285–305 CrossRef CAS.
- L. Gan, C. Cui, S. Rudi and P. Strasser, Top. Catal., 2014, 57(1–4), 236–244 CrossRef CAS.
- European Commission, Hydrogen Energy and Fuel Cells: A Vision of Our Future, Office for Official Publications of the European Communities, Luxembourg, 2003, p. 33, ISBN 92-894-5589-6 Search PubMed.
- H.-Y. Park, T.-Y. Jeon, K.-S. Lee, S. J. Yoo, Y.-E. Sung and J. H. Jang, J. Electrochem. Sci. Technol., 2016, 7(4), 269–276 CrossRef CAS.
- M. Ellner, J. Alloys Compd., 2004, 366(1–2), 222–227 CrossRef CAS.
- T. He, E. Kreidler, L. Xiong and E. Ding, J. Power Sources, 2007, 165(1), 87–91 CrossRef CAS.
- E. Ding, K. L. More and T. He, J. Power Sources, 2008, 175(2), 794–799 CrossRef CAS.
- Y. Kim, S. Xu, J. Park, A. L. Dadlani, O. Vinogradova, D. Krishnamurthy, M. Orazov, D. U. Lee, S. Dull, P. Schindler, H. S. Han, Z. Wang, T. Graf, T. D. Schladt, J. E. Mueller, R. Sarangi, R. Davis, V. Viswanathan, T. F. Jaramillo, D. C. Higgins and F. B. Prinz, Appl. Catal., B, 2022, 300 CrossRef CAS.
- P. C. Jennings, B. G. Pollet and R. L. Johnston, J. Phys. Chem. C, 2012, 116(29), 15241–15250 CrossRef CAS.
- J. Zhang, F. Coms and S. Kumaraguru, J. Electrochem. Soc., 2021, 168(2) Search PubMed.
- W. He, X. Zhang, K. Zheng, C. Wu, Y. Pan, H. Li, L. Xu, R. Xu, W. Chen, Y. Liu, C. Wang, Z. Sun and S. Wei, Angew Chem., Int. Ed., 2023, 62(2), e202213365 CrossRef CAS PubMed.
- J. Kim, S. Yang and H. Lee, Electrochem. Commun., 2016, 66, 66–70 CrossRef CAS.
- H. Duan, Q. Hao and C. Xu, J. Power Sources, 2015, 280, 483–490 CrossRef CAS.
- W. Guo, N. Sun, X. Qin, M. Pei and L. Wang, Biosens. Bioelectron., 2015, 74, 691–697 CrossRef CAS PubMed.
- D. Zhao, G. Yu, K. Tian and C. Xu, Biosens. Bioelectron., 2016, 82, 119–126 CrossRef CAS PubMed.
- Q. Zhou and C. Xu, J. Colloid Interface Sci., 2017, 496, 235–242 CrossRef CAS PubMed.
- H. Abe, F. Matsumoto, L. R. Alden, S. C. Warren, H. D. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2008, 130(16), 5452–5458 CrossRef CAS PubMed.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2014, 136(29), 10206–10209 CrossRef CAS PubMed.
- C.-L. Yang, L.-N. Wang, P. Yin, J. Liu, M.-X. Chen, Q.-Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H.-W. Liang, Science, 2021, 374, 459–464 CrossRef CAS PubMed.
- C. Wang, S. Liu, Y. Duan, Z. Huang and S. Che, Sci. Technol. Adv. Mater., 2015, 16(5), 054206 CrossRef PubMed.
- D. Eder and A. H. Windle, Adv. Mater., 2008, 20(9), 1787–1793 CrossRef CAS.
- C. Zhang, H. He and K.-i. Tanaka, Appl. Catal., B, 2006, 65(1–2), 37–43 CrossRef CAS.
- J. A. Wang, A. Cuan, J. Salmones, N. Nava, S. Castillo, M. Morán-Pineda and F. Rojas, Appl. Surf. Sci., 2004, 230(1–4), 94–105 CrossRef CAS.
- S. Kuhaudomlap, O. Mekasuwandumrong, P. Praserthdam, S.-I. Fujita, M. Arai and J. Panpranot, Catalysts, 2018, 8(2), 87 CrossRef.
- Y. Kim, S. Xu, J. Park, A. L. Dadlani, O. Vinogradova, D. Krishnamurthy, M. Orazov, D. U. Lee, S. Dull, P. Schindler, H. S. Han, Z. Wang, T. Graf, T. D. Schladt, J. E. Mueller, R. Sarangi, R. Davis, V. Viswanathan, T. F. Jaramillo, D. C. Higgins and F. B. Prinz, Appl. Catal., B, 2022, 300, 120741 CrossRef CAS.
- X. Nie, S. Zhuo, G. Maeng and K. Sohlberg, Int. J. Photoenergy, 2009, 2009(1), 294042 CrossRef.
- O. Khatim, M. Amamra, K. Chhor, A. M. T. Bell, D. Novikov, D. Vrel and A. Kanaev, Chem. Phys. Lett., 2013, 558, 53–56 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46(4), 855–874 CrossRef CAS.
- A. V. Kalinkin, M. Y. Smirnov, A. I. Nizovskii and V. I. Bukhtiyarov, J. Electron Spectrosc. Relat. Phenom., 2010, 177(1), 15–18 CrossRef CAS.
- S. Hsieh, T. Matsumoto, M. Batzill and B. E. Koel, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68(20), 205417 CrossRef.
- B. C. Beard and P. N. Ross, J. Phys. Chem., 1986, 90(26), 6811–6817 CrossRef CAS.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11(9), 3714–3719 CrossRef CAS PubMed.
- DOE, Multi-Year Research, Development, and Demonstration Plan - Section 3.4 Fuel Cells, https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_myrdd_fuel_cells.pdf, accessed January, 21, 2019 Search PubMed.
- V. Beermann, M. Gocyla, S. Kühl, E. Padgett, H. Schmies, M. Goerlin, N. Erini, M. Shviro, M. Heggen, R. E. Dunin-Borkowski, D. A. Muller and P. Strasser, J. Am. Chem. Soc., 2017, 139(46), 16536–16547 CrossRef CAS PubMed.
- A. Bonakdarpour, T. R. Dahn, R. T. Atanasoski, M. K. Debe and J. R. Dahn, Electrochem. Solid-State Lett., 2008, 11(11), B208 CrossRef CAS.
- Z. Wu, G. Yang, Q. Zhang, Z. Liu and F. Peng, Energy Environ. Sci., 2024, 17(10), 3338–3346 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information on synthesis, ICP and XRD results including Rietveld refinement, XPS spectra, microscopy analysis (TEM, STEM, EDX, HRTEM, and FFTs) and additional results from electrochemical testing and characterization before and after electrochemical stability tests. See DOI: https://doi.org/10.1039/d4ta00364k |
| ‡ Current address: Research Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA. |
| § Current address: Department of Interface Science, Fritz-Haber Institut der Max Planck Gesellschaft, Faradayweg 4-6, 14195 Berlin. |
| This journal is © The Royal Society of Chemistry 2024 |