
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation between the cationic composition and anionic electrochemical activity of Li2MSeO anti-perovskites†
Mikhail V.
Gorbunov
 *a,
Oleg
Janson
a,
Max
Stöber
a,
Volodymyr
Baran
b and
Daria
Mikhailova
*a
*a,
Oleg
Janson
a,
Max
Stöber
a,
Volodymyr
Baran
b and
Daria
Mikhailova
*a
aLeibniz-Institut für Festkörper- und Werkstoffforschung Dresden e. V. Helmholtzstraße 20, 01069, Dresden, Germany. E-mail: m.gorbunov@ifw-dresden.de; d.mikhailova@ifw-dresden.de
bDeutsches-Elektronen Synchrotron DESY Notkestraße 85, 22607, Hamburg, Germany
First published on 30th April 2024
Abstract
Li2MChO anti-perovskites have lately attracted much attention as potential electrode materials for Li batteries owing to their high and stable specific capacity. It is rather surprising, because in terms of the structure, metal ions share the same crystallographic site. Since the redox potentials of chalcogenide anions S2− or Se2− and transition metal cations are close, anti-perovskites can be classified as bifunctional materials with cationic and anionic electrochemical activity. Their high chemical flexibility combined with redox-active ions make cubic anti-perovskites a rather valuable model system for tuning desired electrochemical properties. Herein, we studied six Li2MSeO compounds with Mn, Fe, and Co and their solid solutions in Li cells and tailed their structural and electrochemical behaviour to the cationic composition. Remarkably, a booster property of Co with the highest Co3+/Co2+ potential with regard to an enhanced electrochemical performance was observed for Li2Fe0.5Co0.5SeO in comparison to Li2FeSeO, although only Fe and Se were involved in the redox process while Co remained inactive. In Li2Fe0.5Mn0.5SeO, less-active Mn facilitated the oxidation of Fe and Se, positively influencing the rate capability in comparison to Li2Co0.5Mn0.5SeO. Thus, we found a direct correlation between the M3+/M2+ standard redox potential and the electrochemical activity of Se2−.
1. Introduction
Although many metal-ion energy-storage systems are being developed nowadays, lithium-ion batteries are still considered an optimal solution for portable electronics. Besides, they are implemented in electric and hybrid vehicles.1,2 This can be primarily explained by their high energy density, coulombic efficiency, and low self-discharge. Moreover, a variety of chemical compositions with intercalation properties offer a broad range of working potentials, meeting various practical application requirements.3,4 Currently, the need for cathode materials with a higher charge and energy density than conventional materials such as Li(Ni,Mn,Co)O2, LiFePO4, and LiMn2O4 is growing.5 Li-rich NMC (Ni, Mn, and Co) oxides are considered some of the most promising next-generation cathodes for LIBs.3 However, their high operating voltage, providing a high energy density, also suggests an electrolyte decomposition issue, which is challenging to overcome.2Among many alternatives, ternary oxide-sulfides of Li and transition metal cations with an anti-perovskite structure and the general formula Li2MChO (M = Fe, Co, or Mn) have recently attracted significant attention owing to the possibility of reversible Li (de)intercalation.6–13 In the anti-perovskite structure (Fig. 1a), Li and M cations share the same crystallographic site, which is occupied by oxygen anions in the conventional CaTiO3 perovskite structure, whereas larger chalcogenide (S2− or Se2−) and smaller O2− anions occupy Ca and Ti sites, respectively.7 Generally, the potential application of cubic anti-perovskites is not limited by battery-related research, since they can deliver many valuable properties, including electrocatalytic activity, superconductivity, and piezomagnetism, and these properties are defined by their composition.6
 | ||
| Fig. 1 (a) Crystal structure of Li2MSeO anti-perovskites (M = 3d transition metals). (b) PXRD patterns of the six studied compounds, λ = 1.78896 Å. | ||
Theoretical calculations predict that a significant amount of lithium can be extracted from the Li2FeSO material before structural collapse occurs, achieving a specific capacity of 275 mA h g−1.8,10 Additionally, it has been shown that electrochemical properties depend significantly on the nature and ratio of transition metal cations.9–12 Further, if applied to anti-perovskites analogically to perovskites as a geometrical stability criterion for a cubic structure, Goldschmidt tolerance factor shows a deviation from 1, highlighting the intrinsic structural instability of S-containing anti-perovskites. From this point of view, the bigger Se2− anion could enhance the structural stability. Previously, the effect of S-substitution by Se in the Li2FeS1−xSexO solid solution series was studied,13 and a non-linear change in the electrochemical performance depending on the Se content was observed, highlighting that the Li2FeS0.7Se0.3O composition offered the best rate capability and long-term cyclability. Therefore, it was suggested that the introduction of Se influences the electrochemical performance ambivalently: it enhances the overall structural stability in the pristine and delithiated states, but on the other hand, materials with a high Se amount show higher anionic redox activity, since the standard redox potential of Se is lower than of S (2.370 V and 2.564 V vs. Li, respectively14). This means that for the same amount of Li extracted, in the materials with more Se, charge compensation not only implies the oxidation of Fe2+ to Fe3+, but also a partial oxidation of Se2−. Surely, in this case, the anti-perovskite structure may be destroyed. During the following cell discharge, Li2Se and polyselenides may be formed, thus initiating a shuttle-effect in the cell during cycling, which is also typical for Li–S15 and Li–Se16 rechargeable batteries.
Although Li2FeSeO has a Goldschmidt factor closer to 1 than its Li2FeSO analogue, and thus should show higher structural stability, it undergoes severe amorphization upon Li extraction, too.17 In the present work, we evaluated the impact of Fe, Mn, and Co on the structural stability and anionic redox activity of Li2MSeO anti-perovskites in Li batteries. Differences in the M3+/M2+ redox potentials of the transition metal cations (0.771, 1.542, and 1.920 V for Fe, Mn, and Co, respectively14) and in the electronic configurations resulted in different structural behaviours and degree of selenium participation in the charge-compensation mechanism upon (de)lithiation. Hence, the compositions with equal amounts of two different transition metals on the cationic site of the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m anti-perovskite structure were studied. The cationic impact on the structural and electrochemical properties of Li2MSeO anti-perovskites was evaluated for six compounds: Li2FeSeO, Li2MnSeO, Li2CoSeO, Li2Fe0.5Co0.5SeO, Li2Fe0.5Mn0.5SeO, and Li2Co0.5Mn0.5SeO.
m anti-perovskite structure were studied. The cationic impact on the structural and electrochemical properties of Li2MSeO anti-perovskites was evaluated for six compounds: Li2FeSeO, Li2MnSeO, Li2CoSeO, Li2Fe0.5Co0.5SeO, Li2Fe0.5Mn0.5SeO, and Li2Co0.5Mn0.5SeO.
2. Results and discussion
2.1. Synthesis and powder X-ray diffraction studies
Powder X-ray diffraction studies (PXRD) proved the high purities of the synthesized materials (Fig. 1b). The unit cell parameters increased linearly with the increase in the average cationic radius of M2+, in accordance with Vegard's law,18 as presented in Table 1. Chemical analysis was performed by applying inductively coupled plasma optical emission spectroscopy (ICP-OES), and revealed Li deficiencies in the obtained samples, which was low in materials with Fe, and more significant in compounds with Mn and Co.m). ICP-OES data were normalized on the Se content, and the oxygen content was calculated from the charge difference
| Desired formula | ICP-OES result | Cell par. a, Å |
|---|---|---|
| Li2FeSeO | Li1.97(2)Fe0.968(5)Se1.0O0.95 | 4.0000(2) |
| Li2CoSeO | Li1.809(9)Co0.92(1)Se1.0O0.82 | 3.9729(3) |
| Li2MnSeO | Li1.88(1)Mn0.99(1)Se1.0O0.93 | 4.0622(3) |
| Li2Fe0.5Co0.5SeO | Li1.95(2)Fe0.516(4)Co0.482(7)Se1.0O0.97 | 3.9865(4) |
| Li2Fe0.5Mn0.5SeO | Li1.868(9)Fe0.463(2)Mn0.502(4)Se1.0O0.90 | 4.0337(5) |
| Li2Co0.5Mn0.5SeO | Li1.79(2)Co0.477(9)Mn0.511(7)Se1.0O0.88 | 4.0196(6) |
The Goldschmidt tolerance factor19 was next evaluated referring to ref. 20, and was found to be higher for the Se compounds (∼0.896 for Li2FeSeO) than for their S counterparts (∼0.851 for Li2FeSO), indicating there was less structural strain in the anti-perovskites with Se. Furthermore, they exhibited a higher chemical flexibility compared to the anti-perovskites with S. Thus, the row of Li2Fe1−xMxSeO compositions with Fe/M cation mixing could easily be synthetized, whereas, for example, the synthesis of the complete Li2Fe1−xCoxSO series was unsuccessful.
Formation of the cubic Pmm anti-perovskites proceeded through intermediates, as seen from the in situ synchrotron diffraction experiments for Li2FeSeO synthesis (Fig. 2). No chemical reactions occurred at temperatures below the melting point of Se at 493 K. Above 500 K, Li2Se was first formed as a crystalline phase. Afterwards, the reflections of Li2FeO2 (ref. 21) appeared, followed by FeSe and Li2FeSeO formation above 612 K, which was significantly lower than the temperature used for the laboratory syntheses (see the ESI†). Between 612 K and 870 K, a continuous growth of Li2FeSeO reflections was observed, while those of the intermediate phases decayed. Therefore, the following reaction mechanism is proposed:
| Li2O + Fe + Se → 1/2Li2Se + 1/2FeSe + 1/2Li2FeO2 → Li2FeSeO. |
 | ||
| Fig. 2 In situ observation of Li2FeSeO formation. | ||
Starting from 900 K, the reaction with the quartz capillary began, turning the system into an open one.
2.2. DFT calculations
First, we addressed the thermodynamic stability of Li2FeSeO. To this end, we calculated the enthalpies of three possible decomposition reactions: (i) into FeSe, Li2Se, and Li2FeO2; (ii) into Li2Se, FeSe, Fe, and LiFeO2; and (iii) into Fe7Se8, Fe, and Li2O. In the first two cases, Li2FeSeO had a lower enthalpy than the sum of the decomposition products (see ESI, S1–S3†). In contrast, the enthalpies of the corresponding reactions for the Li2FeSO indicated that both decompositions were energetically favourable; and the current result agrees with the previous DFT study in ref. 8. However, Li2FeSeO may decompose into Fe7Se8, Fe, and Li2O, since the energy of Li2FeSeO lay at 47 meV per atom above the convex hull. Although this analysis neglected the entropy and kinetic contributions, the sizeable differences in total energies suggest that the substitution of sulfur with selenium significantly stabilizes the crystal structure.To study how the electronic structure of Li2MSeO depends on the nature of the d-metal, we calculated the densities of states (DOS) for three pristine Li2MSeO (M = Fe, Co, Mn) and three mixed (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) compounds (Fig. 3).
:1) compounds (Fig. 3).
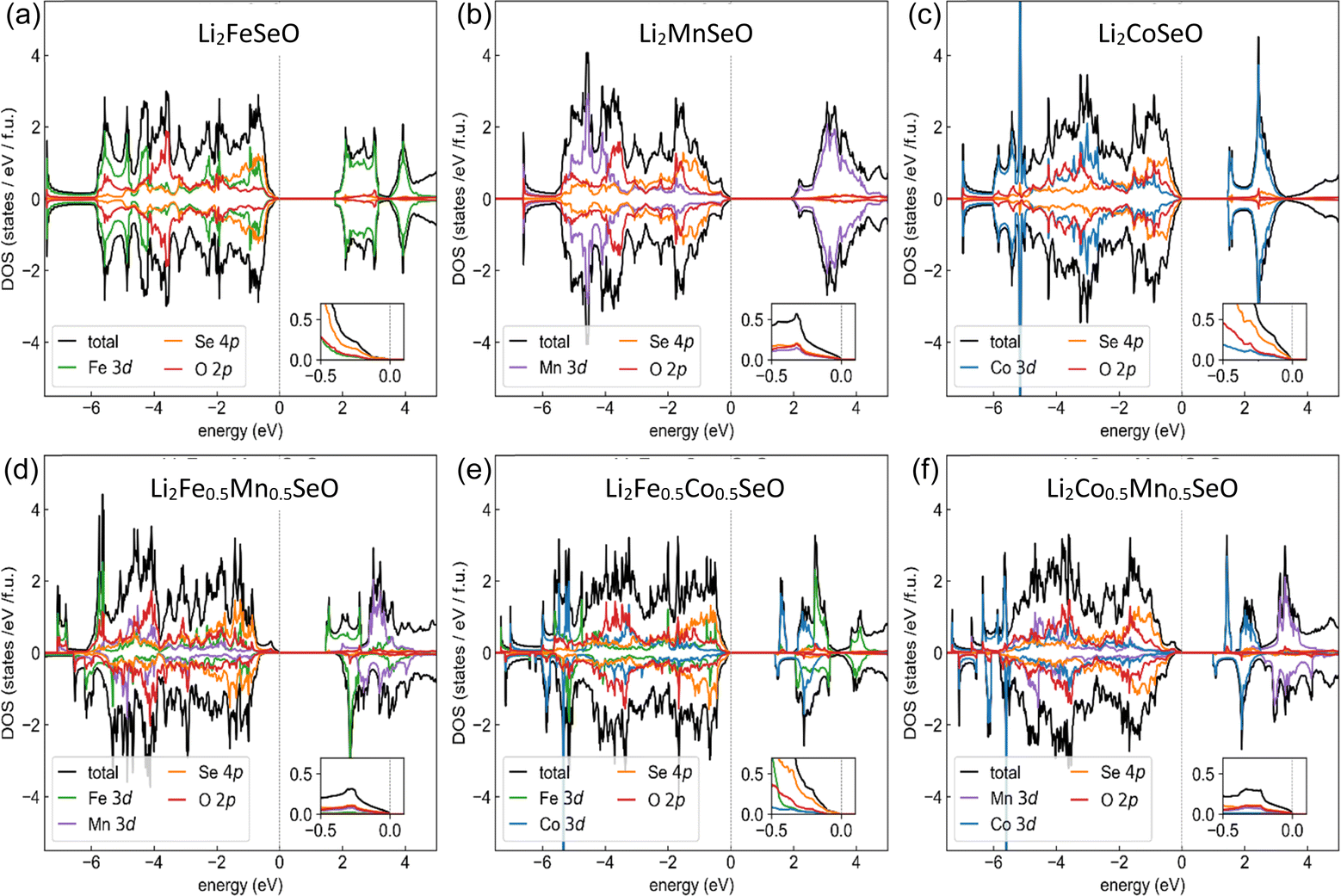 | ||
| Fig. 3 Site- and shell-resolved density of states (DOS) for the lowest-energy antiferromagnetic (except for Li2Fe0.5Mn0.5SeO, ferrimagnetic) configurations of (a–c) Li2MnSeO, Li2FeSeO, and Li2CoSeO and (d–f) compounds with a 1:1 ratio of 3d cations. Positive (negative) DOS values indicate spin-up (spin-down) densities. The Fermi level was at zero energy. Insets highlight the DOS near the Fermi level. All the calculations were done using the GGA+U functional with U = 4 eV and J = 1 eV and the fully localized limit for the double-counting correction. | ||
A straightforward computational analysis was complicated by significant structural and intersite disorder. To keep the problem tractable, we restricted ourselves to a particular arrangement of cations (see ESI† for details), used the experimental unit cell parameters and optimized the internal atomic coordinates by minimizing the total energy as computed with the GGA+U functional. In all the calculations, we kept the Coulomb repulsion and Hund exchange equal to 4 and 1 eV, respectively, and used the fully localized limit for the double-counting correction. The chosen value of U was a compromise between U = 3 eV suggested for Mn2+,22 and U = 5 eV used for Co2+.23 Our analysis shows that for all compositions, the lowest-energy configuration had zero net moment. For all the compounds, we obtained an insulating solution with a band gap ranging from 1.5 to 2 eV. As shown in the insets of Fig. 3, the top edge of the valence band was dominated by the Se 4p contribution. Hence, the occupancy of these states would be altered by the removal of Li atoms, which would be accompanied by a shift in the chemical potential to lower energies. While this behaviour was generally expected for all compositions, the different nature of the d-cations started manifesting itself; if we go deeper into the valence band discussion: the Fe 3d contribution in iron-containing compounds is prominent, and the corresponding Co and Mn contributions in this energy range are much smaller. Based on this observation, we can expect that sizeable delithiation would strongly affect the valence of iron.
While the top of the valence band is always Se-dominated, the bottom of the polarization band is formed by metal d-states and thus depends on the composition: cobalt (if Co is present), iron (if Fe is present and Co is absent), or manganese (only in Li2MnSeO). This indicates that the energy cost of adding an extra electron is smaller for cations with more electrons. This obviously holds only for the shells that are at least half-filled.
For a more detailed analysis, direct DOS calculations of delithiated compositions are necessary. Since the atomic coordinates of such compositions are generally unknown, structural optimizations need to be done prior to DOS calculations. Unfortunately, the sheer amount of possible structural configurations and difficulties in applying DFT+U to doped systems precluded such an analysis here.
2.3. Electrochemical characterization
Galvanostatic cycling of Li2MSeO in Li cells revealed noticeable differences in performance, depending on the cationic composition (Fig. 4, and S4†). For example, Li2FeSeO showed rather high values of specific capacity, in agreement with recently published results,17 reaching 250 mA h g−1 at 0.1C current density. However, the capacity decreased rapidly, and the rate capability was rather poor. Li2FeSeO delivered less than 75 mA h g−1 at a 1C current density; yet its sulfur-based analogue is known for high values around 200 mA h g−1 at the 1C current rate.10 | ||
| Fig. 4 Rate capability tests of Li2MSeO (M = Fe, Co, Mn, or their mixture) in Li cells at 298 K. 1C current density corresponds to the extraction of 1 Li from the formula unit within 1 h. | ||
Li2MnSeO demonstrated stable but moderate characteristics, as well as Li2CoSeO. Although the specific capacity of Li2CoSeO was 55 mA h g−1 at 0.1C, which is the lowest among the studied compositions, it showed a very little dependence on the current density, suggesting enhanced diffusion kinetics.
Mixing the transition metal cations Fe/Co, Fe/Mn, and Co/Mn resulted in capacity values lying between Li2FeSeO and Li2CoSeO at 0.1C, but with an unexpected improvement of the rate capability and a self-healing effect, especially for Li2Fe0.5Co0.5SeO.
The combination of Co and Fe cations in Li2Fe0.5Co0.5SeO resulted in the best electrochemical performance among all the studied compositions. This result is rather surprising, because as it was shown earlier by operando XAS studies of Li2Fe0.9Co0.1SO12 that the cobalt in cubic anti-perovskites presumably remains inactive. Based on this knowledge, we expected a significant decrease in capacity for the composition with a large amount of Co. Moreover, the material showed even higher values of specific capacity at low current densities after being subjected to higher currents, pointing to some activation processes. The replacement of 50% Fe by Mn (Li2Fe0.5Mn0.5SeO) improved the cycling stability, but reduced the absolute capacity, very similar to what was earlier reported for Li2Fe0.5Mn0.5SO.11 However, the Se-material showed a much better rate capability than its S-counterpart. The mixture of Co and Mn led to a weak rate capability and unstable cycling behaviour.
Analysis of the differential capacity plots (Fig. 5) disclosed redox processes at very different cell potentials for these six compositions. The presence of only Fe cations (Li2FeSeO, 5a) resulted in three oxidation and reduction peaks between 1.2 and 2.8 V, and a significant cell polarization. Since the observed features became less pronounced with subsequent cycles (the figures are given for the same cells, subjected to various currents), we may suggest partial decomposition of the material occurred, similarly to for Li2FeSO.10 The presence of only Mn (Li2MnSeO, 5b) reduced the amount of redox processes and decreased the cell polarization: only one redox peak was visible during charge and discharge cycles between 2.2 and 2.4 V. This behaviour was realized in Li2Fe0.5Mn0.5SeO (5d) as well, highlighting Mn as a dominant cation, which strongly influences the properties of the compound. The Li2CoSeO (5c) composition showed two well-defined redox processes at 1.5 and 2 V and two less-pronounced peaks between 2.2 and 2.5 V. In the mixed compounds Li2Fe0.5Co0.5SeO and Li2Co0.5Mn0.5SeO (5e, f), charge and discharge represented multiple processes.
 | ||
| Fig. 5 Differential capacity plots for the first charge–discharge cycle of six Li2MSeO compositions in Li cells at 298 K: (a–c) M = Fe, Mn, Co, respectively, (d) Li2Fe0.5Mn0.5SeO, (e) Li2Fe0.5Co0.5SeO, (f) Li2Co0.5Mn0.5SeO. | ||
2.4. Structural behaviour during (de)lithiation
Operando X-ray diffraction experiments showed drastic differences in the structural response to lithium removal for all the tested materials. In the case of Li2FeSeO, a second isostructural phase was formed upon cell charge (Li removal), which corresponded to the second step of the electrochemical curve. The new phase was electrochemically active, showing continuous change in the lattice parameter (Fig. 6a), while the cell metrics of the initial phase did not change up to the “Li1.5FeSeO” composition, at which the crystallinity of the material abruptly vanished. After lithiation, the crystallinity was not recovered, probably due to an irreversible structural collapse. This behaviour differs from that of its Li2FeSO counterpart, which also became amorphous but did not show any isostructural phase formation upon delithiation.10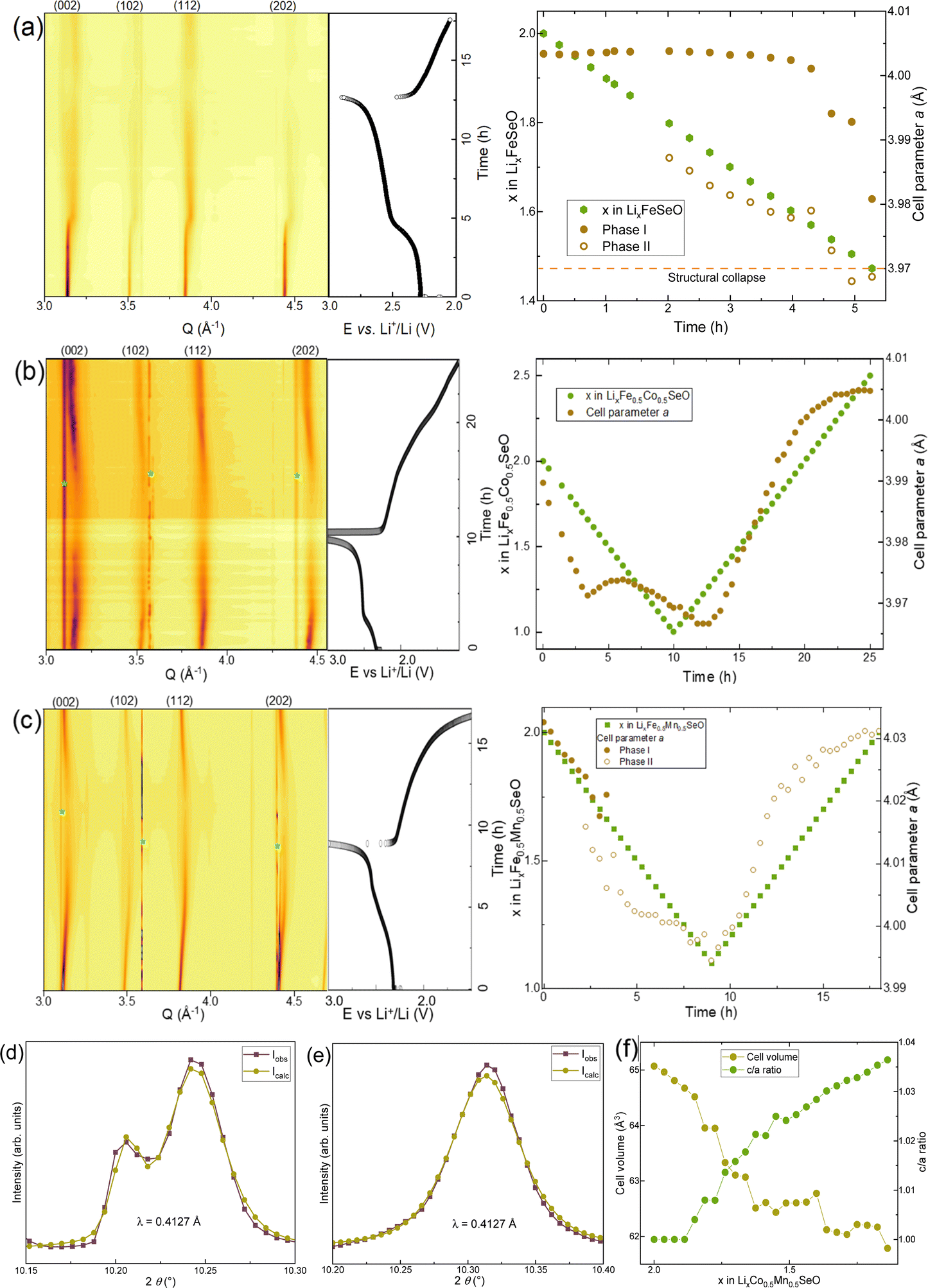 | ||
| Fig. 6 (a–c) Contour-plots of the X-ray diffractograms obtained operando for Li2FeSeO, Li2Fe0.5Co0.5SeO, and Li2Fe0.5Mn0.5SeO together with corresponding galvanostatic curves and cell parameter variation. (d and e) Part of the Le Bail fitted patterns for Li1.75Fe0.5Mn0.5SeO and Li1.75Co0.5Mn0.5SeO, showing the change in (111) Bragg reflection in the partially charged state. (f) Cell volume change for Li2Co0.5Mn0.5SeO. * stands for Al mesh. | ||
The replacement of 50% iron by cobalt changed the situation completely (Fig. 6b). First, Li removal/insertion turns into a single-phase process. Second, the material preserved a certain degree of crystallinity even when a high amount of lithium was extracted, making it possible to estimate the cell parameter evolution throughout the whole electrochemical cycle. The relative change of a cell parameter a of about 1% after removal of 1 Li per formula unit was the smallest among all the studied Pmm anti-perovskites. Additionally, the crystallinity was regained almost completely after discharge, pointing to a stabilizing effect of cobalt introduction into the crystal lattice.
The single-phase reaction mechanism of reversible lithium extraction was shown to be a favourable one for enhancing the electrochemical characteristics in our earlier studies on Li2Fe0.9M0.1SO anti-perovskites.12 The current results confirm this finding for the high degree of cationic substitution. Noteworthily, the lattice parameter a in Li2Fe0.5Co0.5SeO changed non-linearly with the Li amount, which resembles the case for the aforementioned Li2FeSO and Li2Fe0.9Co0.1SO materials. Such non-linearity may imply a competition between the lattice contraction due to Li removal, and some repulsion in the lattice between negatively charged O2− and Se2− anions. Additionally, the non-monotonous redox activity of ions may also contribute to the observed phenomenon.
In summary, we can conclude that the replacement of iron by cobalt in cubic anti-perovskites with S or Se enhances their structural stability against delithiation: while the Li2FeSeO structure collapsed, Li2Fe0.5Co0.5SeO regained its crystallinity after one charge–discharge cycle. The stabilization effect was visible for both a low Co amount of 10% and a high substitution level of 50%, improving the electrochemical performance. However, a complete replacement of Fe by Co resulted in poor capacity values for the material in lithium cells. Yet it provided high stability in the rate capability tests.
The delithiation behaviour of Li2Fe0.5Mn0.5SeO was different as well. Li removal led to the appearance of an isostructural LixFe0.5Mn0.5SeO phase with a significantly smaller cell parameter a, and to a short compositional region with two coexisting phases (Fig. 6c), which was similar to the case for Li removal in its S-counterpart Li2Fe0.5Mn0.5SO.11 There was a hysteresis in the parameter a upon charge and discharge, which probably came from a local structural distortion due to the formation of Jahn–Teller Mn3+ cations,24 and a resulting poor kinetics and impeded cation diffusion. Complete replacement of iron by cobalt and manganese (Li2Co0.5Mn0.5SO) caused anisotropic structural strain during Li extraction, followed by a change of the lattice symmetry from the cubic Pmm to a tetragonal one. This could be concluded by a comparative evaluation of the Bragg reflections of Li2Fe0.5Mn0.5SO and Li2Co0.5Mn0.5SO. Whereas a clear splitting was observed for all reflections of Li2Fe0.5Mn0.5SO (two cubic isostructural phases with different cell metrics), in the case of Li2Co0.5Mn0.5SO, the (hkl) reflections with h = k = l remained non-split, confirming the symmetry change during delithiation. For example, a different behaviour of the (111) reflection at a partially charged state with 1.75 Li is shown in Fig. 6(d)–(f), and S6.† Detailed analysis enabled a conclusion about a group–subgroup relationship with the P4/mmm space group for the delithiated Li2Co0.5Mn0.5SO sample. The symmetry change was probably caused by the emergence of Jahn–Teller Mn3+ cations, as confirmed later by the metal valence sensitive technique. It is worth mentioning that a cubic-to-tetragonal structural transition is well known for Mn-containing materials with a perovskite structure.25
The results of operando XRD together with the electrochemical properties brought us to an assumption that the significant differences in the structural and electrochemical behaviour of Li2MSeO materials may not only originate from the differences in the redox activities of the transition metal cations, but also from the different anionic electrochemical activity of selenium. Since the Se0/Se2− redox couple of around 2.370 V vs. Li14 lay within the studied potential window of the cells with a Li2MSeO cathode and a Li-metal anode, the oxidation of Se could contribute to the entire cathodic specific capacity. Indeed, the redox activity of Se was already directly confirmed in Li2FeS1−xSexO in one of our previous works.13 The presence of transition metal cations with different redox potentials in the structure might influence the anionic redox activity of selenium.
2.5. Evaluation of the charge-compensation mechanism
The redox activities of the cations and Se were tested by applying operando X-ray absorption spectroscopy, using synchrotron radiation.It is well known that an increase in the oxidation state of transition metals leads to a positive shift of the K-edge energy, which depends on the transition metal and its nearest structural surrounding. For Fe, Co, and Mn, the K-edge corresponded to the 1s → 4p electron transition and was strongly correlated with the valence state.26 Using reference materials with well-defined valences and similar structural environments allows doing a reliable quantitative analysis. Transition metal cations in a hetero-atom environment, like oxide–chalcogenides, represent a special case. The covalency of M–O and M–Se bonds is different due to different overlapping of d- and p-orbitals of the transition metal and the small, less-polarized oxygen anion, or the larger and highly polarized selenium anion. Therefore, the K-edge position and the edge shape for the transition metal cations in oxide–chalcogenides may represent rather a superposition of binary oxides and selenides. However, a higher redox activity of cations will similarly result in a higher energy shift of the K-edge. A shift of the K-edge energy must also be observed for selenium anions upon their oxidation.
The K-edge was determined as the position that corresponds to a maximum of the first energy derivative dxμ(E)/dE, coinciding with the inflection point of the absorption edge.27 The oxides FeO, Fe3O4, Fe2O3, CoO, Co3O4, MnO, MnO2, and Mn2O3 were used as references to quantitatively estimate the Fe, Co, and Mn oxidation states. In all the pristine anti-perovskites, the Fe, Co, and Mn K-edge positions were very close to the position of the double-charged metal cations in oxides, which confirmed the presence of Fe2+, Co2+, and Mn2+ in Li2MSeO.
Here, we have to note that evaluation of the oxidation states of transition metal cations using calibration curves is straightforward only for ionic compounds, like oxides. In the case of selenides, a strong overlapping of the Se 4p and M 3d orbitals and lower electronegativity of Se result in a high covalency grade of the M–Se bonding. Moreover, the closeness of the redox potentials of Se2− and the transition metal cations Fe and Mn complicates the situation additionally. Therefore, we could not use the calibration line directly, as was done for sulfur-based anti-perovskites,11 and in our opinion, it is more correct to discuss the shifts of the corresponding transitions on the energy grid.
Upon battery charge and discharge, a reversible oxidation of iron in all three Fe-containing compositions was detected, since the K-edge reversibly shifted to higher energy values and back during (de)lithiation (Fig. 7 and S7–S11†). The energy difference between the Fe K-edges of FeO (Fe2+) and Fe2O3 (Fe3+) was close to 4 eV (a, b). Panel (c) depicts the shift ΔEFe K-edge = Ecurrent − Epristine of the Fe K-edge depending on the Li-amount extracted from the structure during battery charge (as the amount of current flowing through the cell). The ΔEFe K-edge values for Li2FeSeO and Li2Fe0.5Mn0.5SeO were very close, reaching 1.5 eV at a total Li amount x(Li) of 1.5. However, at the same x(Li)-value, the ΔEFe K-edge of Li2Fe0.5Co0.5SeO of 2.5 eV was much higher, pointing to a noticeably stronger oxidation of Fe in this material. The pre-edge feature in the range of 7711–7715 eV, which corresponds to a 1s → 3d electron transition, revealed a local structural distortion of the octahedral Fe environment in all six materials (Fig. 7, S12, and Table S13†). According to the dipole selection rules, this transition is prohibited for the ideal octahedral symmetry of Fe.28 The disorder in local coordination of Fe increased on charge and decreases on discharge. Therefore, the local structure of Fe remained mostly preserved despite the irreversible amorphization of Li2FeSeO material during charge, as observed by operando XRD.
 | ||
| Fig. 7 Operando XAS of Li2FeSeO in the Fe K-edge region on (a) charge and (b) discharge. (c) Relative energy shift of Fe K-edge in iron-containing compounds. Calibration lines for the oxidation states of transition metals are available in the ESI.† | ||
The electrochemical activity of Mn and Co in the materials was evaluated as well. Similar to our previous studies on Li2Fe0.9Co0.1SO,12 Co did not show any redox activity in Li2CoSeO under extraction of about 0.25 Li per formula unit. Cobalt was also inactive in Li2Fe0.5Co0.5SeO and Li2Mn0.5Co0.5SeO, since there was no noticeable shift in the Co K-edge. In contrast, Mn was redox-active in Li2MnSeO, showing a K-edge shift of about 2 eV upon the extraction of 0.5 Li per formula unit, see Fig. S7.† Such an energy shift would correspond to Mn2.5+. However, Mn was less active in Li2Fe0.5Mn0.5SeO, having a shift in energy of only 0.7 eV upon the extraction of 0.6 Li per formula unit, which resembles our previous investigations of its S-counterpart:11 similar to Li2Fe0.5Mn0.5SO, upon Li removal from Li2Fe0.5Mn0.5SeO, Fe was oxidized first, while Mn started to participate in the redox process later, when a significant amount of Li was already extracted. As is known, the Co3+/Co2+ pair has the highest redox potential in various classes of materials, such as LiMPO4 olivines26 or Li2MP2O7 pyrophosphates,27 followed by the Mn3+/Mn2+ and Fe3+/Fe2+ pairs. This tendency is sustained in anti-perovskites as well, because Co with its high redox potential remains inactive in the potential window of 1.2–3.0 V vs. Li, while Fe and Mn are involved in the redox process. Among Co, Fe, and Mn metals, Co has the highest electronegativity (1.88, 1.83, and 1.55 Pauling units, respectively).28 Therefore, Co–Se bonds are more covalent than other M–Se bonds, thus reducing the electron density on Se (negative inductive effect), which may be compensated by metals with a lower electronegativity. With the presence of Co in the compositions Li2Fe0.5Co0.5SeO and Li2Mn0.5Co0.5SeO, both Fe and Mn showed higher redox activity than without it. Therefore, although Co was redox-inactive, it seemed to boost the redox activity of the other transition metal cations.
Further, we analysed the Se K-edge spectra for all the compounds, which confirmed the strong redox activity of Se, visible as an edge shift upon (de)lithiation, see Fig. 8 and S7–S12.† Here, we would like to highlight a note of caution. In the literature, there is a confusion regarding the definition of the Se K-edge. Some authors assume a 1s → 4p electron transition around 12658–12660 eV as the K-edge, as for elemental Se.29 Other works, however, argue for this transition at much higher energy values of 12664–12665 eV, as reported for example for Li2Se or FeSe.30 Lastly, several works discuss a 1s → 5p electron transition as the true K-edge with a 1s → 4p transition as a pre-edge.31 We find this definition as the most reasonable, since this is the only possible transition for negatively charged Se2− in typical ionic compounds. Indeed, the participation of Se2− anions in ionic metal–selenium bonds would result in fully occupied 4p orbitals, suppressing the 1s → 4p transition significantly.31
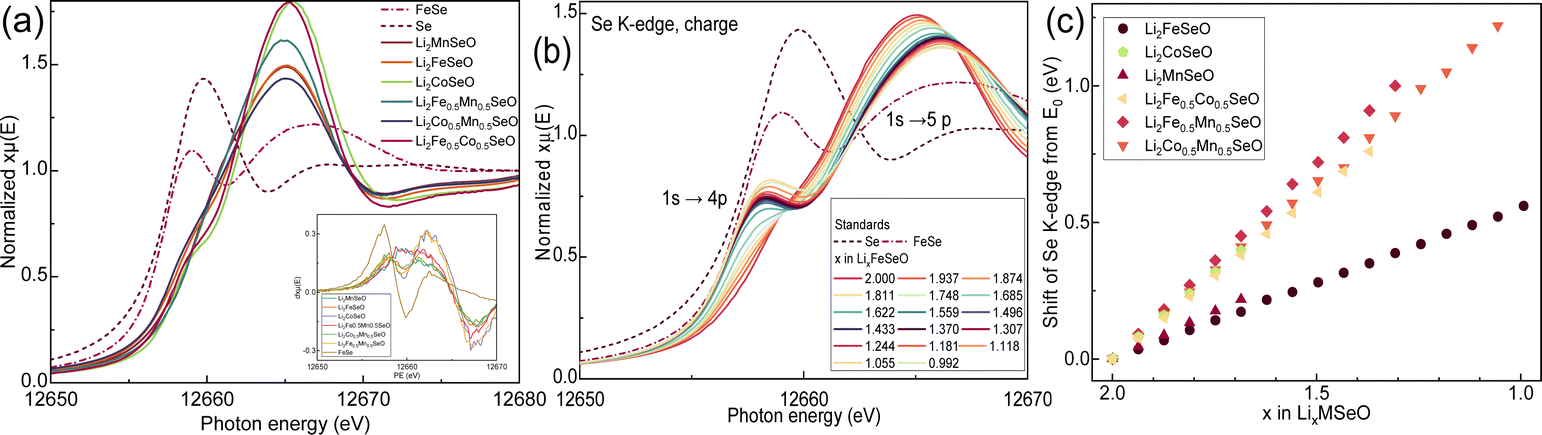 | ||
| Fig. 8 XAS in the Se K-edge region for Li2MSeO compounds: (a) comparative image of Se K-edge for pristine anti-perovskites and Se standards, (b) operando XAS of Li2FeSeO during charge, (c) relative shift of Se K-edge from its initial position for Li2MSeO anti-perovskites during delithiation. | ||
Therefore, the K-edge energy position for Se was set as a maximum of the first derivative dxμ(E)/dE24 at energies around 12662–12664 eV, according to prior work.32 We also recorded a spectrum of FeSe as a reference compound having the same formal oxidation state as Se2−, which however differed from the spectra of the anti-perovskites regarding the shape and position of the K-edge (Fig. 8). A more intense pre-edge feature of FeSe with the maximum at 12658.8 eV is an indication of a higher density of unoccupied states in the Se 4p band and, therefore, more covalent Se–Fe bonding compared to in anti-perovskites. Analysis of the Se K-edge spectra of Li2MSeO led to the following conclusions.
For all materials, a small pre-edge feature was clearly visible at about 12662 eV due to the 1s → 4p electron transition, in agreement with a partially covalent character of M–Se bonds. This tiny pre-edge was also observed in our earlier studies on Li2FeS1−xSexO compounds.13 The intensity of this pre-edge peak correlated with the transition metal electronegativity and the covalency grade of the M–Se bonding. Thereby, Li2MnSeO, Li2Mn0.5Fe0.5SeO, and Li2Co0.5Mn0.5SeO compounds, containing the most electropositive Mn cations (1.55 Pauling units), showed a small pre-edge feature, detectable as a poorly resolved peak in dxμ(E)/dE derivative plots. In contrast, the pre-edge was much more pronounced in Li2CoSeO with less electropositive Co cations (1.88 Pauling units), and, therefore, with a more covalent character of M–Se bonding.
With Li extraction, the pre-edge grew (Fig. 8b, see Li2FeSeO for example). Three effects can contribute here: (i) the amount of ionic Li–Se bonds was decreased, and the covalent M–Se bonds became more and more dominant; (ii) a strengthening of the overlap between Fe 3d and Se 4p orbitals due to the oxidation of Fe2+,31,33 and (iii) the oxidation of selenium itself, making 1s → 4p electron jump more probable. The pre-edge decreased when lithium was re-inserted into the materials (see the ESI†). The oxidation of Se was also proven by the Se K-edge shift to higher energies during charging.34 After a whole charge–discharge cycle, a small energy shift remained, pointing to only a partially reversible Se redox activity.
We compared the relative energy shift of the Se K-edge in all Li2MSeO compounds upon Li extraction (Fig. 8c). Depending on the results, two groups of anti-perovskites could be selected: (1) Li2FeSeO and Li2MnSeO with a low energy shift of the Se K-edge, and (2) Li2Fe0.5Mn0.5SeO, Li2Fe0.5Co0.5SeO, Li2Co0.5Mn0.5SeO, and Li2CoSeO with a significant Se K-edge shift. This finding suggest a lower Se redox activity in Li2FeSeO and Li2MnSeO during delithiation, being in line with the high redox activity of Mn and Fe cations in these materials. In contrast, an equimolar combination of Mn and Fe, Mn and Co, Fe and Co in the structure, or the presence of only Co led to a very high redox activity of Se, visible as a larger shift of the Se K-edge to higher energies for the same amount of Li removed.
The high Se activity in all the Co-containing materials was correlated with the high Co3+/Co2+ standard electrochemical potential and its redox inactivity in anti-perovskites: for the same delithiation state, the charge compensation will not be realized by the second transition metal only, obviously leading to the oxidation of Se as well.
Li2Fe0.5Mn0.5SeO with a high Se activity seemed to be an exception, since the redox activity of Se in compounds with only Fe and with only Mn was low. Probably, a higher redox potential of Mn2+ forced the oxidation of Fe2+ and Se2− similarly as for Co2+ in Co-compositions. Note that there is an intrinsic misfit of metal–oxygen bonds: in Li2Fe0.5Mn0.5SeO, Mn–O became shorter and Fe–O longer than in the individual compounds. Due to the reduced amount of Fe cations in Li2Fe0.5Mn0.5SeO compared to Li2FeSeO, charge compensation upon Li extraction was partially realized by the oxidation of Se anions.
Among Li2MSeO with M as Fe, Mn, Co, or their mixture, only Li2FeSeO became irreversibly amorphous upon delithiation. In order to clarify the reason, we studied the local environment for Fe and Se ions by analysing the Fe and Se K-edge XAFS spectra at different states of charge, see Table 2.
| x in LixFeSeO | 1st coordination shell | 2nd coordination shell |
|---|---|---|
| a Calculations were done for both Fe and Se K-edges. The absorbing element is highlighted. The results for the remaining compounds are available in Table S13. | ||
| 2.00 (pristine) | Fe – O; 1.97(4) Å | Fe – (Li, Fe, Se); 2.8(1) Å |
| 0.77 (charged) | Fe – O; 1.86(4) Å | Fe – Se; 2.31(4) Å |
| Fe – (Li, Fe); 2.64(8) Å | ||
| 1.98 (discharged) | Fe – O; 1.94(7) Å | Fe – Se; 2.85(2) Å |
| Fe – (Li, Fe); 2.79(8) Å | ||
| 2.00 (pristine) | Se – (Li, Fe); 2.79(8) Å | Se – O; 3.5(1) Å |
| 0.99 (charged) | Se – (Li, Fe); 2.46(9) Å | Se – O; 3.3(1) Å |
| 1.99 (discharged) | Se – (Li, Fe); 2.78(8) Å | Se – O; 3.4(1) Å |
| Se – Se; 2.4(1) Å | ||
In the pristine Li2FeSeO, the nearest coordination of Fe consisted of oxygen anions with an Fe–O bond length of 1.97(4) Å, while the second coordination sphere contained Li, Fe, and Se with the same distances to Fe of 2.8(1) Å. In the charged state, the Fe–O distance became shorter (1.86(4) Å), in accordance with the oxidation of Fe. In the second coordination sphere, the distance to Se became significantly shorter, reflecting the oxidation of Fe as well (and eventually of Se). The distance to the neighbouring Fe and Li shortened less. After subsequent Li insertion, the Fe–O bond length returned to the initial value, but the difference between Fe–Se and Fe–(Fe, Li) distances remained, pointing towards a distortion of the crystal structure. The Fe–Se distance of 2.79(8) Å was rather close to the Fe–Se bond length in FeSe of 2.74 Å.35 The changes in the Se local surrounding were more significant and can explain the amorphization phenomenon. The first coordination sphere contained Li and Fe. In the charged state, the distance Se–(Fe, Li) was reduced to 2.46(9) Å, which was higher than the Fe–Se distance of 2.31(4) Å from Fe-EXAFS due to the longer Se–Li bond length.
After discharge, the insertion of an additional Se–Se pathway in the structural model was needed for a proper description of the Fourier-transformed XAFS function. The new Se–Se distance of 2.4(1) Å was close to the bond length of 2.373 Å in elemental Se.36 Thus, a continuous structure decomposition with the formation of Se-islands probably occurred upon the cycling of Li2FeSeO due to the oxidation of both Fe and Se at charging. This process was less pronounced in compounds with other transition metal cations.
Both the cationic and anionic activities of anti-perovskites were confirmed by DFT calculations. There was a big contribution of the Se 4p orbitals to the density of states close to the Fermi level for all the materials. Therefore, delithiation must necessarily result in the oxidation of selenium. Further, we calculated the formal electron densities of M 3d, Se 4p, and O 2p orbitals for the pristine and delithiated states. For the latter, the structural model was the same as for the pristine materials (the cell parameters were taken from the operando data), but the number of electrons per unit cell was reduced accordingly. Upon oxidation, the electron densities decreased for all the orbitals, thus confirming the covalent character of the chemical bonds. However, the electron density loss was different, showing the highest value in Se 4p followed by Mn 3d, Fe 3d, and Co 3d in the compounds with one and two transition metal cations. Calculations of the Li insertion (electron addition) into stoichiometric Li2MSeO led to unexpected conclusions. Thus, Li insertion into Li2Fe0.5Co0.5SeO resulted first in filling the Co 3d orbitals followed by Fe 3d orbitals. In the case of Li2Fe0.5Mn0.5SeO, the Fe 3d orbitals would be filled first, while in Li2Co0.5Mn0.5SeO, the Co 3d would be occupied first, followed by Mn 3d. Therefore, although these values were quite rough estimations, we confirmed the redox bifunctionality of Li2MSeO compounds, and their tunability.
3. Conclusion
Before summing up the outcomes, we would like to specify the following. To make our article more laconic, we put the comprehensive description of theoretical calculations and experimental procedures into the ESI.† Maintaining scientific ethics, we encourage readers to check the works in ref. 37–53 which are directly referred to in the ESI.† The impact of transition metal cations Fe, Mn, and Co in the Li2MSeO structure on the electrochemical behaviour in Li half-cells was evaluated. The redox potential and electronegativity of the transition metal are crucial parameters for the electrochemical properties. Among materials with a single transition metal, Li2FeSeO with the lowest redox potential of the transition metals showed the highest capacity at low currents, but a poor rate capability and was unstable upon cycling. In contrast, a small, but stable and almost current-independent capacity was observed for Li2CoSeO with the highest redox potential of Co, while the properties of Li2MnSeO lay between the Fe- and Co compounds. In Li2FeSeO and Li2MnSeO, both M2+ and Se2− ions participated in the redox process, while in Li2CoSeO, only Se oxidation/reduction compensated the charge during battery operation, and the Co cations remained inactive. Interestingly, pure anionic redox led to a rather stable electrochemical cycling at various current densities, unlike for any cases of simultaneous cationic and anionic oxidation observed for the studied systems.The electrochemical process becomes more complex with two transition metal cations in the structure. In this case, the highest specific capacity and satisfactory rate capability were found for Li2Fe0.5Co0.5SeO. Here, Co served as a structural stabilizer and “booster” for the redox activities of Fe and Se, remaining inactive upon battery operation. The activity of Se was comparable with Li2CoSeO, while the activity of Fe was even higher than in Li2FeSeO.Unfortunately, it was not possible to clarify a direct impact of selenium redox activity on the unit cell metrics, since the materials showed completely different structural behaviours upon delithiation. Thus, Li2FeSeO suffered from structural collapse, while Li2Fe0.5Co0.5SeO and Li2Fe0.5Mn0.5SeO remained crystalline after the first cycle; however, the delithiation mechanisms were different (single-phase vs. two-phase), whereas an operando study of Li2Co0.5Mn0.5SeO revealed tetragonal distortion, causing most likely kinetical complications for lithium (de)intercalation.
The combination of Co and Mn in Li2MSeO is rather unfavourable, since upon Li extraction, besides Se oxidation, Mn2+ was mostly oxidized to Mn3+, leading to a noticeable structural distortion due to the Jahn–Teller effect, and impeded Li diffusion. The mixture of Fe and Mn led to unexpected results: in Li2Fe0.5Mn0.5SeO, the capacity was only moderately aggravated with the current. Probably, due to a higher Mn2+/Mn3+ redox potential than that of Fe2+/Fe3+, whereby Mn cations mainly stabilized the crystal structure upon the oxidation of Fe and Se. We believe that the knowledge of synergistic effects in various cationic compositions may be transferred to other systems with similar crystal structures and relatively low operation voltages, giving some hints for their compositional design. Furthermore, the pure anionic redox activity in anti-perovskites represents an interesting direction for future studies of current-independent electrochemical performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the IFW Dresden excellence program, German Federal Ministry of Education and Research (BMBF) in the project HeNa (03XP0390C), and by German Research Foundation (DFG) in the project KIBSS (448719339). The authors acknowledge Andrea Voss and Anne Voidel (IFW Dresden) for performing the ICP-OES analysis. This research has benefitted from beamtime allocation at beamlines P02.1, P64 and P65 at the PETRA III synchrotron (DESY, Hamburg, Germany), and BL04 at the ALBA Cells synchrotron (Barcelona, Spain).References
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS PubMed.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed.
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942 RSC.
- M. A. Pellow, C. J. M. Emmott, C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2015, 8, 1938 RSC.
- A. Manthiram, J. C. Knight, S. T. Myung, S. M. Oh and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- J. A. Dawson, T. Farmprikis and K. Johnston, J. Mater. Chem. A, 2021, 9, 18746 RSC.
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, J. Am. Chem. Soc., 2017, 139(28), 9645 CrossRef CAS PubMed.
- Z. Lu and F. Ciucci, J. Mater. Chem. A, 2018, 6, 5185 RSC.
- K. T. Lai, I. Antonyshyn, Y. Prots and M. Valldor, Inorg. Chem., 2018, 57(21), 13296 CrossRef CAS PubMed.
- D. Mikhailova, L. Giebeler, S. Maletti, S. Oswald, A. Sarapulova, S. Indris, Z. Hu, J. Bednarick and M. Valldor, ACS Appl. Energy Mater., 2018, 1, 6593 CrossRef CAS.
- M. V. Gorbunov, S. Carocci, S. Maletti, M. Valldor, T. Doert, S. Hampel, I. G. Gonzalez Martinez, D. Mikhailova and N. Gräßler, Inorg. Chem., 2020, 59(21), 15626 CrossRef CAS PubMed.
- M. V. Gorbunov, S. Carocci, I. G. Gonzalez Martinez, V. Baran and D. Mikhailova, Front. Energy Res., 2021, 9, 657962 CrossRef.
- M. A. A. Mohamed, M. V. Gorbunov, M. Valldor, S. Hampel, N. Gräßler and D. Mikhailova, J. Mater. Chem. A, 2021, 9, 23095 RSC.
- Handbook of Chemistry and Physics, ed. W. M. Haynes, D. R. Lide and T. J. Bruno, Boca Raton, London, New York, 2015, pp. 5–80 Search PubMed.
- K. Zhu, C. Wang, Z. Chi, F. Ke, Y. Yang, A. Wang, W. Wang and L. Miao, Front. Energy Res., 2019, 7, 123 CrossRef.
- A. Eftekhari, Sustainable Energy Fuels, 2017, 1, 14 RSC.
- M. A. A. Mohamed, L. Singer, H. Hahn, D. Djendjur, A. Özkara, E. Thauer, I. G. Gonzalez-Martinez, M. Hantusch, B. Büchner, S. Hampel, R. Klingeler, N. Gräßler and J. Pow, Sources, 2023, 558, 232547 CrossRef CAS.
- L. Vegard, Z. Phys., 1921, 5, 17 CrossRef CAS.
- V. M. Goldschmidt, Naturwiss., 1926, 14(21), 477 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751 CrossRef.
- L. Chuan-Jin, Z. Feng and Z. Zi-Zhong, Acta Phys. Sin., 2019, 68(15), 157201 CrossRef.
- M. Aykol and C. Wolverton, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 115105 CrossRef.
- D. Mikhailova, C. Y. Kuo, P. Reichel, A. A. Tsirlin, A. Efimenko, M. Rotter, M. Schmidt, Z. Hu, T. W. Pi, L. Y. Jang, Y. L. Soo, S. Oswald and L. H. Tjeng, J. Phys. Chem. C, 2014, 118(10), 5463 CrossRef CAS.
- I. B. Bersuker, The Jahn–Teller Effect, Cambridge University Press, Cambridge, 2006, p. 35 Search PubMed.
- H. Somaily, S. Kolesnik, J. Mais, D. Brown, K. Chapagain, B. Dabrowski and O. Chmaissem, Phys. Rev. Mater., 2018, 2, 054408 CrossRef CAS.
- M. Newville, Fundamentals of XAFS, Rev. 1.7, University of Chicago, Chicago, IL, 2004 Search PubMed.
- M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, Coord. Chem. Rev., 2017, 345, 182 CrossRef CAS PubMed.
- A. Mauger and C. M. Julien, Batteries, 2018, 4(3), 39 CrossRef.
- M. Tamaru, P. Barpanda, Y. Yamada, S. Nishimura and A. Yamada, J. Mater. Chem., 2012, 22, 24526 RSC.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 3rd edn, 1960 Search PubMed.
- Y. Cui, J. Lu, T. Bolin, Y. Ren, W. Wenig, C. Sun, V. A. Maroni, S. M. Heald and K. Amine, J. Am. Chem. Soc., 2013, 135(21), 8047 CrossRef CAS PubMed.
- B. Joseph, A. Iadecola, L. Simonelli, Y. Mizuguchi, Y. Takano, T. Mizokawa and N. L. Saini, J. Phys.: Condens. Matter, 2010, 22, 485702 CrossRef CAS PubMed.
- J. T. Henthorn, R. J. Arias, S. Koroidov, T. Kroll, D. Sokaras, U. Bergmann, D. C. Rees and S. DeBeer, J. Am. Chem. Soc., 2019, 141(34), 13676 CrossRef CAS PubMed.
- A. L. Ryser, D. G. Strawn, M. A. Marcus, S. Fakra, J. L. Johnson-Maynard and G. Möller, Environ. Sci. Technol., 2006, 40, 46 CrossRef PubMed.
- M. M. Srivastava and O. N. Srivastava, Acta Crystallogr. A, 1975, 31, 82 Search PubMed.
- P. Cherin and P. Unger, Inorg. Chem., 1967, 6, 1589 CrossRef CAS .Following references were cited in the ESI:†.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 38 CrossRef PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- A. M. Abakumov, D. Batuk, J. Hadermann, M. G. Rozova, D. V. Sheptyakov, A. A. Tsirlin, D. Niermann, F. Waschkowski, J. Hemberger, G. Van Tendeloo and E. V. Antipov, Chem. Mater., 2011, 23, 255 CrossRef CAS.
- S. Reschke, A. A. Tsirlin, N. Khan, L. Prodan, V. Tsurkan, I. Kézsmárki and J. Deisenhofer, Phys. Rev. B, 2020, 102, 094307 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. Le Bail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23(3), 447 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229(5), 345 CAS.
- A.-C. Dippel, H.-P. Liermann, J. T. Walter, H. Schulte-Schrepping, O. Seeck and H. Franz, J. Synchrotron Radiat., 2015, 22(3), 675 CrossRef CAS PubMed.
- J. Filik, A. W. Ashton, P. C. Y. Chang, P. A. Chater, S. J. Day, M. Drakopoulos, M. W. Gerring, M. L. Hart, O. V. Magdysyuk, S. Michalik, A. Smith, C. C. Tang, N. J. Terrill, M. T. Wharmby and H. Wilhelm, J. Appl. Crystallogr., 2017, 50, 959 CrossRef CAS PubMed.
- M. Faraday, Philos. Trans. R. Soc., 1834, 124, 77 CrossRef.
- F. Fauth, I. Peral, C. Popescu and M. Knapp., Powder Diffr., 2013, 28, 360 CrossRef.
- M. Herklotz, J. Weiß, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, J. Appl. Crystallogr., 2016, 49, 340 CrossRef CAS.
- W. A. Caliebe, V. Murzin, A. Kalinko and M. Görlitz, AIP Conf. Proc., 2019, 2054, 060031 CrossRef.
- E. Welter, R. Chernikov, M. Herrmann and R. Nemausat, AIP Conf. Proc., 2019, 2054, 040002 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed description of the experimental procedures and additional data. See DOI: https://doi.org/10.1039/d4ta00153b |
| This journal is © The Royal Society of Chemistry 2024 |