
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzobisthiazole-substituted terpolymers for non-halogenated solvent-processed polymer solar cells with enhanced efficiency, thermal stability and mechanical robustness†
Soodeok
Seo‡
a,
Hyesu
Jeon‡
a,
Eun Sung
Oh
b,
Jin-Woo
Lee
a,
Chulhee
Lim
 a,
Trieu
Hoang-Quan Nguyen
a,
Tan
Ngoc-Lan Phan
a,
Dahyun
Jeong
a,
Michael J.
Lee
c,
Taek-Soo
Kim
b and
Bumjoon J.
Kim
*a
a,
Trieu
Hoang-Quan Nguyen
a,
Tan
Ngoc-Lan Phan
a,
Dahyun
Jeong
a,
Michael J.
Lee
c,
Taek-Soo
Kim
b and
Bumjoon J.
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: bumjoonkim@kaist.ac.kr
bDepartment of Mechanical Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
cDepartment of Mechanical Engineering, College of Engineering, Kyung Hee University, Yongin 17104, Republic of Korea
First published on 21st February 2024
Abstract
High power conversion efficiency (PCE), eco-friendly processing, and long-term stability are essential for the commercialization of polymer solar cells (PSCs). In this study, we develop PM6-based terpolymer donors (PM6-DTzBX, where X = 5–20), by substituting the benzodithiophene-dione (BDD) unit with the benzobisthiazole (DTzB) unit, which aims to tune the crystalline properties of the polymers as well as achieving a blend morphology with sufficient intermixed domains. The DTzB-incorporating polymer donors (PDs) demonstrate stronger intermolecular interactions with a Y6-BO small molecule acceptor (SMA) and exhibit more pronounced crystalline properties than the reference PM6 PD. Consequently, PM6-DTzB10 PD-based PSCs achieve a higher PCE of 16.8% compared to that of PM6-based PSCs (15.6%) when processed in a non-halogenated ortho-xylene solvent. Furthermore, under thermal stress at 100 °C for 100 h, PM6-DTzB10-based PSCs maintain 88% of the initial PCE and exhibit enhanced thermal stability compared to PM6-based PSCs, which retain 72% of the initial PCE. Additionally, the PM6-DTzB10:Y6-BO blend films demonstrate a 7-fold increase in stretchability with a crack onset strain (COS) of 11.7%, compared to the PM6:Y6-BO blend films (COS = 1.7%). These enhancements in the PCE, thermal stability, and mechanical robustness can be mainly ascribed to the formation of a well-intermixed PD:SMA blend morphology and enhanced crystalline properties of PM6-DTzB PDs. This study highlights the potential of the terpolymer strategy in developing efficient, thermally stable, and mechanically robust PSCs.
1. Introduction
Polymer solar cells (PSCs) are gaining attention as a promising next-generation energy source due to their advantages including semi-transparency, and cost-effective large-area fabrication through solution processing.1–4 Various polymer donors (PDs) and small-molecule acceptors (SMAs) have been developed to enhance the power conversion efficiency (PCE) of PSCs, with some achieving PCE values exceeding 19%.5–13 However, the commercialization of PSCs necessitates the replacement of hazardous halogenated solvent-based processing, such as the use of chloroform and chlorobenzene, with more eco-compatible alternatives.14–21 However, solution processing based on non-halogenated solvents results in PSCs with lower PCE values than those processed with halogenated solvents, mainly due to unoptimized PD:SMA blend morphologies.The unfavorable blend morphology primarily stems from the lower solubilities of PDs and SMAs and their unoptimized aggregated structures in non-halogenated solvents.14,22,23 In particular, molecular incompatibility between the PD and SMA often leads to severe phase separation between their respective domains during the film formation process. This unoptimized blend morphology adversely affects charge generation and transport in PSCs, resulting in decreased PCEs. In addition, an excessively phase-separated morphology is prone to degrade under external stresses such as light and heat, resulting in decreased photo, thermal, and mechanical stabilities of the PSCs.24–27 Therefore, the design of active layer materials with sufficient solubilities in non-halogenated solvents and improved donor/acceptor interactions is imperative to simultaneously achieve both high performance and stability of the PSCs. For example, pairing donor and acceptor materials with improved molecular compatibility can prevent excessive separation between domains by reducing the thermodynamic penalty for formation of large donor–acceptor interfaces. The presence of sufficient donor–acceptor interfaces and intermixed domains can enhance the exciton dissociation and charge generation efficiency, resulting in higher short-circuit current density (Jsc), fill factor (FF), and PCE values in the PSCs.28 Importantly, the PD:SMA blend film with sufficient intermixed domains can prevent the occurrence of cracks at the PD:SMA interfaces by effectively dissipating mechanical stress, and enhancing the mechanical robustness of the PSCs.29–31
The design of terpolymer-type PDs featuring D–A1–D–A2 type backbones has proven to be an effective strategy for enhancing the molecular compatibility of typical alternating-type PDs (D–A1) with SMAs by replacing the A1 block with a carefully selected A2 block.32–40 A suitable proportion of the A2 block in the terpolymer-type PD can effectively control its surface tension, which determines the enthalpic interaction with the SMA at the donor–acceptor interfaces. For instance, Cao et al. designed a random terpolymer-type PD (JD40-BDD20) by adjusting the molar ratio of dithienobenzothiadiazole (TBT) and benzodithiophene-dione (BDD) units.23 The use of the JD40-BDD20 terpolymer resulted in a reduced phase separation with the PJTVT acceptor compared to the control JD40 PD due to its higher miscibility with the acceptor. As a result, the JD40-BDD20-based PSC demonstrated a higher PCE of 16.35% than that (14.05%) of the JD40-based PSC in ortho-xylene (o-xylene) processing. As another example, our group introduced ethyl thiophene-3-carboxylate units into the D18 PD backbone to yield terpolymer-type PDs (PBET 10–50), which enabled a PD:Y6-BO SMA blend morphology with sufficient intermixed domains compared to that with the reference D18.33 PBET 10–50 enabled solution processing in o-xylene to construct a PSC with a PCE of 15.5%, whereas solution processing of D18 PD was nearly impossible due to its very poor solubility in o-xylene. These studies suggest that developing terpolymer-type PDs can effectively increase their solubility in non-halogenated solvents and improve the molecular compatibility with SMAs. However, most random terpolymers inevitably exhibit reduced crystalline properties compared to alternating copolymers because random incorporation of the third A2 unit into the PD polymer results in the distortion of the molecular conformation and interferes with the effective formation of intermolecular assembly.22,35 This decreased crystallinity of PDs can negatively influence the charge transport properties of the film, compromising the PCE values of the PSCs. Therefore, it is essential to design and incorporate an appropriate third unit into the PD backbone that not only ensures higher crystalline and electrical properties but also maintains its sufficient processability in eco-friendly solvents.
