
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing ion storage and transport in Ti3C2Tz MXene via a “sacrificial cations” strategy †
Xiaodan
Yin‡
,
Wei
Zheng‡
*,
Haifeng
Tang
,
Li
Yang
,
Peigen
Zhang
 * and
ZhengMing
Sun
*
* and
ZhengMing
Sun
*
School of Materials Science and Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: zhengwei22@seu.edu.cn; zhpeigen@seu.edu.cn; zmsun@seu.edu.cn
First published on 7th March 2024
Abstract
MXenes have gained significant attention in supercapacitors (SCs) due to their high electronic conductivity and tunable surface terminations. However, challenges arise during etching such as interlayer restacking and the presence of inactive –F terminations that impede the full utilization of their intrinsic capabilities. To tackle these issues, we propose a “sacrificial cation” strategy involving electrochemistry-driven cation intercalation (ECI) and calcination. Specifically, alkylammonium cations with different chain lengths are intercalated into the interlayers of Ti3C2Tz MXene by ECI, and then these intercalated cations are removed by pyrolysis at 400 °C. This approach aims to augment interlayer spacing and introduce a substantial number of –O surface terminations, thereby enhancing capacitance contributions. The resulting dodecyl-trimethylammonium cation intercalated Ti3C2Tz after calcination (T-C8-C) presents high volumetric capacitances of 1737.6 F cm−3 at 1 A g−1. T-C8-C, when integrated with nitrogen-doped activated carbon (NAC) into an asymmetric SC, achieves outstanding volumetric energy density (56.7 W h L−1 at 0.15 kW L−1), high power densities at elevated energy densities (30.1 kW L−1 at 36.7 W h L−1), and remarkable lifespan (96.2% retention after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 A g−1). This strategy provides valuable insights for developing high-performance 2D materials in energy storage through interlayer spacing adjustment and surface modification.
000 cycles at 10 A g−1). This strategy provides valuable insights for developing high-performance 2D materials in energy storage through interlayer spacing adjustment and surface modification.
Introduction
Efficient energy storage and conversion technology with good reliability and availability is highly sought after.1,2 Supercapacitors (SCs) have the advantages of high power densities and long cycle life.3 However, low energy densities limit their further applications.4 MXenes, as two-dimensional (2D) transition metal carbides/nitrides, with a formula of Mn+1XnTz (M represents transition metal, X denotes C or N, n = 1, 2, 3, or 4, and Tz stands for the terminations, namely –OH, –O, –F, etc.), have achieved great progress in SCs due to their high electrical conductivity and abundant terminations.5–7 Currently, a common method for the large-scale production of MXene involves utilizing F-containing etching solutions, such as HF, NH4HF2, LiF–HCl, etc. Such solutions can effectively break the weaker M–A bonds in the MAX phase, selectively etching away the A-layer atoms, while leaving the stronger M–X bonds unaffected.8,9 However, during etching preparation, interlayer restacking caused by van der Waals forces10,11 and the attachment of inactive –F terminations limit their potential applications.12–18To resolve the above issues, various strategies including compositing, atomic doping, and intercalation have been proposed. Compositing MXene with materials such as carbon, metal oxides or conducting polymers enhances the ion penetration but increases the preparation complexity.19–22 Atomic doping introduces nitrogen, sulfur or boron, optimizing MXene's electronic structure and improving conductivity, though it may introduce impurities.23–26 Intercalation is widely employed to pillar up the collapsed layers and modify the surface terminations, thereby facilitating ion transport and enhancing the availability of active sites for ion storage. Intercalators such as metal ion,27 polymers,28 nanocarbons,27etc., have been used. Particularly, polymers with different chain lengths can finely tune the interlayer spacing of MXenes, resulting in enhanced SC performance.29,30 However, these organic cations have shown difficulty in full intercalation due to their long chain lengths. More time was required, even though these organic cations can be intercalated into MXene interlayers. The electrochemistry-driven cation intercalation (ECI) method with rapid intercalation time has been developed to address this issue.31,32 ECI involves utilizing the charging/discharging process to intercalate cations into the Ti3C2Tz interlayers, which not only preserves the integrity of the Ti3C2Tz matrix but also opens up the obstructed channels that cannot participate in reactions.33,34 Importantly, various voltages can be utilized to determine the quantity of cations intercalated into MXenes, thereby enabling precise control over the intercalation process.32,35
Literature reports have shown that polymers such as PMMA can be removed by pyrolysis at an elevated temperature,36–38 leaving more space for ion accommodation. Similarly, if the intercalation organic cations decompose at high temperatures, it can also have more space left. Furthermore, the surface terminations such as active –O terminations can be formed, and –F terminations will be reduced during calcination.27,39–41 Therefore, this “sacrificial cations” strategy is expected to enhance SC performance.
In this paper, we choose Ti3C2Tz as the electrode and alkyl materials with different chain lengths (C4, C8, C12, and C16) as the intercalators. Together with ECI and calcination, it is found that the T-C8-C electrode has more uniformed interlayer spacing and more favorited –O terminations, leading to high specific capacitances and exceptional rate performance, together with a good cycling life. To improve the energy densities of the device, an asymmetric SC is assembled with the positive electrode (nitrogen-doped activated carbon, NAC) and the negative electrode (T-C8-C), which has high energy density up to 56.7 W h L−1 at 0.15 kW L−1 and excellent cycling stability over 30000 cycles at 10 A g−1. Impressively, high power densities are maintained even at elevated energy densities (30.1 kW L−1 at 36.7 W h L−1). The combination of high energy densities at high powers is attractive in the SC field.
Results and discussion
Fig. 1 shows the preparation process of Ti3C2Tz-Carbon X-Calcination (T-CX-C, X represents carbon chain length). The synthesis process of the Ti3C2Tz freestanding films via Ti3AlC2 etching, sonication, and filtration is similar to our previous work.42 The XRD patterns of Ti3AlC2 and Ti3C2Tz are shown in Fig. S1.† The absence of the (104) peak and the downshift of the (002) peak indicate the successful synthesis of MXene. Ti3C2Tz aqueous suspension has the Tyndall effect shown in Fig. S2,† meaning the good dispersion of MXene nanosheets. Ti3C2Tz-Carbon X (T-CX) films were obtained by intercalating different organic cations (butyl-, octyl-, dodecyl- and hexadecyl-trimethylammonium, marked as C4, C8, C12 and C16, respectively) into Ti3C2Tz using ECI. Finally, calcination was used to remove the intercalated organic cations by pyrolysis and modify T-CX's terminations simultaneously, obtaining T-CX-C. | ||
| Fig. 1 Schematic diagram showing the preparation of T-CX-C. | ||
Fig. 2(a) shows the X-ray diffraction (XRD) patterns of the T-CX-C electrodes, and Fig. 2(b) shows its magnification at the 3–20° range, from which the (002) peak of pristine Ti3C2Tz (abbreviated to T) is at 7.05°. Ti3C2Tz-Calcination (abbreviated to T-C) is shifted to the right to 7.3° after calcination due to the part removal of absorbed water and –F terminations.27 Compared to T-C, the (002) characteristic peaks of T-CX-C electrodes are all shifted left, meaning the expansion of the interlayer spacing. Furthermore, the (002) peaks for T-C4-C and T-C8-C electrodes are observed at 5.98° with no additional peaks before this angle, indicating that the removal of C4 and C8 has left uniform space in Ti3C2Tz interlayers. T-C12-C exhibits a minor peak at 4.66°, which means only a portion of C12 successfully intercalated into the Ti3C2Tz interlayers, which is likely ascribed that the longer chain length hinders complete intercalation and causes different interlayer distances. This phenomenon is also reflected in T-C16-C.
 | ||
| Fig. 2 X-ray diffraction (XRD) patterns from (a) 3°–70° and (b) 3°–20° of T-CX-C. The XRD patterns T and T-C are not shown. The purple and cyan lines show the (002) peak of T and T-C, respectively. (c)–(f) Scanning electron microscopy (SEM) images of T-C4-C, T-C8-C, T-C12-C, and T-C16-C, respectively. The magnified crystal lattice stripes in Transmission electron microscopy (TEM) images of (g) Ti3C2Tz and (h) T-C8-C. | ||
The SEM images in Fig. 2(c)–(f) show the cross-section morphology of the flexible T, T-C, and T-CX-C electrodes, respectively. It can be clearly seen that freestanding electrodes are composed of layers stacked sequentially, and their thickness is about 5 μm. Fig. S3† shows their optical photographs, and it has been observed that the freestanding films maintain their integrity well after calcination. Fig. 2(g) and (h) display lattice stripes in TEM images of both Ti3C2Tz and T-C8-C, respectively. It can be clearly seen that there is more uniform layer spacing, and the obstructed channels are reduced in the T-C8-C interlayers compared with the pristine Ti3C2Tz. Furthermore, no residues can be observed in T-C8-C, meaning the “sacrificial cations” have been removed totally.
XPS was utilized to analyze the surface chemistries of the Ti3C2Tz and T-CX-C electrodes in Fig. 3(a). The T-CX-C electrodes do not display new peaks which is consistent with FTIR spectra (Fig. S4†) but show a decrease in F 1s peaks and an intensification of O 1s peaks, likely ascribed to the reduced –F and increased –O terminations after the ECI and calcination processes. The elemental composition of the electrodes is depicted from XPS (Fig. 3(b)). Notably, the percentage of the F element on the T-C8-C is lowest and the O element is highest. For a quantitative analysis of the O 1s peak in Ti3C2Tz and T-C8-C (Fig. 3(c)), the enhancement of Ti–O bonds on the T-C8-C's surface is observed. After ECI and calcination treatment, the intensity of Ti–O peaks rises from 52.4% to 68.3%, compared with the pristine Ti3C2Tz. The underlying reason for this change is still unknown. The contact angle serves as a measure of electrolyte and electrode wettability, impacting the kinetics of the electrochemical process (Fig. S5†). Ti3C2Tz was prepared through the LiF and HCl etching process, resulting in the incorporation of terminations such as –O, –OH, –F, and –Cl. The negatively charged surface terminations enable Ti3C2Tz nanosheets to form a stable colloidal dispersion in water. As a result, Ti3C2Tz films exhibited wettability in sulfuric acid electrolytes with a contact angle of 81.6°. T-C8-C is much smaller than that of pure Ti3C2Tz, which is only 59.3°. This is primarily attributed to ion intercalation enhancing the active sites on the electrode. Simultaneously, the calcination process reduces the hydrophobic –F content and increases the hydrophilic –O, effectively modifying the electrode surface terminations. The electrodes prepared by the “sacrificial cation” method have strong hydrophilicity with aqueous electrolyte, which is favorable for energy storage.43 However, it is well known that alkylammonium cations easily decompose into NH3 and acid during calcination (will be discussed in Fig. 3(f)). These factors should be accountable for alterations in terminations and are expected to be beneficial in demonstrating improved electrochemical performance.14,44,45 The Raman spectrum of Ti3C2Tz (red line in Fig. 3(d)) is categorized into two regions: the modes at 199 and 725 cm−1 represent A1g symmetry out-of-plane vibrations of Ti and C atoms. Meanwhile, the modes at 288, 375, 569, and 628 cm−1 are Eg group vibrations, encompassing in-plane (shear) modes of Ti, C, and surface terminations.46,47 All the T-CX-C films show the signals of A1g and Eg peaks, evidencing the well structure maintained of Ti3C2Tz after treatment. Additionally, the T-CX-C films have a comparable intensity ratio between D and G (ID/IG), indicating their similar structures. The pronounced increase of D and G peaks of the T-CX-C films, compared with pristine Ti3C2Tz, is likely ascribed to the increased carbon species at the edges or defects after calcination.10,48 The TG results in Fig. 3(e) demonstrate that C8 undergoes a decomposition started at 180 °C and completed at 270 °C. Therefore, calcination at 400 °C is enough to remove all C8. Clearly, Ti3C2Tz gains a 0.6 wt% increase, while T-C8-C has a 10.1 wt% loss after calcination. Therefore, 10.7 wt% of C8 has been intercalated into Ti3C2Tz. The removal of organic cations results in larger interlayer space and more –O terminations for ion storage and transport, thereby leading to enhanced SC performance, which will be discussed in the electrochemical test part.
 | ||
| Fig. 3 (a) X-ray photoelectron spectroscopy (XPS) survey of Ti3C2Tz and T-CX-C. (b) The count of C 1s, O 1s, and F 1s for Ti3C2Tz and T-CX-C electrodes. (c) O 1s XPS spectra of Ti3C2Tz and T-C8-C. (d) Raman spectra of Ti3C2Tz and T-CX-C electrodes. (e) TG analysis of Ti3C2Tz, T-C8-C, and C8. (f) TG-MS spectra of T-C8. | ||
To investigate the effect of the pyrolysis products of the organic cation on the electrode, thermo-gravimetry-mass spectrometry (TG-MS) under an Ar atmosphere was employed to monitor gas evolution during calcination in Fig. 3(f). Based on the alkylammonium cations intercalation, coupled with MS detection of amu signals, a gradual mass loss is observed at around 50 °C, indicating the evaporation of water adsorbed between Ti3C2Tz layers. Subsequently, a noticeable mass loss and some gas products appear around 200 °C, indicating that the alkylammonium cations occur in pyrolysis. Due to the constraints imposed by the Ti3C2Tz layers, the alkylammonium cations experience an extended duration for complete pyrolysis. It was observed that alkylammonium cations readily decomposed into NH3 (m/z 17), N2 (m/z 28), and HCl (m/z 36) during calcination. The detection of HCl is attributed to the use of hydrochloric acid as an etchant, and the remaining Cl between the Ti3C2Tz layers is thermally decomposed at elevated temperatures. NH3 and N2 undergo pyrolysis at elevated temperatures facilitated by the incorporation of alkylammonium cations. The release of NH3 and N2 gases is most prominent upon reaching 277 °C. The gases play a crucial role in altering surface terminations, holding the potential for enhancing the electrochemical performance of electrodes.
To study the electrochemical performance of the T-CX-C electrodes, 3 M sulfuric acid (H2SO4) was chosen as the electrolyte for testing, which is suitable for MXene to exhibit superior performance due to the fast surface redox reactions between H+ and –O terminations.49–56 To investigate the optimal calcination temperatures for conducting electrochemical tests, the T-C8 electrodes were subjected to different calcination temperatures, specifically at 300, 400, 500, and 600 °C (Fig. S6†). The specific capacitances exhibit a trend of increasing and then decreasing with rising calcination temperature. The maximum specific capacitance is 1737.6 F cm−3 at 400 °C and the minimum specific capacitance recorded is only 358.7 F cm−3 at 600 °C. The reaction kinetics was investigated by electrochemical impedance spectroscopy (EIS) in Fig. S6(d).† T-C8-C calcinated 400 °C has the lowest charge transfer resistance, with the imaginary part of the impedance nearly vertical to the real part, indicating its ideal capacitive behavior.57,58 The main reason is that with the increase in calcination temperature, oxidation is severe, and there will be more TiO2 generation, which destroys the structure of MXene and decreases the electrode conductivity.27,59 Therefore, 400 °C was optimized as the calcination temperature.
Fig. 4(a) compares the CV curves for Ti3C2Tz and T-CX-C electrodes at 20 mV s−1. These electrodes show a pair of broad redox peaks, where reversible intercalation/deintercalation of ions occurs during the redox reaction between H+ and –O terminations.49–56 The GCD curves of Ti3C2Tz, T-C, and T-CX-C electrodes are compared in Fig. 4(b). All the GCD curves show symmetric charge/discharge processes, indicating good reaction reversibility.53,60 Fig. S7† illustrates the Nyquist plots of Ti3C2Tz, T-C, and T-CX-C electrodes. T-C8-C exhibits the smallest internal resistance (Rs) with a value of 1.31 Ω. Compared to other T-CX-C electrodes, T-C8-C shows a steeper slope in the low-frequency region, indicating a more capacitive process. The volumetric capacitances of Ti3C2Tz, T-C, and T-CX-C electrodes are displayed in Fig. 4(c) as a function of current densities from 1 to 50 A g−1. When considering both specific capacitances and capacitance retention, T-C8-C demonstrates the best performance. It not only achieves 1737.6 F cm−3 at 1 A g−1 but also maintains 708.5 F cm−3 when the current density increases to 50 A g−1. The charge storage kinetics of T-CX-C is assessed by analyzing the relationship between peak current (ip) and scan rate (v), according to the following eqn (1):
| ip = avb | (1) |
 | ||
| Fig. 4 (a) Cycle voltammetry (CV) curves at 20 mV s−1 for the Ti3C2Tz, T-C, and T-CX-C electrodes. (b) Galvanostatic charge/discharge (GCD) curves at 1 A g−1 for the Ti3C2Tz and T-CX-C electrodes. (c) Capacitance comparison of Ti3C2Tz and T-CX-C electrodes at different current densities. (d) The CV curves from 1 mV s−1 to 50 mV s−1 and (e) GCD curves from 1 A g−1 to 50 A g−1 for the T-C8-C electrode. (f) Cycling stability of T-C8-C at 10 A g−1. Inset of (f) shows the GCD curves of the first and the last 5 cycles. | ||
Due to the highest volumetric capacitances and impressive rate performance of the T-C8-C electrodes, we conducted further investigations into its electrochemical properties. The CV curves of T-C8-C exhibit one pair of broad peaks at different scan rates (Fig. 4(d)), indicating intercalated pseudocapacitance (PC) behavior, as reported for Ti3C2Tz-based electrodes previously.49–56Fig. 4(e) represents the GCD curves of T-C8-C between −0.5 to 0.2 V (vs. Ag/AgCl) at different current densities from 1 to 50 A g−1. The GCD curves exhibit nearly equal charge and discharge times at any current density, signifying excellent electrochemical reaction reversibility.52,61
From Fig. S9,† it can be observed that the capacitive-controlled contribution surpasses the diffusion-controlled contribution at any scan rate from 1 to 50 mV s−1, which indicates T-C8-C possesses rapid charge storage kinetics and is advantageous for achieving excellent rate performance. When conducting 10000 cycles at 10 A g−1, the T-C8-C electrode maintains 102.8% of its initial capacitance and the coulombic efficiency is nearly 100% (Fig. 4(f)). Additionally, compared to other Ti3C2Tz-based electrodes, the T-C8-C maintains a leading position in specific capacitances, capacitance retention, and long-cycling performance, as shown in Table S1.†
Post cycling characterization is typically conducted to assess the stability of electrodes. Herein, the T-C8-C electrode after 10000 cycles at 10 A g−1 was characterized with XRD, Raman, and SEM. Fig. S10(a) and (b)† show the XRD comparison of the T-C8-C before and after long cycling tests. By comparing the (002) characteristic peak, it is observed that the peak shifts from 6.68° to 6.42° (Fig. S10(b)†). This shift is primarily attributed to the increased interlayer spacing from trapped ions during cycling. Meanwhile, the electrode maintains the characteristic peaks of Ti3C2Tz, and its layered structure is reserved after cycling (inset in Fig. S10(a)†). Furthermore, the Raman spectra of the electrodes still maintain the characteristic peaks of the pristine Ti3C2Tz after long cycling tests (Fig. S10(c)†). Therefore, it is confidently claimed that the T-C8-C has good cycling stability.
To further investigate the electrochemical mechanism of the T-C8-C electrodes during the charging/discharging processes, ex situ XRD, HRTEM, XPS, and Raman were conducted. During the charging process from −0.5 V to 0.2 V (Fig. 5(a) point A to C), the (002) peak shifts to the right which is attributed to the H+ deintercalation.60,62 Conversely, the (002) peak shifts to the left during discharging (Fig. 5(a) point C to E). Ex situ HRTEM was applied in T-C8-C in Fig. 5(d). Lattice fringes with lattice spacings of 2.02 Å, corresponding to (101) of Ti3C2Tz, were found when the electrode was completely charged to 0.2 V vs. Ag/AgCl. When completely discharged to −0.5 V vs. Ag/AgCl, the lattice spacing of 2.02 Å corresponds to (101) of Ti3C2Tz, which has been increased due to the embedding of protons from the electrolyte. These phenomena have been observed in ex situ XRD, as shown in Fig. 5(b) and (c).
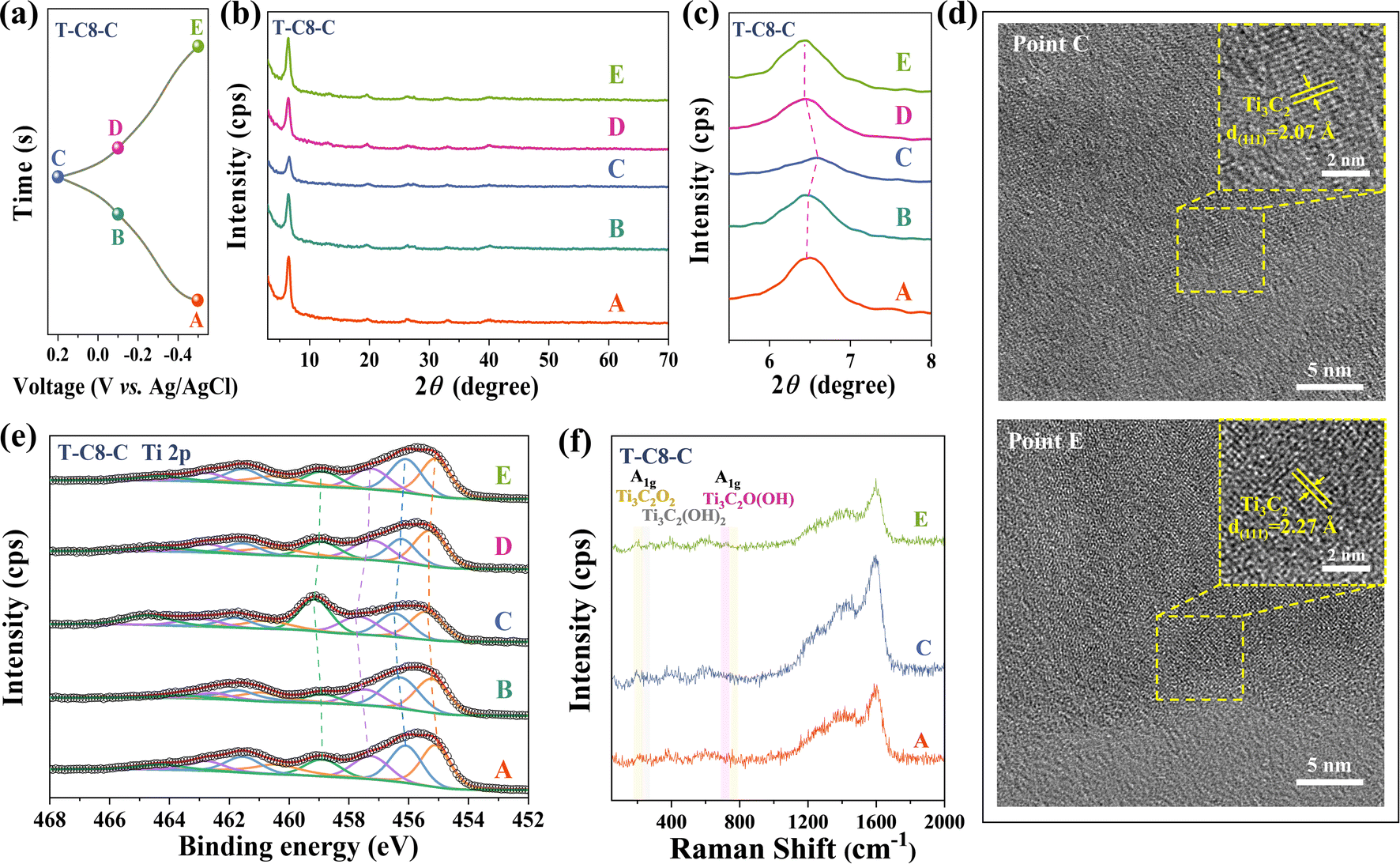 | ||
| Fig. 5 (a) GCD curve of the T-C8-C electrode at 1 A g−1. The corresponding ex situ XRD patterns (b) and (c). (d) Crystal lattice stripes of T-C8-C electrode at point C and E. Ex situ XRD (e) and Raman patterns (f) of T-C8-C electrode. | ||
Combined with the ex situ XPS, it is observed that the Ti peaks shift towards higher binding energies during charging which means oxidization. In comparison, the Ti peaks shift to lower binding energies during discharging, indicating reduction. Raman spectra were obtained from electrodes representing three different charge and discharge states (corresponding to points A, C, and E marked on the GCD curve, as shown in Fig. 5(a)). In Fig. 5(f), the reversible peak intensity changes of the A1g band were observed with different potentials. The A1g peaks represent the symmetric out-of-plane vibrations of Ti, C, and surface terminations, respectively.63 The peak at 720 cm−1 is attributed to the out-of-plane vibration of a C–Ti bond surrounded by –O terminations, as seen in Ti3C2O2, while the peak at 708 cm−1 corresponds to C–Ti bond in a Ti3C2O(OH) environment.64 During the charging process (point A to C), the peak intensity of A1g weakens. This is attributed to the gradual transformation of the environment of Ti from –OH to –O, causing the peak to shift downward. Conversely, the A1g peak intensity strengthens during the discharge process. Similar phenomena have also been observed in related literature.63,65 Additionally, a similar phenomenon was observed in the 150–300 cm−1. Around 200 cm−1 for A1g (Ti, O, C) such as Ti3C2O2, during the charging process (point A to C), the Raman peak at 208 cm−1 for A1g gradually intensifies, while the peak at 265 cm−1 originating from A1g (Ti, OH, C) such as Ti3C2(OH)2 gradually weakens. The discharge process is also reversible. Therefore, the variation of peak intensity in the Raman is attributed to the (de)embedding of protons from the electrolyte, coupled with –O termination bond/de-bond processes.66 The reaction process in MXene can be expressed as the eqn (2):67
| Ti3C2Ox(OH)yFz + δH+ + δe− ↔ Ti3C2Ox−δ(OH)y+δFz | (2) |
Therefore, a combination of ex situ XRD, HRTEM, XPS, and Raman can illustrate that the (de)embedding of H+ is involved in the T-C8-C electrode.
To demonstrate the practical application of the T-C8-C electrodes, an ASC was assembled, where T-C8-C was used as the negative electrode coupling with the NAC positive electrode, as illustrated in Fig. 6(a). The electrochemical performance of the ASC was tested using CV to determine the potential window. Fig. 6(b) displays the CV curves of T-C8-C and NAC electrodes separately at 20 mV s−1. The former exhibits a pair of redox peaks and has a potential window from −0.5 to 0.2 V (vs. Ag/AgCl), and the NAC's potential window is from 0 to 1 V (vs. Ag/AgCl). Hence, the ASC has a 1.5 V voltage. Fig. 6(c) shows the CV curves at different scan rates ranging from 1 mV s−1 to 50 mV s−1. The deviation from the ideal rectangular shape of the CV shape is due to the different charge storage mechanisms of the negative and positive electrodes. The shape of the CV can still be maintained when the scan rate increases, indicating a good rate performance. The GCD curves at the current densities of 0.1 A g−1 to 20 A g−1 are shown in Fig. 6(d). With the increase of current densities, the discharge curves gradually become straight, indicating that the contribution of capacitance is transformed from the pseudocapacitance (PC) to the electrical double-layer capacitance (EDLC). Fig. 6(e) compares the volumetric capacitances of T-C8-C//NAC in terms of the current densities, and the ASC has a very excellent capacitance retention of 70.1% when the scan rate is increased from 1 mV s−1 to 100 mV s−1. The Ragone plot of the T-C8-C//NAC is shown in Fig. 6(f). At a power density of 0.2 kW L−1, T-C8-C//NAC can achieve a high energy density of 56.7 W h L−1. Even when the power density increases to 30.1 kW L−1, T-C8-C//NAC can still deliver 36.7 W h L−1. This combination of energy densities at high powers is excellent, surpassing other reports.68–76 The self-discharge performance of T-C8-C//NAC was tested in Fig. 6(g). After 10 hours, the voltage of the cells is observed to stabilize at 0.57 V, indicating a retention rate of 38.0%. The T-C8-C//NAC exhibits good cycling performance with a capacitance retention of 96.2% after 30000 cycles at 10 A g−1 (Fig. 6(h)), and 88.6% retention after 2000 cycles at 1 A g−1 (Fig. S11†), which is still competitive compared to previous reports, as shown in Table S1.†
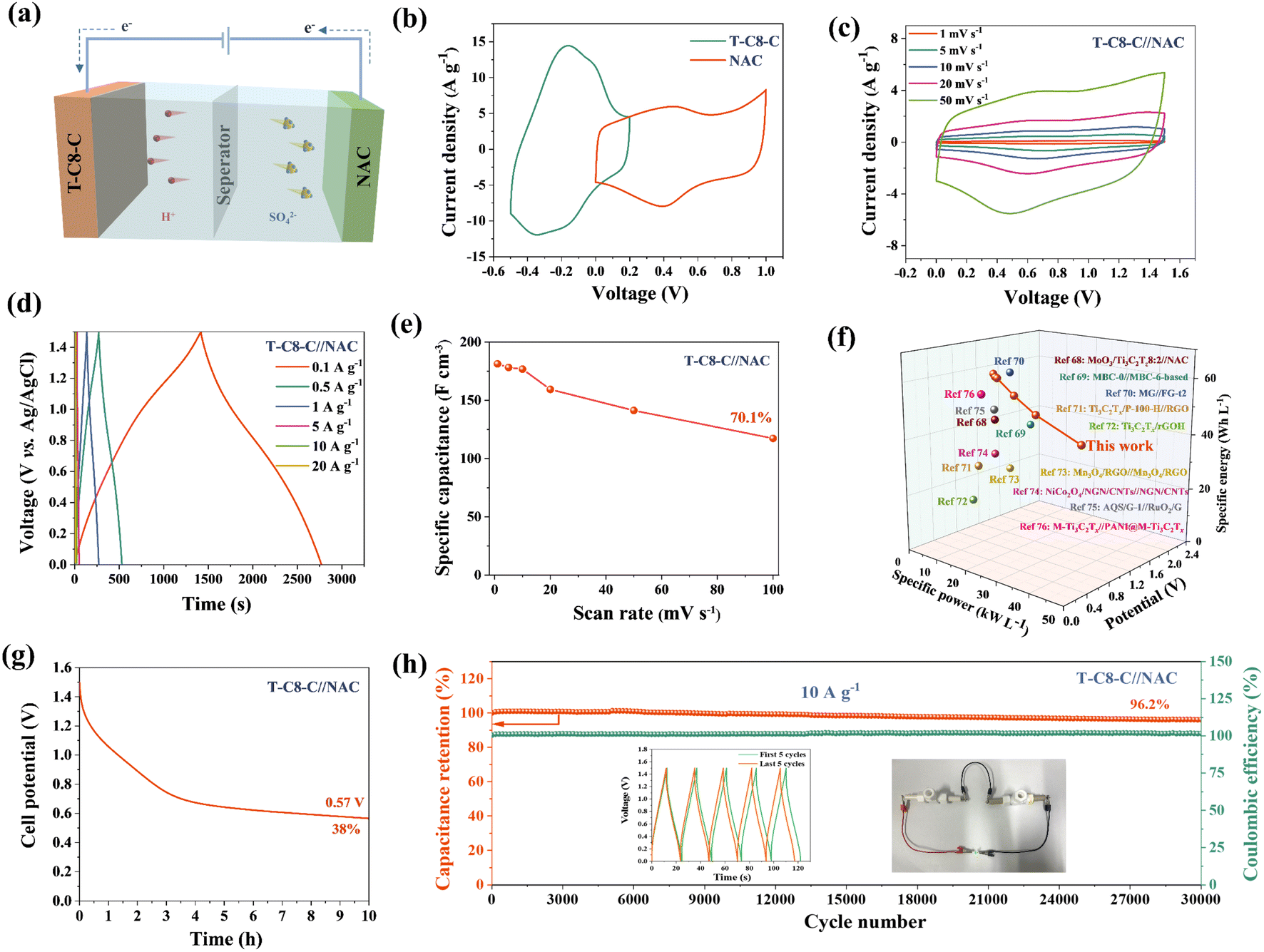 | ||
| Fig. 6 Asymmetric supercapacitor (ASC) electrochemical performance of T-C8-C//NAC. (a) Schematic representation of T-C8-C//NAC. (b) CV curves of T-C8-C and NAC electrode at 20 mV s−1. (c) CV curves at different scan rates. (d) GCD curves at different current densities. (e) Specific capacitance comparison at different current densities. (f) Ragone plots (volumetric values). (g) Self-discharge performance. (h) Cycling performance at 10 A g−1 for 30000 cycles (insets show GCD curves of the first and last 5 cycles and photographs showing the ASC device to light up a green LED). | ||
Conclusion
MXene-based freestanding electrodes with high SC performance via a “sacrificial cations” method combined with an electrochemistry-driven cation intercalation and calcination process have been designed. As a result, the MXene nanosheet restacking is effectively addressed and the surface terminations are modified. When applied as the electrodes for SCs in a three-electrode system, the freestanding T-C8-C electrodes show high volumetric capacitances, excellent rate capability, and impressive cycling stability. Furthermore, the T-C8-C//NAC asymmetric SCs exhibit extraordinary volumetric energy densities (56.7 W h L−1 at 0.15 kW L−1, and 36.7 W h L−1 at 30.1 kW L−1), and excellent cycling stability with stable performance of over 30000 cycles. Due to its straightforward fabrication process and remarkable electrochemical performance, we believe this design strategy can find widespread application in other 2D materials, enabling the fabrication of SCs with high energy densities and power densities.
Experimental methods
With the exception of dodecyl-trimethylammonium (Macklin Biochemical company, China), all chemical agents were purchased from Bidepharm (China) without any further treatment.Preparation of Ti3C2Tz films
The procedure for synthesizing Ti3C2Tz freestanding films is detailed in our prior research.42 To begin, lab-made Ti3AlC2 (∼48 μm) was introduced into a solution of HCl and LiF for 24 h at 40 °C. Next, the as-obtained mixture was washed with deionized (DI) water, followed by ultrasonication and centrifugation to obtain Ti3C2Tz suspensions. Subsequently, the Ti3C2Tz suspension was filtered through a membrane (Celgard 3501, with a pore size of 0.22 mm) by using a Buchner funnel equipped with a sand core (40 mm in diameter). Finally, the membrane was freeze-dried for 24 hours, after which the freestanding Ti3C2Tz films peeled off from the filter membrane.Preparation of Ti3C2Tz-CX (T-CX) and Ti3C2Tz-CX-C (T-CX-C) films
To accommodate the glassy carbon electrode test, Ti3C2Tz film was punched into discs with a 6 mm diameter. Initially, 1 M alkylammonium salts aqueous solutions were papered as electrolytes. The employed alkylammonium salts have a formula of [(CH3)3NR]+, where R represents CnH2n+1. The carbon content in R was named C4, C8, C12 and C16, corresponding to butyl-, octyl-, dodecyl- and hexadecyl-trimethylammonium, respectively. Next, Ti3C2Tz discs were used as working electrodes, platinum (Pt) was used as counter electrodes, and Ag/AgCl in saturated KCl was used as reference electrodes. In order to prevent fragmentation of the Ti3C2Tz discs in the electrolytes, they were fixed with carbon paper on both sides. Subsequently, organic cations with different chain lengths were intercalated into the Ti3C2Tz layer using a cyclic voltammetry (CV) program in the electrochemical workstation (CHI 660E, China). The scan rate was 20 mV s−1 from −0.8 V to 0.4 V (vs. Ag/AgCl) and was cycled 50 times. The obtained T-CX discs were washed with DI water and dried naturally at room temperature for 24 h. To obtain the T-CX-C electrodes, the T-CX discs were calcinated at 400 °C for 2 h at a temperature increase rate of 5 °C min−1 under an Ar gas atmosphere with a flow of 100 mL min−1. For comparison, pure Ti3C2Tz films were also calcined to obtain T-C electrodes.Preparation of AC and NAC films
The preparation method refers to previous work.77 Briefly, the N source (urea) and activated carbon (AC) solutions were thoroughly mixed. Then it was transferred to a Teflon-lined stainless-steel autoclave and the hydrothermal reaction was held at 180 °C for 24 h. Next, the reaction products were cleaned by centrifuge and dried to obtain nitrogen-doped AC (NAC).To make freestanding electrodes for AC or NAC, the powder was mixed with Poly(tetrafluoroethylene) (PTFE) in a mass ratio of 9:1. Then DI water was added to make the mixture into a slurry which was rolled into a flat film and dried at 70 °C for 24 h. The AC or NAC freestanding films were punched into 6 mm discs to be used as counter or positive electrodes.
Materials characterizations
XRD patterns were recorded by a DX-2700BH (Haoyuan) diffractometer with Cu Kα radiation in the range of 2θ = 3°–70°. SEM images were obtained using a Nova Nano SEM450 microscope with an acceleration voltage of 15 kV. TEM and HRTEM images were acquired by a Talos F200X microscope with an acceleration voltage of 200 kV. XPS spectra were recorded by a Thermo Scientific K-Alpha spectrometer with Al Kα radiation. Raman spectra were obtained by a Thermo Fisher Renishaw inVia spectrometer with λ = 532 nm. TG measurements were performed on STA 449F5 in the dry nitrogen atmosphere at the heating rates of 5 °C min−1. FTIR spectra were recorded at room temperature by using a Nicolet iS10 spectrometer. TG-MS patterns were recorded by thermo plus EVO2 in an Ar atmosphere at the heating rates of 10 °C min−1.Electrochemical measurements
The 6 mm Ti3C2Tz-based discs were tested directly in a plastic Swagelok T-shaped cell. Before testing, it was necessary to pre-cycle all electrodes at a scan rate of 20 mV s−1 for 50 cycles to stabilize performance. For all testing, glassy carbon (CHI Instruments, China) was used as current collectors, polypropylene membranes (Celgard 3501) were used as separators, and 3 M H2SO4 was used as electrolytes. All electrochemical measurement data were recorded on a CHI 660E electrochemical workstation, except the long-cycling testing and self-discharge testing, which was recorded on the SP-150 electrochemical workstation (Bio-logic). Techniques employed included cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS). The frequency range for EIS measurements was from 10 mHz to 100 kHz. All electrochemical measurements were conducted at RT.The volumetric capacitance (Cv) of the electrode was calculated from the CV curves following eqn (3)–(5):
| ρ = m/Ad | (3) |
 | (4) |
| Cv = ρCg | (5) |
The volumetric energy (Ev) and power densities (Pv) were calculated assuming (6) and (7):
| Ev = CvU2/7.2 | (6) |
| Pv = 3.6 × Evv/U | (7) |
The self-discharge testing was conducted by first holding the cells at specific voltages for 0.5 h and then tracking the cell voltage as a function of time for 10 h.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52301263, 52171033, and U23A20574).References
- M. S. Ziegler, J. M. Mueller, G. D. Pereira, J. Song, M. Ferrara, Y. M. Chiang and J. E. Trancik, Joule, 2019, 3, 2134–2153 CrossRef.
- N. Kittner, F. Lill and D. M. Kammen, Nat. Energy, 2017, 2, 1–6 CrossRef.
- K. Naoi, S. Ishimoto, J. Miyamoto and W. Naoi, Energy Environ. Sci., 2012, 5, 9363–9373 RSC.
- K. Naoi, W. Naoi, S. Aoyagi, J. Miyamoto and T. Kamino, Acc. Chem. Res., 2013, 46, 1075–1083 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, Acs Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- A. K. Tareen, K. Khan, M. Iqbal, S. Golovynskyi, Y. Zhang, A. Mahmood, N. Mahmood, J. Long, A. Al-Ghamdi, C. Li and H. Zhang, Mater. Today Chem., 2022, 26, 101205 CrossRef CAS.
- K. Khan, A. K. Tareen, M. Iqbal, I. Hussain, A. Mahmood, U. Khan, M. F. Khan, H. Zhang and Z. J. Xie, J. Mater. Chem. A, 2023, 11, 19764–19811 RSC.
- F. C. Cao, Y. Zhang, H. Q. Wang, K. Khan, A. K. Tareen, W. J. Qian, H. Zhang and H. Ågren, Adv. Mater., 2022, 34, 2107554 CrossRef CAS PubMed.
- D. B. Xiong, X. F. Li, Z. M. Bai and S. G. Lu, Small, 2018, 14, 1703419 CrossRef PubMed.
- X. W. Hu, Q. C. Zhang, N. Gong, X. F. Chen, L. Wang, W. C. Peng, Y. Li, F. B. Zhang and X. B. Fan, Energy Storage Mater., 2022, 50, 802–809 CrossRef.
- Q. Tang, Z. Zhou and P. W. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- M. R. Lukatskaya, S. M. Bak, X. Q. Yu, X. Q. Yang, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1500589 CrossRef.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- Y. Guo, T. R. Wang, Q. Yang, X. L. Li, H. F. Li, Y. K. Wang, T. P. Jiao, Z. D. Huang, B. B. Dong, W. J. Zhang, J. Fan and C. Y. Zhi, Acs Nano, 2020, 14, 9089–9097 CrossRef CAS PubMed.
- T. Zhong, Z. B. Yu, R. H. Jiang, Y. P. Hou, H. J. Chen, L. Ding, C. F. Lian and B. S. Zou, Sol. RRL, 2022, 6, 2100863 CrossRef CAS.
- S. T. Xiao, Y. J. Zheng, X. Z. Wu, M. Zhou, X. Rong, L. Y. Wang, Y. J. Tang, X. K. Liu, L. Qiu and C. Cheng, Small, 2022, 18, 2203281 CrossRef CAS PubMed.
- L. Li, J. Meng, X. R. Bao, Y. P. Huang, X. P. Yan, H. L. Qian, C. Zhang and T. X. Liu, Adv. Energy Mater., 2023, 13, 2203683 CrossRef CAS.
- Z. Otgonbayar, S. H. Y. Yang, I. J. Kim and W. C. Oh, Chem. Eng. J., 2023, 472, 144801 CrossRef CAS.
- N. S. Shaikh, S. B. Ubale, V. J. Mane, J. S. Shaikh, V. C. Lokhande, S. Praserthdam, C. D. Lokhande and P. Kanjanaboos, J. Alloys Compd., 2022, 893, 161998 CrossRef CAS.
- Y. J. Luo, W. X. Que, X. Q. Bin, C. J. Xia, B. S. Kong, B. W. Gao and L. B. Kong, Small, 2022, 18, 2201290 CrossRef CAS PubMed.
- Y. Yoon, M. Lee, S. K. Kim, G. Bae, W. Song, S. Myung, J. Lim, S. S. Lee, T. Zyung and K. S. An, Adv. Energy Mater., 2018, 8, 1703173 CrossRef.
- M. Cai, X. C. Wei, H. F. Huang, F. L. Yuan, C. Li, S. K. Xu, X. Q. Liang, W. Z. Zhou and J. Guo, Chem. Eng. J., 2023, 458, 141338 CrossRef CAS.
- Y. P. Tian, M. M. Ju, Y. J. Luo, X. Q. Bin, X. J. Lou and W. X. Que, Chem. Eng. J., 2022, 446, 137451 CrossRef CAS.
- K. Nasrin, V. Sudharshan, K. Subramani, M. Karnan and M. Sathish, Small, 2022, 18, 2106051 CrossRef CAS PubMed.
- J. Li, X. T. Yuan, C. Lin, Y. Q. Yang, L. Xu, X. Du, J. L. Xie, J. H. Lin and J. L. Sun, Adv. Energy Mater., 2017, 7, 1602725 CrossRef.
- Z. Ling, C. E. Ren, M. Q. Zhao, J. Yang, J. M. Giammarco, J. S. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS PubMed.
- M. Ghidiu, S. Kota, J. Halim, A. W. Sherwood, N. Nedfors, J. Rosen, V. N. Mochalin and M. W. Barsoum, Chem. Mater., 2017, 29, 1099–1106 CrossRef CAS.
- K. Liang, R. A. Matsumoto, W. Zhao, N. C. Osti, I. Popov, B. P. Thapaliya, S. Fleischmann, S. Misra, K. Prenger, M. Tyagi, E. Mamontov, V. Augustyn, R. R. Unocic, A. P. Sokolov, S. Dai, P. T. Cummings and M. Naguib, Adv. Funct. Mater., 2021, 31, 2104007 CrossRef CAS.
- Y. P. Yan, H. M. Tang, Z. Qu, W. L. Zhang, O. G. Schmidt and M. S. Zhu, Adv. Mater. Technol., 2023, 8, 2200466 CrossRef CAS.
- J. Y. Wan, S. D. Lacey, J. Q. Dai, W. Z. Bao, M. S. Fuhrer and L. B. Hu, Chem. Soc. Rev., 2016, 45, 6742–6765 RSC.
- Z. J. Li, J. Dai, Y. R. Li, C. L. Sun, A. L. Meng, R. F. Cheng, J. Zhao, M. M. Hu and X. H. Wang, Nano Res., 2022, 15, 3213–3221 CrossRef CAS.
- S. L. Lyu, C. X. Guo, J. N. Wang, Z. J. Li, B. Yang, L. C. Lei, L. P. Wang, J. P. Xiao, T. Zhang and Y. Hou, Nat. Commun., 2022, 13, 6171 CrossRef CAS PubMed.
- M. S. Stark, K. L. Kuntz, S. J. Martens and S. C. Warren, Adv. Mater., 2019, 31, 1808213 CrossRef PubMed.
- M. Q. Zhao, X. Q. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. X. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef PubMed.
- D. Seo, M. R. Kim, J. K. Song, E. Kim, J. Koo, K. C. Kim, H. Han, Y. Lee and C. W. Ahn, Chemelectrochem, 2022, 9, E202101344 CrossRef CAS.
- T. T. Li, L. P. Liang, Z. Y. Chen, J. L. Zhu and P. K. Shen, Chem. Eng. J., 2023, 474, 145970 CrossRef CAS.
- R. Ma, X. J. Zhang, J. T. Zhuo, L. Y. Cao, Y. T. Song, Y. J. Yin, X. F. Wang, G. W. Yang and F. Yi, Acs Nano, 2022, 16, 9713–9727 CrossRef CAS PubMed.
- X. Zhao, Z. Wang, J. Dong, T. Huang, Q. H. Zhang and L. L. Zhang, J. Power Sources, 2020, 470, 228356 CrossRef CAS.
- W. F. Liu, Y. F. Zheng, Z. Zhang, Y. N. Zhang, Y. H. Wu, H. X. Gao, J. Su and Y. H. Gao, J. Power Sources, 2022, 521, 230965 CrossRef CAS.
- X. D. Yin, W. Zheng, H. F. Tang, P. G. Zhang and Z. M. Sun, Nanoscale, 2023, 15, 10437–10446 RSC.
- H. S. Li, K. Xu, P. H. Chen, Y. Y. Yuan, Y. Qiu, L. G. Wang, L. Zhu, X. G. Wang, G. H. Cai, L. M. Zheng, C. Dai, D. Zhou, N. A. Zhang, J. X. Zhu, J. L. Xie, F. H. Liao, H. L. Peng, Y. Peng, J. Ju, Z. F. Lin and J. L. Sun, Natl. Sci. Rev., 2022, 9, Nwac079 CrossRef CAS PubMed.
- X. B. Zang, J. L. Wang, Y. J. Qin, T. Wang, C. P. He, Q. G. Shao, H. W. Zhu and N. Cao, Nano-Micro Lett., 2020, 12, 77 CrossRef CAS PubMed.
- J. Z. Chen, H. Chen, M. F. Chen, W. J. Zhou, Q. H. Tian and C. P. Wong, Chem. Eng. J., 2022, 428, 131380 CrossRef CAS.
- M. Naguib, O. Mashtalir, M. R. Lukatskaya, B. Dyatkin, C. F. Zhang, V. Presser, Y. Gogotsi and M. W. Barsoum, Chem. Commun., 2014, 50, 7420–7423 RSC.
- T. Z. Zhou, C. Wu, Y. L. Wang, A. P. Tomsia, M. Z. Li, E. Saiz, S. L. Fang, R. H. Baughman, L. Jiang and Q. F. Cheng, Nat. Commun., 2020, 11, 2077 CrossRef CAS PubMed.
- H. Y. Wang, X. B. Sun, S. H. Yang, P. Y. Zhao, X. J. Zhang, G. S. Wang and Y. Huang, Nano-Micro Lett., 2021, 13, 1–15 CrossRef PubMed.
- J. Xu, X. H. Hu, X. H. Wang, X. Wang, Y. F. Ju, S. H. Ge, X. L. Lu, J. N. Ding, N. Y. Yuan and Y. Gogotsi, Energy Storage Mater., 2020, 33, 382–389 CrossRef.
- M. Boota and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1802917 CrossRef.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1703043 CrossRef.
- P. Zhang, J. P. Li, D. Y. Yang, R. A. Soomro and B. Xu, Adv. Funct. Mater., 2023, 33, 2209918 CrossRef CAS.
- H. J. Xu, J. X. Fan, H. Su, C. F. Liu, G. Chen, Y. Dall'agnese and Y. Gao, Nano Lett., 2023, 23, 283–290 CrossRef CAS PubMed.
- W. Zheng, J. Halim, P. O. Å. Persson, J. Rosen and M. W. Barsoum, Mater. Rep.: Energy, 2022, 2, 100078 CAS.
- Y. P. Tian, W. X. Que, Y. Y. Luo, C. H. Yang, X. T. Yin and L. B. Kong, J. Mater. Chem. A, 2019, 7, 5416–5425 RSC.
- M. R. Lukatskaya, S. Kota, Z. F. Lin, M. Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P. L. Taberna, M. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 1–6 Search PubMed.
- J. Tang, T. S. Mathis, N. Kurra, A. Sarycheva, X. Xiao, M. N. Hedhili, Q. Jiang, H. N. Alshareef, B. M. Xu, F. Pan and Y. Gogotsi, Angew. Chem., Int. Ed., 2019, 58, 17849–17855 CrossRef CAS PubMed.
- F. Yi, H. Y. Ren, K. R. Dai, X. F. Wang, Y. Z. Han, K. X. Wang, K. Li, B. L. Guan, J. Wang, M. Tang, J. Y. Shan, H. Yang, M. S. Zheng, Z. You, D. Wei and Z. F. Liu, Energy Environ. Sci., 2018, 11, 2016–2024 RSC.
- C. J. Shi, Z. J. Liu, Z. Tian, D. Li, Y. J. Chen, L. Guo and Y. Z. Wang, Appl. Surf. Sci., 2022, 596, 153632 CrossRef CAS.
- Y. M. Wang, X. Wang, X. F. Li, X. L. Li, Y. Liu, Y. Bai, H. H. Xiao and G. H. Yuan, Adv. Funct. Mater., 2021, 31, 2008185 CrossRef CAS.
- S. K. Hwang, S. J. Patil, N. R. Chodankar, Y. S. Huh and Y. K. Han, Chem. Eng. J., 2022, 427, 131854 CrossRef CAS.
- H. Shao, K. Xu, Y. C. Wu, A. Iadecola, L. Y. Liu, H. Y. Ma, L. T. Qu, E. Raymundo-Piñero, J. X. Zhu, Z. F. Lin, P. L. Taberna and P. Simon, ACS Energy Lett., 2020, 5, 2873–2880 CrossRef CAS.
- Y. Y. Zhu, S. H. Zheng, P. F. Lu, J. X. Ma, P. Das, F. Su, H. M. Cheng and Z. S. Wu, Natl. Sci. Rev., 2022, 9, Nwac024 CrossRef CAS PubMed.
- P. Salles, D. Pinto, K. Hantanasirisakul, K. Maleski, C. E. Shuck and Y. Gogotsi, Adv. Funct. Mater., 2019, 29, 1809223 CrossRef.
- H. Li, K. Fan, P. Xiong, H. M. Zhou, Z. Z. Lin, K. Y. Tao, T. C. Liu, X. Y. Guo, Y. Zhu, L. Y. C. Zhuang, W. Han, C. Yang, Y. Liu, M. M. J. Li, M. W. Fu, J. H. Wang and H. T. Huang, J. Mater. Chem. A, 2024, 12, 3449–3459 RSC.
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS.
- X. P. Mu, D. S. Wang, F. Du, G. Chen, C. Z. Wang, Y. J. Wei, Y. Gogotsi, Y. Gao and Y. Dall'agnese, Adv. Funct. Mater., 2019, 29, 1902953 CrossRef.
- W. Zheng, J. Halim, A. El Ghazaly, A. S. Etman, E. N. Tseng, P. O. A. Persson, J. Rosen and M. W. Barsoum, Adv. Sci., 2021, 8, 2003656 CrossRef CAS PubMed.
- Y. H. Cui, Q. S. Shi, Z. Liu, J. C. Lv, C. Wang, X. B. Xie and S. H. Zhang, Chem. Eng. J., 2023, 472, 144701 CrossRef CAS.
- Y. T. Hu, C. Guan, Q. Q. Ke, Z. F. Yow, C. W. Cheng and J. Wang, Chem. Mater., 2016, 28, 7296–7303 CrossRef CAS.
- L. Li, N. Zhang, M. Y. Zhang, X. T. Zhang and Z. G. Zhang, Dalton Trans., 2019, 48, 1747–1756 RSC.
- A. M. Navarro-Suarez, K. L. Van Aken, T. Mathis, T. Makaryan, J. Yan, J. Carretero-Gonzalez, T. Rojo and Y. Gogotsi, Electrochim. Acta, 2018, 259, 752–761 CrossRef CAS.
- T. Xiong, W. S. V. Lee, X. L. Huang and J. M. Xue, J. Mater. Chem. A, 2017, 5, 12762–12768 RSC.
- S. H. Yue, H. Tong, L. Lu, W. W. Tang, W. L. Bai, F. Q. Jin, Q. W. Han, J. P. He, J. Liu and X. G. Zhang, J. Mater. Chem. A, 2017, 5, 689–698 RSC.
- L. N. Zhang, D. L. Han, Y. Tao, C. J. Cui, Y. Q. Deng, X. M. Dong, W. Lv, Z. F. Lin, S. C. Wu, Z. Weng and Q. H. Yang, J. Mater. Chem. A, 2020, 8, 461–469 RSC.
- K. Li, X. H. Wang, S. Li, P. Urbankowski, J. M. Li, Y. X. Xu and Y. Gogotsi, Small, 2020, 16, 1906851 CrossRef CAS PubMed.
- W. Zheng, J. Halim, A. S. Etman, A. El Ghazaly, J. Rosen and M. W. Barsoum, Electrochim. Acta, 2021, 370, 137665 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07867a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |