
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the role of a ground state charge transfer complex in carbon nanoparticles for highly efficient metal-free solar hydrogen production†
Subhajit
Kar
a,
Amit
Kumar
b,
Ramesh
Mandal
a,
Sakshi
Chawla
b,
Shanti Gopal
Patra
c,
Arijit K.
De
 *b and
Santanu
Bhattacharyya
*a
*b and
Santanu
Bhattacharyya
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Berhampur, Berhampur, Odisha-760010, India. E-mail: santanub@iiserbpr.ac.in
bCondensed Phase Dynamics Group, Department of Chemical Sciences, Indian Institute of Science Education and Research Mohali, SAS Nagar, Punjab 140306, India. E-mail: akde@iisermohali.ac.in
cDepartment of Chemistry, National Institute of Technology Silchar, Silchar, 788010, India
First published on 16th January 2024
Abstract
Carbon-based nanoparticles (CNPs) are exciting metal-free photocatalysts for solar hydrogen production. However, significant challenges remain in elucidating their complex structures and identifying the potential catalytic sites. Herein, we report a specific ground-state charge-transfer complex between nitrogen, phosphorus-co-functionalized carbon-based nanoparticles (N, P-CNPs) and a sacrificial electron donor (SED) namely triethanolamine (TEOA), defined as N, P-CNPs (TEOA). Optical and surface analytical investigations show the initial snapshot of the ground-state charge-transfer complex, which is further clarified through femtosecond transient absorption spectroscopy, electrochemical study, and computational analysis. Results suggest the crucial role of phosphorus functionalization in establishing the ground-state charge-transfer complex. The long-lived free carriers in the N, P-CNP (TEOA) charge transfer state stimulate highly efficient photocatalytic solar hydrogen production rates of 1.02 mmol g−1 h−1 and 651 μmol g−1 h−1 from normal (mili-Q) and natural sea water, respectively, without any metal co-catalyst. Notably, TEOA plays a dual role; it forms a unique ground-state charge-transfer state with N, P-CNPs, and the excess TEOA molecules act as a potential hole-scavenger to boost the photocatalytic hydrogen generation process.
Introduction
The urgency to resolve the world's energy concerns while minimizing fossil fuel emissions has spurred renewable energy research.1–3 In this context, photocatalytic and photoelectrochemical water splitting has emerged as a desirable approach.4–8 Green hydrogen through photocatalysis is one of these fuels that has gained serious interest due to its excellent energy density and minimum greenhouse emissions.9–11 Over the past few decades, research on HER active photo-catalysts has primarily focused on metal oxides, organic–inorganic hybrids, and polymeric carbon nitride systems, which have drawbacks like metal-based toxicity, water insolubility, a wide band gap, and high charge carrier recombination limiting their overall efficiency.12 Moreover, most of these photo-catalysts mentioned above require expensive noble metal-based (e.g., Pt, Au, etc.) co-catalysts.13In this regard, highly water-soluble carbon nanoparticles have gained potential interest as a true metal-free photo-catalyst for H2 generation by eliminating the necessity of a noble metal co-catalyst.14–18 As reported before, these carbon nanoparticles have numerous advantages, including tunable optical properties, nontoxicity, and exceptional solubility in diverse solvent media.19 Furthermore, these carbon-based nanoparticles can be easily produced from a broad spectrum of precursors, including small molecules, biomass, and organic compounds, using reasonably inexpensive synthetic pathways.20 Additionally, the ease of heteroatom functionalization in carbon-based nanoparticles permits effortless tuning of their electronic, morphological, structural, and optical behaviors.21 These intriguing traits of carbon-based nanoparticles lead to their widespread applicability.19,22
However, employing these materials in the realm of photo-catalytic hydrogen production has proven to be exceedingly difficult due to their intricate development process and the lack of a robust structure–property correlation.23 It is essential to highlight that conventional carbon-based nanoparticles are often synthesized via a bottom-up approach, creating many defects in the nanostructure and decreasing the number of free carriers.24 Furthermore, the active site determination and charge carrier dynamics of carbon-based nanoparticles, essential for photo-catalytic hydrogen generation, continue to be an obstacle.25 Most importantly, prior to photocatalysis, the interactions of carbon-based nanoparticles with a specific sacrificial electron donor (SED), followed by their electronic structure modification, were largely overlooked. Consequently, up to this point, carbon nanoparticles have mainly been employed as co-catalysts or sensitizers alongside metal complexes or metal oxide catalysts for photo-catalytic hydrogen generation.26,27 Lastly, the reported photo-catalytic hydrogen efficiency in carbon-based nanoparticles without metal co-catalysts is exceptionally poor. Therefore, developing a metal-free, effective carbon nanoparticle-based HER active photocatalyst with in-depth structure–property correlation is highly appealing.
Herein, we represent a novel type of nitrogen and phosphorus-co-functionalized carbon-based layered nanoparticle (N, P-CNP) developed from biomass chitosan and phosphoric acid. We have identified a specific ground-state charge-transfer (CT) complex between N, P-CNPs and sacrificial electron donor (SED) triethanolamine (TEOA), designated as N, P-CNPs (TEOA). A comprehensive assessment confirms that the N, P-CNP (TEOA) adduct induces long-lived free carriers and dramatically alters the N, P-CNP electronic environment. This N, P-CNP (TEOA) adduct is exceptionally soluble in both normal (mili-Q) and natural seawater, and demonstrated record photo-catalytic hydrogen production rates of 1.02 mmol g−1 h−1 and 651 μmol g−1 h−1, respectively, without any noble metal co-catalyst. The N, P-CNP (TEOA) adduct revealed its photo-catalytic H2 production selectivity by producing significantly more H2 with TEOA than any other SED environments, and it outperforms N-CDs under identical TEOA conditions by a factor of 31. Typically, the larger size of the phosphorus atom compared to carbon and nitrogen could offer unoccupied orbitals, which allows Lewis acid-base type interaction between TEOA and N, P-CNPs, resulting in the formation of a ground-state charge-transfer state.28,29 Furthermore, the weaker electro-negativity of P atoms compared to other neighbouring atoms in carbon nanoparticles makes them relatively positive. As a result, phosphorus can act as a bridge for transferring electrons between the two counterparts of the adduct.29,30 Electron injection from TEOA via the P atom to neighbouring atoms (O and C) causes overall negative charges on N, P-CNPs, and it helps N, P-CNPs (TEOA) to take the form of {(N, P-CNPs˙−–TEOA˙+)} in their excited, long-lived state. Moreover, HRTEM investigation indicates that a bigger sp2-domain of N, P-CNPs catalyzed by phosphoric acid further boosts the charge transport process of N, P-CNPs (TEOA).14 Furthermore, XPS, NMR, and zeta potential studies accessed the fundamental features of N, P-CNPs (TEOA) at the molecular level. Time resolved spectroscopy up to an ultrafast time scale further unveils the nature of the free carriers in N, P-CNP (TEOA) adducts. TD-DFT-based computational studies support the experimental findings and reveal that the water adsorption occurs due to N, P-CNP (TEOA) adduct formation, which eventually facilitates the photocatalytic HER activities. Overall, the current findings will be highly beneficial for future research on metal-free solar hydrogen generation using innovative heteroatom-functionalized carbon-based nanomaterials.
Results and discussion
In the present study, we have developed nitrogen and phosphorus co-functionalized unique carbon nanoparticles (N, P-CNPs) employing chitosan gel and phosphoric acid in an aqueous medium31 (depicted in Fig. 1(a) and Experimental methods). The high-resolution TEM images of N, P-CNPs, and the overall morphological information at low resolution are shown in Fig. 1(b) and S1,† respectively. The HRTEM studies show that the N, P-CNPs possess more or less spherical shapes embedded in a distinct layered type morphology (inset of Fig. 1(b)).20,31 The estimated distance between these layers (inset of Fig. 1(b)) is a bit larger (d002 = 0.38) than a traditional graphitic plane (002) because of their enhanced interfacial repulsion.31,32 On top of that, the HRTEM images (Fig. 1(b)) and the SAED patterns (inset of Fig. 1(b)) imply that N, P-CNPs have complimentary quantities of sp3 and conjugated π-domains. As a reference, we have also prepared pure N-CDs solely from chitosan gel using a standard protocol mentioned elsewhere, and these N-CDs (Fig. S2†) are mostly dot-like structures with moderate crystallinity.31 The typical surface group analysis (XPS and FTIR) of our current study reveals that nitrogen of N, P-CNPs is typically found (Fig. S3 and S4(a)†) in the interior of highly graphitized sp2–π domains.33 Meanwhile the surface states of the N, P-CNPs primarily consist (Fig. S3 and S4(b)†) of oxygenated phosphorus or phosphates, accompanied by the existence of a small number of pyrrolic/amine-based functional groups.34,35 Furthermore, the Raman study (Fig. S5†) confirms the presence of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (890 cm−1) and P–OH (1076 cm−1) surface functionalities in N, P-CNPs, consistent with our XPS and IR studies.36 Raman analysis (Fig. S5†) further reveals the presence of typical D (1320 cm−1) and G (1530 cm−1) bands as common features of carbon-based materials, along with prominent disorder bands (D′, D + D′′ and 2D band) that support the layered structure of N, P-CNPs obtained from the TEM study.37 These results nicely match with previous reports in this direction.14,34 As depicted in Fig. 1(c), the as-synthesized N, P-CNPs exhibit excellent solar light absorption, featuring substantial absorption in the UV region (270–380 nm) and a pronounced absorption band in the visible spectrum (∼456 nm). Significant absorption of N, P-CNPs in the near-UV region (∼290 nm) can be attributed to the strongly aromatized π-domain (π–π* transition).38 Meanwhile the n–π* transition from CO/CN containing moieties in N, P-CNPs are accountable for the low-intensity absorption in the far-UV region (320–400 nm).39 The absorption characteristics at 456 nm most likely originate from the phosphorus containing surface states of N, P-CNPs.14,31 Notably, after the addition of TEOA, along with the characteristic peaks of N, P-CNPs at 456 nm, a new absorption peak arises at a longer wavelength (∼506 nm), which indicates the formation of a new charge-transfer complex N, P-CNPs (TEOA). The absorption features of N, P-CNPs (TEOA) are depicted in Fig. 1(c). Moreover, an intermediate state (as shown in Fig. 1(c)) has been observed between N, P-CNPs and N, P-CNPs (TEOA) at a significantly low concentration (0.4 V% or 30.12 mM) of TEOA.
O (890 cm−1) and P–OH (1076 cm−1) surface functionalities in N, P-CNPs, consistent with our XPS and IR studies.36 Raman analysis (Fig. S5†) further reveals the presence of typical D (1320 cm−1) and G (1530 cm−1) bands as common features of carbon-based materials, along with prominent disorder bands (D′, D + D′′ and 2D band) that support the layered structure of N, P-CNPs obtained from the TEM study.37 These results nicely match with previous reports in this direction.14,34 As depicted in Fig. 1(c), the as-synthesized N, P-CNPs exhibit excellent solar light absorption, featuring substantial absorption in the UV region (270–380 nm) and a pronounced absorption band in the visible spectrum (∼456 nm). Significant absorption of N, P-CNPs in the near-UV region (∼290 nm) can be attributed to the strongly aromatized π-domain (π–π* transition).38 Meanwhile the n–π* transition from CO/CN containing moieties in N, P-CNPs are accountable for the low-intensity absorption in the far-UV region (320–400 nm).39 The absorption characteristics at 456 nm most likely originate from the phosphorus containing surface states of N, P-CNPs.14,31 Notably, after the addition of TEOA, along with the characteristic peaks of N, P-CNPs at 456 nm, a new absorption peak arises at a longer wavelength (∼506 nm), which indicates the formation of a new charge-transfer complex N, P-CNPs (TEOA). The absorption features of N, P-CNPs (TEOA) are depicted in Fig. 1(c). Moreover, an intermediate state (as shown in Fig. 1(c)) has been observed between N, P-CNPs and N, P-CNPs (TEOA) at a significantly low concentration (0.4 V% or 30.12 mM) of TEOA.
 | ||
| Fig. 1 Schematic representation of the fabrication of N, P-CNPs from chitosan gel and phosphoric acid (a); HRTEM image and SAED pattern of N, P-CNPs (b); UV-vis absorption of N, P-CNPs, the intermediate state and N, P-CNPs (TEOA) respectively (c). | ||
Such a transient state contains an identical visible absorption peak (∼456 nm) of bare N, P-CNPs, and an additional peak at ∼486 nm. At an optimum concentration of TEOA (1 vol% or 75.35 mM), the red-shifted absorption band becomes predominant at 506 nm. The longer wavelength absorption maxima did not shift further after increasing the TEOA concentration beyond the optimum concentration (ESI, Fig. S6†). In addition, the visual change in the color of N, P-CNPs from light yellow to reddish upon introducing TEOA is displayed in the inset of Fig. 1(c). This result further indicates a possible charge-transfer complex formation in the ground state. To get further insight into the role of general SED agents, we have investigated the possible interaction between other available SEDs with N, P-CNPs. However, no specific interactions between N, P-CNPs and other SEDs were found (Fig. S7†). Furthermore, as shown in Fig. S8,† no such ground-state charge-transfer complex has been observed in the case of pure N-CDs (the reference sample). Therefore, the emergence of a newly red-shifted absorption peak (∼506 nm) due to the N, P-CNP (TEOA) adduct is most likely originated from specific ground state charge-transfer complexation between N, P-CNPs and TEOA (displayed in Fig. 1(c)).
The PL emission spectra (Fig. 2(a) and S9(a)†) of N, P-CNPs reveal that the PL emission intensity is predominantly concentrated at around 430 nm. The overall PL spectra (Fig. 2(a) and S9(a)†) of N, P-CNPs show excitation wavelength-dependent behaviour similar to common fluorescent CNPs. However, at longer excitation wavelengths, the PL spectra of N, P-CNPs become almost excitation wavelength independent (Fig. 2(a) and S9(a)†). This excitation wavelength independent PL emission nature is attributed to the integration of phosphorus into the carbon nanostructure. In contrast, pure N-CDs do not exhibit significant PL emission at longer wavelengths (Fig. S9b†). Interestingly, at the intermediate state (0.4 V% TEOA), the emission band intensity at a longer wavelength excitation becomes predominant with peak maxima at ∼507 nm (Fig. 2(b) and S10(a)†). However, the emission band at a shorter wavelength region remains unaltered. The above observations suggest the possible interactions between the phosphorus-containing moieties of N, P-CNPs and TEOA. Finally, as evidenced by PL emission features (depicted in Fig. 2(c) and S10(b)†) at the optimum complexation concentration (1 V% TEOA), the PL emission maxima completely shifted to the more extended wavelength region (∼525 nm), confirming the establishment of a stable N, P-CNP (TEOA) charge-transfer state. In a comparable circumstance, pure N-CDs show no change in their PL characteristics (depicted in Fig. S11†). The finding implies that the charge-transfer interaction between N, P-CNPs and TEOA is quite particular. Interestingly, phosphorus functionalization in CNPs initiates and stabilizes such a specific emissive state through charge-transfer complex formation. After attaining the stable N, P-CNP (TEOA) charge-transfer state, upon further addition of TEOA, the absorption and emission features and their position barely change (Fig. S6 and S12†). This suggests the N, P-CNP (TEOA) complexation was successfully retained at increased TEOA content. Furthermore, we have implemented a time-resolved PL study to understand the photophysical properties of N, P-CNPs and the respective charge transfer complex N, P-CNPs (TEOA). The time-resolved PL decay kinetics are evaluated by using a 450 nm laser as the excitation source and probing the emissive states of N, P-CNPs and N, P-CNPs (TEOA) at 507 and 525 nm, respectively (Fig. 2(d)). The average lifetimes for both N, P-CNPs and N, P-CNPs (TEOA) were fitted by using bi-exponential decay kinetics (details in Table S1†). The calculated average lifetime of N, P-CNPs is 590 ps; however, upon complexation with TEOA, the average lifetime of N, P-CNPs increases up to 2.70 ns (depicted in Fig. 2(d)). The significant enhancement in the lifetime upon complex formation between N, P-CNPs and TEOA suggests the formation of long-lived free carriers.40,41
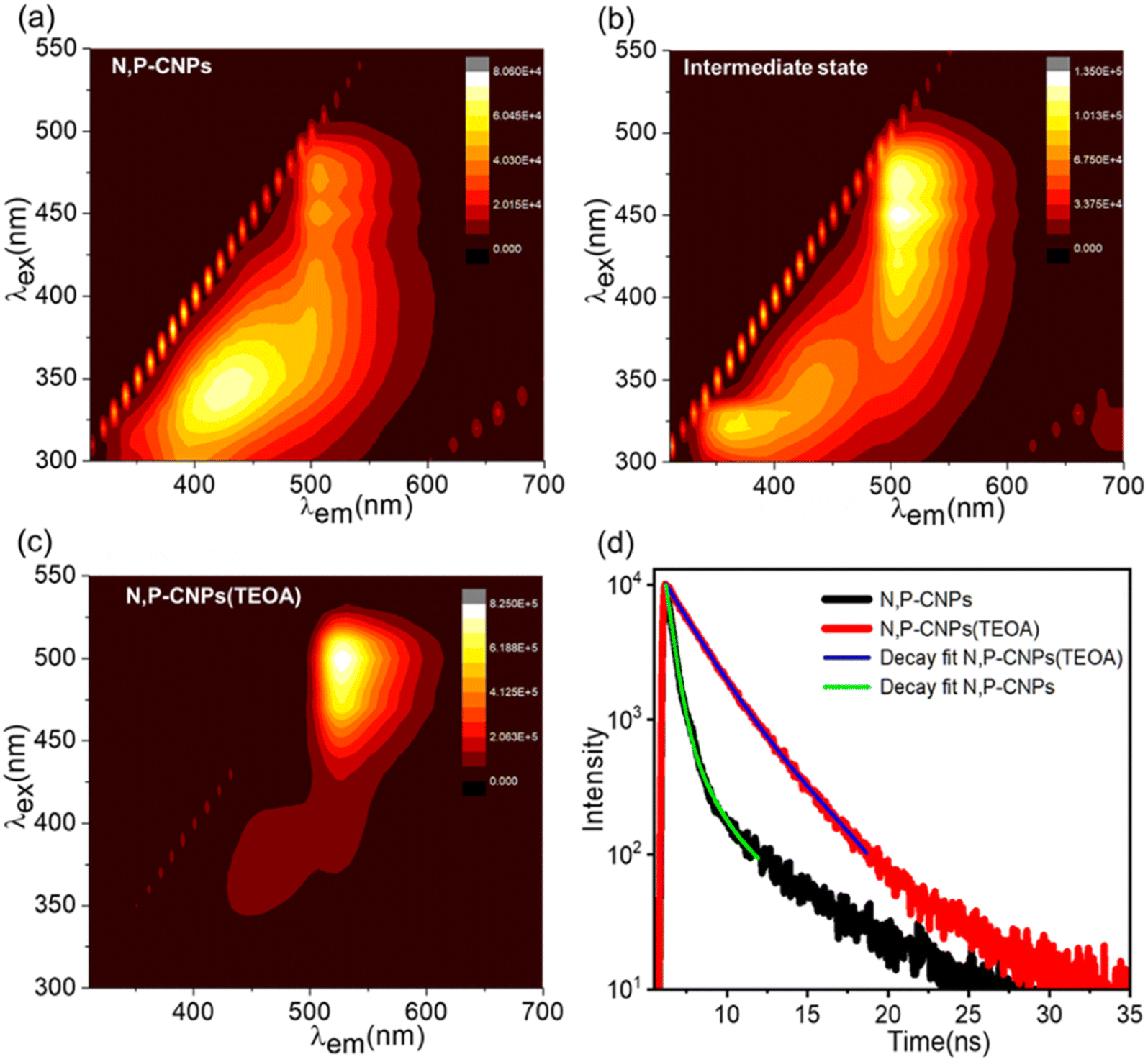 | ||
| Fig. 2 Monitoring the gradual evolution of the N, P-CNP (TEOA) complex starting from pure N, P-CNPs using the steady-state PL emission spectra (a–c). Time-resolved Photoluminescence decay measurements of N, P-CNPs and N, P-CNPs (TEOA) (d). | ||
Therefore, the fundamental absorption and photoluminescence studies mentioned above are convincing enough to support the formation of a stable, long-lived charge-transfer state between N, P-CNPs and TEOA.
To attain an in-depth grasp of the molecular mechanism underlying the N, P-CNP (TEOA) charge-transfer complex, we have employed a variety of surface and elemental analytical techniques. The electrokinetic zeta potential (shown in Fig. 3(a)) of bare N, P-CNPs exhibits a positive surface charge (ζ = +21.8 mV). The positive surface charge indicates that N, P-CNPs possess protonated phosphorus and a few amino functionalities on the surface, but there is no evidence of negatively charged (e.g., –COOH or –OH) species (Fig. S3, S4 and S5†).42 Meanwhile the zeta potential of the N, P-CNP (TEOA) adduct revealed a significantly negative surface potential (ζ = −32.2 mV), as depicted in Fig. 3(a). This negative surface charge suggests the formation of a large number of negatively charged reduced phosphorus (phosphonate or phosphinate) functionalities upon N, P-CNP (TEOA) adduct formation.42,43 The development of these abundant negative charges on the surface promotes the hydrophilic properties and stability of the N, P-CNP (TEOA) adduct in an aqueous medium.44 Moreover, Pure N, P-CNP and N, P-CNP (TEOA) adducts have been studied by 31P NMR (D2O solvent), which further supports our hypothesis. Pure N, P-CNPs have a 31P NMR signal near −0.19 ppm (Fig. 3(b)), consistent with protonated phosphorus functionalities on their surface.45 However, upon formation of the N, P-CNP (TEOA) adduct, the 31P NMR signal de-shielded by approximately 2.4 ppm compared to pure N, P-CNPs. It strongly suggests the emergence of positively charged phosphorus nuclei, suggesting the negatively charged reduced phosphorus groups over the N, P-CNP (TEOA) complex.46,47 These results are further verified through the surface elemental composition by employing X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of pure N, P-CNP and N, P-CNP (TEOA) complexes are depicted in the ESI (Fig. S3(a) and S13†). The deconvoluted P2P XPS spectra of pure N, P-CNPs around 134.3 eV and 135.2 eV are attributed (Fig. 3(c)) to the presence of C–P–O(OH)2/CPO3− and C–O–P(OH)2 containing surface moieties.14,48,49 The P2P XPS spectrum for N, P-CNPs (TEOA) is centered at lower binding energy (Fig. 3(d)) than pure N, P-CNPs. The deconvoluted XPS spectra of N, P-CNPs (TEOA) at 130.9 eV indicate visible P–C bonding due to electron cloud displacement from the core and diminished protonated phosphorus functional groups (Fig. 3(d)) as a result of the adduct.50,51 However, the XPS spectrum at 133.7 eV is primarily attributable to the development of a significant number of CPO32− functionalities (Fig. 3(d)).50,52,53 Overall, the aforementioned results strongly suggest that negatively charged reduced phosphorus species (mostly phosphonate) populate over the bare N, P-CNPs upon N, P-CNPs (TEOA) adduct formation. Furthermore, based on the structural, elemental, and surface charge analysis, we have chosen a perylene-based model (Fig. S14†) system with a combination of protonated phosphate and negatively charged phosphonate containing surface functionalities for the TD-DFT study.34 B3LYP/Def2SVP level of theory is utilized to optimize the model and further computation calculations.54,55 The computational details and coordinates of the optimized geometries of the N, P-CNP (TEOA) model are provided in the Experimental methods and Table S2† (ESI) respectively. The simulated UV-visible spectra of model N, P-CNP (TEOA) absorption closely match our experimental findings (Fig. S15†). Furthermore, as depicted in Fig. 3(e), the HOMO of the model system N, P-CNPs (TEOA) reveals that the HOMO resides on PO32− moieties. In contrast, the LUMO is centered on the π* orbitals of the perylene moiety (Fig. S16†).
 | ||
| Fig. 3 (a) Zeta potential, (b) 31P NMR(D2O), and (c and d) deconvoluted P2P XPS spectra of N, P-CNPs and N, P-CNPs (TEOA). Representation of HOMO (e) and ESP mapping (f) of the plausible model for the state of N, P-CNPs after complexation with TEOA. | ||
The fact that the HOMO is centered on PO32− moieties rather than π-orbitals (carbon core) or protonated phosphorus moieties implies that charge accumulation occurs over PO32− moieties as a result of the N, P-CNP (TEOA) adduct formation in the ground state. These findings validate our experimental data regarding the abundance of negatively charged reduced phosphorus species over the N, P-CNP nanostructures owing to the ground-state charge-transfer complexation. The ESP mapping shown in Fig. 3(f) further confirms the presence of an intense electron cloud (red region) on the negatively charged PO32− moiety. The experimental and theoretical analyses indicate that phosphorus plays a crucial role in the adduct formation between TEOA and N, P-CNPs by introducing their empty orbitals. Following that, phosphorus's higher electrical conductivity (than C or N atoms) serves as a pathway for the transfer of electrical charge to neighboring atoms, allowing the formation of a stable N, P-CNP (TEOA) ground-state charge transfer complex, which can eventually increase the photocatalytic activities.28,29,40
Furthermore, we have combined both optical and electrochemical approaches to obtain an in-depth grasp of the electronic level shifts from bare N, P-CNPs to the N, P-CNP (TEOA) adduct. Detailed electrochemical measurement techniques are given in the Experimental methods section. The optical band gaps (Eg) of the samples have been investigated from tauc plots.56 The primary optical band gap for bare N, P-CNP and N, P-CNP (TEOA) adducts are found to be 3.91 and 3.43 eV, respectively (Fig. 4(a)). The flat band potential (Ef) of N, P-CNPs is measured by extrapolating Mott–Schottky plots to 1/C2 = 0, which reveals that the flat band potential (Ef) of N, P-CNPs moved from 0.11 to 0.19 V (vs. RHE) upon N, P-CNP (TEOA) adduct formation (Fig. 4(b)).14 Furthermore, the valence band XPS study of both samples showed a significant difference in the edge location of their valence bands (Fig. 4(c)), suggesting a change in their electronic state and band position. The relative location of a material's valence band with respect to its Fermi level (Ef) is indicated by its valence band edge position (VBE) or valence band maximum (VBM).4,57
 | ||
| Fig. 4 Tauc plot (a), Mott–Schottky analysis (b), Valence band XPS spectra (c), and pictorial representation of the modification of the CB and VB band positions of N, P-CNPs upon N, P-CNP (TEOA) complex formation (d). | ||
The valence band maximum (VBM) of N, P-CNPs and N, P-CNPs (TEOA) is determined to be 3.68 eV and 1.76 eV, respectively, derived by linear extrapolation of the onset of VB-XPS spectra to the baseline (Fig. 4(c)). The upshift in the VBM of N, P-CNPs (TEOA) clearly suggests that the addition of TEOA to N, P-CNPs entirely changes the ground state electronic structure of N, P-CNPs, further confirming the N, P-CNP (TEOA) ground state adduct formation.57 Depending on the above studies, Fig. 4(d) depicts the overall band structure of N, P-CNPs and N, P-CNPs (TEOA). According to the overall band structure estimates, the electronically modified N, P-CNP (TEOA) charge transfer state has VB and CB locations at 1.95 and −1.48 V, respectively (Fig. 4(d)). Meanwhile the VB and CB of bare N, P-CNPs are positioned at 3.79 and −0.12 V, respectively (Fig. 4(d)). Therefore, the improved electronic energy level alignment of the N, P-CNP (TEOA) charge-transfer complex strongly indicates that it is ideally positioned to accomplish effective photocatalytic solar H2 production.58
To obtain more profound insights into the excited state dynamics of pure N, P-CNPs and the ground-state charge-transfer complex upon interaction with TEOA, femtosecond transient absorption spectroscopy (fs-TAS) is employed (depicted in Fig. 5). A detailed description of fs-TAS (ESI†) can be found elsewhere.59,60 We primarily utilized a 400 nm pump pulse excitation for pure N, P-CNPs and a 530 nm pump pulse excitation for the N, P-CNP (TEOA) adduct, corresponding to their steady-state absorption spectra. Additionally, we have employed 400 nm excitation to investigate the dynamics associated with the blue-edge excitation of the CT state in the N, P-CNP (TEOA) adduct. In the case of pure N, P-CNPs, Fig. 5((a) and (b)) illustrate the color contour plots and the corresponding spectral traces derived from fs-TAS data using a 400 nm pump pulse. Spectral traces at different time delays reveal a negative signal from 430 nm to 480 nm corresponding to ground-state bleach (GSB) overlapping with the stimulated emission (SE). In addition, a positive signal related to excited state absorption (ESA) has been observed. It ranges from 480 to 660 nm with a maximum of 500 nm. Apart from multiexponential fits to select wavelengths (ESI†), global lifetime analysis of fs-TAS data (Experimental methods) has been performed to clearly understand excited state dynamics after photoexcitation.61 The three exponential state model A → B → C → D (Fig. S17(a)†) is used to extract the significant time component and corresponding evolution-associated difference spectra (EADS), which provide information about the spectral evolution corresponding to different states with increasing time. The EADS obtained after global fitting are shown in Fig. 5(c) for pure N, P-CNPs. The detailed global analysis procedure is discussed in the Experimental methods section. Since the excitation energy is higher than the absorption maxima (455 nm), initially, the charge carrier is transferred deep into the valence and conduction bands, so first EADS (2.59 ps) corresponds to intra-band hot charge carrier relaxation (τ1) to the band edge. The second EADS is related to the transfer of population (τ2) to the emissive state formed due to the presence of the phosphorus containing surface state of pure N, P-CNPs, and the final EADS corresponds to charge recombination (τ3). The population kinetics and the kinetic fit for pure N, P-CNPs are shown in Fig. S18((a) and (b)).† Additionally, a single wavelength kinetic fit is performed at the maxima of each signal (430 nm and 519 nm); the details of the obtained time component and the fitting equation can be found in the ESI (Table S3).† In the case of the N, P-CNP (TEOA) adduct (1 V% TEOA), upon 530 nm excitation, distinct spectral features are obtained, which includes an ESA signal ranging from 385 nm to 460 nm with maxima around 430 nm with a decay of 500 ps. In addition, a broad negative signal ranging from 460 nm to 660 nm is spotted due to the overlapping of GSB and SE feature maxima at 519 nm, which decay up to 2.5 ns. The respective color contour and spectral traces are shown in Fig. 5((d) and (e)). After excitation into the deep valence and conduction bands, hot charge carriers dissipate their energy through intra-band relaxation within the initial hundred femtoseconds. This process results in an enhanced population of charge carriers at the band edge, which leads to a rise in the intensity of the GSB signal.62 However, in our measurements, we did not observe the growth in the GSB signal, which could be due to faster dissociation of the exciton into free charge carriers at a faster time scale, which could not be resolved within our instrumental limit.59 It has been shown earlier that the initial hot charge carrier relaxation dynamics for the ESA signal are primarily attributed to electrons, while for GSB, they are primarily attributed to holes.63 Here, in the case of N, P-CNPs (Table S3†) and N, P-CNPs (TEOA) (Table S4†), the difference in the initial time scale obtained from ESA and GSB signals suggests that the presence of free charge carriers predominantly drives the intra-band relaxation. Due to the overlapping features of TA signals, it is challenging to capture the true dynamics through single wavelength kinetics (see details in the Experimental methods and Sections SI and SII of the ESI†). Therefore, global analysis is conducted on fs-TAS data to enhance the comprehension of the charge-transfer (CT) state dynamics of the N, P-CNP (TEOA) adduct. As depicted in the model (Fig. S17(b)†), a two-exponential decay model is utilized to obtain a suitable fit for the data. The time components obtained from the global analysis are tabulated in Table S5.† The first EADS in N, P-CNPs (TEOA) [1 V% TEOA] (Fig. 5(f)) corresponds to charge separation (τ2). However, the final EADS can be related to the charge recombination with a time scale of 1.21 ns (τ3). The kinetic trace and fit at different probe wavelengths of N, P-CNPs (TEOA), [1 V% TEOA] are depicted in Fig. S19,† which presents the population kinetics. The charge recombination times align well with the TCSPC lifetime measurements (Section SIII, Table S6 of the ESI†). Further insights into the charge carrier dynamics are obtained at blue edge excitation of the ground N, P-CNP (TEOA) CT state using a 400 nm pump (depicted in model Fig. S17(c)†). The respective color contour and spectral traces for N, P-CNPs (TEOA) are depicted in Fig. 5((g) and (h)). Two ESA signals emerge in N, P-CNPs (TEOA), with maxima at 460 and 690 nm, respectively. The first ESA signal ranges from 440 to 540 nm and decays up to 50 ps. The second ESA signal, which emerges after the addition of TEOA in pure N, P-CNPs, indicates the progression of the initial excited state toward the charge-separated state obtained from the oxidized form of TEOA after donating one electron to the N, P-CNPs. Consequently, the CT complex can be defined as the (N, P-CNPs)˙−–(TEOA)˙+, charge separated form in their excited long-lived state.16,42 The second ESA signal exhibits a rise within 0.5 ps (Fig. S20†). At a later probe time delay (within 10 ps), the weak, negative features started appearing from 490 nm to 540 nm with maxima around 520 nm, which can be correlated with the overlapping parts of both the GSB and SE signal. The obtained EADS is shown in Fig. 5(i). The first EADS corresponds to the initial intra-band hot charge carrier relaxation (τ1 = 0.79 ps). The second EADS is related to the transfer of charge carriers to a charge-separated state (τ2 = 4.45 ps), and the third EADS corresponds to charge recombination (τ3 = 1940 ps). The population kinetics and fit to the data are shown in Fig. S21.† The final time component closely matched the fluorescence lifetime obtained at 405 nm excitation (Table S6†). At higher concentrations of TEOA (>1 V% TEOA), similar traces like N, P-CNPs (TEOA) are obtained for each excitation (530 and 400 nm pump). The result implies that the nature of the N, P-CNPs (TEOA) remains intact at elevated TEOA concentration, which supports the results obtained from PL data. Detailed kinetic parameters obtained from single wavelength fit for varying TEOA content at the different probe wavelengths for 530 and 400 nm excitation are provided in Table S7.† A global analysis (530 and 400 nm pump) of other samples with higher concentrations of TEOA was performed using a similar model (Fig. S17†) like N, P-CNPs (TEOA). The transient contour plots and spectral traces (530 nm pump) for N, P-CNPs with 2 V% TEOA and 5 V% TEOA are shown in Fig. S22((a) and (b)) and S22((c) and (d)),† respectively. The obtained EADS for 2 V% TEOA and 5 V% TEOA are depicted in Fig. S23(a and b),† respectively. The EADSs at higher TEOA concentrations appear identical to those of the N, P-CNPs (TEOA). However, with further increasing TEOA content, the decay dynamics become faster. Population kinetic and global fit at assigned wavelength is shown in Fig. S24† for 2 V% (a and b) and for 5 V% TEOA concentration (c and d). With increasing concentrations of TEOA, faster decay dynamics (1940 ps to 558.4 ps) have been observed (depicted in Table S8†), which is in consistent with the trends observed in TCSPC studies (Table S9†). Interestingly, the valence band location (Fig. 4(d)) of the N, P-CNP (TEOA) complex (1.95 V) is well suited for the hole transfer process to free TEOA molecules (SED) in aqueous environments (–0.57 to 0.85 V).64
 | ||
| Fig. 5 Femtosecond transient absorption studies of pure N, P-CNP (top row) and N, P-CNP (TEOA) adducts (middle and bottom rows): (a, d and g) transient colour contour maps, (b, e and h) spectral traces at different probe delays, and (c, f and i) evolution-associated decay spectra obtained from global analysis. | ||
Likewise, it is well known that the longer time scale event (τ3) is rarely influenced by the electron-transfer process, indicating that the faster decay rate at elevated TEOA concentration can be attributed to the interfacial hole transfer from the photo-excited N, P-CNP (TEOA) charge-transfer (CT) state to free TEOA molecules (SED).64,65 For the 400 nm pump-pulse, the contour plots and spectral traces are shown for 2 V% TEOA in Fig. S25 (a and b) and 5 V% TEOA in Fig. S25 (c and d),† respectively. Global analysis is conducted to extract the time component from TA data for different TEOA contents, and the data are fitted with three exponential kinetic models (Fig. S17(c)†). The EADS, population kinetics, and global fits at the selected wavelength (to show the quality of fit) are shown at higher concentrations of TEOA, as depicted in Fig. S26 (a, b, and c) and S26 (d, e, and f)† respectively. Kinetic parameters obtained for single wavelength fit for TEOA content variation are given in Table S7.† A faster decay rate (at 400 nm pump) at a higher TEOA content (>1 V%) can again be correlated to the interfacial hole transfer to the free TEOA molecules in the system.64,65 Although higher TEOA concentrations show similar traces (400 nm excitation) like N, P-CNPs (TEOA), a close inspection revealed significant differences in free-carrier populations, most likely in the form of photo-excited electrons. A representative plot of ESA and GSB maxima at 1 ps time delay, with varying TEOA concentration, is shown in Fig. S27.† The steady increase in the intensity of the ESA and GSB signals at higher TEOA concentrations (Fig. S27†) suggests an increase of photo-excited populations in the excited charge-separated state {(N, P-CNPs˙−–TEOA˙+)}.65 This enhanced charge carrier accumulation in the form of photo-excited electrons can potentially increase the photo-catalytic reduction efficiency of pure N, P-CNPs (TEOA) in their excited state.66
Following an in-depth understanding of the nature of the unique N, P-CNP (TEOA) charge-transfer complex, we have utilized this modified photocatalyst for solar hydrogen production (under 1-sun illumination) without any metal-based co-catalyst (Experimental methods). Therefore, it allows us to investigate the critical role of solely the ground-state charge transfer complex of N, P-CNPs for photocatalytic H2 production. The N, P-CNP (TEOA) ground-state charge transfer complex shows highly efficient photo-catalytic hydrogen generation activity from regular (mili-Q) and natural sea water under full solar light illumination, with H2 production rates of ≈1.02 mmole g−1h−1 and ≈ 651 μmole g−1 h−1, respectively (Fig. 6(a)). These results are one of the highest photo-catalytic hydrogen generation activity reported to date for solely a carbon-based nanoparticle without a metal-based co-catalyst. N, P-CNPs exhibit the highest photo-catalytic activity when 20% (V/V) TEOA is used in water (Fig. S28†). It suggests that excess TEOA preserves the N, P-CNP (TEOA) adduct and maintains an uninterrupted supply of molecular TEOA (SED) during photocatalysis. To maximize the efficiency of photocatalytic hydrogen generation and preserve the N, P-CNP (TEOA) charge-transfer (CT) complex, a constant TEOA concentration (20 V%) has been used. The H2 generation rate of N, P-CNPs (TEOA) grows linearly with time without any decline over 24 hours of continuous photo-catalytic reaction (Fig. 6(b)). After 24 hours of steady light illumination, the N, P-CNP (TEOA) adduct shows an excellent H2 generation activity of ≈ 20.02 mmol g−1 (Fig. 6(b)), which clearly suggests the superior stability of the catalyst. Furthermore, we observed that the photo-catalytic H2 generating activities of N, P-CNPs are 31-fold higher than that of pure N-CDs (≈32.7 μmol g−1 h−1) at the same level of TEOA (Fig. S29†). The time-dependent H2 generation activities of N-CDs are also provided in Fig. S30.† We have also examined the photo-catalytic hydrogen generation activities of N, P-CNPs with other sacrificial electron donors of different molecular structures: methanol (MeOH), EDTA, Na2SO3, and ascorbic acid (AA). At the optimum concentration of all SEDs, N, P-CNPs have hydrogen generation rates of 156.3, 40.2, 90.1, and 46 μmol g−1 h−1 for MeOH, EDTA, Na2SO3, and AA, respectively (Fig. S31†). The result clearly indicates that, in contrast to TEOA, N, P-CNPs have comparatively poor photo-catalytic activity when other sacrificial agents are present. These results further prove the specificity of the N, P-CNP (TEOA) adduct formation and their precise selectiveness towards photo-catalytic hydrogen generation. Furthermore, the N, P-CNPs (TEOA) demonstrate an exceptionally high STH efficiency, achieving a value of 15.3% in regular (mili-Q) water and 9.7% in natural seawater. On the other hand, N-CDs have a very poor STH efficiency of 0.49% at the same amount of TEOA concentration. The apparent quantum efficiency (A.Q.Y) of N, P-CNPs and N-CDs for hydrogen generation has been determined to be approximately 3.37% and 0.12%, respectively, while irradiating 360 nm light with an intensity of 25 mW cm−2. The Experimental methods section explains the methods for calculating STH and AQE. To ensure the consistency and reliability of hydrogen production from N, P-CNPs (TEOA), we have reused (recycled) (Fig. S32†) the catalyst for at least five consecutive cycles. The N, P-CNP (TEOA) adduct produces hydrogen at a very stable rate during each cycle (Fig. S32†), indicating the system's robustness for long-term metal-free solar hydrogen production.
 | ||
| Fig. 6 Photo-catalytic solar hydrogen generation rate of the N, P-CNP (TEOA) complex in normal water (mili-Q) and natural seawater (a) and time-dependent (hours) hydrogen production of the N, P-CNP (TEOA) complex (b). A plausible mechanism for hydrogen production using the N, P-CNP (TEOA) charge transfer complex (c). Transient photocurrent response and EIS studies (d and e) of N, P-CNPs and N, P-CNPs (TEOA) respectively. | ||
According to the current finding, the N, P-CNP (TEOA) adduct modifies the electronic band structure of pure N, P-CNPs (Fig. 4(d)) by effectively reducing the band gap (ΔEg = 0.5 eV) and elevating the reduction potential (−1.59 V). The lowering of the band gap increases the overall solar light absorption and the strong reduction potential of N, P-CNPs, which eventually promotes the effective photo-reduction of water to form molecular hydrogen. Furthermore, the excited state dynamics of N, P-CNPs (TEOA) confirm the existence of a long-lived charge-separated state (Fig. 5) in the form of {N, P-CNPs˙−–(TEOA)˙+}. The charge recombination rate calculation from the corresponding ESA signal (Fig. 5 and Table S5†) of N, P-CNPs (TEOA) demonstrates a much slower charge recombination rate of 1940 ps (400 nm pump) and 1180 ps (530 nm pump) compared to pure N, P-CNPs (870 ps and 400 nm pump). The time-resolved photoluminescence decay (TRPL) study further supports our fs-TAS spectroscopy investigation. The average lifespan of the free carriers in N, P-CNPs (TEOA) gets significantly prolonged compared to pure N, P-CNPs (Table S1†). Therefore, both fs-TAS and TRPL hint at the potential development of a charge accumulation center (long-lived state) in N, P-CNPs (TEOA), which generates durable free carriers and ultimately improves the photo-catalytic hydrogen generation.40,67,68 Furthermore, fs-TAS analysis also depicts that a higher amount of TEOA (above the stable N, P-CNP (TEOA) complex) increases the free carrier population in terms of photoexcited electrons (Fig. S27†) in the excited charge-transfer (CT) state of N, P-CNPs (TEOA) explaining the enhanced photocatalytic hydrogen generation activities at elevated TEOA concentration.66 The plausible photo-catalytic hydrogen generation mechanism using the N, P-CNP (TEOA) charge-transfer (CT) state is shown in Fig. 6(c). An elevated TEOA concentration with a fixed amount of N, P-CNPs creates a unique scenario where a portion of TEOA (marked in red) is involved with N, P-CNPs to form a stable N, P-CNP (TEOA) complex (Fig. 6(c)). The rest of the free TEOA molecules in the systems (marked in blue) can act as a potential hole scavenger. Overall, the N, P-CNP (TEOA) adduct performs two key roles in improving the photocatalytic activities: (i) upon photoexcitation, the free carriers in a long-lived state accumulate on the catalytic center, namely the carbon core (Fig. S16†) of N, P-CNPs (TEOA), and efficiently enable the photo-reduction of H+ to H2. Secondly, (ii) the free TEOA molecules limit the photo-generated holes in the system, both vital for the photo-catalytic hydrogen production process (Fig. 6(c)). This is why N, P-CNPs and TEOA in their adduct form work so well for solar hydrogen production. Photoelectric and electrochemical studies provided further insight into the charge separation and transfer behaviors of N, P-CNPs and N, P-CNPs (TEOA). The instantaneous photocurrent response (30 s pulse duration) under full solar-light irradiation demonstrates a significantly increased photocurrent density (17-fold) (Fig. 6(d)) of the N, P-CNP (TEOA) adduct (17.88 μA cm−2) compared to bare N, P-CNPs (1.02 μA cm−2). Likewise, the photocurrent density of the N, P-CNP (TEOA) adduct experiences a modest rise over time, suggesting the persistent supply of photoinduced free carriers in the long run.
The high and stable photocurrent density of N, P-CNPs (TEOA) provides further evidence for the increased photoinduced free carrier and charge separation, promoting the photo-catalytic hydrogen evolution activities.41,60 The anodic photocurrent nature and the positive slope in the Mott–Schottky analysis collectively depict (Fig. 6(d) and 4(c)) the n-type characteristics of both N, P-CNPs and N, P-CNPs (TEOA).69 The assertive n-type behavior (Fig. 6(d)) of N, P-CNPs (TEOA) indicated a higher electron density than bare N, P-CNPs, which speeds up the photocatalytic HER activity. Moreover, EIS measurements indicate a smaller semicircle in the Nyquist plot (Fig. 6(e)) for N, P-CNPs (TEOA), unlike N, P-CNPs, suggesting a reduced charge transfer resistance of the free carriers at the interface of the N, P-CNP (TEOA) complex.70 Mott–Schottky analysis further strengthens the finding from the Nyquist measurements. Mott–Schottky analysis (Fig. 4(c)) demonstrates that N, P-CNPs (TEOA) have a positive Fermi-level potential shift of 8 mV compared to N, P-CNPs, which helps to mitigate the band bending in N, P-CNPs (TEOA) facilitating the free carrier transfer and boosting the photocatalytic HER activity.71 Post-catalytic structural characterization of purified N, P-CNPs through TEM and XPS studies (Fig. S33 and S34†) shows the negligible changes of the photocatalysts during experiments. It indicates the high stability of N, P-CNPs during the photocatalytic experiments. Furthermore, we have computed the free energies of water adsorption (ΔGads, (kcal mol−1)) at six different sites of our N, P-CNP model compound (depicted in Fig. S14†). Table S10† shows the possible active sites and the corresponding Gibbs free energies for H2O adsorption (ΔGads, (kcal mol−1)). The lowering of ΔGads is found to be maximum in the case of the PO32− containing group (site_5: – 67.8 kcal mol−1, i.e.-single H-bonding site, and site_6: – 68.1 kcal mol−1, i.e.– double H-bonding site) and moderate in the case of two H-bonding sites of PO3H2 groups (site_2: – 54.4 kcal mol−1 and site_4: – 60.0 kcal mol−1). However, the lowest reduction of ΔGads was found in the H-bonding site originating due to edge nitrogen (site_3: – 56.3 kcal mol−1) and weak vdW interaction dominated aromatic ring (from top) (site_1: – 52.4 kcal mol−1). As mentioned above, our detailed optical, elemental, and ultrafast analyses suggest that the N, P-CNPs in TEOA environments possess a significant population of PO32− containing groups alongside other primary moieties (PO3H2, pyridyl nitrogen, and sp2 carbon core). Therefore, during photocatalysis, a large population of PO32− groups owing to the N, P-CNP (TEOA) adduct formation facilitates the water adsorption and significantly boosts the solar hydrogen production. Moreover, phosphorus imparts enhanced electrical conductivity and rapidly transports the photo-generated free carriers at the interfacial catalytic center of the N, P-CNP (TEOA) complex, promoting the overall photocatalytic process.29,72 Thus, the current study suggests that the highly efficient metal-free photo-catalytic hydrogen generation activities arise due to the formation of a unique ground-state charge-transfer complex between N, P-CNPs and TEOA.
Conclusion
In conclusion, we have observed a particular ground-state interaction (Lewis acid–base type) between the P-functional group of N, P-CNPs and TEOA (SED), leading to a stable ground-state charge-transfer complex formation [defined as N, P-CNPs (TEOA)]. In contrast, no interaction for pure N-CDs with TEOA suggests the crucial role of P-functionality, which is further validated by detailed molecular level analysis. The N, P-CNP (TEOA) adduct showed highly efficient photocatalytic hydrogen generation activities of 1.02 mmol g−1 h−1 from regular water (mili-Q) and 651 μmol g−1 h−1 from natural seawater without any metal co-catalyst. In-depth structure–property correlation indicates the dual role of TEOA for improved photocatalytic activity. TEOA helps to create long-lived free carriers by forming charge-transfer complexes with N, P-CNPs and acting as hole scavengers to complete the photoredox cycle efficiently. Furthermore, the modification of electronic energy levels and facile water adsorption process in N, P-CNPs (TEOA) further facilitate the overall photocatalytic HER process. Overall, the current work will benefit the fundamental development of metal-free carbon based nanomaterials for cost effective solar energy to chemical energy conversions through a completely green approach.Experimental methods
Materials
Chitosan powder (deacetylated chitin) and acetic acid (glacial, ∼99%) were supplied by Sigma Aldrich. Ortho-phosphoric acid (85%) and triethanolamine (TEOA) were purchased from LOBA Chemie. The reagents were all used exactly as received. Milli-Q water was collected from a UV water purifying system from Merck Synergy, and natural seawater was collected from Gopalpur Beach (Odisha, India).Synthesis of heteroatom functionalized carbon nanoparticles (CNPs)
Chitosan gel, the primary precursor (carbon and nitrogen source), was initially prepared following a standard protocol.31 In the conventional method, 20 ml of 1% glacial acetic acid and 1 g of chitosan powder were rapidly swirled at 60 °C for 30 minutes. The N, P-co-functionalized carbon nanoparticles (N, P-CNPs) were developed by thermolyzing 1 g of chitosan gel and 300 μL of phosphoric acid (dropwise addition) for 8 hours at 180 °C in a hydrothermal autoclave with a Teflon container. Following the natural cooling of the hydrothermal container, the products were collected and centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) for 10 minutes to eliminate larger particles. Ultimately, a solution containing N, P-CNPs (marked in a pale-yellow color) was acquired by eliminating black aggregates using a PTFE syringe filter. Subsequently, purification was done using a dialysis membrane with a 1000-D cutoff to eradicate any remaining unreacted molecules. A similar method was employed to produce bright yellow N-CDs using only chitosan gel.
000 rpm) for 10 minutes to eliminate larger particles. Ultimately, a solution containing N, P-CNPs (marked in a pale-yellow color) was acquired by eliminating black aggregates using a PTFE syringe filter. Subsequently, purification was done using a dialysis membrane with a 1000-D cutoff to eradicate any remaining unreacted molecules. A similar method was employed to produce bright yellow N-CDs using only chitosan gel.
Computational details
All the density functional theory-based calculations (optimization, frequency, and UV-vis) were performed using the Gaussian16 suite of the program73 (Gaussian, Inc., Wallingford CT 2016). B3LYP, a hybrid exchange-correlation function developed by Becke and Lee, Yang, and Parr, was utilized in all the calculations.54,55 The geometry optimization was performed using the def2-SVP basis set and for all other calculations the def2-TZVP basis set was used.74,75 To account for the dispersion effect the Grimme's dispersion correction (GD3), including the parameters of Becke–Johnson, was utilized.76 Time-dependent density functional theory (TDDFT) methods were used to calculate absorption spectra. GaussView5.0 was used to draw the input geometries. The highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) were drawn using GaussView5.0.Electrochemical measurements
Mott–Schottky analysis, transient photocurrent response, and Nyquist (EIS) measurements are all performed on a CHI 760e electrochemical workstation. We have done our experiments using a three-electrode setup, a common practice in all electrochemical measurements. The present study utilizes a working electrode composed of fluorine-doped tin oxide (FTO) decorated with carbon nanoparticles. The reference electrode used in this experiment was Ag/AgCl (saturated KCl), whereas a Pt wire was utilized as the counter electrode. A 0.1 M aqueous Na2SO4 solution was used as a supporting electrolyte to maintain adequate conductivity during electrochemical studies. Carbon nanoparticle-coated working electrodes were developed by drop-casting an ink made of 1 mg of CNPs dispersed in 500 μL of acetonitrile and 10 μL of Nafion-117 binding agent on a 1cm × 1cm FTO substrate. Mott–Schottky analysis was performed at 3 kHz in the potential range of 0.0 to 0.90 V (vs. Ag/AgCl). Transient photocurrent response (i–t curve) was obtained at 0.6 V (vs. Ag/AgCl) under the entire solar light spectrum for nine on–off cycles with a 30 second pulse width. Electrochemical impedance spectra (EIS) were recorded between 0.1 and 100 kHz. For referencing the recorded potential (vs. sat. Ag/AgCl electrode) to the reversible hydrogen electrode, the following equation was followed4| E (vs. RHE) = E (vs. Ag/AgCl) + E0 (sat. Ag/AgCl) + 0.0591 × pH | (1) |
Global analysis of fs-TAS data
To extract the temporal component of pure N, P-CNPs with varying concentrations of TEOA, global analysis is conducted using GLOTARAN (1.5.1) software. GLOTARAN is a Java-based graphical user interface for the R package TIMP Singular value decomposition (SVD) and applied to the time-resolved absorption (TA) dataset matrix ΔA (t, λ) to determine the number of major components that should be fitted in the global analysis.61 In the global analysis, a sequential, unbranched, and unidirectional model is utilized to capture the spectral evolution of the excited-state species, resulting in the generation of evolution-associated difference spectra (EADS). Furthermore, the global analysis accounts for the correction of probe pulse dispersion and instrument response function (IRF) | (2) |
 | (3) |
Photo-catalytic measurements
Each photo-catalytic measurement was carried out in a 15.5 ml transparent quartz vial. The quartz container contains 11 ml of an aqueous solution with certain quantities of carbon nanoparticles (N, P-CNPs and N-CDs) and 20% TEOA. Before the light illumination, the quartz containers were sealed with a rubber septum and kept for 30 minutes in an argon atmosphere in the dark. A xenon lamp of 300 W (Newport-model-66902) with a total intensity of 1 sun (110 mW cm−2) was used for photocatalysis. The samples were kept at a suitable distance from the xenon light to prevent damage to the samples from the lamp's heat. The generation of hydrogen from fresh seawater was conducted in the same manner, with mili-Q water replaced in the system. Gas chromatography (SHIMADZUGC-2030, TCD detector) was used to evaluate the amount of hydrogen gas evolved during photo-catalytic experiments by injecting the gas from the headspace of the quartz vials into the GC. Before the final reporting of hydrogen generation for all samples, a minimal amount of hydrogen gas produced by only TEOA photocatalysis was eliminated in each case. Each photo-catalytic hydrogen producing activity was examined at least four times, and the relative error was found to be less than 10%.Photo-catalytic efficiency calculation (STH and AQE)
STH conversion efficiency is defined here as the hydrogen gas (chemical energy) ratio produced to total solar energy irradiation on the system.4,14Eqn (1) gives the efficiency of STH. The chemical energy is defined as the rate of hydrogen production multiplied by the Gibbs free energy (at 25 °C, G = 237 kJ mol−1) per mole of H2. The incident light intensity (Ptotal in mW cm−2) multiplied by the sample's lit effective area (cm2) equals the total solar energy given to the system. | (4) |
The apparent quantum yield (A.Q.Y) for photo-catalytic hydrogen generation is calculated using the following equation.4
 | (5) |
Now, we can determine the total energy delivered to the system and the energy of each photon as follows
| Etotal = P × S × t, | (6) |
 | (7) |
So, the total number of incident photons is obtained using
 | (8) |
Therefore, the final equation for apparent quantum yield becomes as follows
 | (9) |
Data availability
The data supporting the current findings is available from the corresponding authors upon appropriate request.Author contributions
Subhajit Kar: experiment, methodology, investigation, conceptualization, writing – original draft and editing, and visualization; Amit Kumar: fs-TAS spectroscopy experiment and methodology, and writing; Ramesh Mandal: experiment and methodology; Sakhshi Chawla: fs-TAS spectroscopy experiment; Shanti Gopal Patra: computational analysis; Arijit K. De: conceptualization, visualization, writing – review and editing, and supervision; Santanu Bhattacharyya: conceptualization, visualization, writing – review and editing, and supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. K. acknowledges IISER Berhampur for awarding a fellowship for his PhD studies. S. B. acknowledges IISER Berhampur for the start-up grant (IG/21082018/B0035). The authors thank DST-SERB (SRG/2019/000026) for financial support and for using the IISER Berhampur Central Advanced Instrument Facility (CAIF) for characterization. A. K. D. thanks IISER Mohali for the start-up grant and SERB and DST (CRG/2021/003981) for funding. A. K. thanks CSIR-UGC, and S. C. thanks IISER Mohali for the graduate fellowship.References
- L. Ran, S. Qiu, P. Zhai, Z. Li, J. Gao, X. Zhang, B. Zhang, C. Wang, L. Sun and J. Hou, J. Am. Chem. Soc., 2021, 143(19), 7402–7413 CrossRef CAS PubMed.
- X. Zhang, P. Zhai, Y. Zhang, Y. Wu, C. Wang, L. Ran, J. Gao, Z. Li, B. Zhang, Z. Fan, L. Sun and J. Hou, J. Am. Chem. Soc., 2021, 143(49), 20657–20669 CrossRef CAS PubMed.
- Y. Wu, Y. Zhao, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Mater., 2022, 34(29), 2202523 CrossRef CAS PubMed.
- Y.-B. Li, T. Li, X.-C. Dai, M.-H. Huang, Y. He, G. Xiao and F.-X. Xiao, J. Mater. Chem. A, 2019, 7(15), 8938–8951 RSC.
- Y. Song, Y. Ren, H. Cheng, Y. Jiao, S. Shi, L. Gao, H. Xie, J. Gao, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2023, 62(32), e202306420 CrossRef CAS PubMed.
- C. Wang, P. Zhai, M. Xia, Y. Wu, B. Zhang, Z. Li, L. Ran, J. Gao, X. Zhang, Z. Fan, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2021, 60(52), 27126–27134 CrossRef CAS PubMed.
- C. Wang, P. Zhai, M. Xia, W. Liu, J. Gao, L. Sun and J. Hou, Adv. Mater., 2023, 35(6), 2209307 CrossRef CAS PubMed.
- Y. Song, X. Zhang, Y. Zhang, P. Zhai, Z. Li, D. Jin, J. Cao, C. Wang, B. Zhang, J. Gao, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2022, 61(16), e202200946 CrossRef CAS PubMed.
- S. Kundu, B. Kommula and S. Bhattacharyya, Nanoscale Adv., 2020, 2, 5130–5151 RSC.
- Y.-B. Li, T. Li, X.-C. Dai, M.-H. Huang, S. Hou, X.-Y. Fu, Z.-Q. Wei, Y. He, G. Xiao and F.-X. Xiao, ACS Appl. Mater. Interfaces, 2020, 12(4), 4373–4384 CrossRef CAS PubMed.
- X. Yan, X.-Y. Fu and F.-X. Xiao, Adv. Funct. Mater., 2023, 33(48), 2303737 CrossRef CAS.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8(4), 3602–3635 CrossRef CAS.
- D. Wang and X.-Q. Gong, Nat. Commun., 2021, 12(1), 158 CrossRef CAS PubMed.
- S. Kar, L. Bishwal, B. Kommula and S. Bhattacharyya, Adv. Opt. Mater., 2022, 10(14), 2102641 CrossRef CAS.
- S. Bhattacharyya, F. Ehrat, P. Urban, R. Teves, R. Wyrwich, M. Döblinger, J. Feldmann, A. S. Urban and J. K. Stolarczyk, Nat. Commun., 2017, 8(1), 1401 CrossRef PubMed.
- B. Jana, Y. Reva, T. Scharl, V. Strauss, A. Cadranel and D. M. Guldi, J. Am. Chem. Soc., 2021, 143(48), 20122–20132 CrossRef CAS PubMed.
- J. Fang, T. Debnath, S. Bhattacharyya, M. Döblinger, J. Feldmann and J. K. Stolarczyk, Nat. Commun., 2020, 11(1), 5179 CrossRef CAS PubMed.
- J. Fang, Y. Wang, M. Kurashvili, S. Rieger, W. Kasprzyk, Q. Wang, J. K. Stolarczyk, J. Feldmann and T. Debnath, Angew. Chem., 2023, 135(33), e202305817 CrossRef.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- G. Speranza, Nanomaterials, 2021, 11(4), 967 CrossRef CAS PubMed.
- J. Alom, Md. S. Hasan, Md. Asaduzaman, M. T. Alam, D. Belhaj, R. Selvaraj, Md. A. Hossain, M. Zargar and M. B. Ahmed, Catalysts, 2023, 13(5), 823 CrossRef CAS.
- A. Ferrer-Ruiz, T. Scharl, L. Rodríguez-Pérez, A. Cadranel, M. Á. Herranz, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2020, 142(48), 20324–20328 CrossRef CAS PubMed.
- G. Ragazzon, A. Cadranel, E. V. Ushakova, Y. Wang, D. M. Guldi, A. L. Rogach, N. A. Kotov and M. Prato, Chem, 2021, 7(3), 606–628 CAS.
- Y. Choi, Y. Choi, O. Kwon and B. Kim, Chem. - Asian J., 2018, 13(6), 586–598 CrossRef CAS PubMed.
- Y. Yu, Q. Zeng, S. Tao, C. Xia, C. Liu, P. Liu and B. Yang, Adv. Sci., 2023, 10(12), 2207621 CrossRef CAS PubMed.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55(32), 9402–9406 CrossRef CAS PubMed.
- H. Luo, Y. Liu, S. D. Dimitrov, L. Steier, S. Guo, X. Li, J. Feng, F. Xie, Y. Fang, A. Sapelkin, X. Wang and M.-M. Titirici, J. Mater. Chem. A, 2020, 8(29), 14690–14696 RSC.
- R. Hoffmann, J. M. Howell and E. L. Muetterties, J. Am. Chem. Soc., 1972, 94(9), 3047–3058 CrossRef CAS.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55(5), 1830–1834 CrossRef CAS PubMed.
- M. Latorre-Sánchez, A. Primo and H. García, Angew. Chem., 2013, 125(45), 12029–12032 CrossRef.
- S. Kar, K. Bramhaiah, N. S. John and S. Bhattacharyya, Chem. – Asian J., 2021, 16(9), 1138–1149 CrossRef CAS PubMed.
- M. E. Plonska-Brzezinska, ChemNanoMat, 2019, 5(5), 568–580 CrossRef CAS.
- A. D. Martínez-Iniesta, A. Morelos-Gómez, E. Muñoz-Sandoval and F. López-Urías, RSC Adv., 2021, 11(5), 2793–2803 RSC.
- L. Bishwal, S. Kar and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2023, 15(12), 15907–15916 CrossRef CAS PubMed.
- J. Zhou, X. Shan, J. Ma, Y. Gu, Z. Qian, J. Chen and H. Feng, RSC Adv., 2014, 4(11), 5465–5468 RSC.
- E. M. Oliveira, M. Rogero, E. C. Ferreira and J. A. Gomes Neto, Spectrochim. Acta, Part A, 2021, 246, 119025 CrossRef CAS PubMed.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12(8), 3925–3930 CrossRef CAS PubMed.
- E. M. Villota, H. Lei, M. Qian, Z. Yang, S. M. A. Villota, Y. Zhang and G. Yadavalli, ACS Sustainable Chem. Eng., 2018, 6(1), 1318–1326 CrossRef CAS.
- B. Shi, Y. Su, L. Zhang, M. Huang, R. Liu and S. Zhao, ACS Appl. Mater. Interfaces, 2016, 8(17), 10717–10725 CrossRef CAS PubMed.
- C. Wang, W. Wang, H. Fan, N. Zhao, J. Ma, M. Zhang and A. K. Yadav, ACS Appl. Mater. Interfaces, 2020, 12(5), 5234–5243 CrossRef CAS PubMed.
- L. Jing, R. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27(46), 1703484 CrossRef.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136(49), 17308–17316 CrossRef CAS PubMed.
- J. L. Havlin and A. J. Schlegel, Soil Syst., 2021, 5(3), 52 CrossRef CAS.
- X. Gong, Q. Zhang, Y. Gao, S. Shuang, M. M. F. Choi and C. Dong, ACS Appl. Mater. Interfaces, 2016, 8(18), 11288–11297 CrossRef CAS PubMed.
- P. Sannigrahi and E. Ingall, Geochem. Trans., 2005, 6(3), 52 CrossRef CAS PubMed.
- C. Godinot, M. Gaysinski, O. P. Thomas, C. Ferrier-Pagès and R. Grover, Sci. Rep., 2016, 6(1), 21760 CrossRef CAS PubMed.
- B. Cademenun, Talanta, 2005, 66(2), 359–371 CrossRef CAS PubMed.
- B. Peng, Y. Xu, K. Liu, X. Wang and F. M. Mulder, ChemElectroChem, 2017, 4(9), 2140–2144 CrossRef CAS.
- A. R. MacIntosh, G. Jiang, P. Zamani, Z. Song, A. Riese, K. J. Harris, X. Fu, Z. Chen, X. Sun and G. R. Goward, J. Phys. Chem. C, 2018, 122(12), 6593–6601 CrossRef CAS.
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11(5), 1287–1298 RSC.
- A. García-Mulero, A. Rendón-Patiño, A. M. Asiri, A. Primo and H. Garcia, ACS Appl. Mater. Interfaces, 2021, 13(41), 48753–48763 CrossRef PubMed.
- C. Ge, Z. Fan, J. Zhang, Y. Qiao, J. Wang and L. Ling, RSC Adv., 2018, 8(60), 34682–34689 RSC.
- F. J. García-Mateos, R. Berenguer, M. J. Valero-Romero, J. Rodríguez-Mirasol and T. Cordero, J. Mater. Chem. A, 2018, 6(3), 1219–1233 RSC.
- A. D. Becke, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- Y. Yang, W. Kong, H. Li, J. Liu, M. Yang, H. Huang, Y. Liu, Z. Wang, Z. Wang, T.-K. Sham, J. Zhong, C. Wang, Z. Liu, S.-T. Lee and Z. Kang, ACS Appl. Mater. Interfaces, 2015, 7(49), 27324–27330 CrossRef CAS PubMed.
- L. Wang, C.-S. Tsang, W. Liu, X. Zhang, K. Zhang, E. Ha, W.-M. Kwok, J. H. Park, L. Y. Suk Lee and K.-Y. Wong, J. Mater. Chem. A, 2019, 7(1), 221–227 RSC.
- X. Chen, S. Shen, L. Guo and S. Mao, Chem. Rev., 2010, 110(11), 6503–6570 CrossRef CAS PubMed.
- S. Mishra, S. Sapru, S. N. Upadhyay, A. Singh, S. Pakhira and A. K. De, J. Phys. Chem. C, 2023, 127(4), 1881–1890 CrossRef CAS.
- B. Kommula, P. Durairaj, S. Mishra, S. Kar, A. Sury, A. Kumar, A. K. De, S. Sarkar and S. Bhattacharyya, ACS Appl. Nano Mater., 2022, 5(10), 14746–14758 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, 2004, 1657(2–3), 82–104 CrossRef CAS PubMed.
- V. I. Klimov, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(19), 13740–13749 CrossRef CAS.
- B. Yang, J. Chen, S. Yang, F. Hong, L. Sun, P. Han, T. Pullerits, W. Deng and K. L. Han, Angew. Chem., Int. Ed., 2018, 57(19), 5359–5363 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20(3), 283–295 CrossRef CAS.
- R. A. Scheidt, E. Kerns and P. V. Kamat, J. Phys. Chem. Lett., 2018, 9(20), 5962–5969 CrossRef CAS PubMed.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141(28), 11219–11229 CrossRef CAS PubMed.
- L. Jing, R. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27(46), 1703484 CrossRef.
- X. Tian, Y. Sun, J. He, X. Wang, J. Zhao, S. Qiao and F. Li, J. Mater. Chem. A, 2019, 7(13), 7628–7635 RSC.
- R. Beranek and H. Kisch, Angew. Chem., Int. Ed., 2008, 47(7), 1320–1322 CrossRef CAS PubMed.
- X.-X. Fang, L.-B. Ma, K. Liang, S.-J. Zhao, Y.-F. Jiang, C. Ling, T. Zhao, T.-Y. Cheang and A.-W. Xu, J. Mater. Chem. A, 2019, 7(18), 11506–11512 RSC.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134(38), 15720–15723 CrossRef CAS PubMed.
- L. Kong, Y. Ji, Z. Dang, J. Yan, P. Li, Y. Li and S. Liu, Adv. Funct. Mater., 2018, 28(22), 1800668 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97(4), 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100(8), 5829–5835 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, IR, XPS, UV-vis absorption and PL emission spectra, computational analysis, TCSPC, and fs-TAS analysis and photocatalytic hydrogen evolution data of N, P-CNPs, N-CDs and N, P-CNPs (TEOA), and detailed description of characterization techniques. See DOI: https://doi.org/10.1039/d3ta07895g |
| This journal is © The Royal Society of Chemistry 2024 |