In this study, we develop a new series of terpolymer-type PDs (PM6-DTzBX, X = 5, 10, and 20) by incorporating the benzo[1,2-d:4,5-d′]bis(thiazole) (DTzB) unit into the PM6 PD backbone, replacing the BDD unit. Then, we utilize them to produce efficient and stable PSCs using non-halogenated solvent-based processing. We select the DTzB unit as the third component in the PD for the following reasons: (1) the DTzB unit, containing nitrogen atoms, offers higher polarity and hydrophilicity than the BDD unit, thereby enhancing the molecular compatibility of the PDs with SMAs.41–43 (2) The thiazole units in the DTzB block facilitate strong molecular interactions with adjacent thiophenes through a N–S noncovalent bonding.44–47 This improved intra- and inter-molecular interaction can overcome the inherent limitations of random terpolymers, resulting in PDs with higher crystallinity and superior charge transport properties. As a result, with o-xylene solvent-based processing, PM6-DTzB10 PD-based PSCs achieve a higher PCE of 16.8% compared to PM6-based PSCs (PCE = 15.6%). This increase is mainly attributed to higher crystallinity and hole mobility of the PM6-DTzB10. In addition, the PM6-DTzB10:SMA blend exhibits decreased domain size and purity compared to those of the reference PM6:SMA blend. This improved morphology of the PM6-DTzB10:SMA blend contributes to enhanced thermal stability and mechanical robustness through the formation of large donor–acceptor interfaces. Under continuous heating at 100 °C for 100 h, PM6-DTzB10-based PSCs maintain 88% of the initial PCE and exhibit enhanced thermal stability compared to PM6-based PSCs retaining 72% of the initial PCE. Moreover, the PM6-DTzB10:Y6-BO blend film exhibits remarkably enhanced stretchability with a higher crack onset strain (COS) value of 11.7%, compared to the PM6:Y6-BO blend film (COS = 1.7%). This study highlights the importance of selecting the third unit in terpolymer PDs to simultaneously enhance the PD crystallinity and PD–SMA interaction for achieving highly efficient and stable PSCs.
2. Results and discussion
2.1. Synthesis and characterization of materials
To explore the impact of the incorporated DTzB unit in the PD chain on polymer properties and photovoltaic performance, PM6-DTzBX (X = 5, 10, and 20) terpolymers were synthesized. Here, X represents the molar fraction of the DTzB unit relative to the benzodithiophene (BDT) unit in the PD backbone. The planar-structured DTzB unit was chosen to yield the PM6-DTzBX terpolymers for several reasons. (1) The nitrogen atoms in the DTzB unit can effectively enhance the polarity of the resulting polymers, reducing the thermodynamic immiscibility between the PM6-DTzBX terpolymers and SMAs.41,48 (2) The thiazole units in the DTzB block promote strong N–S noncovalent bonding with thiophene, enhancing intra- and intermolecular interactions between PDs.42 (3) Importantly, the DTzB unit has a fused ring structure without an alkyl solubilizing group, which helps to reinforce the backbone planarity of the PM6-DTzBX terpolymers.49,50 These factors can synergistically improve the crystallinity of the PD, thereby enhancing its charge-transporting capability.Fig. 1a presents the chemical structures of the PD and 2,2′-((2Z,2′Z)-((12,13-bis(2-butyloctyl)-3,9-diundecyl-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (Y6-BO) SMA. Y6-BO was selected as a SMA due to its excellent light absorption ability, electron mobility, and sufficient solubility in processing solvents including o-xylene. The synthetic details of the DTzB monomer and PDs are provided in the Experimental section of the ESI (Fig. S1 and S2†). All PDs were synthesized to have similar number-average molecular weight (Mn) values ranging from 46 to 49 kg mol−1 (Table 1). We also confirmed that all PDs exhibited sufficient solubility in o-xylene (>40 mg mL−1).
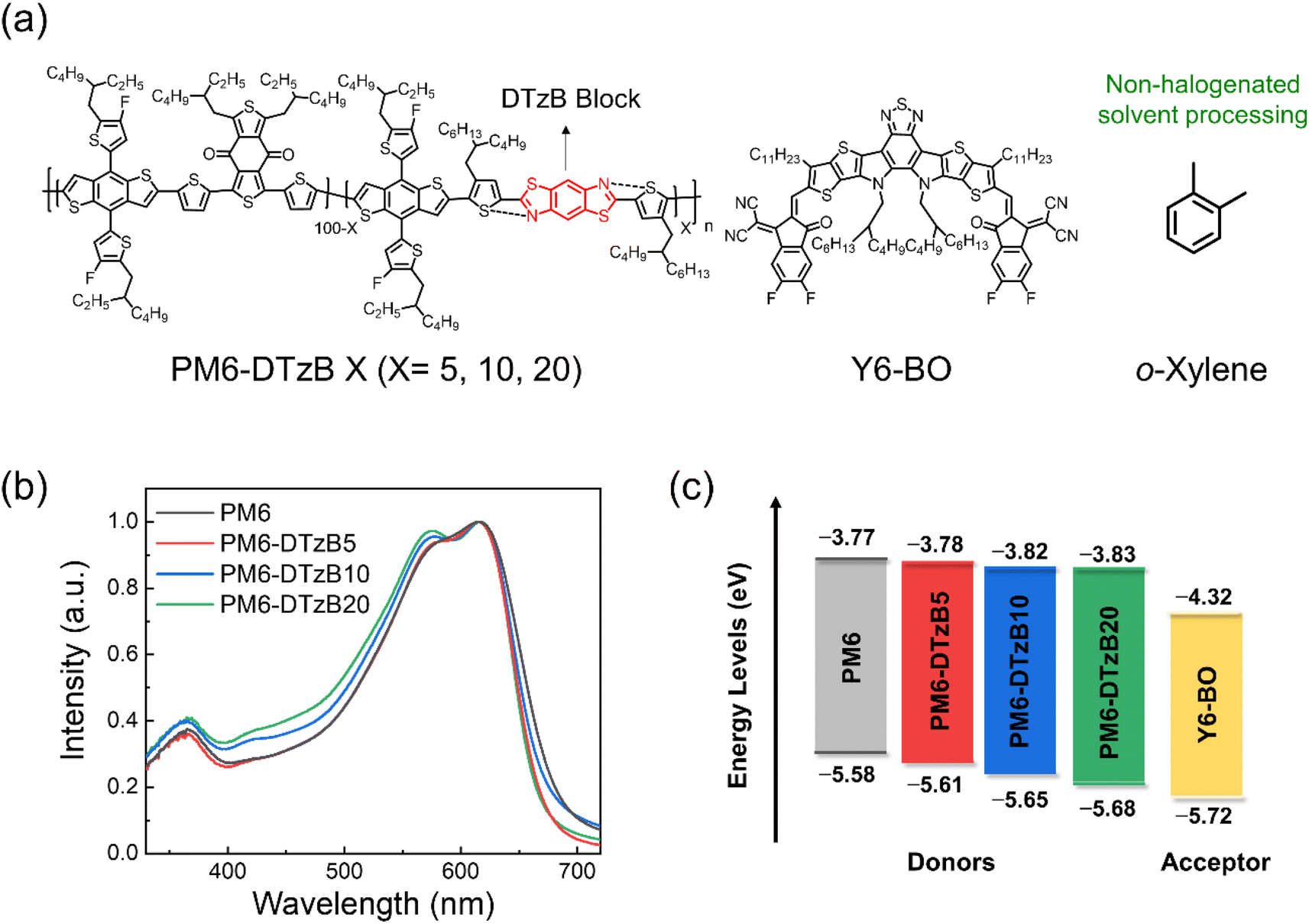 | ||
| Fig. 1 (a) Chemical structures of the materials and a processing solvent used in this study. (b) UV-vis absorption spectra of PD films. (c) Energy levels of the materials used in this study. | ||
| Material | M n(Ð)a [kg mol−1] | λ filmmax [nm] | ε filmmax [×105 cm−1] | λ edgefilm [nm] | E optg [eV] | E HOMO [eV] | E LUMO [eV] |
|---|---|---|---|---|---|---|---|
| a Determined by gel permeation chromatography eluting in 1,2,4-trichlorobenzene relative to polystyrene standards. b Estimated from the UV-vis spectra of the thin film state spin-coated from the o-xylene solution. c Estimated from the absorption onsets in thin films spin-coated from the o-xylene solution using Egopt = 1240/λedgefilm. d Calculated from the cyclic voltammetry spectra. e LUMO = HOMO + Eoptg. | |||||||
