
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile electrochemical manufacturing of mixed metal sulfide/N-doped rGO composites as bifunctional catalysts for high power rechargeable Zn–air batteries†
Jaime S.
Sanchez
ag,
Zhenyuan
Xia
 *bf,
Keyvan
Mirehbar
a,
Sankar
Sasidharan
b,
S. Assa
Aravindh
c,
Andrea
Liscio
d,
Jinhua
Sun
b,
Meganne
Christian
e,
Jesus
Palma
a,
Vincenzo
Palermo
*bf and
Rebeca
Marcilla
*a
*bf,
Keyvan
Mirehbar
a,
Sankar
Sasidharan
b,
S. Assa
Aravindh
c,
Andrea
Liscio
d,
Jinhua
Sun
b,
Meganne
Christian
e,
Jesus
Palma
a,
Vincenzo
Palermo
*bf and
Rebeca
Marcilla
*a
aElectrochemical Processes Unit, IMDEA Energy Institute, Avenida Ramón de la Sagra 3, Móstoles, Madrid, 28935, Spain. E-mail: rebeca.marcilla@imdea.org
bIndustrial and Materials Science, Chalmers University of Technology, Hörsalsvägen 7B, Göteborg, 41296, Sweden. E-mail: zhenyuan@chalmers.se
cSustainable Chemistry & MME, Faculty of Technology, University of Oulu, 90014, Oulu, Finland
dConsiglio Nazionale delle Ricerche, Istituto per la Microelettronica e Microsistemi (IMM) – Roma Unit, Via del fosso del cavaliere 100, Roma, 00133, Italy
eConsiglio Nazionale delle Ricerche, Istituto per la Microelettronica e i Microsistemi (IMM), Via Gobetti 101, Bologna, 40129, Italy
fConsiglio Nazionale delle Ricerche, Istituto per la Sintesi Organica e la Fotoreattività (ISOF), Via Gobetti 101, Bologna, 40129, Italy. E-mail: palermo@isof.cnr.it
gR&D Department, China Three Gorges (Europe) S.A, C. del Príncipe de Vergara, 112, Planta 7, Madrid, 28002, Spain
First published on 19th March 2024
Abstract
The development of rechargeable zinc–air batteries requires air cathodes capable of performing both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) with high performance and an extended operational lifespan. Here, we present a cost-effective and versatile electrochemical method for the direct assembly of such electrocatalysts, consisting of nitrogen-doped reduced graphene oxide (NrGO) and mixed transition metal sulfides (NiCoMnSx or NCMS). To this end, we use a small electric bias to electro-deposit both NrGO and NCMS directly on conductive graphene foam, resulting in a perfect porous network and two interpenetrated paths for the easy transport of electrons and ions. The NCMS/NrGO composite shows one of the highest limiting currents reported so far for a non-noble metal catalyst. Additionally, it exhibits outstanding bifunctional performance for the ORR/OER, superior to both mixed transition metal compounds and noble metals from previous reports. Thus, it serves as a highly efficient air cathode for practical zinc–air batteries featuring high power densities (124 mW cm−2) and long catalyst durability (1560 cycles, around 260 h). We attribute the excellent performance to the synergistic effect between hetero-structured metallic sites and nitrogen dopants. Our approach can be used for preparing efficient zinc–air cathodes on conductive 3D carbon substrates with arbitrary shapes and good performance.
1. Introduction
The ever-growing energy demand and environmental concerns have attracted unprecedented interest in developing sustainable energy storage and conversion systems capable of satisfying the current and near-future energy needs without damaging our planet. Lithium-ion (Li-ion) batteries have emerged as the primary energy storage choice for portable electronics and one of the most viable options for electric vehicles (EVs) and grid-scale energy storage.1 Regardless of the commercial success, current lithium-ion technologies still face challenges such as low energy density (<350 W h kg−1), relatively high cost (currently $150 kW−1 h−1) and inherent safety risk (due to flammable organic electrolytes). Several possible alternative energy storage and conversion systems have been suggested, focusing on utilizing eco-friendly aqueous electrolytes. These options include rechargeable alkaline batteries,2 water electrolyzers,3 fuel-cells,4 and metal–air batteries.5 Among metal–air battery technologies, rechargeable zinc–air batteries (ZABs) have spurred extensive attention owing to their remarkable features. They possess a high theoretical energy density (1086 W h kg−1, surpassing that of Li-ion technology), excellent safety (aqueous electrolytes are employed), natural abundance of zinc reserves (ranked 24th in terms of material abundance on earth), peculiar open structure (consuming atmospheric oxygen as a cathodic active material), and lower cost (currently <$100 kW−1 h−1) than other current technologies.6,7 However, current ZABs suffer from poor cycling stability, moderate round-trip efficiencies, and inferior charging/discharging current densities to those of LIBs. These drawbacks are mainly due to the lack of high efficiency and durable bifunctional electrocatalysts, which in turn catalyze the oxygen evolution reaction (OER) during charging and the oxygen reduction reaction (ORR) during the discharging process.8,9 To date, Pt-based materials remain the most commonly employed commercial catalysts for the ORR, while RuO2 and IrO2 are widely used as catalysts for the OER. However, the high cost and scarcity, poor bifunctional oxygen activity and inferior stability of these noble metals hindered the widespread commercialization of rechargeable ZABs.10 Therefore, the pursuit of highly active, durable, and cost-effective bifunctional electrocatalysts (ORR/OER) derived from earth-abundant elements is imperative.11Transition metal compounds (TMCs) including oxides,12,13 hydroxides,14 phosphates,15 nitrides16 and sulfides17 represent a class of promising non-precious metal oxygen electrocatalysts due to their inherent advantages of abundant resources, low price and eco-friendliness. Previous studies showed that, compared to monovalent TMCs, multi-valent/mixed transition metals deliver superior performance and increased structural stability, providing chemisorption sites with donor–acceptor characteristics for the reversible adsorption of oxygen molecules.18 The coupling between various metals enhances the electronic occupancy status and thus raises the electrocatalytic activity compared to monometallic materials, as proved by advanced characterizations such as electron paramagnetic resonance (EPR) and X-ray absorption spectroscopy studies.19,20 Furthermore, recent studies report that hybridizing mixed TMCs with heteroatom-doped (e.g., nitrogen) carbonaceous materials produces enhanced bifunctional ORR/OER properties. This enhancement is attributed to fast interfacial electron transfer and the synergistic effects, arising from chemical coupling between multi-valent metallic atoms and N-doped carbons.21,22 Additionally, carbon-based materials such as graphene can enhance electrical properties and mitigate the volume expansion of TMCs, thereby increasing the lifespan of the catalyst. In previous studies, we effectively synthesized different TMCs/N-doped carbon composites, designed to enhance the catalytic activity, that were successfully used as bifunctional air cathodes in ZABs.23–25 More recently, rare-earth materials, mainly the lanthanide ones, also show their huge potential in OER/ORR catalyst materials due to their unique electronic configuration with valence 4f–3d–2p orbital coupling. The combination of rare-earth oxides with transition metals, such as Pd-Gd2O3 carbon composites, has been proved to produce excellent cathodes for ZABs.20,26,27
However, these electrocatalyst materials are commonly synthesized via sol–gel, sputtering, microemulsion, spray pyrolysis or hydrothermal methods, resulting in random and uncontrolled morphology structures. Furthermore, their fabrication often requires high-temperature treatments, time-consuming and multistep processes, and the addition of substantial amounts of conductive agents during ink preparation. These inks are typically coated on a gas diffusion layer (GDL), a practice that is undesirable for industrial applications. In addition, those conventional methods commonly imply the use of ink additives and generate impurities, which can merge in the catalyst reducing its catalytic activity. Electrocatalysis is a surface-sensitive process, and thus catalysts deposited on hierarchical, porous nanostructures can give improved performance by increasing the specific surface area as well as the homogeneity and the density of catalytic sites.28
Compared to the above traditional synthetic approaches, electrochemical deposition techniques hold great promise in one step safe and green synthesis of TMCs directly onto the target current collector substrates. Electrochemical deposition, or electrodeposition, is a process that involves passing an electric current through an electrolyte solution containing ions of the material intended for deposition. It has been widely used in various industries like electronics, automotive, and aerospace for metal plating, surface finishing, and the functional coating of thin films with specific properties like conductivity and corrosion resistance. This technique provides several benefits such as precise control of the amount of deposited coating by easily adjusting the plating parameters in aqueous solution, rapid synthesis of uniformly dispersed electrocatalysts without the need for any binding agents, and the potential for simple and scalable synthesis of nanostructured composite materials under ambient conditions.29
In previous studies, we effectively used electrodeposition to achieve functional coating of structural substrates with TMCs and graphene additives. Notably, this approach was successfully applied to carbon fibers,30 as well as 3-dimensional foams,31 allowing us to achieve precise control over the architecture, composition, and properties of the resulting materials. Subsequently, these compounds were utilized as electrode materials for asymmetric micro-supercapacitors,32 Li-ion batteries,31 or rechargeable alkaline batteries,2 demonstrating high performances.
Herein, we use this electrochemical approach for a more challenging system, i.e. air cathodes for rechargeable ZABs. We successfully prepared Zn–air-cathodes made of NiCoMnSx (NCMS) nanosheets and nitrogen-doped reduced graphene oxide (NrGO) (NCMS/NrGO) deposited onto a macroscopic 3D graphene foam. In some of our previous studies we already demonstrated that N-doping of rGO layers can modify the electron density of carbon atoms, positively influencing the adsorption of O2 and the cleavage of O–O bonds in the ORR process.23,24 The synthetic strategy combines two consecutive steps consisting of electrophoretic and cyclic voltammetry deposition (Scheme 1). Electrophoresis is a purely physical process, with the motion of dispersed particles relative to a fluid under the influence of a spatially electric field (Scheme S1a†). It allows uniform and conformal coating of two-dimensional nanosheets like NrGO onto arbitrary three-dimensional substrates with high efficiency.33 On the other hand, cyclic voltammetry deposition is driven by electrochemical reactions, allowing for the direct growth of mixed metal sulfides from the electrochemically generated sulfur anions and dissolved metal cations onto any conductive substrates (Scheme S1b†).2,30 Our two-step deposition approach enables the facile deposition of mixed-sulfide compounds/2D material composites with tailored stoichiometry and enhanced adhesion of the carbon/metal sulfide interface. We then obtain large electroactive surfaces/interfaces and plenty of exposed catalytic active sites in the form of NCMS nanosheets, exhibiting higher ORR/OER activities than those traditionally synthesized at higher temperature via hydro/solvothermal methods. Moreover, by encapsulating NCMS nanostructures with N-doped graphene layers, a synergistic effect was realized, providing more active sites and enhancing interfacial charge transfer.34 Consequently, the observed synergy culminated in exceptional electrochemical performance in both the ORR and OER (ΔE of 0.75 V). This composite sample surpassed even the investigated state-of-the-art bifunctional noble-metal catalyst (PtRuC 20%) and distinguished itself as one of the most efficient bifunctional catalysts reported to date. When assembled in practical rechargeable ZABs with NCMS/NrGO-GF as the air cathode catalyst, it endows them with a high power density of 124 mW cm−2, surpassing the performance of the investigated state-of-the-art bifunctional noble-metal catalyst (PtRuC 20%) and demonstrating a long-life catalyst durability.
 | ||
| Scheme 1 Schematic illustration of the preparation process for NCMS/NrGO graphene foam air-cathode architectures. | ||
2. Results and discussion
2.1 Structural and morphological investigations
Scheme 1 illustrates the preparation procedure for the free-standing NCMS/NrGO-GF air cathode. We first fabricated a 3D graphene foam using CVD on a sacrificial nickel template. After etching away the nickel in an acid solution, we obtained an all-graphene free-standing substrate, mimicking the shape of the interconnected 3D structure of the nickel template (Fig. S1†). Then we used electrophoretic deposition (EPD) to coat NrGO from the DMF solution onto the graphene foam (GF), and cyclic voltammetry electrodeposition (CV) to deposit NCMS from a water/EtOH co-solvent solution (Scheme S1†). Fig. 1 shows the scanning electron microscope (SEM) images of the nanostructured morphology of the samples synthesized. Fig. 1a shows that large amounts of NCMS nanosheets are uniformly coated and vertically aligned on the surface of NrGO all over the graphene foam. The phenomenon of vertically oriented growth of sulfides was consistent with other work with binary metal-sulfides although the exact mechanism underlying this phenomenon remains not fully understood.35,36 The interconnected NrGO nanosheets form a three-dimensional (3D) hierarchical network (inset in Fig. 1a) with vertical nanosheets spaced at ∼50–100 nm, ensuring proper accessibility of the electrolyte to the metal active sites, to ensure optimal performance of the catalyst for the oxygen reactions.37 In addition, some nanosheets seem to grow on others (see the white arrow in Fig. 1a), which also reveals the interconnected feature of the NCMS hierarchical structure. | ||
| Fig. 1 SEM images of (a) NCMS/NrGO (coated onto GF); inset shows the SEM image obtained with high magnification. (b) Cross-sectional SEM image of NCMS/NrGO-GF showing a schematic representation of the different layers in the layer-by-layer structure corresponding to GF, NrGO and NCMS indicated by false coloring. The original SEM image with no color is available in Fig. S3a.† (c) Cross-sectional SEM image of the NCMS/NrGO composite on GF (marked by the yellow arrow) (d) TEM micrograph of the NCMS/NrGO composite showing anchored sulfide nanosheets on thin NrGO layers, and (e) corresponding HR-TEM image. (f) Line scanning EDS profile of the NCMS/NrGO-GF layer-by-layer sample prepared via cryogenic fracture. | ||
We used EDS elemental mapping to define the chemical composition of the NCMS/NrGO-GF sample, observing the presence of Ni, Co, Mn, S, N and C uniformly distributed throughout the composite sample (Fig. S2†). To demonstrate that the Mn, Co, and Ni mixed metal sulfides are deposited on top of thin NrGO layers, we recorded cross-sectional FE-SEM images of the NrGO and NCMS/NrGO samples deposited on GF (Fig. 1b, see also ESI, Fig. S3a and b†). Fig. S3b† demonstrates that, following the EPD of NrGO, the surface of the graphene foam is entirely coated with thin NrGO sheets, as identified by the increased surface roughness. The average thickness of the NrGO layers was less than 60 nm. The cross-sectional SEM image of NCMS/NrGO-GF in Fig. 1c and the corresponding SEM-energy dispersive X-ray spectrometry (EDS) line profile mapping in Fig. 1f allow the relative elemental concentrations for each element along with its position in the multilayered structure to be distinguished. It is noticed that the peak width in the EDS line profile cannot be used to accurately estimate the NrGO thickness. This discrepancy is much larger than the actual layer thickness due to the complex, tortuous shape of the foam, which hinders obtaining straight cross-section information. The transmission electron microscopy (TEM) image (Fig. 1d) clearly reveals that tiny interconnected nanosheets are randomly dispersed on the edges of a denser N-doped rGO nanoarchitecture matrix, without obvious detachments and aggregation. The HR-TEM image in Fig. 1e reveals lattice fringes with inter-planar distances of 0.186 nm, 0.261 nm and 0.259 nm, corresponding to the (511), (200) and (101) planes of the MTMS structure. For the sake of comparison, a control sample of NCMS directly grown by CV onto GF (no intermediate layer of NrGO) was prepared and tested. Fig. S3c† shows that the cathodic electrodeposition of MTMSs directly on GF results in an NCMS layer of ∼150–200 nm after 1 CV cycle, with some irregularities caused by the curved structure of the foam.30 As demonstrated in previous publications, initial attempts to deposit MTMSs directly on the GF surface proved unsuccessful, leading to inadequate adhesion of the NCMS material, with detached flakes or cracks.2 The preliminary coating of NrGO serves as a primer, interacting with metal sulfides owing to the oxygen functional groups on its surface. Additionally, NrGO acts as a binder and a conductive spacer, facilitating subsequent deposition of the NCMS layer.
The XRD pattern in Fig. S4a† shows well-defined peaks corresponding to the crystalline phase(s) of the NCMS/NrGO, NCMS and NrGO confirming the good quality of the coatings on GF. The control samples, produced by direct deposition of NCMS on graphene foam (NCMS-GF), exhibited diffraction peaks indexed to different crystallographic structures of cubic phases. Specifically, the (311), (400), (422), (440) and (511) peaks were associated with the CoNi2S4 (JCPDS 43-1477), NiCo2S4 (JCPDS 20-0782) or Co3S4 phase (JCPDS 47-1738), while the (101) peak was attributed to the Ni3S2 heazlewoodite phase (JCPDS 98-000-6248). In addition, the (200) and (222) peaks were identified with the α-MnS phase (JCPDS 88-2223).38–40 The diffraction peaks observed at 26°, 44°, and 55° in all samples were attributed to the graphitic carbon matrix used as a conductive substrate (JCPDS 12-0212).31 This polycrystallinity is noteworthy, as previous work has indicated the difficulty in obtaining highly crystalline samples through electrosynthesis methods. The observed polycrystallinity is notable, as it is consistent with the previous work demonstrating the difficulty in achieving highly crystalline samples through the electrodeposition method.41 Raman spectroscopy (Fig. S4b†) confirmed the successful formation of the NCMS/NrGO composite. Two peaks centered at 1357 cm−1 (D band) and 1582 cm−1 (G band), typical of graphitic materials, were observed in both the NrGO-GF control sample and the NCMS/NrGO composite sample. The D band is related to the disorder-induced breathing mode of carbon atoms, while the G band is a characteristic signature of the in-plane vibration of the sp2 hybridized carbon structure. We observed that the relative intensity of D and G bands in NCMS/NrGO is significantly lower than that of the NrGO sample. This is mainly due to the thick NCMS layer, which affects the measured Raman scattered light and results in a reduced intensity of the NrGO peaks underneath the NCMS film. Moreover, the vibrational modes of M–S (M = Ni, Co, or Mn) were observed in the range of 200 to 750 cm−1, corresponding to those detected in the NCMS-GF control sample.42–44 Overall, NCMS/NrGO-GF exhibited a combination of peaks with no significant changes compared with the individual components. Our observation confirms that the bulk structure of the two components remained intact during the formation of the composite. Brunauer–Emmett–Teller (BET) surface area values measured on NCMS/NrGO-GF, NCMS-GF and NrGO-GF were 132, 96 and 151 m2 g−1 respectively (Fig. S5a†), evidencing that NCMS/NrGO features a higher surface area than the NCMS control sample, facilitating infiltration of the electrolyte and enhancing the contact area during catalytic processes. Increasing the surface area corresponds to a narrowing of the pore size distribution – calculated by Barrett–Joyner–Halenda (BJH) analysis – and a corresponding decrease in the average pore diameter close to the experimental cut-off close to 4 nm (see Fig. S5b†).45 Micropores with the potential to host active sites can contribute to the majority of catalytic activity, while mesopores can effectively facilitate the transport of reactants and products to and from the catalytic active sites within the micropores. All three materials show mesoporosity (pore diameter within 2 nm and 50 nm). However, although the cut-off prevents the direct measurement, the presence of micropores (pore diameter < 2 nm) cannot be excluded, particularly in NCMS/NrGO-GF and NrGO-GF. Thermogravimetric analysis (TGA) in air (Fig. S6†) was employed to determine the weight percentage of mixed sulfides in the samples. The overall weight loss for control NCMS-GF, NCMS/NrGO-GF and NrGO-GF samples up to 700 °C was 5.2 wt%, 34.5 wt%, and 98.9 wt%, respectively. Since the weight of GF and the loading amount of NCMS and NrGO are similar in all samples, we can estimate the weight composition of our NCMS/NrGO-GF nanocomposite: ca. 65 wt% NCMS and 35 wt% carbon (3.4 wt% GF and 31.5 wt% NrGO).
To gain a deeper insight into the surface elements and chemical oxidation states of the NCMS/NrGO composites, we used near ambient pressure X-ray photoelectron spectroscopy (NAPXPS) coupled with an electrochemical cell to monitor the sample composition. An ex situ accelerated durability test (ADT) was employed using a cyclic voltammetry method (1.2–1.6 V, 1000 cycles). Sample changes before and after the ADT were recorded by NAPXPS measurement in a low vacuum (ca. 10−4 mbar), as shown in Fig. 2 and S7.† The Ni 2p spectrum of the initial NCMS/NrGO exhibits two main peaks at 856.5 and 874.2 eV (Fig. 2a), which are associated with Ni 2p3/2 and Ni 2p1/2 of Ni3+.46 The predominant Ni3+ peaks clearly demonstrate that Ni exhibited an oxidation state of ca. +3, possibly with the formation of Ni–S–OH intermediates. The formation of Ni–O–OH intermediates can be excluded, as the two satellite peaks are located at +6 eV (862.4 and 880.6 eV), corresponding to the presence of surface plasmon loss features rather than shake-up peaks.47 After ADT cycling, there is almost no difference in the XPS spectrum, indicating stable electrochemical behavior from the Ni cation. Fig. 2b shows the high-resolution spectra of the Co 2p core level before and after the ADT. The initial Co spectrum contains two peaks that can be deconvoluted into three doublets. The dominant doublet at 782.3 and 797.4 eV should be assigned to Co2+ 2p1/2 and 2p3/2 species, with the formation of Co–S. The presence of peak satellites at ca. +5.5 eV suggests the presence of Co–O–OH,46 in agreement with the peaks of the right shoulder which indicate the presence of Co3+, as a result of the inevitable oxidation in the air, and the possible formation of compounds such as Co–S–OH.24 After the ADT, the Co3+ content was slightly increased compared to the initial one, indicating the further oxidation of Co element during the OER cycling process. In the Mn 2p spectra, a broad peak with three doublets at 640.8, 642.2, and 643.7 eV represents the mixed oxidation states of Mn2+, Mn3+, and Mn4+ at Mn 2p3/2, while Mn3+ was the dominant species on the surface of the composites.48 These three contributions are not resolved in the Mn 2p1/2 peak located at 653.6 eV and are assigned to the Mn 2p1/2 region. Similar to the XPS data of Co, the Mn4+ content shows a slight increase after the ADT.
 | ||
| Fig. 2 NAPXPS surface analysis of the NCMS/NrGO composite structure: (a) Ni 2p, (b) Co 2p, (c) Mn 2p, (d) S 2p, (e) O 1s and (f) N 1s core levels before and after the accelerated degradation test (ADT, 1000 cycles). | ||
Besides the metal component, the existence of heteroatoms such as S, N, and O was also determined by NAPXPS. Fig. 2d displays the high-resolution S 2p spectrum with the peaks at 161.3 and 163.4 eV, corresponding to S 2p3/2 and S2p1/2. The high intensity ratio from S 2p1/2 might be attributed to the presence of other chemical bonds besides metal sulfides, such as C-S/C-Sx moieties. The other two peaks at 166.4 and 167.7 eV were from the metal sulfate (SOx) moiety due to surface oxidation.49 Interestingly, the sulfur content increased after ADT cycling, and the chemical state remained unchanged. It is known that sulfur based hetero-structured catalysts can improve the redox stability for metal ions during water electrolysis. Our XPS results confirm the stable formation of trimetallic sulfides prepared by the electrochemical deposition method. In the high-resolution spectrum of the O 1s core level (Fig. 2e), a broad peak can be deconvoluted into three components, centered at 529.4, 531.1 and 532.9 eV, respectively. The first peak emerged from the O2− ions of the surface lattice oxygen in a mixed M–O (M: Ni, Co, Mn) structure.50 The middle one, at around 531.1 eV, was assigned to the oxygen defects originating from the metal oxide on the surface and is associated with oxygen vacancies. These oxygen vacancy sites usually play an important role in the catalytic behavior of the OER process.50 The last peak at the highest binding energy is from the chemisorbed hydroxyl group, in the form of OH−. Fig. 2f shows the N 1s envelope featuring a broad peak comprising three main components: pyridinic nitrogen (398.5 eV), pyrrolic nitrogen (399.5 eV), and graphitic nitrogen (400.7 eV), which were from the NrGO matrix.24 The graphitic N can improve the electric conductivity of the rGO nanosheets, thus enhancing the transfer of electrons from the carbon atoms to the antibonding orbitals of O2. Meanwhile, the pyridinic and pyrrolic N can facilitate oxygen adsorption and hydroperoxide decomposition, thus improving the onset potential for the ORR. After the ADT, the chemical bond states of O remain consistent. However, there is a slight increase of the graphitic and pyrrolic N. It was reported that the attachment of OH- (the intermediate products during the ADT process) to the pyridinic N in graphene will result in an upshift of the N binding energy from 398.5 to 399.8 eV in XPS.51 Thus, the increase of pyrrolic N is from the increased amount of the OH- attached pyridinic N. However, we cannot exclude the possibility of nitrogen loss during the ADT cycling due to the nitrogen oxidation process on both pyrrolic and pyridinic N, which might further explain the increase of graphitic N.
The C 1s spectrum (Fig. S7b†) with an asymmetric peak was deconvoluted into four distinguishable components at 284.5 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.5 eV (C–N/C–OH), 286.9 eV (C–O–C), and 288.5 eV (CO).24 Furthermore, we also combined Auger electron spectroscopy (AES) with X-ray absorption spectroscopy (XAS) to monitor the Ni, Co, and Mn L3-edge and S K-edge XAS. We noticed that there is a huge amount of oxygen (51.7%) detected by XPS (Table S5†). This is because the NCMS surface phase is highly reactive due to a high density of defects, thus resulting in a large excess of oxygen being always present. However, we have to keep in mind that XPS probes the first 10 nm underneath the surface, which is why we didn't observe the oxide or hydroxide components either from XRD, Raman or TEM measurements. Meanwhile, the amount of Mn (1.0%) is much less than that of the Ni (9.5%) and Co (8.6%) elements, which is due to the low electrodeposition efficiency of Mn2+ according to the electrochemical reactions of precursors under aqueous conditions.52 The 2D-XAS mapping data, as depicted in Fig. S8,† do not show substantial variations in the main features confirming that the electronic structures of trimetallic sulfides are well preserved after the ADT cycling process.
C), 285.5 eV (C–N/C–OH), 286.9 eV (C–O–C), and 288.5 eV (CO).24 Furthermore, we also combined Auger electron spectroscopy (AES) with X-ray absorption spectroscopy (XAS) to monitor the Ni, Co, and Mn L3-edge and S K-edge XAS. We noticed that there is a huge amount of oxygen (51.7%) detected by XPS (Table S5†). This is because the NCMS surface phase is highly reactive due to a high density of defects, thus resulting in a large excess of oxygen being always present. However, we have to keep in mind that XPS probes the first 10 nm underneath the surface, which is why we didn't observe the oxide or hydroxide components either from XRD, Raman or TEM measurements. Meanwhile, the amount of Mn (1.0%) is much less than that of the Ni (9.5%) and Co (8.6%) elements, which is due to the low electrodeposition efficiency of Mn2+ according to the electrochemical reactions of precursors under aqueous conditions.52 The 2D-XAS mapping data, as depicted in Fig. S8,† do not show substantial variations in the main features confirming that the electronic structures of trimetallic sulfides are well preserved after the ADT cycling process.
2.2 Electrocatalytic properties of NCMS/NrGO
The electrocatalytic performance of our materials towards oxygen reaction was analyzed using rotating disk/ring-disk electrodes (RDE/RRDE). In order to do that, the prepared catalyst was gently scratched from the GF surface and subsequently drop cast on a glassy carbon electrode (see the experimental in the ESI†). The electrocatalytic performance of the samples toward the ORR was investigated in O2-saturated 0.1 M KOH. For comparison, NCMS and NrGO control samples prepared in the same way as NCMS/NrGO were also measured. Commercial PtRu/C 20% (Fuel Cell Store, USA) powder catalyst was also tested as a benchmark. Fig. 3a displays the typical cyclic voltammograms (CVs) in 0.1 M KOH saturated with Ar (dotted line) or O2 (solid line) between +1.2 and +0.2 V vs. RHE with a scan rate of 20 mV s−1. | ||
| Fig. 3 (a) CV curves of NCMS, NrGO, NCMS/NrGO, and commercial PtRuC catalysts on glassy carbon electrodes in O2-saturated (solid line) or Ar-saturated (dashed line) 0.1 M KOH with a scan rate of 20 mV s−1. (b) Rotating disk LSV profiles of the prepared samples in O2-saturated 0.1 M KOH solution at a rotation rate of 1500 rpm with a scan rate of 20 mV s−1. (c) Corresponding Tafel plots. | ||
Without O2, all samples demonstrated almost rectangular CV curves with no clear cathodic redox peak. In contrast, an intense redox peak appeared in the cathodic scan for all samples in the presence of O2, indicating the catalytic activity of samples toward the ORR. Pure NCMS and NrGO showed poor ORR catalytic activity, with oxygen reduction peak potentials of 0.73 and 0.72 V vs. RHE, respectively, and a cathodic peak current of ∼0.6 mA cm−2. Remarkably, the NCMS/NrGO composite material showed significantly better catalytic activity than the individual components, exhibiting a higher reduction peak potential of 0.83 V vs. RHE and almost two-fold peak current density of ∼1 mA cm−2. It should be emphasized that the reduction peak potential of the PtRuC commercial catalyst, which is used as a benchmark, is 0.79 V vs. RHE. Fig. 3a illustrates that, in contrast to the individual components, our synthesized NCMS/NrGO catalyst surpasses this value (0.83 V vs. RHE), demonstrating the excellent ORR catalytic activity of the composite material.
The improved electrocatalytic activity of NCMS/NrGO, as opposed to those of the individual components and commercially available catalyst, can be attributed to faster reaction kinetics, resulting in a more substantial transfer of electrons per oxygen molecule during the ORR. To explain the ORR mechanism and kinetics in various samples, linear sweep voltammetry (LSV) measurements were conducted on a rotating-disk electrode (RDE) in O2-saturated 0.1 M KOH solutions. Fig. 3b shows the linear sweep voltammetry (LSV) polarization curves of the ORR for different catalysts, including PtRuC as the state-of-the-art benchmark, along with the individual components NCMS and NrGO. The parameters derived from LSV curves are summarized in Table S1† which also compiles recently reported values for TMC-based electrocatalysts. Both mixed NCMS and NrGO exhibited comparable and moderate ORR catalytic activity, with an oxygen reduction onset potential (Eonset) of 0.81 V vs. RHE and a small limiting current (JLevich) of 3.9 mA cm−2. Remarkably, the NCMS/NrGO composite surpassed the individual examined catalysts and demonstrated ideal catalytic activity towards the ORR, with Eonset of 0.94 V vs. RHE. This value is superior to that exhibited by a PtRuC commercial catalyst (Eonset = 0.89 V) and other recently reported TMC-based electrocatalysts (Table S1†). Moreover, the NCMS/NrGO composite exhibited a significantly higher limiting current (JLevich) than that of the pure compounds (∼30% higher than NCMS and NrGO control samples), presenting one of the highest limiting currents reported so far for a non-noble metal catalyst, clearly suggesting that the electronic coupling of NCMS nanosheets with NrGO layers increased the electrocatalytic activity of the sample. The superior ORR kinetics of the NCMS/NrGO can be further confirmed by the Tafel slope obtained from ORR polarization curves (Fig. 3c). Specifically, the Tafel slope of the NCMS/NrGO is 27 mV dec−1, which is smaller than those of NCMS (139 mV dec−1), NrGO (97 mV dec−1), and even PtRuC (46 mV dec−1). These results indicate that the notable synergistic effect between NCMS and NrGO, combined with the precisely controlled nanostructure determined by the electrosynthesis parameters, contributes to the enhanced ORR activity within the NCMS/NrGO composite.
The slopes of the Koutecky–Levich plots, extracted from the respective LSV curves at different rotating rates, were employed to ascertain the number of electrons (n) participating in the ORR (Fig. S9†). This analysis reveals the 4e− ORR process for all the catalysts, except for the NrGO control sample. RRDE measurements in Fig. S10a† show the ring current, associated with the reduction of the peroxide produced in the event of a 2-electron reaction, for NCMS/NrGO and PtRuC benchmark catalysts. These results were used to calculate the peroxide yields and the number of transferred electrons (n), leading to values of below 5% and 3.99 for the NCMS/NrGO composite, and around 10% and 3.94 for PtRuC, respectively (Fig. S10b†). These findings further confirm the four-electron transfer pathway associated with the ORR in the NCMS/NrGO composite (Fig. S10b†). Chronoamperometry at a constant potential was used to evaluate the durability of the NCMS/NrGO catalysts. Notably, a small decay (12.2%) of current density at a potential of 0.6 V vs. RHE was obtained for NCMS/NrGO after 50 h (Fig. S11†). This decay is significantly lower than that of PtRuC (39.5%), demonstrating the excellent long-term stability of the NCMS/NrGO. It is important to note that the PtRuC commercial catalyst, apart for being expensive, is highly susceptible to surface poisoning. This constitutes an additional drawback for employing PtRuC in energy storage/conversion devices. Fig. S12† shows that our NCMS/NrGO exhibited a much higher tolerance to methanol poisoning compared to commercial PtRuC. This robust toxicity resistance was evidenced by the absence of any noticeable change in current density upon adding methanol, in contrast to the observed current drop in PtRuC (Fig. S12†).
Encouraged by the outstanding ORR activity and durability, the catalytic behavior of NCMS/NrGO towards the OER was also assessed. Fig. 4a provides a comparison of the OER polarization curves. In our NCMS/NrGO, a clear wide anodic peak at ∼1.33 V was observed during the initial cycling, corresponding to the anodic peaks from the redox conversion of the metal cations present in the composite (nickel and/or manganese), associated with the in situ formed intermediates such as metal hydroxyl sulfides. Once a stable interface with a thin hydroxide layer was formed, the relative peak disappeared. This phenomenon has also been reported in other works with similar spinel-type mixed metal compounds.23 The anodic peak is followed by a considerable current increase associated with the generation of oxygen. Accordingly, an OER onset potential of ∼1.41 V vs. RHE was obtained for the NCMS/NrGO sample, which is slightly lower than that of the state-of-the-art PtRuC catalyst (∼1.44 V vs. RHE). Moreover, NCMS/NrGO exhibited an OER potential of 1.59 V vs. RHE at the current density of 10 mA cm−2 (denoted as Ej10), being slightly lower than that of commercial PtRuC (1.63 V vs. RHE). NCMS/NrGO also exhibited a much smaller Tafel slope compared to the commercial catalyst, illustrating its better OER kinetics (Fig. 4b). Interestingly, the Tafel slopes observed at the higher potential for the muti-metal sulfide (similar to the commercial PtRu/C) could be attributed to the change in the adsorption of reaction intermediates (i.e., hydroxyl sulfide, oxo-hydroxides) during the OER steps and also related to the change in rate determining steps,53 which makes a mechanistic understanding of the OER process difficult. When it comes to the long-term stability of NCMS/NrGO, a substantial current retention of 84.2% was observed following an extended stability test for the OER (∼3 hours). This retention rate surpassed the commercial PtRuC (34.7%) performance which was also confirmed by the accelerated stability/degradation test as shown in Fig. 4d. During the study, the OER curve of NCMS/NrGO almost overlays the initial one after 1000 cyclic voltammetry (CV) cycles, providing additional evidence of the catalytic stability under alkaline conditions.
 | ||
| Fig. 4 (a) Evaluation of the OER catalytic activity of NCMS/NrGO and commercial PtRuC in Ar-saturated 0.1 M KOH. (b) Corresponding OER Tafel plots. (c) Durability of OER catalytic activity. (d) Polarization curves before and after 1000 OER cycles. | ||
The overall bifunctional electrocatalytic performance was evaluated by the potential difference (ΔE) between Ej10 for the OER and E1/2 for the ORR. A reduced ΔE value signifies improved performance of NCMS/NrGO as a bifunctional catalyst. Fig. S13† shows the electrochemical performances for both the OER and ORR of the NCMS/NrGO in comparison with commercial PtRuC (Fig. S13a†) and other representative reported materials synthesized by hydrothermal methods (Fig. S13b†). Fig. 5 shows a more extensive comparison of OER/ORR performance including also other representative catalysts previously reported in the literature.23–25,54–63 The potential difference of our NCMS/NrGO is 0.75 V, smaller than that of the commercial PtRuC catalyst (0.81 V), with this performance being comparable or superior to those of most bifunctional non-noble metal catalysts reported to date, even those using more expensive or energy consuming synthesis techniques, suggesting the superior advantages of using NCMS/NrGO as air cathodes in rechargeable Zn–air batteries. The exceptional catalytic activity of the electrosynthesized NCMS/NrGO can be linked to the well-organized nanoporous structure, the stochastic dispersion of active sites, synergistic interactions among various components, and abundant diffusion channels that efficiently boost the mass transport of ionic species within the foam matrix.
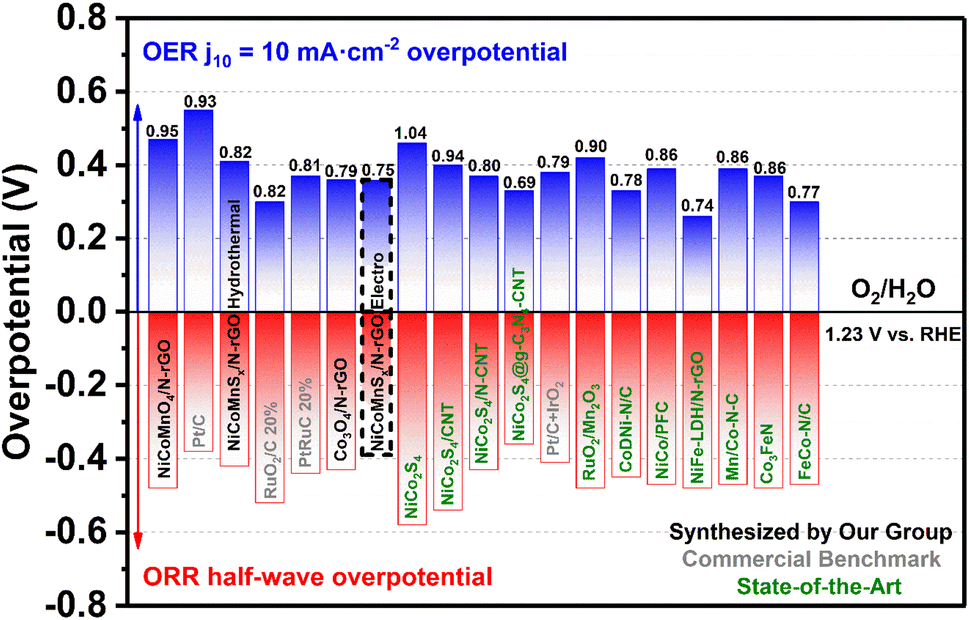 | ||
| Fig. 5 Comparison of the oxygen bifunctional electrocatalytic performance of catalysts in this work with previously reported catalysts in our group and in the literature.23–25,54–63 Potential difference (ΔE) between Ej10 for the OER and E1/2 for the ORR is indicated on top of each bar. | ||
2.3 Density functional theory (DFT) calculations
It is difficult to correlate the various intermediate adsorption states on the catalyst surfaces with reaction kinetics. To support our experimental observations, we performed density functional theory (DFT) calculations to study the OER and ORR kinetics and the role of M–N interactions in NrGO-supported NiCoMnSx (NCMS/NrGO). We also tried to correlate the experimental findings and theoretical information to understand the oxygen electrochemistry over NCMS with respect to the possible active sites. Initially, a graphene sheet with appropriate X and Y dimensions was optimized using DFT, and the NCMS cluster was attached to it afterwards. For accuracy of atomic environments and coordination, a cluster was cut out from bulk NiCoS and optimized. Further, Mn was alloyed to this cluster to create an NCMS model and optimized again. The free-standing cluster was placed on the surface of pristine rGO as well as on N doped rGO sheets (Fig. S14†). The distance of the NCMS cluster from the rGO and NrGO surfaces was 5.81 Å and 5.47 Å respectively, and this reduced distance was due to the N doping influencing the charge transfer between the surface and NCMS clusters. The calculated binding energy of NCMS with NrGO substrates was calculated to be −0.457 eV compared to a positive value of 0.507 eV for the NCMS over rGO. This clearly indicates that the N doping strategy favors a stronger binding of the catalyst layer with the reaction being exothermic in the NrGO substrate, which is in good agreement with the experimental observations. The OER and ORR features of the optimized NCMS/NrGO structures were analyzed based on H2O and O2 adsorption characteristics.64 When we analyzed the ORR characteristics of the NCMS + NrGO surface through the adsorption energy of O2 over various metal sites (Fig. 6a) the binding energy obtained at the Ni site was −1.75 eV over the NrGO surface. The O2 molecule is spontaneously broken while attached over Co and Mn, unlike the Ni site. When attached to S, it was adsorbed with an energy of −1.18 eV and at a longer S–O distance (Table S2†). This clearly suggests Ni as the main active site in NCMS during the ORR, with higher adsorption energies than all other metal sites. We have plotted the projected density of states (PDOS) of the atoms for the O2 adsorbed at the Ni site for NrGO as shown in : the Ni orbitals show a significant overlap with that of O, indicating the strong adsorption characteristics. A closer look at the PDOS indicates that in the presence of N atoms on the graphene surface, the O atom density of states (mainly contributed by the p orbitals) shows an upward shift towards positive energies, indicating a transition from antibonding to bonding orbitals. This agrees with the enhanced adsorption of O2. The Bader charges for the rGO and NrGO surfaces with O2 adsorbed on the Ni atom (most energetically favorable configuration) show clearly that the O2 molecule gains −0.43 electrons in both cases. The S atoms also gain charges, −0.34 and −0.78 respectively. On the other hand, the Ni atom that is directly bonded to O2 loses charge as well as Mn and Co which are bonded to this Ni atom. The charge redistribution by attachment of O2 molecules on top of the NCMS clusters does not vary much between the rGO and NrGO surfaces. | ||
| Fig. 6 DFT calculation based (a) optimized geometry of NCMS with an O2 adsorption configuration over N-rGO during the ORR process and (b) optimized geometry of NCMS with an H2O adsorption configuration over N-rGO during the OER process; (c and d) projected density of states of the adsorbed molecule and the Ni atoms in the NCMS/N-rGO with O2 adsorption and H2O adsorption over the Ni site. | ||
In the case of the OER based on H2O molecule adsorption energies, the Ni site showed a favorable binding energy of −0.464 eV (Fig. 6b). This is due to a very strong Ni–N interaction as we observe a decrease in Ni–O lengths, in the N-doped sample (2.10 Å) compared to the undoped one (2.3 Å). In the case of the Co site, the binding energy of the H2O molecule is −0.415 eV and that for the Mn site is −1.10 eV (Table S3†). The more negative binding energy for the H2O in Ni sites indicates that H2O adsorbs more strongly there, and with a reduced Ni–O distance (2.10 Å) compared to other metal sites; therefore, this configuration is more favorable for the OER activity. When H2O is attached to the sulfide part of NCMS, the binding energy is 5.81 eV. Thus, it is clear that Ni acts as the main active site in the mixed metal sulfides favoring bifunctional oxygen electrochemistry. Our calculation results are in agreement with the previous PDOS studies on single metal sulfite Ni3S2, indicating that mixed transition metal sulfides can alter the electronic status and improve the catalytic activity of NiSx.65 The Bader charges were calculated to understand the quantitative charge transfer between the cluster and adsorbates for the most favorable configurations. For the H2O adsorbed on the rGO surface, the net Bader charge for the H2O molecule is 0.0294 and that of the Ni atom to which it is bonded is 0.3464, indicating that both H2O and Ni lose charge. Furthermore, the Mn and Co atoms bonded to this Ni atom also lose charge and the net Bader charges are 0.9637 and 0.5989 respectively, while the S atoms, on the other hand, gain charge (−0.385, −0.8575). The Bader charge calculated for the water molecule on top of the NrGO surface is 0.0185 and that of the N atom bonded to the H2O is 0.4103. In this case also, the S atoms bonded to this Ni atom gain charge (−0.3992, −0.7630). This charge distribution ensures that the Ni site is favorable for the water adsorbate in both rGO and NrGO surfaces. The projected density of states analysis showed that the Ni orbitals hybridize with the H2O molecule (Fig. 6d), improving the attachment. Fig. S15† shows the density of states for different sites of NCMS/NrGO-GF during the O2 and H2O adsorption.
2.4 Tests as air cathodes in rechargeable Zn–air batteries (ZABs)
The superior bifunctional ORR/OER capability and durability of NCMS/NrGO motivated us to investigate its practical applicability and performance in homemade rechargeable 3D printed Zn–air batteries (Fig. 7a). The directly assembled free-standing electro-synthesized NCMS/NrGO-GF can serve as an air cathode for rechargeable ZABs without the need for additional manufacturing, using a Zn plate used as the anode. The alkaline ZABs catalyzed by NCMS/NrGO-GF exhibited a consistently stable open-circuit voltage (OCV) of 1.42 V (Fig. 7b), higher than those using PtRuC supported on a gas diffusion layer (GDL) as the benchmark catalyst (1.39 V). The ZABs catalyzed by NCMS/NrGO-GF have a high specific capacity of 815.5 mA h gZn−1 when discharged at 10 mA cm-2 (Fig. 7c). The combination of high voltage and capacity values leads to an impressive energy density of 1001 W h kgZn−1 which is ca. 92% of the theoretical energy density (1086 W h kgZn−1). Remarkably, these values obtained surpass those of batteries using PtRuC-GDL (766 mA h gZn−1 and 822 W h kgZn−1). | ||
| Fig. 7 (a) Schematic illustration of a 3D-printed Zn–air cell. (b) Open circuit voltage of NCMS/NrGO-GF cells and PtRuC cells. (c) Discharge curve of Zn–air batteries with NCMS/NrGO and PtRuC as oxygen electrode catalysts at 10 mA cm−2. (d) Discharge profiles of the Zn–air battery based on NCMS/NrGO-GF and PtRuC-GDL catalyst electrodes at various current densities. (e) Galvanodynamic discharge profile and power density curve of NCMS/NrGO-GF and PtRuC-GDL air cathodes. | ||
Fig. 7d shows the discharge profiles of both ZABs at various current densities; the battery using NCMS/NrGO-GF maintains stable plateaus for different current densities (e.g., 1.25 V at 10 mA cm−2), surpassing the performance of the PtRuC-GDL based battery. Even at high current densities, the battery could provide stable plateaus (e.g., 1.01 V at 40 mA cm−2) confirming the superior catalytic activity of the composite towards the ORR. It should be mentioned here that operating at a discharging current density as high as 40 mA cm−2 has been rarely reported for Zn–air batteries. Additionally, a study of both ZABs at different charge conditions is shown in Fig. S16.† As observed, the battery using NCMS/NrGO-GF exhibits consistently stable plateaus under various current loads (e.g., 2.06 V at 5 mA cm−2), outperforming the battery assembled with PtRuC-GDL, indicating their superior rate performance. Fig. 7e displays the galvanodynamic discharge profiles along with their corresponding power density curves. The ZAB based on NCMS/NrGO-GF achieved a peak power density of 124 mW cm−2, much better than that of PtRuC-GDL (81 mW cm−2), indicating that NCMS/NrGO-GF is a potential candidate to replace noble-metal based materials in commercial ZABs.
The long-term rechargeability of the ZABs with the NCMS/NrGO-GF air cathode was evaluated by continuous galvanostatic discharging/charging at different current densities of 2 and 10 mA cm−2, with 10 min per cycle in both cases (discharge 5 min and charge 5 min). The battery was continuously and steadily operated for more than 1560 cycles (i.e., 260 h) at 2 mA cm−2 without any obvious decay in performance, whereas the ZAB with the PtRuC-GDL air cathode exhibited serious degradation after only 170 cycles (i.e., 30 h) (Fig. 8). The improved performance and enhanced stability of the NCMS/NrGO-GF air electrode compared to PtRuC-GDL is attributed to the unique morphology and characteristics of the multilayer NCMS/NrGO structure which facilitates continuous oxygen flow into the air electrode and provides ideal triphase regions for the oxygen reactions in metal–air batteries.
 | ||
| Fig. 8 Charge–discharge profiles of Zn–air batteries with NCMS/NrGO-GF and PtRuC oxygen catalysts at 2 mA cm−2. | ||
In addition, we observed a small round-trip voltage increase (difference between charging and discharging voltages) of only 0.64 V to 1.02 V, equal to a voltage efficiency decrease from 65.2% to 56.1% for the NCMS/NrGO-GF and PtRuC-GDL. On the other hand, while both batteries initially showed similar charge/discharge profiles, the performance of the battery using PtRuC-GDL as the air cathode quickly degraded, evidenced by an increased round trip voltage from 0.67 V to 1.51 V after only 30 h of cycling. Moreover, when NCMS/NrGO-GF is used as an air cathode the ZAB is repeatedly discharged/recharged even at 10 mA cm−2 for a 60 h cycling period (Fig. S17†) with almost no significant degradation either in discharge or charge segments. Pictures of the electrodes after cycling at 2 mA cm−2 are also included to observe the possible degradation suffered (see the inset of Fig. 8). ZnO in black can be clearly observed in the zinc anode after cycling, indicating the formation of irreversible byproducts during the cycling, deteriorating the cell kinetics and limiting the battery lifetime. Furthermore, the cycled air-cathode was subjected to post-mortem analysis using XRD and SEM. As shown Fig. S18a,† the XRD pattern of the GF air-cathode reveals that the main composition of the NCMS/NrGO-GF catalyst remained unchanged, which is consistent with the previous XPS analysis after the ADT. The SEM micrograph of the scratched sample after cycling (Fig. S18b†) demonstrated that, by letting aside the appearance of some wrinkles or cracks due to the cycling, NCMS/NrGO-GF retained its original morphology, providing solid evidence for the stability of the sample during the catalytic reaction. These findings collectively demonstrate the effectiveness of NCMS/NrGO-GF as an excellent bifunctional catalyst for metal–air batteries.
Finally, Table S4† shows that the ZABs based on the electrosynthesized NCMS/NrGO-GF catalyst exhibited performance that is either superior or comparable to those of recently reported highly active non-noble metal materials. However, these materials were obtained through more energy consuming processes, such as pyrolysis or hydrothermal/solvothermal methods. This indicates that our composite can be inexpensive and show high performance, fostering the progress of electrochemical metal–air technologies.
3. Conclusions
In summary, we designed and successfully synthesized a multilayer NCMS/NrGO composite catalyst on a 3D conductive graphene foam substrate via a facile, controllable, versatile and fast reductive combination of electrosynthesis approaches.The innovative electrochemical synthetic pathway allowed the creation of complex, hierarchical structures in the form of mesoscopic foams coated by porous, vertically ultrathin nanosheets, featuring a high density of catalytic active sites such as oxygen vacancies, heteroatom doping, and intrinsic defects. This structure favors oxygen adsorption which boosts the electrocatalytic activity of the material. The produced multifunctional composites showed excellent bifunctional electrocatalytic performance for the OER and ORR (ΔE = 0.75 V) compared to previously reported transition metal and noble-metal bifunctional electrocatalysts. This material exhibited exceptional catalytic behavior in Zn–air batteries, which is superior to the performance of its individual components and existing commercial benchmark catalysts like PtRuC. Meanwhile, DFT calculations allowed the identification of active centers in NCMS oxygen electrochemistry during the OER and ORR. Theoretical simulation identifies the mechanism where N species help in coactivating metallic active sites in NCMS, especially Ni, through a Ni, Co, Mn–N moiety center and actively supports the Faraday redox reaction of metal ions, thereby fostering reversible oxygen electrocatalysis. Summarizing the experimental and simulation data, the remarkable performances and cycling stability of the Zn–air battery can be attributed to several key factors: (1) the intricate nanoporous 3D structure of the self-standing NCMS/NrGO-GF air cathodes provides catalytically active sites that are easily accessible to the electrolyte, facilitating the rapid escape of O2 bubbles; (2) the exceptional stability of the NCMS/NrGO-GF ensures long-term catalytic activity for the OER/ORR; (3) the NCMS/NrGO composite displays superior bifunctional OER/ORR activity and stability, with the mesoporous trimetallic sulfide driving the OER and the electron-donating quaternary N sites in N-doped graphene governing the ORR; (4) the NrGO-GF heterostructure acts as a three-dimensional (3D) diffusion and conductive channel, promoting efficient mass and electron transfer on the electrode surface.
We expect that the flexibility of this layer-by-layer and cost-effective electrochemical fabrication approach will extend beyond the reported materials, enabling the creation of various 2D composite electrode structures. Moreover, these findings contribute to a more profound comprehension of the systematic design of effective and long-lasting nano-catalysts through electrochemical tuning of interfaces, applicable to a range of energy storage and conversion devices.
4. Experimental section
All the chemicals utilized for synthesis and experiments were of analytical grade and used as received. GO was synthesized by oxidizing natural graphite powder (Sigma Aldrich, <20 μm, San Luis, USA) with a modified Hummers method.664.1 Preparation of 3D graphene foam (GF)
Nickel foams served as 3D templates for the chemical vapor deposition (CVD) growth of graphene. A Ni foam piece (60 × 30 mm) underwent ultrasonic washing in dilute HCl (3 wt%), DI water, and acetone sequentially to eliminate the organic contaminants. It was then positioned in a standard quartz tube furnace, heated to 1000 °C under a H2 gas flow (50 s.c.c.m.), and annealed for 30 minutes to clean the surface and etch the thin nickel oxide layer. Subsequently, CH4 was introduced into the tube with low concentration (50 s.c.c.m.), while the H2 flow was increased to 500 s.c.c.m. for a 10 minute growth time. The sample was quickly cooled to room temperature with a cooling rate of ca. 100 °C min−1 under an argon atmosphere. The resulting sample was immersed in an FeCl3 solution (4.5 wt% in DI water) at 80 °C overnight, followed by etching in 10 wt% HCl for 4 hours at 80 °C to completely eliminate the nickel metal and salt residues.4.2 Electrophoretic deposition (EPD) of NrGO on GF
Graphene oxide was used to prepare the nitrogen doped GO (NrGO) for the EPD over the GF substrate. The GO was initially dispersed in ethanol followed by addition of 25% ammonia solution drop by drop and water using ultrasonication. The GO dispersion was then heated to 80 °C under stirring overnight. This solution was transferred to a Teflon lined hydrothermal reactor, heated to 150 °C and held for 3 h. After the reaction and cooling down, the NrGO particles were centrifugally washed with ethanol and dried at 70 °C in an air oven. For the EPD, 20 mg of NrGO powder was dispersed in 50 mL of N,N′-dimethylformamide (DMF) under ultrasonication for 20 min. Then, 500 μL of poly(diallyldimethylammonium chloride) (PDDA; 20 wt% in H2O, Sigma Aldrich) was added to the NrGO dispersion to tune the surface charge of NrGO nanosheets positively.33 Subsequently, we immersed the graphene foam (GF) in the above-mentioned suspension, and the deposition proceeded through cathodic electrophoresis. The working electrode (WE) consisted of a graphene foam (30 × 10 mm), while a Pt foil (30 × 10 mm) served as a counter electrode (CE) located parallel to the graphene foam at a distance of 40 mm. EPD coating of NrGO was achieved by applying a DC potential of −20 V for 10 minutes.2 A very thin layer (∼60 nm) of NrGO with a mass loading of ∼0.3 mg cm−2 was obtained on a GF matrix. Then, the NrGO-GF electrodes were dried at 70 °C for 30 minutes to remove the solvents.4.3 Cyclic voltammetry (CV) electrodeposition of NCMS nanostructures
Since the chemical vapor deposited graphene is chemically inert and possesses a hydrophobic surface, obtaining uniformly deposited thin films of metal compounds through electrochemical deposition is extremely difficult. To improve the surface adhesion of the metallic compounds over the CVD graphene, we start by applying a thin NrGO layer to create NrGO/GF. Following this process, the graphene foam (30 × 10 mm dimensions) having NrGO (NrGO/GF) was immersed in a water/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) electrolyte having three metal salts: 5 mM NiCl2·6H2O, 5 mM CoCl2·6H2O, and 10 mM MnCl2·6H2O dispersed along with the sulfur source, and 0.1 M thiourea at room temperature. The Ar saturated solution was then used for electrodeposition of NCMS nanostructures via cyclic voltammetry (CV) with a three-electrode system, where the 3D NrGO-GF was the WE, a platinum mesh served as the CE (located parallel to the rGO-GF, at ∼40 mm distance) and Ag/AgCl served as the RE (see Scheme 1). The electrodeposition was performed at a scan rate of 5 mV s−1 between −1.2 V and 0.3 V. With the process, a very thin layer (∼150–200 nm) was deposited on the surface of NrGO/GF, corresponding to a mass loading of ∼0.6–0.8 mg cm−2. After coating, the electrodes were washed and dried at 70 °C for 1 h. The cyclic voltammogram during the process exhibited broad oxidation and reduction peaks centered at 0.2 and −0.50 V vs. Ag/AgCl, respectively (see Fig. S19†), and these peaks are attributed to the one-electron oxidation of thiourea to formamidine disulfide and its corresponding reduction according to the following mechanism already reported in the literature as described below:67,68
:1 volume ratio) electrolyte having three metal salts: 5 mM NiCl2·6H2O, 5 mM CoCl2·6H2O, and 10 mM MnCl2·6H2O dispersed along with the sulfur source, and 0.1 M thiourea at room temperature. The Ar saturated solution was then used for electrodeposition of NCMS nanostructures via cyclic voltammetry (CV) with a three-electrode system, where the 3D NrGO-GF was the WE, a platinum mesh served as the CE (located parallel to the rGO-GF, at ∼40 mm distance) and Ag/AgCl served as the RE (see Scheme 1). The electrodeposition was performed at a scan rate of 5 mV s−1 between −1.2 V and 0.3 V. With the process, a very thin layer (∼150–200 nm) was deposited on the surface of NrGO/GF, corresponding to a mass loading of ∼0.6–0.8 mg cm−2. After coating, the electrodes were washed and dried at 70 °C for 1 h. The cyclic voltammogram during the process exhibited broad oxidation and reduction peaks centered at 0.2 and −0.50 V vs. Ag/AgCl, respectively (see Fig. S19†), and these peaks are attributed to the one-electron oxidation of thiourea to formamidine disulfide and its corresponding reduction according to the following mechanism already reported in the literature as described below:67,68| 2[SC(NH2)2] → [SC(NH)NH2]2 + 2H+ + 2e− | (1) |
The overall reactions involved in the deposition of the NCMS films are given below:
| Generation of hydrogen: 2H2O + 2e− → 2OH− + H2 | (2) |
| Decomposition of thiourea: SC(NH2)2 + 2OH− → S2− + OC(NH2)2 + H2O | (3) |
| Formation of MTMS: Ni2+ + Co2+ + Mn2+ + xS2− → NiCoMnSx | (4) |
The final mass loading of the NCMS/NrGO electrode was ∼1 mg cm−2, with an average sulfide to carbon weight ratio of 7:3. For comparison sake, pure NCMS on GF and NrGO on GF samples were also synthesized using the same procedure, including no NrGO in the pure sulfide sample and no metal precursors + thiourea in the NrGO sample. Each sample was produced and characterized twice, and each electrochemical measurement was repeated 5 times. The experimental details for the microstructural and electrochemical measurements can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under GrapheneCore3 881603-Graphene Flagship and the HySolChem project grant agreement No. 101017928. Furthermore, funding from VINNOVA (project GO-FOR-WATER 2019-05353), the Swedish Innovation Agency, and the Swedish Åforsk Foundation (project no. 222-263) is gratefully acknowledged. The authors also thank grant RED2022-134552-T, funded by MICIN/AEI/10.13039/501100011033 and the HySolChem project, which has received funding from the European Unions’s Horizon 2020 research and innovation programme (101017928). We acknowledge MAX IV Laboratory for time on Beamline HIPPIE under Proposal 20230905. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. Z. Y. Xia gratefully acknowledges support by Dr Robert Temperton and Dr Andrey Shavorskiy for the XPS and XAS measurements at MAX IV. The CSC-IT Center for Science, Finland is acknowledged for computational resources.References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. S. Sanchez, Z. Xia, N. Patil, R. Grieco, J. Sun, U. Klement, R. Qiu, M. Christian, F. Liscio, V. Morandi, R. Marcilla and V. Palermo, Small, 2022, 18, 2106403 CrossRef CAS PubMed.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 CrossRef CAS PubMed.
- X. L. Tian, X. Zhao, Y. Q. Su, L. J. Wang, H. M. Wang, D. Dang, B. Chi, H. F. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- Y. P. Deng, Y. Jiang, R. Liang, S. J. Zhang, D. Luo, Y. Hu, X. Wang, J. T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed.
- K. W. Leong, Y. Wang, M. Ni, W. Pan, S. Luo and D. Y. C. Leung, Renewable Sustainable Energy Rev., 2022, 154, 111771 CrossRef CAS.
- C. Zhou, X. Chen, S. Liu, Y. Han, H. Meng, Q. Jiang, S. Zhao, F. Wei, J. Sun, T. Tan and R. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704 CrossRef CAS PubMed.
- H. Yang, S. Gao, D. Rao and X. Yan, Energy Storage Mater., 2022, 46, 553–562 CrossRef.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- Z.-F. Huang, J. Wang, Y. Peng, C.-Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700544 CrossRef.
- M. Mechili, C. Vaitsis, N. Argirusis, P. K. Pandis, G. Sourkouni and C. Argirusis, Renewable Sustainable Energy Rev., 2022, 156, 111970 CrossRef CAS.
- Y. R. Ying, K. Fan, X. Luo, J. L. Qiao and H. T. Huang, J. Mater. Chem. A, 2021, 9, 16860–16867 RSC.
- L. Lu, Y. Zheng, R. Yang, A. Kakimov and X. Li, Mater. Today Chem., 2021, 21, 100488 CrossRef CAS.
- L. Song, T. Zheng, L. Zheng, B. Lu, H. Chen, Q. He, W. Zheng, Y. Hou, J. Lian, Y. Wu, J. Chen, Z. Ye and J. Lu, Appl. Catal., B, 2022, 300, 120712 CrossRef CAS.
- Z. Cui, G. Fu, Y. Li and J. B. Goodenough, Angew Chem. Int. Ed. Engl., 2017, 56, 9901–9905 CrossRef CAS PubMed.
- X. Y. Wu, S. M. Li, J. H. Liu and M. Yu, ACS Appl. Nano Mater., 2019, 2, 4921–4932 CrossRef CAS.
- X. Y. Yu and X. W. David Lou, Adv. Energy Mater., 2018, 8, 1701592 CrossRef.
- J. Y. Xue, C. Li, F. L. Li, H. W. Gu, P. Braunstein and J. P. Lang, Nanoscale, 2020, 12, 4816–4825 RSC.
- X. Wang, J. Zhang, P. Wang, L. C. Li, H. Y. Wang, D. M. Sun, Y. F. Li, Y. W. Tang, X. F. Lu, Y. Wang and G. T. Fu, Energy Environ. Sci., 2023, 16, 5500–5512 RSC.
- D. Zhou, Z. Cai, X. Lei, W. Tian, Y. Bi, Y. Jia, N. Han, T. Gao, Q. Zhang, Y. Kuang, J. Pan, X. Sun and X. Duan, Adv. Energy Mater., 2017, 8, 1701095 Search PubMed.
- G. Li, Y. Tang, T. Fu, Y. Xiang, Z. Xiong, Y. Si, C. Guo and Z. Jiang, Chem. Eng. J., 2022, 429, 132174 CrossRef CAS.
- A. Pendashteh, J. Palma, M. Anderson and R. Marcilla, Appl. Catal., B, 2017, 201, 241–252 CrossRef CAS.
- A. Pendashteh, J. S. Sanchez, J. Palma, M. Anderson and R. Marcilla, Energy Storage Mater., 2019, 20, 216–224 CrossRef.
- J. S. Sanchez, R. R. Maca, A. Pendashteh, V. Etacheri, V. A. D. O'Shea, M. Castillo-Rodriguez, J. Palma and R. Marcilla, Catal. Sci. Technol., 2020, 10, 1444–1457 RSC.
- S. W. Ning, M. Li, X. Wang, D. Zhang, B. Y. Zhang, C. K. Wang, D. M. Sun, Y. W. Tang, H. Li, K. Sun and G. T. Fu, Angew. Chem., Int. Ed., 2023, 62, 202314565 CrossRef PubMed.
- C. Fan, X. Wang, X. R. Wu, Y. S. Chen, Z. X. Wang, M. Li, D. M. Sun, Y. W. Tang and G. T. Fu, Adv. Energy Mater., 2023, 13, 2203244 CrossRef CAS.
- B. Liu, S. X. Qu, Y. Kou, Z. Liu, X. Chen, Y. T. Wu, X. P. Han, Y. D. Deng, W. B. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 30433–30440 CrossRef CAS PubMed.
- M. B. Kale, R. A. Borse, A. Gomaa Abdelkader Mohamed and Y. Wang, Adv. Funct. Mater., 2021, 31, 2101313 CrossRef CAS.
- Z. Xia, V. Mishukova, S. Sollami Delekta, J. Sun, J. S. Sanchez, J. Li and V. Palermo, Nanoscale, 2021, 13, 3285–3294 RSC.
- Z. Y. Xia, M. Christian, C. Arbizzani, V. Morandi, M. Gazzano, V. Quintano, A. Kovtun and V. Palermo, Nanoscale, 2019, 11, 5265–5273 RSC.
- Z. Y. Xia, D. Wei, E. Anitowska, V. Bellani, L. Ortolani, V. Morandi, M. Gazzano, A. Zanelli, S. Borini and V. Palermo, Carbon, 2015, 84, 254–262 CrossRef CAS.
- J. S. Sanchez, J. Xu, Z. Y. Xia, J. H. Sun, L. E. Asp and V. Palermo, Compos. Sci. Technol., 2021, 208, 108768 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- Z. H. Li, X. Li, L. Xiang, X. Xie, X. Li, D. R. Xiao, J. Shen, W. Q. Lu, L. Lu and S. Y. Liu, J. Mater. Chem. A, 2016, 4, 18335–18341 RSC.
- H. S. Lee, J. Pan, G. S. Gund and H. S. Park, Adv. Mater. Interfaces, 2020, 7, 2000138 CrossRef CAS.
- X. Wu, X. Han, X. Ma, W. Zhang, Y. Deng, C. Zhong and W. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 12574–12583 CrossRef CAS PubMed.
- M. G. Park, J. W. Choi, I. W. Ock, G. H. Kim and J. K. Kang, Adv. Energy Mater., 2021, 11, 2003563 CrossRef CAS.
- Y. Ma, Y. J. Ma, G. T. Kim, T. Diemant, R. J. Behm, D. Geiger, U. Kaiser, A. Varzi and S. Passerini, Adv. Energy Mater., 2019, 9, 1902077 CrossRef CAS.
- Q. W. Tian, M. H. Tang, F. R. Jiang, Y. W. Liu, J. H. Wu, R. J. Zou, Y. G. Sun, Z. G. Chen, R. W. Li and J. Q. Hu, Chem. Commun., 2011, 47, 8100–8102 RSC.
- C. Y. Zhang, X. Y. Cai, Y. Qian, H. F. Jiang, L. J. Zhou, B. S. Li, L. F. Lai, Z. X. Shen and W. Huang, Advanced Science, 2018, 5, 1700375 CrossRef PubMed.
- C. C. Xiang, W. J. Sheng, P. F. Zhang, S. J. Zhang, J. T. Li, Y. Zhou, L. Huang and S. G. Sun, Sci. China Mater., 2021, 64, 2397–2408 CrossRef CAS.
- X. Q. Cai, X. P. Shen, L. B. Ma, Z. Y. Ji and L. R. Kong, RSC Adv., 2015, 5, 58777–58783 RSC.
- S. H. Li, J. W. Chen, J. Q. Xiong, X. F. Gong, J. Ciou and P. S. Lee, Nano-Micro Lett., 2020, 12, 34 CrossRef CAS PubMed.
- Y. Jiang, Y. P. Deng, R. Liang, J. Fu, R. Gao, D. Luo, Z. Bai, Y. Hu, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 5858 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- J. Liu, X. Meng, J. H. Xie, B. Liu, B. Tang, R. Y. Wang, C. Wang, P. Gu, Y. D. Song, S. C. Huo and J. L. Zou, Adv. Funct. Mater., 2023, 33, 2300579 CrossRef CAS.
- M. M. Sabri, J. Jung, D. H. Yoon, S. Yoon, Y. J. Tak and H. J. Kim, J. Mater. Chem. C, 2015, 3, 7499–7505 RSC.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. M. Huang and Y. Chen, ACS Nano, 2014, 8, 6856–6862 CrossRef CAS PubMed.
- Y. Ahmed and R. Salman, Case Stud. Chem. Environ. Eng., 2023, 8, 100386 CrossRef CAS.
- L. Negahdar, F. Zeng, S. Palkovits, C. Broicher and R. Palkovits, Chemelectrochem, 2019, 6, 5588–5595 CrossRef CAS.
- H. S. Park, E. Seo, J. Yang, Y. Lee, B. S. Kim and H. K. Song, Sci. Rep., 2017, 7, 7150 CrossRef PubMed.
- X. Han, W. Zhang, X. Ma, C. Zhong, N. Zhao, W. Hu and Y. Deng, Adv. Mater., 2019, 31, 1808281 CrossRef PubMed.
- Q. Lu, J. Yu, X. H. Zou, K. M. Liao, P. Tan, W. Zhou, M. Ni and Z. P. Shao, Adv. Funct. Mater., 2019, 29, 1904481 CrossRef.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- X. D. Duan, S. S. Ren, F. Y. Ge, X. K. Zhu, M. D. Zhang and H. G. Zheng, Nanoscale, 2021, 13, 17655–17662 RSC.
- G. T. Fu, Y. F. Chen, Z. M. Cui, Y. T. Li, W. D. Zhou, S. Xing, Y. W. Tang and J. B. Goodenough, Nano Lett., 2016, 16, 6516–6522 CrossRef CAS PubMed.
- T. R. Zhan, X. L. Liu, S. S. Lu and W. G. Hou, Appl. Catal., B, 2017, 205, 551–558 CrossRef CAS.
- L. C. Wei, L. J. Qiu, Y. Y. Liu, J. M. Zhang, D. S. Yuan and L. Wang, ACS Sustainable Chem. Eng., 2019, 7, 14180–14188 CrossRef CAS.
- H. P. Guo, X. W. Gao, N. F. Yu, Z. Zheng, W. B. Luo, C. Wu, H. K. Liu and J. Z. Wang, J. Mater. Chem. A, 2019, 7, 26549–26556 RSC.
- X. D. Duan, S. S. Ren, N. Pan, M. D. Zhang and H. G. Zheng, J. Mater. Chem. A, 2020, 8, 9355–9363 RSC.
- W. J. Liu, D. W. Rao, J. Bao, L. Xu, Y. C. Lei and H. M. Li, J. Energy Chem., 2021, 57, 428–435 CrossRef CAS.
- C. Han, W. J. Li, C. Z. Shu, H. P. Guo, H. K. Liu, S. X. Dou and J. Z. Wang, ACS Appl. Energy Mater., 2019, 2, 5363–5372 CrossRef CAS.
- J. Chen, B. W. Yao, C. Li and G. Q. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- A. Irshad and N. Munichandraiah, ACS Appl. Mater. Interfaces, 2017, 9, 19746–19755 CrossRef CAS PubMed.
- J. Y. Lin, J. H. Liao and S. W. Chou, Electrochim. Acta, 2011, 56, 8818–8826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07765a |
| This journal is © The Royal Society of Chemistry 2024 |