| PM6 | 48.8 (2.6) | 615 | 0.64 | 685 | 1.81 | −5.58 | −3.77 |
| PM6-DTzB5 | 46.0 (2.5) | 615 | 0.63 | 678 | 1.83 | −5.61 | −3.78 |
| PM6-DTzB10 | 47.8 (3.6) | 617 | 0.65 | 677 | 1.83 | −5.65 | −3.82 |
| PM6-DTzB20 | 45.9 (3.2) | 617 | 0.62 | 673 | 1.84 | −5.68 | −3.83 |
| Y6-BO | — | 829 | 1.13 | 886 | 1.40 | −5.72 | −4.32 |
Ultraviolet-visible (UV-vis) absorption spectra of the PDs in film and solution are shown in Fig. 1b and S3,† respectively. Interestingly, with an increase in the quantity of DTzB units within the PD backbone, PDs exhibited enhanced light absorption in a broad range spectrum, including wavelengths from 400 to 550 nm. While the maximum absorption wavelengths (λfilmmaxs) of the PDs were similar in a range of 615–617 nm, the wavelengths at their absorption edges (λedgemaxs) gradually blue-shifted with increasing DTzB content in the PDs. For example, the λedgemaxs of PM6, PM6-DTzB5, PM6-DTzB10, and PM6-DTzB20 were 685, 678, 677, and 673 nm, respectively. As a result, the optical bandgap (Eoptg) of the PDs gradually increased with increasing content of the DTzB units (Table 1). The frontier molecular orbital energy levels of the PDs were measured by cyclic voltammetry (Fig. 1c and S4†). Increasing the content of DTzB units gradually down-shifted the highest occupied molecular orbital (HOMO) energy levels of the PDs. For example, the HOMO energy levels of PM6, PM6-DTzB5, PM6-DTzB10, and PM6-DTzB20 were −5.58, −5.61, −5.65, and −5.68 eV, respectively. Energy levels, which were estimated from density functional theory (DFT) calculations performed at the B3LYP/6-31G(d,p) level, supported the energy level trend observed in CV measurements (Fig. S5†). For example, the BDT-DTzB structure possessed a lower HOMO energy level (−5.24 eV) compared to that of the BDT-BDD structure (HOMO = −5.15 eV) in the simulation.41,44 In addition, the BDT-DTzB structure exhibited a larger bandgap value (Eg = 2.92 eV) than the BDT-BDD structure (2.71 eV). Therefore, the downshifted HOMO energy levels of the PDs are attributed to the strong electron-withdrawing properties of DTzB units and an increased bandgap of the polymers. The down-shifted HOMO energy levels of the PDs are advantageous in achieving high Voc of the PSCs.
The aggregation properties of the PDs were investigated by measuring temperature-dependent UV-vis absorption spectra in o-xylene solution (Fig. S6†). The degree of aggregation was quantified by comparing the ratio of maximum intensity at 100 °C to that at 20 °C (I100 °Cmax/I20 °Cmax). PM6-DTzB PDs showed stronger aggregation (I100 °Cmax/I20 °Cmax = 0.84–0.88) compared to PM6 PD (I100 °Cmax/I20 °Cmax = 0.77) (Table S1†). This result suggests that introducing the DTzB unit into the PD backbone enhances the intermolecular interaction between PD chains, leading to a higher degree of aggregation.41
The crystalline properties of neat PD films were investigated using grazing incidence X-ray scattering (GIXS). In the GIXS 2D image and line-cut profiles, all PDs predominantly exhibited a face-on packing orientation, as evidenced by distinct (100) peaks in the in-plane (IP) direction and (010) peaks in the out-of-plane (OOP) direction (Fig. S7 and S8†).51 For a quantitative comparison of the relative crystallinity of the PDs, their coherence length (Lc) values for the (010) scattering peaks in the OOP direction (Lc(010)) were estimated using Scherrer's equation (Table S2†).52 Interestingly, PDs containing DTzB units exhibited higher Lc(010) values (1.2 nm for PM6-DTzB10 and 1.3 nm for PM6-DTzB20) than PM6 (Lc(010) = 0.6 nm), suggesting relatively larger crystal sizes in the film. The higher crystallinity of PM6-DTzB PDs compared to PM6 may be attributed to their stronger aggregation in solution, which helps forming well-ordered intermolecular assemblies during the film formation by solution processing.53
The charge transport abilities of PD films were assessed by measuring the space charge limited current (SCLC) (Table S3†). The hole mobilities (μhs) of neat PDs gradually increased with increasing DTzB content. For example, the μhs of PM6, PM6-DTzB10, and PM6-DTzB20 were 1.36 × 10−4, 1.63 × 10−4, and 1.72 × 10−4 cm2 V−1 s−1, respectively. The enhanced electrical properties of the DTzB-containing PDs compared to PM6 correlate with their enhanced aggregation and crystalline properties. It is notable that, while most random terpolymer-type PDs exhibited reduced crystalline and electrical properties compared to the alternating copolymers owing to reduced structural regularity,23,36,54 incorporation of the DTzB units does not compromise those properties.
2.2. Photovoltaic properties
The photovoltaic properties of all PD:Y6-BO systems were investigated by fabricating conventional-type PSCs. The device architecture and fabrication procedure of PSCs are described in the Experimental section of the ESI.† All PD:Y6-BO PSCs were processed in the o-xylene solvent. The current density–voltage (J–V) curves are shown in Fig. 2, and the corresponding photovoltaic parameters of the PSCs are presented in Table 2. Notably, the introduction of the DTzB unit instead of the BDD unit into PM6 PD sequentially increased the open-circuit voltage (Voc) of PSCs (0.830 V for PM6, 0.839 V for PM6-DTzB5, 0.842 V for PM6-DTzB10, and 0.856 V for PM6-DTzB20). This Voc trend is correlated with the lower HOMO levels of DTzB-incorporating PDs than PM6 (Table 1). As a result, PM6-DTzB10-based PSCs exhibited a higher PCE (16.8%) than that of the control PD (PM6, 15.6%), mainly owing to a higher Voc value (0.842 V) and Jsc of 25.14 mA cm−2, in comparison to PM6-based PSCs (Jsc = 24.03 mA cm−2). However, a further increase in the content of DTzB units led to a decreased PCE value (16.1% for PM6-DTzB20:Y6-BO). The PCE distributions of PSCs fitted with Gaussian functions are displayed in Fig. 2b, indicating that all systems showed consistent PCEs. The external quantum efficiency (EQE) spectra of the PSCs are shown in Fig. 2c. The PM6-DTzB-based PSCs exhibited higher EQE responses in both the PD absorption range (400–650 nm) and SMA absorption range (650–900 nm). This result supports higher Jsc values in DTzB-incorporating PD:Y6-BO PSCs than PM6:Y6-BO PSCs. The calculated Jsc values matched well with the Jscs measured from the PSCs with errors within 2% (Table 2). | ||
| Fig. 2 (a) J–V curves, (b) PCE distributions, (c) EQE spectra, and (d) Jph–Veff curves of PD:Y6-BO PSCs. | ||
| P D | V oc [V] | J sc [mA cm−2] | Cal. Jscb [mA cm−2] | FFa | PCEmax (avg)a [%] |
|---|---|---|---|---|---|
| a Average values were obtained from 10 independent devices. b Calculated from EQE profiles. | |||||
| PM6 | 0.830 | 24.03 | 23.53 | 0.76 | 15.60 (15.13 ± 0.18) |
| PM6-DTzB5 | 0.839 | 24.59 | 24.33 | 0.73 | 15.37 (15.04 ± 0.21) |
| PM6-DTzB10 | 0.842 | 25.14 | 25.19 | 0.76 | 16.81 (16.14 ± 0.27) |
| PM6-DTzB20 | 0.856 | 25.02 | 24.65 | 0.74 | 16.08 (15.73 ± 0.18) |
To elucidate the origin of the different photovoltaic properties, we investigated the charge generation, dissociation, transport, and recombination behaviors of the PD:Y6-BO blends. To investigate charge generation behaviors, the free charge carrier generation rate (G(E,T)) of the PSCs was evaluated. The G(E,T) can be determined from the maximum exciton generation rate (Gmax) and exciton dissociation probability (P(E,T)).55 The calculation procedure of Gmax, P(E,T) and G(E,T) is described in the Experimental section of the ESI.† The incorporation of an appropriate proportion of DTzB in PDs increased the Jsat and Gmax of the blend film. For example, the Gmax values of the PM6-DTzB5-, PM6-DTzB10-, and PM6-DTzB20-based blends were 1.89, 1.97 and 1.94 cm−3 s−1, respectively, surpassing that of the PM6 based blend (1.81 cm−3 s−1). Therefore, the G(E,T) values of the PM6-DTzB-based blends were higher than that of the PM6-based blend (Fig. 2d and Table S4†). This result indicates that the incorporation of the DTzB unit in the PD facilitates the charge generation and contributes to a higher Jsc during PSC operation.
The charge recombination properties of the PSCs were investigated by measuring their light intensity (P)-dependent Jsc and Voc values (Fig. S9†).56 All the blends exhibited similar slopes (α) in the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Jsc–logP plots, indicating similar bimolecular recombination properties. The slope (S) in the Voc–logP plots exhibited comparable values in PM6- (1.10 kT q−1) and PM6-DTzB10-based PSCs (1.09 kT q−1), indicating that the introduction of an appropriate amount of DTzB does not cause monomolecular/trap-assisted recombination.57 The SCLC hole and electron mobilities of the PD:Y6-BO blend films were measured to evaluate their charge transport abilities (Table S5†). All the blends showed similar μh values (2.1–2.3 × 10−4 cm2 V−1 s−1). Interestingly, the electron mobility (μe) was the highest in the PM6-DTzB10 blend (1.55 × 10−4 cm2 V−1 s−1) among all blends, resulting in the most balanced mobility ratio (μh/μe = 1.50) compared to other blends.58
Jsc–logP plots, indicating similar bimolecular recombination properties. The slope (S) in the Voc–logP plots exhibited comparable values in PM6- (1.10 kT q−1) and PM6-DTzB10-based PSCs (1.09 kT q−1), indicating that the introduction of an appropriate amount of DTzB does not cause monomolecular/trap-assisted recombination.57 The SCLC hole and electron mobilities of the PD:Y6-BO blend films were measured to evaluate their charge transport abilities (Table S5†). All the blends showed similar μh values (2.1–2.3 × 10−4 cm2 V−1 s−1). Interestingly, the electron mobility (μe) was the highest in the PM6-DTzB10 blend (1.55 × 10−4 cm2 V−1 s−1) among all blends, resulting in the most balanced mobility ratio (μh/μe = 1.50) compared to other blends.58
2.3. Structural and morphological properties
The morphological properties of the PD:Y6-BO blends were analyzed using GIXS, resonant soft X-ray scattering (RSoXS), and in situ UV-vis spectroscopy. We first measured the GIXS profiles of a neat Y6-BO film (Fig. S10 and S11†). Interestingly, the GIXS 2D image and line-cut profile of the Y6-BO neat film exhibited a much sharper and more distinct (010) peak along the OOP direction after annealing, which is attributed to its strong crystalline property. Subsequently, the GIXS profiles of the PD:Y6-BO blend films were compared (Fig. 3a, b and S12†). All PD:Y6-BO blend films displayed IP (100) and OOP (010) peaks around qxy = 0.28–0.29 Å−1 and qz = 1.45–1.95 Å−1, respectively. Therefore, we assessed the relative degree of crystallinity (rDoC) of the (010) peaks of the PD:Y6-BO blend films in the blend GIXS profiles (Fig. 3b and Table 3). The r-DoC(010) values increased in the following order: PM6:Y6-BO (0.65) < PM6-DTzB10:Y6-BO (0.87) < PM6-DTzB20:Y6-BO (1.00).59 | ||
| Fig. 3 GIXS line-cut profiles of PD:Y6-BO blends in (a) the IP and (b) OOP directions. (c) RSoXS profiles of PD:Y6-BO blends. (d) Time evolution of the UV-vis absorption intensity change of PD:Y6-BO blends at 615 and 800 nm, respectively, during spin-coating. | ||
| P D | r-DOC(010)a | Domain sizeb [nm] | r-DPb | t sat at λmax of PDc [s] | t sat at λmax of Y6-BOc [s] |
|---|---|---|---|---|---|
| a Estimated from the OOP (010) peak of GIXS line-cut profiles in the range of 1.45–1.95 Å−1. b Estimated from the RSoXS profiles. c Estimated from the in situ UV-vis spectroscopy profiles of PD:Y6-BO blends. | |||||
| PM6 | 0.65 | 37.2 | 1 | 13.2 | 12.8 |
| PM6-DTzB10 | 0.87 | 33.1 | 0.58 | 12.6 | 12.4 |
| PM6-DTzB20 | 1 | — | 0.50 | 12.4 | 12.2 |
To investigate the degree of phase separation between the PD domain and Y6-BO domain, RSoXS measurements were performed, using a beam energy of 285.0 eV to maximize the contrast between the components in the blend films (Fig. 3c). The domain size and relative domain purity (r-DP) of the blends were estimated from their RSoXS profiles. Both the PM6:Y6-BO blend (37.2 nm) and PM6-DTzB10:Y6-BO blend (33.1 nm) exhibited appropriate domain sizes for charge transport, while the PM6-DTzB20:Y6-BO blend showed no distinguishable peak. The r-DP was significantly higher for the PM6:Y6-BO blend (1.00) compared to the PM6-DTzB10-based blend (0.58) and PM6-DTzB20-based blend (0.50) (Table 3). A large interfacial area between donor and acceptor domains in a blend film is advantageous for charge generation and exciton dissociation. Therefore, the well-intermixed domain in the PM6-DTzB10:Y6-BO blend facilitates both higher Jsc and FF values compared to other blends.60–63 At the same time, the incorporation of the DTzB unit into the PM6-DTzB PDs increased their crystalline properties, enhancing the charge transport properties and the PCE values in the PSCs. However, the PM6-DTzB20:Y6-BO blends exhibited an excessive amount of intermixed domains with lower domain size and purity, resulting in decreases of charge transport properties and photovoltaic performance.
The in situ UV-vis absorption measurements during the spin coating process were performed to investigate the film formation kinetics of different PD:SMA blend systems and understand its impact on the blend morphology (Fig. 3d, S13,† and Table 3). Extended quenching time in film solidification generally leads to a higher degree of liquid–liquid phase separation, typically resulting in more distinct phase separation between PD and SMA domains.64 The saturation times (tsats) in the absorption intensities at the maximum absorption wavelengths (λmax) of PDs and Y6-BO were tracked. At the λmax of PDs, tsat values decreased in the following sequence: PM6 (13.2 s) > PM6-DTzB10 (12.6 s) > PM6-DTzB20 (12.4 s). These reduced tsat values of PM6-DTzB PDs could be attributed to the stronger aggregation of PM6-DTzB PDs in o-xylene solvent compared to PM6, as shown in Fig. S6.† Similarly, the tsat value of Y6-BO in the blend solution also followed a decreasing trend: PM6 (12.8 s) > PM6-DTzB10 (12.4 s) > PM6-DTzB20 (12.2 s). These results suggest that the incorporation of DTzB into the PDs accelerated the solidification of both the PD and Y6-BO domains during film formation. This acceleration mitigates excessive phase separation and promotes the development of sufficiently intermixed domains.
To further investigate the changes in the molecular compatibility between the PD and SMA depending on the terpolymer structure, we conducted contact angle measurements to estimate the interfacial tensions between the PD and SMA (Fig. S14 and Table S6†).65,66 The contact angles of water droplets on neat PD films decreased in the order of PM6 (103.1°) > PM6-DTzB10 (100.9°) > PM6-DTzB20 (99.5°). Correspondingly, the surface tension values of PDs linearly increased from PM6 (19.2 mN m−1) to PM6-DTzB10 (20.5 mN m−1) and finally to PM6-DTzB20 (21.2 mN m−1). These results are consistent with the previous studies where polymers containing DTzB units, such as PBB1 and PBB2 polymers, exhibited higher hydrophilicity than the reference polymers PM6 and PM7.41,43 The surface tension value of the Y6-BO SMA film was measured to be 24.8 mN m−1. Thus, interfacial tension (γD–A) values between the PD and Y6-BO decreased sequentially in the order of PM6:Y6-BO (0.79), PM6-DTzB10:Y6-BO (0.50), and PM6-DTzB20:Y6-BO blends (0.38). This result supports the view that the inclusion of DTzB units into the PDs reduces molecular incompatibility with the Y6-BO SMA, thereby thermodynamically facilitating the formation of larger donor–acceptor interfaces and intermixed domains.
2.4. Thermal stability
Next, to assess the influence of the polymer structure on the thermal stability of PSCs, the changes in the PCEs of both PM6- and PM6-DTzB10-based PSCs were monitored under continuous heating at 100 °C (Fig. 4a). The difference in PCE changes between the two blend systems was evident. This degradation was mainly attributed to the unstable initial morphology associated with the poor miscibility between the PM6 PD and SMA.67,68 PM6:Y6-BO-based PSCs showed a notable PCE decrease within 24 hours, maintaining less than 80% of their initial efficiency (PCE = 12.0%). In contrast, PM6-DTzB10:Y6-BO-based PSCs retained 93% of their initial PCE (PCE = 15.6%) under identical conditions.33,69,70 | ||
| Fig. 4 (a) Thermal stability of PD:Y6-BO PSCs under a 100 °C heating condition. (b) RSoXS profiles of the PD:Y6-BO blend in the initial state (solid line) and after (dotted line) thermal annealing at 100 °C for 72 h. (c) AFM height images of PD:Y6-BO blend films before (left) and after (right) thermal annealing at 100 °C for 24 h. Each image size is 3 μm × 3 μm. | ||
To elucidate the difference in thermal stability, morphological changes during the thermal annealing were compared through RSoXS and atomic force microscopy (AFM) measurements (Fig. 4 and Table S7†). RSoXS profiles revealed significant phase separation in the PM6:Y6-BO blend under thermal stress, with r-DP increasing from 0.65 to 1.00 (Fig. 4b). In contrast, the PM6-DTzB10:Y6-BO blend maintained a relatively consistent domain purity (∼0.4), both before and after thermal annealing.71,72 This consistency suggests that the PM6-DTzB10:Y6-BO blend preserved its initial morphology mainly due to enhanced PD–SMA interaction and more strongly developed crystalline domains. Also, a similar trend was observed in their AFM height images. The root-mean-square averaged roughness (Rq) value of the PM6:Y6-BO blend increased from 1.3 to 1.9 nm after 24 h of heating. The increased Rq values and the formation of agglomerates on the PM6:Y6-BO blend are indicative of its unstable initial blend morphology. In contrast, the PM6-DTzB10:Y6-BO blend exhibited almost the same Rq value from 1.2 to 1.3 nm under identical heating conditions. These results confirm that the PM6-DTzB10:Y6-BO-based PSCs maintain their initial PSC performance under thermal stress by preserving their blend morphology.73
2.5. Mechanical properties
The mechanical robustness of the PSCs is a critical factor for their application as power suppliers in wearable devices.74,75 At first, we evaluated the stretchability of neat PD films using a pseudo free-standing tensile test (Fig. S15 and Table S8†).76,77 Interestingly, both the COS value and toughness were enhanced with the incorporation of the DTzB unit into the PD backbone. Specifically, the COS values increased in the following order: PM6 (16.9%) < PM6-DTzB5 (20.7%) < PM6-DTzB10 (23.1%) < PM6-DTzB20 (28.0%) films. This improvement in the mechanical properties in the PM6-DTzB PD could be attributed to its random terpolymer structure and increased intermolecular interaction between PDs, compared to PM6 PD.Next, we compared the stretchability of the PD:Y6-BO blend films (Fig. 5a and Table S9†). All active layers were spin-coated using the same process for device fabrication. Since the SMA molecules typically exhibit unconnected brittle domains due to their inherent rigid structure, it is important to dissipate mechanical stress to prevent crack propagation in the SMA domains and at the interfaces between the PD and SMA domains.78 For example, the PM6:Y6-BO exhibited a relatively low COS of only 1.7%. In contrast, well-intermixed PD:SMA domains have advantages in enhancing the film stretchability.23 With an increasing content of the DTzB unit, the COS value increased from 5.5% for PM6-DTzB5:Y6-BO, to 11.7% for PM6-DTzB10:Y6-BO, and 17.0% for PM6-DTzB20:Y6-BO. Similarly, the toughness values also increased in the order of 0.2 MJ m−3 for PM6:Y6-BO, 1.0 MJ m−3 for PM6-DTzB5:Y6-BO, 2.5 MJ m−3 for PM6-DTzB10:Y6-BO, and 3.7 MJ m−3 for PM6-DTzB20:Y6-BO. Fig. 5b shows the optical microscope (OM) images of the PM6:Y6-BO and PM6-DTzB10:Y6-BO blend films upon stretching during a tensile test. The PM6:Y6-BO blend showed distinct crack formation even at a small strain of 3%. In contrast, the PM6-DTzB10:Y6-BO blend displayed plastic deformation without any observable cracks at a strain of 10%. This improvement in mechanical properties of DTzB-incorporated PD-based blends is mainly attributed to the presence of a larger fraction of intermixed PD:SMA domains, which effectively dissipates mechanical stress and suppresses crack propagation. Especially, the PM6-DTzB10:Y6-BO blend and PM6-DTzB20:Y6-BO blend contained more intermixed blend domains than the PM6:Y6-BO blend, as shown in RSoXS profiles (Fig. 3c). These well-intermixed PD:SMA blend domains can facilitate the dissipation of mechanical stress on the brittle Y6-BO domains and at fragile PD:SMA interfaces.23
 | ||
| Fig. 5 (a) Stress–strain curves of PD:Y6-BO blend films. Optical microscope (OM) images of the blend films (b) during the tensile test and (c) on TPU substrates under stretching. | ||
Additionally, the crack formation behavior while stretching the PD:Y6-BO blend films on the thermoplastic polyurethane (TPU) substrate was compared to assess the potential of the DTzB-incorporated PDs in stretchable PSC applications (Fig. 5c).79–81 For the PM6:Y6-BO blend, cracks were clearly observed at only 10% strain and rapidly propagated under 30% strain, despite the stress dissipation from the elastomer support. In contrast, the PM6-DTzB10:Y6-BO blend film did not show any visible cracks during stretching up to 30% strain.
3. Conclusions
We developed a series of PM6-DTzB terpolymers by introducing the planar structured DTzB unit into the PM6 PD backbone and achieved efficient, thermally-stable, and mechanically robust PSCs by non-halogenated solvent processing. The introduction of an appropriate amount of the DTzB unit into the PD backbone resulted in improved molecular compatibility with the Y6-BO SMA as well as enhanced crystalline and electrical transport abilities of the PD film. This led to the formation of the blend morphology with sufficient PD:SMA intermixed domains as well as well-developed crystalline domains of PDs and SMAs, enhancing both the charge generation and transport properties in the PSCs. Therefore, PM6-DTzB10:Y6-BO PSCs exhibited a higher PCE of 16.8% compared to that of PM6:Y6-BO PSCs (PCE = 15.6%) with solution processing in o-xylene. The optimized blend morphology of the PM6-DTzB10:Y6-BO was also beneficial in suppressing the burn-in degradation of PSCs while significantly improving the stretchability of the active layer. The PM6-DTzB10-based PSCs showed a higher thermal stability against annealing at 100 °C than the PM6-based PSCs. Moreover, the PM6-DTzB10:Y6-BO blend films demonstrated superior mechanical properties, exhibiting 7 times higher stretchability (COS = 11.7%) compared to the PM6-based blend (COS = 1.7%).Author contributions
S. S and H. J. contributed equally to this work. S. S. and H. J. conceptualized this research, performed the investigation, and wrote the original draft. E. S. O., C. L., T. H-Q. N, and T. N-L. P. supported the investigation. J.-W. L, D. J, M. J. L, and T.-S. K. supported editing the draft. B. J. K. led data curation, project administration, funding acquisition, and editing the draft.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (2020M3H4A1A02084906 and RS-2023-00283244). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.References
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- E. Dauzon, X. Sallenave, C. Plesse, F. Goubard, A. Amassian and T. D. Anthopoulos, Adv. Mater., 2021, 33, 2101469 CrossRef CAS.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- X. Dong, Y. Jiang, L. Sun, F. Qin, X. Zhou, X. Lu, W. Wang and Y. Zhou, Adv. Funct. Mater., 2022, 32, 2110209 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- C. Han, J. Wang, S. Zhang, L. Chen, F. Bi, J. Wang, C. Yang, P. Wang, Y. Li and X. Bao, Adv. Mater., 2023, 35, 2208986 CrossRef CAS PubMed.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K.-Y. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS.
- G. Zhang, H. Ning, H. Chen, Q. Jiang, J. Jiang, P. Han, L. Dang, M. Xu, M. Shao, F. He and Q. Wu, Joule, 2021, 5, 931–944 CrossRef CAS.
- J. Yi, M. Pan, L. Chen, Y. Chen, I. C. Angunawela, S. Luo, T. Zhang, A. Zeng, J. Chen, Z. Qi, H. Yu, W. Liu, J. Y. L. Lai, H. K. Kim, X. Zhu, H. Ade, H. Lin and H. Yan, Adv. Energy Mater., 2022, 12, 2201850 CrossRef CAS.
- B. Pang, C. Liao, X. Xu, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2300631 CrossRef CAS PubMed.
- H. Liang, H. Chen, P. Wang, Y. Zhu, Y. Zhang, W. Feng, K. Ma, Y. Lin, Z. Ma, G. Long, C. Li, B. Kan, Z. Yao, H. Zhang, X. Wan and Y. Chen, Adv. Funct. Mater., 2023, 33, 2301573 CrossRef CAS.
- X. Yang, B. Li, X. Zhang, S. Li, Q. Zhang, L. Yuan, D.-H. Ko, W. Ma and J. Yuan, Adv. Mater., 2023, 35, 2301604 CrossRef CAS PubMed.
- S. Dong, T. Jia, K. Zhang, J. Jing and F. Huang, Joule, 2020, 4, 2004–2016 CrossRef CAS.
- Z. Zhong, S. Chen, J. Zhao, J. Xie, K. Zhang, T. Jia, C. Zhu, J. Jing, Y. Liang, L. Hong, S. Zhu, D. Ma and F. Huang, Adv. Energy Mater., 2023, 13, 2302273 CrossRef CAS.
- Z. U. Rehman, M. Haris, S. U. Ryu, M. Jahankhan, C. E. Song, H. K. Lee, S. K. Lee, W. S. Shin, T. Park and J.-C. Lee, Adv. Sci., 2023, 10, 2302376 CrossRef CAS.
- H. Xia, Y. Zhang, W. Deng, K. Liu, X. Xia, C.-J. Su, U. S. Jeng, M. Zhang, J. Huang, J. Huang, C. Yan, W.-Y. Wong, X. Lu, W. Zhu and G. Li, Adv. Mater., 2022, 34, 2107659 CrossRef CAS PubMed.
- H. Lu, G. Ran, Y. Liu, Z. Pei, W. Liu, Y. Liu, Z. Tang, W. Zhang and Z. Bo, Adv. Funct. Mater., 2023, 33, 2301866 CrossRef CAS.
- C. Wang, X. Ma, Y.-f. Shen, D. Deng, H. Zhang, T. Wang, J. Zhang, J. Li, R. Wang, L. Zhang, Q. Cheng, Z. Zhang, H. Zhou, C. Tian and Z. Wei, Joule, 2023, 7, 2386–2401 CrossRef CAS.
- S. Kim, H. Choi, M. Lee, H. Jung, Y. Shin, S. Lee, K. Kim, M. H. Kim, K. Kwak and B. Kim, Polymers, 2023, 15, 1354 CrossRef CAS PubMed.
- T. Dai, P. Lei, B. Zhang, A. Tang, Y. Geng, Q. Zeng and E. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 21556–21564 CrossRef CAS PubMed.
- S. Lee, D. Jeong, C. Kim, C. Lee, H. Kang, H. Y. Woo and B. J. Kim, ACS Nano, 2020, 14, 14493–14527 CrossRef CAS PubMed.
- J.-W. Lee, D. Jeong, D. J. Kim, T. N.-L. Phan, J. S. Park, T.-S. Kim and B. J. Kim, Energy Environ. Sci., 2021, 14, 4067–4076 RSC.
- K. Zhou, J. Xin and W. Ma, ACS Energy Lett., 2019, 4, 447–455 CrossRef CAS.
- L. Ye, M. Gao and J. Hou, Sci. China Chem., 2021, 64, 1875–1887 CrossRef CAS.
- K. Wang, Y. Li and Y. Li, Macromol. Rapid Commun., 2020, 41, 1900437 CrossRef CAS PubMed.
- W. Yang, Z. Luo, R. Sun, J. Guo, T. Wang, Y. Wu, W. Wang, J. Guo, Q. Wu, M. Shi, H. Li, C. Yang and J. Min, Nat. Commun., 2020, 11, 1218 CrossRef CAS PubMed.
- B. Li, Q. Zhang, S. Li, X. Yang, F. Yang, Y. Kong, Y. Li, Z. Wu, W. Zhang, Q. Zhao, Y. Zhang, H. Young Woo, J. Yuan and W. Ma, Chem. Eng. J., 2022, 438, 135543 CrossRef CAS.
- J.-W. Lee, C. Sun, D. J. Kim, M. Y. Ha, D. Han, J. S. Park, C. Wang, W. B. Lee, S.-K. Kwon, T.-S. Kim, Y.-H. Kim and B. J. Kim, ACS Nano, 2021, 15, 19970–19980 CrossRef CAS PubMed.
- E. Van Hemelrijck, P. Van Puyvelde, S. Velankar, C. W. Macosko and P. Moldenaers, J. Rheol., 2004, 48, 143–158 CrossRef CAS.
- Y.-A. Su, N. Maebayashi, H. Fujita, Y.-C. Lin, C.-I. Chen, W.-C. Chen, T. Michinobu, C.-C. Chueh and T. Higashihara, ACS Appl. Mater. Interfaces, 2020, 12, 12083–12092 CrossRef CAS PubMed.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge and J. Hou, Adv. Mater., 2019, 31, 1903441 CrossRef.
- J. Kim, M. Kyeong, J.-W. Ha, H. Ahn, J. Jung, S. Seo, T. N.-L. Phan, C. Lee, S. C. Yoon, B. J. Kim and S.-J. Ko, J. Mater. Chem. A, 2021, 9, 27551–27559 RSC.
- C. Lim, S. Lee, D. Han, C. Lee and B. J. Kim, Macromolecules, 2022, 55, 10395–10404 CrossRef CAS.
- J.-W. Lee, C. Lim, S.-W. Lee, Y. Jeon, S. Lee, T.-S. Kim, J.-Y. Lee and B. J. Kim, Adv. Energy Mater., 2022, 12, 2202224 CrossRef CAS.
- H. Lu, H. Wang, G. Ran, S. Li, J. Zhang, Y. Liu, W. Zhang, X. Xu and Z. Bo, Adv. Funct. Mater., 2022, 32, 2203193 CrossRef CAS.
- J. Kim, G.-U. Kim, D. J. Kim, S. Lee, D. Jeong, S. Seo, S.-J. Ko, S. C. Yoon, T.-S. Kim and B. J. Kim, J. Mater. Chem. A, 2023, 11, 4808–4817 RSC.
- X. Jing, Y. Zhao, Q. Wang, X. Kang, T. Zhuang, X. Liu, X. Wang, L. Yu and M. Sun, Polymer, 2022, 254, 125089 CrossRef CAS.
- H. Jung, G. Yu, J. Kim, H. Bae, M. Kim, K. Kim, B. Kim and Y. Lee, Sol. RRL, 2021, 5, 2100513 CrossRef CAS.
- S. Chen, H. J. Cho, J. Lee, Y. Yang, Z.-G. Zhang, Y. Li and C. Yang, Adv. Energy Mater., 2017, 7, 1701125 CrossRef.
- J. Wang, C. Han, F. Bi, D. Huang, Y. Wu, Y. Li, S. Wen, L. Han, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2021, 14, 5968–5978 RSC.
- J. Wang, C. Han, S. Wen, F. Bi, Z. Hu, Y. Li, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2023, 16, 2327–2337 RSC.
- J. Wang, C. Han, J. Han, F. Bi, X. Sun, S. Wen, C. Yang, C. Yang, X. Bao and J. Chu, Adv. Energy Mater., 2022, 12, 2201614 CrossRef CAS.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CrossRef CAS PubMed.
- X. Guo, Q. Fan, J. Wu, G. Li, Z. Peng, W. Su, J. Lin, L. Hou, Y. Qin, H. Ade, L. Ye, M. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 2322–2329 CrossRef CAS PubMed.
- T. Zhang, C. An, Y. Cui, J. Zhang, P. Bi, C. Yang, S. Zhang and J. Hou, Adv. Mater., 2022, 34, 2105803 CrossRef CAS PubMed.
- G. Conboy, R. G. D. Taylor, N. J. Findlay, A. L. Kanibolotsky, A. R. Inigo, S. S. Ghosh, B. Ebenhoch, L. Krishnan Jagadamma, G. K. V. V. Thalluri, M. T. Sajjad, I. D. W. Samuel and P. J. Skabara, J. Mater. Chem. C, 2017, 5, 11927–11936 RSC.
- L. Xu, W. Tao, M. Guan, X. Yang, M. Huang, H. Chen, J. Zhang, B. Zhao and S. Tan, ACS Appl. Energy Mater., 2021, 4, 11624–11633 CrossRef CAS.
- E. Ahmed, S. Subramaniyan, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2011, 44, 7207–7219 CrossRef CAS.
- L. Zhou, L. Meng, J. Zhang, C. Zhu, S. Qin, I. Angunawela, Y. Wan, H. Ade and Y. Li, Adv. Funct. Mater., 2022, 32, 2109271 CrossRef CAS.
- L.-H. Chou, T. Mikie, M. Saito, C.-L. Liu and I. Osaka, ACS Appl. Mater. Interfaces, 2022, 14, 14400–14409 CrossRef CAS PubMed.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- S. Seo, J. Kim, H. Kang, J.-W. Lee, S. Lee, G.-U. Kim and B. J. Kim, Macromolecules, 2021, 54, 53–63 CrossRef CAS.
- J. Zhang, Q. Huang, K. Zhang, T. Jia, J. Jing, Y. Chen, Y. Li, Y. Chen, X. Lu, H. Wu, F. Huang and Y. Cao, Energy Environ. Sci., 2022, 15, 4561–4571 RSC.
- M. Mohan, V. Nandal, S. Paramadam, K. P. Reddy, S. Ramkumar, S. Agarwal, C. S. Gopinath, P. R. Nair and M. A. G. Namboothiry, J. Phys. Chem. C, 2017, 121, 5523–5530 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B, 2010, 82, 245207 CrossRef.
- H. D. Kim, Y. Horiuchi, S. Iwasaki, T. Fukuhara and H. Ohkita, ACS Appl. Mater. Interfaces, 2021, 13, 39322–39330 CrossRef CAS.
- D. K. Tran, A. Robitaille, I. J. Hai, X. Ding, D. Kuzuhara, T. Koganezawa, Y.-C. Chiu, M. Leclerc and S. A. Jenekhe, J. Mater. Chem. A, 2020, 8, 21070–21083 RSC.
- Y. Cai, Y. Li, R. Wang, H. Wu, Z. Chen, J. Zhang, Z. Ma, X. Hao, Y. Zhao, C. Zhang, F. Huang and Y. Sun, Adv. Mater., 2021, 33, 2101733 CrossRef CAS PubMed.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- D. R. Kozub, K. Vakhshouri, S. V. Kesava, C. Wang, A. Hexemer and E. D. Gomez, Chem. Commun., 2012, 48, 5859–5861 RSC.
- J.-W. Lee, C. Sun, C. Lee, Z. Tan, T. N.-L. Phan, H. Jeon, D. Jeong, S.-K. Kwon, Y.-H. Kim and B. J. Kim, ACS Energy Lett., 2023, 8, 1344–1353 CrossRef CAS.
- W. Wang, Q. Wu, R. Sun, J. Guo, Y. Wu, M. Shi, W. Yang, H. Li and J. Min, Joule, 2020, 4, 1070–1086 CrossRef CAS.
- L. J. Richter, D. M. DeLongchamp and A. Amassian, Chem. Rev., 2017, 117, 6332–6366 CrossRef CAS PubMed.
- L. Zhang, X. Xu, B. Lin, H. Zhao, T. Li, J. Xin, Z. Bi, G. Qiu, S. Guo, K. Zhou, X. Zhan and W. Ma, Adv. Mater., 2018, 30, 1805041 CrossRef PubMed.
- K.-H. Kim, H. Kang, H. J. Kim, P. S. Kim, S. C. Yoon and B. J. Kim, Chem. Mater., 2012, 24, 2373–2381 CrossRef CAS.
- N. Gasparini, M. Salvador, S. Strohm, T. Heumueller, I. Levchuk, A. Wadsworth, J. H. Bannock, J. C. de Mello, H.-J. Egelhaaf, D. Baran, I. McCulloch and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1700770 CrossRef.
- R. Ma, Q. Fan, T. A. Dela Peña, B. Wu, H. Liu, Q. Wu, Q. Wei, J. Wu, X. Lu, M. Li, W. Ma and G. Li, Adv. Mater., 2023, 35, 2212275 CrossRef CAS PubMed.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- J. Lee, J. W. Kim, S. A. Park, S. Y. Son, K. Choi, W. Lee, M. Kim, J. Y. Kim and T. Park, Adv. Energy Mater., 2019, 9, 1901829 CrossRef.
- J.-W. Lee, C. Sun, T. N.-L. Phan, D. C. Lee, Z. Tan, H. Jeon, S. Cho, S.-K. Kwon, Y.-H. Kim and B. J. Kim, Energy Environ. Sci., 2023, 16, 3339–3349 RSC.
- C. Zhang, J. Song, J. Xue, S. Wang, Z. Ge, Y. Man, W. Ma and Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202308595 CrossRef CAS PubMed.
- B. Li, X. Yang, S. Li and J. Yuan, Energy Environ. Sci., 2023, 16, 723–744 RSC.
- J. S. Park, G.-U. Kim, S. Lee, J.-W. Lee, S. Li, J.-Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, 2201623 CrossRef CAS PubMed.
- D. J. Lipomi, B. C.-K. Tee, M. Vosgueritchian and Z. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed.
- J.-H. Kim, A. Nizami, Y. Hwangbo, B. Jang, H.-J. Lee, C.-S. Woo, S. Hyun and T.-S. Kim, Nat. Commun., 2013, 4, 2520 CrossRef PubMed.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- Q. Fan, W. Su, S. Chen, W. Kim, X. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. Ma, T. Yang, W. Zhuang, Y. Li, Y. Li, T.-S. Kim, L. Hou, C. Yang, H. Yan, D. Yu and E. Wang, Joule, 2020, 4, 658–672 CrossRef CAS.
- Q. Wan, S. Seo, S.-W. Lee, J. Lee, H. Jeon, T.-S. Kim, B. J. Kim and B. C. Thompson, J. Am. Chem. Soc., 2023, 145, 11914–11920 CrossRef CAS PubMed.
- S. Seo, J.-W. Lee, D. J. Kim, D. Lee, T. N.-L. Phan, J. Park, Z. Tan, S. Cho, T.-S. Kim and B. J. Kim, Adv. Mater., 2023, 35, 2300230 CrossRef CAS PubMed.
- J.-W. Lee, S. Seo, S.-W. Lee, G.-U. Kim, S. Han, T. N.-L. Phan, S. Lee, S. Li, T.-S. Kim, J.-Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, 2207544 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00117f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |