
Pyridinic-N exclusively enriched CNT-encapsulated NiFe interfacial alloy nanoparticles on knitted carbon fiber cloth as bifunctional oxygen catalysts for biaxially flexible zinc–air batteries†
Milan Babu
Poudel
ab,
Subramanian
Vijayapradeep
a,
Karthikeyan
Sekar
c,
Jong Seok
Kim
d and
Dong Jin
Yoo
 *ab
*ab
aDepartment of Energy Storage/Conversion Engineering (BK21 FOUR) of Graduate School, Hydrogen and Fuel Cell Research Center, Jeonbuk National University, 567 Baekje-daero, Deokjin-gu, 54896, Jeonju-si, Jeollabuk-do, Republic of Korea. E-mail: djyoo@jbnu.ac.kr; Fax: +82-63-270-3909; Tel: +82-63-270-3608
bDepartment of Life Science, Jeonbuk National University, 567 Baekje-daero, Deokjin-gu, 54896, Jeonju-si, Jeollabuk-do, Republic of Korea
cDepartment of Chemistry, Faculty of Engineering and Technology, SRM institute of Science and Technology, Kattankulathur, Tamil Nadu 603 203, India
dSchool of Semiconductor and Chemical Engineering, Jeonbuk National University, 567 Baekje-daero, Deokjin-gu, 54896, Jeonju-si, Jeollabuk-do, Republic of Korea
First published on 1st March 2024
Abstract
The electrocatalytic oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) are the core reactions in reversible zinc–air batteries but are kinetically challenging because of their complex multi-electron transfer process. In this case, the exploration and rational design of non-precious bifunctional oxygen electrocatalysts with dense active sites and optimized electronic structures can facilitate favorable 4e− transfer. In this study, we report a highly reversible bifunctional electrocatalyst for flexible Zn–air batteries featuring pyridinic-N exclusively enriched carbon-nanotube-encased nickel–iron (NiFe) interfacial alloy nanoparticles derived from an LDH template on knitted carbon fiber cloth. The NiFe nanoparticles were catalytically released from NiFe-MOFs to form CNT tentacles when pyrolyzed in an inert atmosphere. XPS and XAS studies revealed the dominant presence of pyridinic-N, which reduces electron localization around NiFe centers and improves the interaction with oxygenated species. As a result, NiFe–N-CNT-KCC catalysts exhibited a low operating overpotential (η10) of 173 mV for the OER and a half-wave potential (E1/2) of 0.87 V for the ORR, which are superior to benchmark electrocatalysts. As an air cathode for zinc–air batteries, the NiFe–N-CNT-KCC-based battery showed an excellent electrochemical performance, with an open circuit voltage (OCV) of 1.55 V, high power density of 153 mW cm−2, excellent specific capacity of 793.2 mA h g−1, and long-term stability. Impressively, a solid-state flexible zinc–air battery with the NiFe–N-CNT-KCC cathode showed an admirable rate performance and exceptional mechanical stability under arbitrary bending and twisting conditions, showing great potential for practical implementation in next-generation high-power and high-energy-density batteries wearable applications.
1. Introduction
Zinc–air batteries offer a compelling solution for next-generation energy systems, with the potential to expand the use and improve the performances of portable devices and emerging smart electronics owing to their high specific energy density, reliable safety, low cost, and environmental friendliness.1–5 The electrochemical oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) governed by 4e− transfer play a critical role in the charge/discharge process in zinc–air batteries.6 However, the typically slow 4e− transfer oxygen kinetics at the cathode have a negative impact on their potential for commercial applications. Thus, to overcome these barriers, the exploration of highly efficient OER/ORR electrocatalysts is necessary.7,8 In this context, numerous studies have revealed that the reversible OER/ORR catalytic performances of materials depend on their physicochemical structures, including active sites, morphology, specific surface area, and electronic properties.9 One successful strategy for producing efficient heterogeneous catalysts for O2 oxidation and reduction is to create synergistic effects between precious metals (Pt, Pd, Au, etc.) and active metal oxides (RuO2, IrO2, etc.).10 However, another method is to encase nanocatalysts in porous and chemically resistive conductive layers as an affordable and effective approach for designing stable cathode electrocatalysts for the OER, ORR, and metal–air batteries.11,12Transition metals and their compounds, particularly 3d transition metals, have emerged as promising catalysts to replace noble metals because of their low cost, high abundance, and superior catalytic activity.13–15 Among them, nickel–iron (Ni–Fe) bimetallic catalysts have attracted attention for their low overpotential and durability under alkaline conditions.16 The combination of metallic NiFe alloys can enhance electrocatalysis with added synergistic properties. This is due to the lattice strain difference between Ni and Fe, leading to varying redox potentials and structural organization.17 When alloyed with semiconductor metal oxides, the coupling of NiFe alloys is predicted to enhance their catalytic activity by creating a Schottky barrier, which aids in charge separation.18 Thus, integrating NiFe alloys and their oxides can be an efficient approach to design electrocatalysts for the ORR and OER, leading to improved performance. However, bare-alloy catalysts that come into contact with liquid electrolytes are prone to erosion and particle aggregation during electrochemical cycling, resulting in the degradation of their catalytic activity.2,19 Alternatively, multi-component/metallic catalysts typically exhibit composition-dependent activity and selectivity, and their optimized structures often outperform single-component catalysts.9,20,21 Thus, to address the above-mentioned issues and enhance the catalytic activity of Ni–Fe nanomaterials, researchers have explored various strategies, including carbon-based coating. Encapsulating Ni–Fe nanoparticles with nitrogen-doped carbon or N-doped graphene layers has been shown to improve their catalytic performance by protecting the metal core from the harsh electrolyte and increasing their electroconductivity. For example, Zhang and coworkers obtained N-doped carbon supported Fe–Ni alloy via the pyrolysis of polyphthalocyanine.22 However, their thick carbon layers and small specific surface area limited the exposure of the active sites, leading to an unsatisfactory catalytic performance. Newly reported NiFe–N–C materials displayed superior ORR performances, which are even better than that of commercial precious group metals, under alkaline conditions by improving the density of FeNx and NiNx units for the ORR by tailoring the local carbon structure (the N and C atoms are located in the first and second coordination sphere of the transition metal centers, respectively).23 In the reverse process of the ORR, the OER of NiFe–N–C is kinetically sluggish, which is mainly due to the slow O2 formation because of the strong O* binding strength on the central Fe/Ni structure. Fe and Ni are normally the active sites for the ORR and OER in N-doped carbon hybrid catalyst systems. Specifically, Fe is the active site and Ni provides conductive support for catalysis.18 The introduction of M–Nx causes Fe3+ electron delocalization and transforms the spin state of the Fe3+ transition from a low spin (t2g5eg0) state to an intermediate spin (t2g4eg1) state, readily penetrating the antibonding π-orbital of oxygen and allowing excellent ORR activity.24 Thus, by alloying Fe with Ni, its intrinsic activity can be altered by inducing lattice strain in the alloy, which can be modulated by adjusting the content of the materials, with an anticipated increase in OER/ORR activity. However, a research bottleneck is that only particular configurations of N dopants will help to synergistically promote reversible oxygen electrocatalysts.11,25 In this case, it is anticipated that the introduction of more targeted pyridinic-N (pyri-N)-defected carbon will allow reversibility and 4 e− transfer.26 Wang et al. reported the synthesis of pyri-N-enriched 3D defective graphene, exhibiting a superior reversible OER/ORR process.27 Several theoretical studies revealed that the pyridinic-N-doped carbon site has a synergistic configuration with the distortion in the active sites, with a significantly minimal overpotential.9 Liu and coworkers reported that the chemisorption of O2 molecules and O-containing intermediates on electron-deficient carbon atoms can be facilitated by heteroatom doping, improving the oxygen reaction activity and the resulting Zn–air batteries.28 There are fewer concerns regarding the synthesis of layered double hydroxide (LDH) templates of 2D metal–organic frameworks (MOFs) with adequate metal-nitrogen coordination derivatives to alloy nanoparticles within pyridinic-N-enriched defective carbon. In principle, a synergistically coupled bimetallic alloy with a 2D MOF-derived carbon matrix is a practical ideal that can allow the tuning of reverse oxygen activity. However, the facile synthesis of this type of catalyst has not been achieved to date.
Herein, we rationally designed a flexible and stretchable air cathode utilizing some of the most abundant and affordable metals found on Earth. The global annual extraction of Fe and Ni is 2537 and 2.5 million tons, respectively, while the total extraction of Ir is less than 7 tons (global nickel mining industry-statistics and facts (https://www.statista.com), and iron ore facts, https://www.natural-resources.canada.ca). Furthermore, bulk Ir costs more than $196 million per ton, while Fe costs $113 per ton and Ni costs around $23800 per ton.29 Thus, the raw market price for NiFe–N-CNT-KCC catalyst is about 3.1 × 104 times less than that of conventional Ir-based catalysts. In this case, a strategy is proposed to synthesize hybrid electrocatalysts composed of a pyridinic-N-enriched carbon nanotube matrix. Herein, we report the in situ transformation of bimetallic MOFs into bimetallic NiFe-alloy nanoparticles confined in a pyridinic-N-enriched sp2 carbon skeleton on biaxially stretchable carbon cloth, which exhibited bifunctional ORR and OER. Their outstanding OER activity can be attributed to the large number of Ni/Fe–Nx active sites and the large amounts of graphitic and pyridinic-N derived from the MOF, in which graphitic-N facilitated the charge transfer efficiency and pyridinic-N aided in O2 adsorption. Further, aqueous and solid-state Zn–air batteries were assembled with the NiFe–N-CNT-KCC cathode, delivering an excellent power density output and cycling performance, as well as commendable stretchability and flexibility.
2. Experimental
2.1. Synthesis of NiFe-LDHs nanosheets
In the typical synthesis of NiFe-LDH nanosheets on knitted carbon fiber cloth (KCC), 2 mmol of Ni(NO3)2·6H2O, 1 mmol of Fe(NO3)3·6H2O, 15 mmol of (NH2)2CO, and 8 mmol NH4F were dissolved in 30 mL of ultrapure water under continuous magnetic stirring. The resultant solution was transferred to a Teflon-lined stainless-steel autoclave with a precleaned KCC immersed at an incline in the reaction system and heat treated at 120 °C for 6 h. After cooling to room temperature, the resultant KCC covered with NiFe-LDH was rinsed with ultrapure water and ethanol and dried in an electric oven at 80 °C overnight. For optimization, NiFe-LDHs were prepared with varying molar contents of Ni2+ and Fe3+. Also, Ni(OH)2 and FeO(OH) were prepared using the same process but different precursors.2.2. Synthesis of NiFe-MOF-KCC
NiFe-MOF-KCC was synthesized through a solvothermal method. In detail, 2 mmol 2-aminoterapthalic acid was dissolved in 30 mL ethanol under high-speed magnetic stirring. The previously obtained NiFe-LDHs-KCC was immersed in the as-prepared solution and transferred to a stainless-steel autoclave for thermal treatment (130 °C for 6 h). After the reaction was complete, NiFe-MOF-KCC was washed with ethanol and dried for further modifications. Ni-MOF-KCC and Fe-MOF-KCC were also prepared using the same procedure.2.3. Synthesis of bimetallic NiFe alloy encapsulated in pyridinic-N-doped carbon nanotubes on knitted carbon fiber cloth
The NiFe alloy encapsulated in pyridinic-N-doped carbon nanotubes was denoted as NiFe–N-CNT-KCC. The typical synthetic process of NiFe–N-CNT-KCC is as follows. NiFe-MOF-KCC was kept in a quartz glass boat and placed in the center of a tubular furnace. The furnace was heated at 900 °C for 2 h at a heating rate of 2 °C min−1 under a constant Ar flow and was naturally cooled to room temperature to obtain the final NiFe–N-CNT-KCC catalyst. For comparison, Ni–N-CM-KCC and Fe–N-CM-KCC were also prepared via the pyrolysis of Ni-MOF-KCC and Fe-MOF-KCC under the same conditions, respectively.3. Results and discussion
3.1. Physicochemical and morphological properties
A series of NiFe–N-CNT-KCC catalysts for the OER and ORR was achieved via a simultaneous hydrothermal and pyrolysis process of MOFs, which were in situ transformed from NiFe-LDH nanosheets grown directly on carbon cloth, as presented in Scheme 1. The reaction proceeded by the initial nucleation of the NiFe-LDH nanosheets as a precursor for the in situ transformation of NiFe-MOF, followed by the self-templated assembly of O-bridged Ni and Fe clusters with an ethanolic solution of 2-amino terephthalic acid. After pyrolysis in an Ar atmosphere, the NiFe-MOF nanosheets were transferred to hierarchical porous structures, where the carbon matrices, composed of sheets, and CNTs were retained on the surface of the carbon cloth. During pyrolysis, Ni2+ and Fe3+/2+ were reduced to NiFe-alloy nanoparticles, which further catalyzed the formation of N-doped CNTs. According to the field-emission scanning electron microscopy (FESEM) images, ultrafine quasi-vertically arranged NiFe-LDH nanosheet array structures with high aspect ratios were observed and retained their morphology without significant change after crystallization of the MOF (Fig. 1(A, D, B, and E), S8 and S9†). However, due to carbonization, the coordination bond between the metal and ligand in MOFs was broken and the overall structure gradually decomposed. Instantly, the FeNi ions/clusters were reduced to nanoclusters and catalyzed the formation of N-doped CNTs, forming FeNi nanoparticles at the tips of the hollow CNTs. This avoided the dissolution of the NiFe nanoparticles, Ostwald ripening, and agglomeration during the long-term electrocatalytic process.30 As displayed in Fig. 1(C and F), a large number of N-doped CNTS was extended homogeneously on the outside surface of the knitted carbon fiber cloth and interwoven with each other, forming a multidimensional interconnected network. The HR-TEM images indicated that metallic NiCo-alloy nanoparticles (∼10 nm) were covered by several-layered graphitic carbon with an interlayer spacing of 0.37 nm, congruent to the d(002) plane of graphitic carbon (Fig. 1(G–I)).17 Some randomly arranged graphitic layers on the carbon matrix can be ascribed to the mismatch in the atomic size of the host guest atom while doping. As shown in Fig. 1(H(i) and (ii)), the NiFe alloy encapsulated in the N-doped CNTs was identified by the lattice spacing of 0.20 nm and 0.17 nm, which is assigned to the d(111) and d(200) planes of NiFe, respectively.31 There were numerous dislocations and fragmented fringes within the graphitic carbon layers, which are advantageous for exposing additional edges and openings. Thus, the doped nitrogen level was enhanced, supporting the dynamic electrochemical process by increasing the number of active sites and improving their intrinsic activity. Interestingly, the NiFe alloy nanoparticles distinguished by the HAADF-STEM image and the TEM EDS color mapping further support the homogenous distribution of Ni, Fe, N, and C elements (Fig. 1(J)). In essence, the highly porous walls of the CNT-covered bimetallic alloy can expose abundant edges, promote the catalytic activity, and ensure long-term stability, enhancing the electrical conductivity. Compared to the pyrolysis product of bimetallic NiFe-MOF, the microstructure and morphological studies of monometallic Ni-MOF and Fe-MOF indicate no evolution of carbon nanotubes. The monometallic Ni and Fe nanoparticles were encapsulated in the nitrogen-doped graphitic carbon ring-like structure, suggesting that only bimetallic ions/clusters enable the catalytic growth of carbon nanotubes (Fig. S10 and S11†). The HRTEM image and TEM EDS mapping confirm the presence of the constituent elements.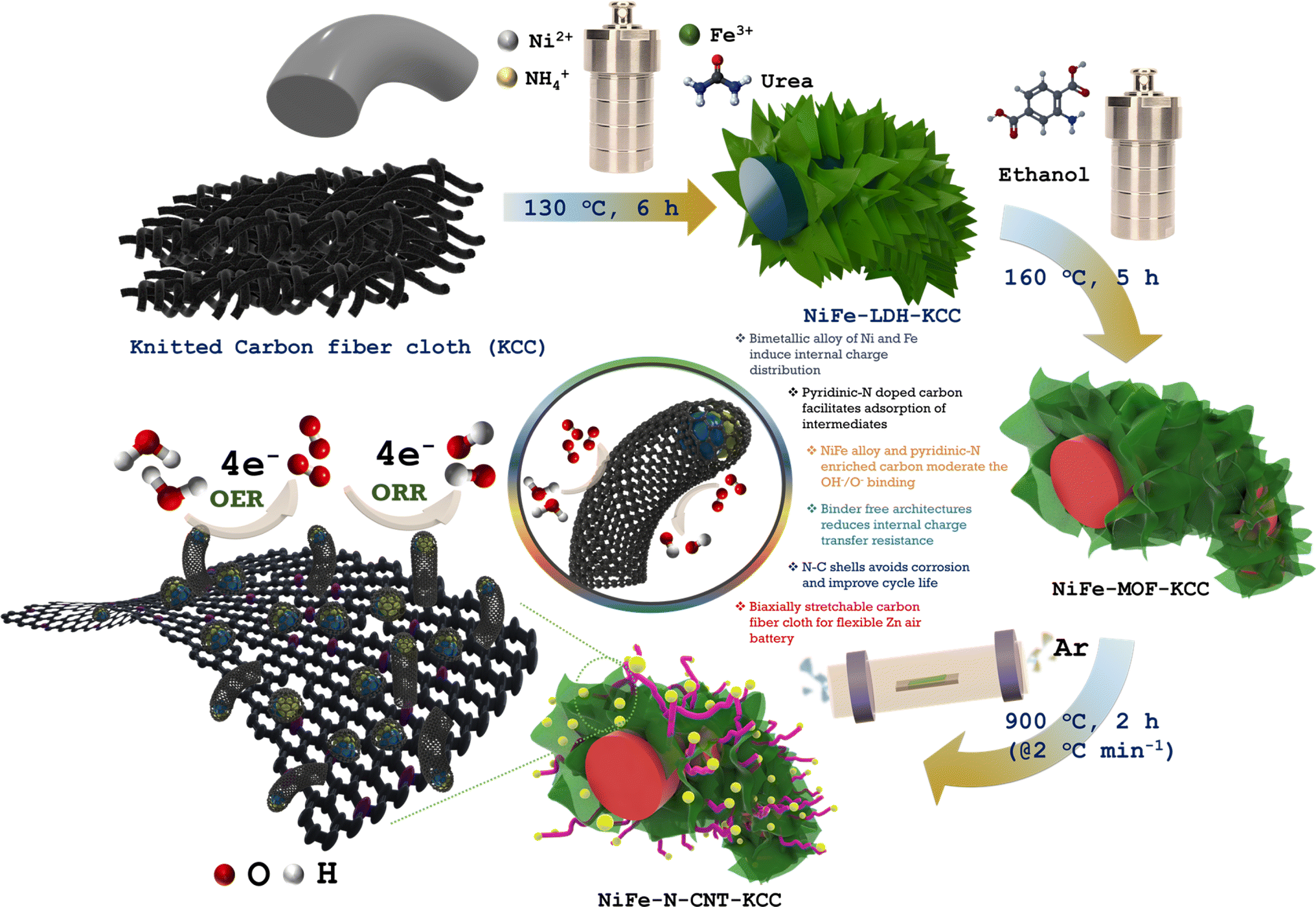 | ||
| Scheme 1 Fabrication scheme for NiFe–N-CNT-KCC catalysts. | ||
 | ||
| Fig. 1 Morphological and microstructural studies: FESEM images of (A and D) NiFe-LDH-KCC, (B and E) NiFe-MOF-KCC, and (C and F) NiFe–N-CNT-KCC at different magnifications. (G–I) TEM and HRTEM images indicating lattice spacing. (J) HAADF-STEM images and analogous elemental mapping of Fe, Ni, C and N on NiFe–N-CNT-KCC. | ||
The crystal phases of NiFe–N-CNT-KCC, NiFe-MOF-KCC, NiFe-LDH-KCC were identified by XRD, and the results are shown in Fig. S2–S4,† respectively. The characteristic diffraction peaks of NiFe-LDH well matched the rhombohedral nickel iron hydrotalcite structure (CCDC#040-0215).32,33 As expected, the XRD patterns of NiFe-MOF contained several peaks in the 2θ range of 5–50°, indicating the complete transformation of LDH to NiFe-MOF. In the XRD patterns of NiFe–N-CNT-KCC, Ni–N-CM-KCC, and Fe–N-CM-KCC, the wide diffraction peak at ∼25° can be indexed to the d(003) plane of graphitic carbon, whereas the featured peaks at 2θ angles of 44.4° and 51.62° can be attributed to the (111) and (200) planes of the NiFe alloy, respectively. Interestingly, the XRD patterns of the Ni–N-CM-KCC and Fe–N-CM-KCC showed no changes in the crystal structure and crystal plane spacing because of the almost equivalent atomic radii of Ni and Fe. However, in all the XRD patterns, an intense peak centered at around 2θ = ∼26° is related to the carbon cloth.
XPS was further used to probe the surface chemistry of NiFe–N-CNT-KCC, Ni–N-CM-KCC, and Fe–N-CM-KCC. The existence of the respective elements was evidenced by the XPS survey spectra (Fig. 2(A), S5 and S6†). Analogously, the high-resolution Ni 2p XPS also displayed two regions of Ni 2p3/2 and Ni 2p1/2. The dominant Ni0 peaks at 851.59 eV and 867.23 eV were clear over the oxidized Ni and satellite peaks (Fig. 2(B) and S5(c)†).34,35 The high-resolution Fe 2p XPS spectrum of NiFe–N-CNT-KCC, as shown in Fig. 2(C) and S6(B),† confirms the presence of Fe0 (Fe 2p3/2 at 706.092 and Fe 2p1/2 at 719.14 eV) and Fe2+ (Fe 2p3/2 at 709.7 and Fe 2p1/2 at 723.1 eV), with a pair of shakeup satellite humps.36,37 The presence of Ni2+/3+ and Fe2+/3+ in the NiFe alloy can be ascribed to the surface oxidation upon long standing in air. Notably the Fe0 2p3/2 peak position of Fe–N-CM-KCC shifted to a lower binding energy than that of NiFe–N-CNT-KCC, and the fitting Ni0 2p3/2 in Ni–N-CM-KCC showed negative shifts compared to that of NiFe–N-CNT-KCC, indicating the synergistic effect of Ni on the electronic configuration of Fe and vice versa. The negative shifts observed in the corresponding peaks indicated an electron interaction between the N atoms in the carbon shell and NiFe alloy; the difference in electronegativity facilitates the electron transfer.38Fig. 2(D) shows an intense peak in the C1s HRXPS at 284.5 eV, revealing the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (sp2 carbon).39 Subsequently, the C–C (285.02 eV, N-sp2-C), C–N (286.59 eV, N-sp3-C), and CO (280.13) groups are associated with the knitted carbon cloth.40 The N 1s spectra contained five peaks of pyridinic-N, Fe/Ni–Nx, pyro-N, graphitic-N, and partially oxidized N at the respective binding energies as major components (Fig. 2(D)). NiFe–N-CNT-KCC contained 42% pyridinic-N compared to pyrrolic-N and graphitic-N. Pyridinic-N and Fe/Ni–Nx had the richest distribution of active sites for enhancing the selectivity efficiency for the OER and ORR (especially regarding 4e− transfer). The XPS spectra of Ni–N-CM-KCC and Fe–N-CM-KCC show similar trends, indicating the dominance of the pyridinic-N groups. Meanwhile, Raman analysis revealed two intense peaks at 1349.48 and 1576.30 cm−1, which are ascribed to the disordered carbon atoms and sp2-hybridized graphitic carbon, respectively (Fig. S7†).
C (sp2 carbon).39 Subsequently, the C–C (285.02 eV, N-sp2-C), C–N (286.59 eV, N-sp3-C), and CO (280.13) groups are associated with the knitted carbon cloth.40 The N 1s spectra contained five peaks of pyridinic-N, Fe/Ni–Nx, pyro-N, graphitic-N, and partially oxidized N at the respective binding energies as major components (Fig. 2(D)). NiFe–N-CNT-KCC contained 42% pyridinic-N compared to pyrrolic-N and graphitic-N. Pyridinic-N and Fe/Ni–Nx had the richest distribution of active sites for enhancing the selectivity efficiency for the OER and ORR (especially regarding 4e− transfer). The XPS spectra of Ni–N-CM-KCC and Fe–N-CM-KCC show similar trends, indicating the dominance of the pyridinic-N groups. Meanwhile, Raman analysis revealed two intense peaks at 1349.48 and 1576.30 cm−1, which are ascribed to the disordered carbon atoms and sp2-hybridized graphitic carbon, respectively (Fig. S7†).
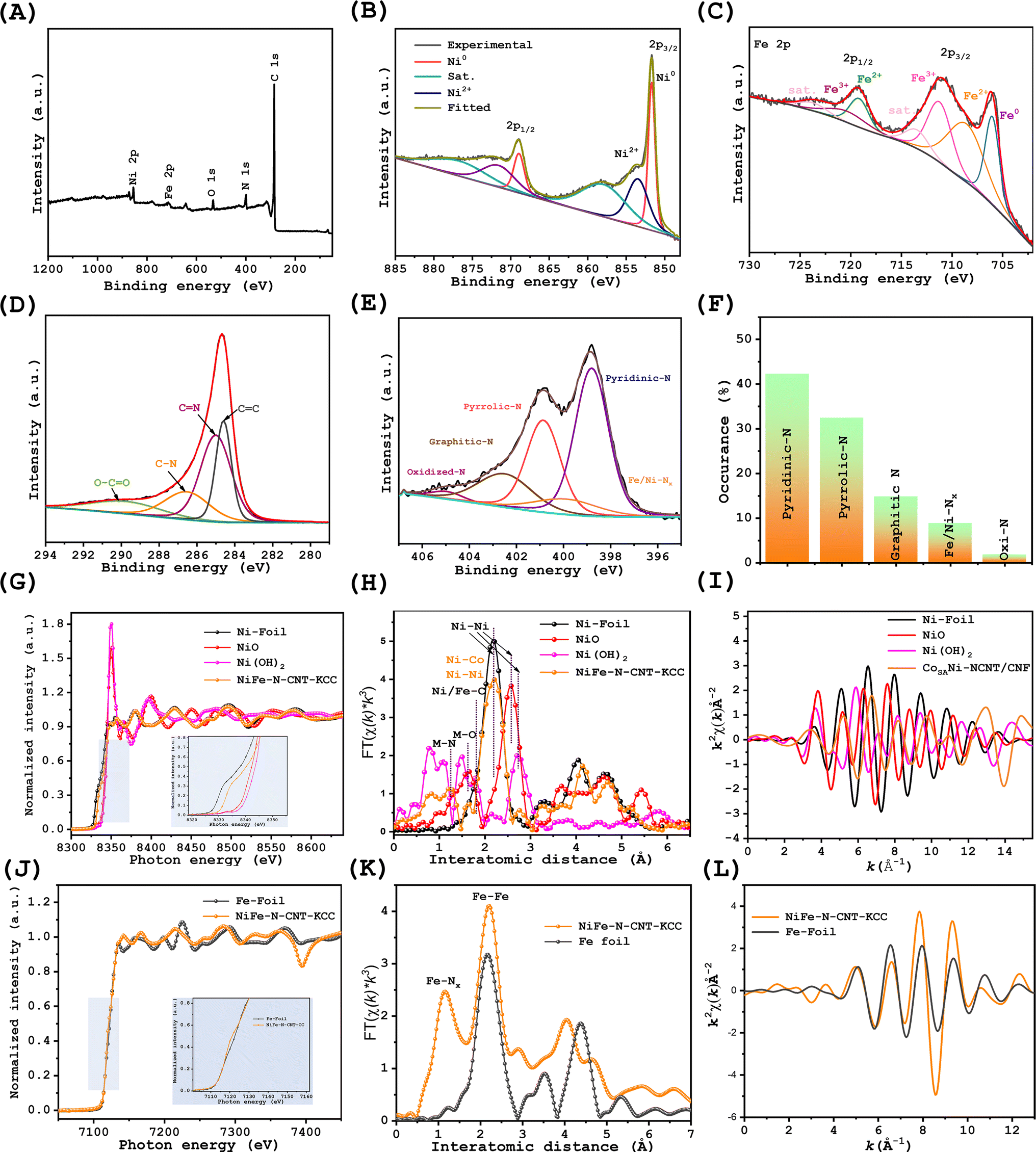 | ||
| Fig. 2 Electronic and atomic coordination measurements. (A) XPS survey spectrum. (B–E) High-resolution Ni 2p, Fe 2p, C 1s, and N 1s spectra and (F) corresponding N species content in NiFe–N-CNT-KCC, (G) Ni K-edge XANES spectra of NiFe–N-CNT-KCC with respect to NiO, Ni(OH)2, and Ni-foil; (H) k3-weighted FT-EXAFS spectra of NiFe–N-CNT-KCC with respect to NiO, Ni(OH)2, and Ni-foil. (I) Ni K-edge oscillation function k3X(k) of NiFe–N-CNT-KCC and Fe-foil. (J) Fe K-edge XANES spectra of NiFe–N-CNT-KCC with respect to Fe-foil; (K) k3-weighted FT-EXAFS spectra of NiFe–N-CNT-KCC and Fe-foil. (L) Fe K-edge oscillation function k3X(k) of NiFe–N-CNT-KCC and Fe-foil. | ||
The XPS results indicated the surface oxidation of the catalysts, although this method was not sufficient to probe the alloy core. Instead, X-ray absorption spectroscopy was conducted to determine the bonding configuration and coordination environment of Ni in NiFe–N-CNT/KCC. Fig. 2(G) shows the Ni K-edge X-ray absorption near-edge structure (XANES) spectra of NiFe–N-CNT-KCC, NiO, Ni(OH)2, and Ni foil. The near-edge absorption energy positions of NiFe–N-CNT-KCC were close to that of Ni-foil, with a slight positive shift due to the positive valence between the dominant Ni0 feature and the partial oxidation to Ni2+. The Fourier transfer extended absorption fine structure (FT-EXAFS, Fig. 2(H)) analysis showed three intense peaks located at 1.23, 1.6, and 2.2 Å.41 In contrast, the peak at 2.2 Å is assigned to the typical Ni–Ni and Ni–Fe pair, the peak at around 1.8 Å to the Ni–C sites, and peak at 1.3 Å to the Ni–N coordination. The presence of a peak at around 1.6 Å is ascribed to the Ni–O interactions at the NiO sites. This verifies the low N coordination number and presence of binuclear sites. Further, Ni k-edge extended XANES showed equal oscillations ascribed to Ni-foil, confirming its high metallicity (Fig. 2(I)). Fe k edge XANES was also conducted for NiFe–N-CNT-KCC, and its results were compared with that of Fe-foil and Fe–O to understand the coordination of Fe in the NiFe–N-CNT/KCC catalysts (Fig. 2(J–L)), respectively.
3.2. OER and ORR activities
The electrocatalytic OER performances of NiFe–N-CNT-KCC, Ni–N-CM-KCC, Fe–N-CM-KCC, and their precursors were measured in 1.0 M KOH solution. Fig. 3(A) and S13(G)† display the OER polarization curves together with that of the benchmark RuO2. Interestingly, there were substantial variations in the anodic peak potentials of the nanocatalysts, which can be ascribed to the redox reactions on the surfaces of the active Ni and Fe sites.42 The OER activity of the bimetallic NiFe–N-CNT-KCC greatly surpassed the performance of the individual metals and their precursors, indicating that the combination of metallic Ni and Fe generated highly active sites and boosted the active catalytic sites for the adsorption of oxygen intermediates, generating molecular O2. For example, NiFe–N-CNT-KCC required an overpotential of only 173 mV to reach 10 mA cm−2, demonstrating its suitable catalytic activity. This overpotential is significantly lower than that of its Ni–N-CM-KCC, Fe–N-CM-KCC, and NiFe-LDH-KCC counterparts (Fig. 3(B)). The ratio of Ni and Fe in the synthesis of NiFe-LDH-KCC was optimized, and Fig. S12† shows that the optimal catalyst was synthesized at an Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratio of 1:1. The overpotential for NiFe–N-CNT-KCC was ∼57 mV lower than that of the benchmark RuO2 and comparable to that of metal-based carbon composite catalysts. The NiFe–N-CNT-KCC catalyst also showed a lower Tafel slope of 37.87 mV dec−1 than the commercial RuO2 catalyst, suggesting a faster OER rate in NiFe–N-CNT-KCC. In addition, the highest electron transfer efficiency was evidenced by the electrochemical impedance spectroscopy (EIS) plot of NiFe–N-CNT-KCC compared with the other prepared samples (Fig. 3(D)). Furthermore, the electrochemically active surface areas were investigated by analyzing the double-layer capacitance (Cdl), as shown in Fig. S13 and S14.† Surprisingly, the comparison of the Cdl of NiFe–N-CNT-KCC, Ni–N-CM-KCC, NiFe-LDH-KCC, Ni(OH)2, and FeO(OH) implied that NiFe–N-CNT-KCC has the most abundant active sites. The Nyquist plots of the NiFe–N-CNT-KCC electrocatalyst further manifest a much smaller charge transfer resistance than the other electrodes, indicating the significantly facilitated electron transfer and enhanced OER kinetics of NiFe–N-CNT-KCC. This result indicates that the lethargic OER kinetics of transition metals can be significantly accelerated by alloying nanoparticles and enriching the carbon matrix with pyridinic-N species. The superior OER activity of NiFe–N-CNT-KCC compared to the single-atom-centered Ni–N-CM-KCC and Fe–N-CM-KCC originated from the synergistic effect of the alloy nanoparticles and high contents of pyridinic-N and NiFe-Nx species. Further, it is widely recognized that the electron-accepting pyridinic-N can impart relatively high positive charge density on the sp2-hybridized C atoms, facilitating the generation of the reaction intermediates and promoting electron transfer between the catalysts and oxygen intermediates. NiFe–N-CNT-KCC retained its OER activity even with a sudden change in current density (Fig. 3(E)). The NiFe–N-CNT-KCC electrode exhibited admirable long-term stability with a minimal potential increase after 50 h of continuous chronopotentiometric operation at 10 mA cm−2 (Fig. 3(H)). After the long-term I/t test, the catalyst maintained its original morphology and contained all its original elements (Fig. S18†). However, its XPS spectrum (Fig. S19†), revealed its partial transformation to oxides and oxyhydroxides. The stability for the OER is attributed to the stable core–shell structure, where the NiFe-alloy nanoparticles can be effectively protected by the pyridinic-N-rich few-layer graphitic carbon shell.
:Fe ratio of 1:1. The overpotential for NiFe–N-CNT-KCC was ∼57 mV lower than that of the benchmark RuO2 and comparable to that of metal-based carbon composite catalysts. The NiFe–N-CNT-KCC catalyst also showed a lower Tafel slope of 37.87 mV dec−1 than the commercial RuO2 catalyst, suggesting a faster OER rate in NiFe–N-CNT-KCC. In addition, the highest electron transfer efficiency was evidenced by the electrochemical impedance spectroscopy (EIS) plot of NiFe–N-CNT-KCC compared with the other prepared samples (Fig. 3(D)). Furthermore, the electrochemically active surface areas were investigated by analyzing the double-layer capacitance (Cdl), as shown in Fig. S13 and S14.† Surprisingly, the comparison of the Cdl of NiFe–N-CNT-KCC, Ni–N-CM-KCC, NiFe-LDH-KCC, Ni(OH)2, and FeO(OH) implied that NiFe–N-CNT-KCC has the most abundant active sites. The Nyquist plots of the NiFe–N-CNT-KCC electrocatalyst further manifest a much smaller charge transfer resistance than the other electrodes, indicating the significantly facilitated electron transfer and enhanced OER kinetics of NiFe–N-CNT-KCC. This result indicates that the lethargic OER kinetics of transition metals can be significantly accelerated by alloying nanoparticles and enriching the carbon matrix with pyridinic-N species. The superior OER activity of NiFe–N-CNT-KCC compared to the single-atom-centered Ni–N-CM-KCC and Fe–N-CM-KCC originated from the synergistic effect of the alloy nanoparticles and high contents of pyridinic-N and NiFe-Nx species. Further, it is widely recognized that the electron-accepting pyridinic-N can impart relatively high positive charge density on the sp2-hybridized C atoms, facilitating the generation of the reaction intermediates and promoting electron transfer between the catalysts and oxygen intermediates. NiFe–N-CNT-KCC retained its OER activity even with a sudden change in current density (Fig. 3(E)). The NiFe–N-CNT-KCC electrode exhibited admirable long-term stability with a minimal potential increase after 50 h of continuous chronopotentiometric operation at 10 mA cm−2 (Fig. 3(H)). After the long-term I/t test, the catalyst maintained its original morphology and contained all its original elements (Fig. S18†). However, its XPS spectrum (Fig. S19†), revealed its partial transformation to oxides and oxyhydroxides. The stability for the OER is attributed to the stable core–shell structure, where the NiFe-alloy nanoparticles can be effectively protected by the pyridinic-N-rich few-layer graphitic carbon shell.
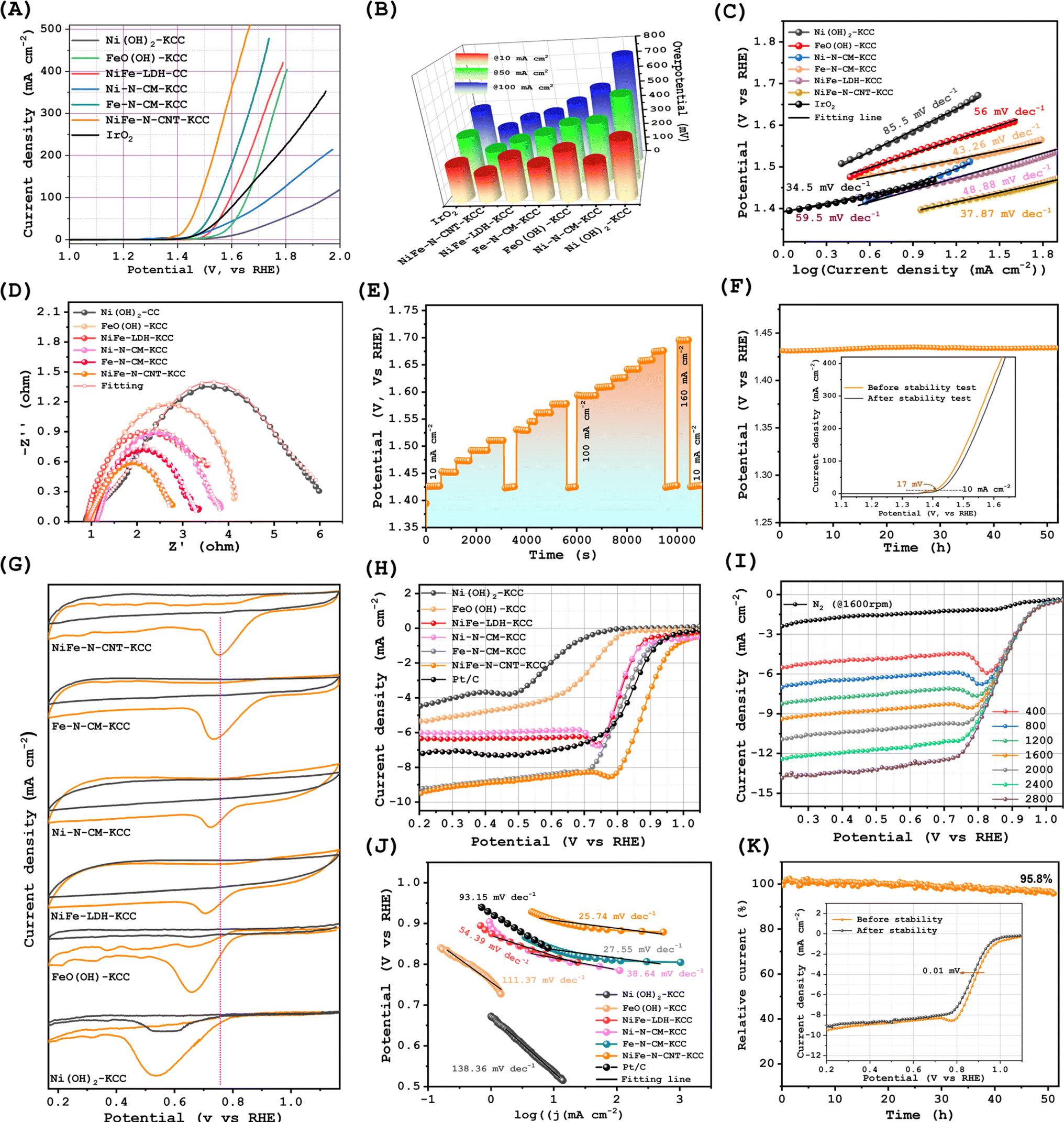 | ||
| Fig. 3 Oxygen bifunctional electrochemical activities: (A) LSV profiles for the OER and (B) overpotentials of all the catalysts at different current densities. (C) OER Tafel slopes, (D) Nyquist plots, (E) multistep chronopotentiometry plot for the NiFe–N-CNT-KCC catalyst, (F) Long-term chronopotentiometry analysis for NiFe–N-CNT-KCC (inset; LSV profiles of NiFe–N-CNT-KCC after 52 h), (G) cyclic voltammetry of all the samples in N2 and O2 saturated 0.1 M KOH, (H) LSV profiles under continuous O2 flow, (I) comparison of E1/2 and Jk, (J) ORR Tafel plots, and (K) chronoamperometric test for NiFe–N-CNT-KCC with a minimum degradation of its halfwave potential. | ||
To access the electrocatalytic performance of the as-prepared Ni–N-CM-KCC, Fe–N-CM-KCC, NiFe-LDH-KCC, and NiFe–N-CNT-KCC, their ORR activities were evaluated in an alkaline electrolyte. CV measurements were first performed in O2- and N2-saturated 0.1 M KOH solutions. All the samples demonstrated reduction peaks, and the peak potential for NiFe–N-CNT-KCC was more highly positive (Fig. 3(G)). As shown in Fig. 3(H), the LSV measurements showed that the NiFe–N-CNT-KCC electrode exhibited a better ORR performance compared to the other electrodes, with a limiting kinetic current density of around 9.66 mA cm−2, which is even higher than that of the expensive commercial Pt/C catalyst (Fig. S17†). As expected, NiFe–N-CNT-KCC exhibited a more highly positive half-wave potential (E1/2) of 0.87 V compared with that of Fe–N-CM-KCC, Ni–N-CM-KCC, NiFe-LDH-KCC, Ni(OH)2, FeO(OH), and even Pt/C catalysts (Fig. 3(I)). Additionally, the lower Tafel values of NiFe–N-CNT-KCC exemplified its outstanding ORR kinetics. These results support the construction of binder-free pyridinic-N-enriched carbon nanostructure-encapsulated bimetallic NiFe alloy interfaces, with hierarchical porosity that provides additional structural advantages. Most importantly, the presence of pyridinic-N bonds dominantly contribute to the ORR process and induced charge transfer from the π orbital to antibonding orbitals in O2, resulting in significantly weakened O–O bonds and further dissociation of the *OOH intermediate into *O and *OH, which is consistent with previous studies.43 The thin pyridinic-N-doped carbon matrix-encased NiFe interface alloy avoided the isolation of the active sites and reduced the dead mass, exposing abundant active sites. Meanwhile, to validate the superior ORR activities, the electron transfer number (n) was calculated using RRDE polarization curves at different rpm in the potential range of 0.2 to 0.8 V (Fig. 3(I), S15 and S16†). The electron transfer number for the NiFe–N-CNT-KCC catalysts during the ORR was ∼3.93, approaching that of the commercial Pt/C catalyst and satisfying the desirable 4e− reduction pathway. Ni–N-CM-KCC and Fe–N-CM-KCC showed the same trends, although their precursors of NiFe-LDH-KCC, Ni(OH)2-KCC, and FeO(OH)-KCC exhibited different n values. Pyridinic-N contributed to the 4e− transfer. Chronoamperometric measurements were performed for 50 h and confirmed the robust ORR stability of NiFe–N-CNT-KCC (Fig. 3(K)). In addition, there were no distinct morphological and microstructural changes in NiFe–N-CNT-KCC after the stability test, validating its outstanding long-term stability (Fig. S20†). To ascertain the positive effect of pyridinic-N, XPS analysis was performed for NiFe–N-CNT-KCC after the ORR (Fig. S21†). The pyridinic-N content was marginally converted to pyrrolic/pyridinic-N. This difference in N content post-ORR indicates that carbon atoms next to pyridinic-N can easily interact with the oxygen intermediates, which then improve the overall ORR activity.26,44 The overall bifunctional oxygen electrocatalytic activities of the NiFe–N-CNT-KCC catalysts were estimated by calculating the potential differences (ΔE) between E1/2 and η10 under the same operating conditions. Remarkably, the NiFe–N-CNT-KCC catalysts exhibited a substantially lower ΔE than that of Pt/C + IrO2, which is comparable to most of the state-of-art bifunctional electrocatalysts. This suggests the superior advantages of NiFe–N-CNT-KCC in the charge/discharge process for rechargeable Zn–air batteries. Thus, it can be concluded that the fundamental synergistic electronic interaction between the bimetallic alloys and pyridinic-N-enriched carbon contributes the high oxygen activity, which is expected to be a feasible alternative to commercial expensive catalysts.
3.3. Zinc–air battery performance
Additionally, the excellent overall OER/ORR performance of NiFe–N-CNT-KCC exhibited a narrow potential gap (ΔE) between the half-wave potential of the ORR and OER at η10, which outperforms the commercial Pt/C and IrO2 analogs. Leveraging the results of this investigation, the prepared NiFe–N-CNT-KCC catalyst is a robust catalyst for the production of a metal–air cathodes for rechargeable Zn–air batteries. Accordingly, a homemade Zn–air battery was assembled using a Zn plate as the anode, NiFe–N-CNT-KCC as the air cathode, and 6.0 M KOH + 0.2 M Zn(CH2COO)2 as the electrolyte, as represented in Fig. 4(A). For comparison, NiFe–N-CNT-KCC and Pt/C were tested under identical conditions. As shown in Fig. 4(B), the NiFe–N-CNT-KCC-based Zn–air battery exhibited a stable open-circuit voltage of 1.55 V, which was superior to that of a Pt/C + IrO2-based battery (1.42 V). As expected, the NiFe–N-CNT-KCC–air cathode delivered a high-power density of 153.02 mW cm−2 and a larger specific capacity of 793.2 mA h gZn−1, which are superior to that of its noble Pt/C + IrO2 counterparts (Fig. 4(C and D)), respectively. The reliable performance of NiFe–N-CNT-KCC was validated by illuminating a pair of light-emitting diode (LED) indicators connected in series. Most interestingly, when two aqueous NiFe–N-CNT-KCC-based Zn–air batteries were connected in series, the output was 3.0 V and water splitting occurred, producing O2 and H2 gas in an H-cell consisting of a Pt/C anode and NiFe–N-CNT-KCC cathode. As illustrated in Fig. 4(E), the NiFe–N-CNT-KCC–air cathode presented a minor increase in the trip voltage during 180 h long-term operation at 10 mA cm−2. In contrast, the Pt/C + IrO2-based battery demonstrated a very poor cycling performance, which is likely caused by the corrosion of Pt/C in strong electrolyte conditions. Overall, our NiFe–N-CNT-KCC electrode outperformed commercial electrolysis devices and current state-of-the-art aqueous Zn–air batteries in terms of peak power density, specific capacity, and long-term durability (Fig. 4(F)). The unique electronic structure and abundantly exposed active sites obtained by the LDH template synthesis method contributed to the excellent bifunctional OER and ORR activities during the charge/discharge process in Zn–air batteries. | ||
| Fig. 4 Zn–air battery performance: (A) schematic representation of the NiFe–N-CNT-KCC-based aqueous Zn–air battery, (B) open circuit potentials for NiFe–N-CNT-KCC with respect to commercial Pt/C + IrO2, (C) specific capacity plot for NiFe–N-CNT-KCC and Pt/C + IrO2, (D) discharging and power density profiles of assembled Zn–air batteries, (E) cycling durability of the NiFe–N-CNT-KCC-based aqueous Zn–air battery, (F) comparison of the NiFe–N-CNT-KCC-based aqueous Zn–air battery with some recently reported catalysts, (G) schematic illustration of the NiFe–N-CNT-KCC-based solid and flexible Zn–air battery, (H) open circuit potentials for NiFe–N-CNT-KCC, (I) discharging and power density profiles of the flexible Zn–air battery, (J) specific capacity plot, and (K) cycling durability of the NiFe–N-CNT-KCC-based solid and flexible Zn–air battery. | ||
To demonstrate practical applications and inspire future research, a solid-state Zn–air battery was assembled using NiFe–N-CNT-KCC as a cathode and Zn plate as an anode, sandwiching PVA-KOH/(0.2 M)Zn(CH3COO)2 as the electrolyte (Fig. 4(G)). This battery delivered a higher OCV of 1.42 V and reached the same voltage when twisted and bent in different orientations (Fig. 4(H) and S22†). This excellent flexibility is ascribed to the direct growth of bifunctional NiFe–N-CNTs on the biaxially stretchable carbon fiber cloth without using a potentially resistive polymer additive, allowing the strong adhesion of the active materials on the conductive current collector. In addition, the NiFe–N-CNT-KCC-based flexible Zn–air battery showed a peak power density of 112.3 mW cm−2 and a specific capacity of 556.54 mA h g−1 (Fig. 4(I and J)). Most importantly, the NiFe–N-CNT-KCC-based flexible Zn–air battery continuously and steadily operated for 500 charge–discharge cycles at 10 mA cm−2 without obvious degradation in its performance, demonstrating excellent long-term stability in both aqueous and solid-state conditions (Fig. 4(K) and S21†), respectively. In addition, two batteries connected in series could simultaneously power two blue LED lights and an electric fan (Fig. 4(H); (inset) and Supplementary Video VS1 and VS2†). The NiFe–N-CNT-KCC-based Zn–air battery exhibited the advantages of binder-free hierarchical growth of pyridinic-N-rich carbon and bimetallic alloy nanoparticles due to its bifunctional OER and ORR performances.
4. Conclusions
In conclusion, we demonstrated a facile approach for the fabrication of a free-standing and well-defined NiFe–N-CNT-KCC bifunctional electrocatalyst for high-energy aqueous and solid-state flexible Zn air batteries using LDH-template-derived MOFs followed by pyrolysis. During pyrolysis, the bimetallic MOFs catalyze the formation of carbon nanotubes and covalent interactions between the optimal active sites (bimetal NiFe nanoparticles are induced by the pyridinic-N-rich and disordered CNTs), enabling efficient mass transport for excellent OER/ORR activity compared to the expensive benchmark catalysts. The NiFe–N-CNT-KCC-based aqueous Zn–air battery exhibited a higher power density and specific capacity than that of its Pt/C + IrO2 counterparts. The battery cycles worked steadily within a small charge/discharge voltage over 140 h, indicating its robust stability. Moreover, our solid-state Zn–air battery revealed an excellent performance and mechanical strength, which demonstrates its potential as a candidate for use in flexible devices. This class of flexible catalysts may pave the way to cost-effective, efficient, and durable electrocatalysts for energy storage and conversion systems.Conflicts of interest
There is no conflict of interests.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by Ministry of Education (NRF-2022R1I1A1A01073572). This work was supported by “Regional Innovation Strategy (RIS)” through National Research Foundation of Korea (NRF) funded by Ministry of Education (MOE) (2023RIS-008).References
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- M. Wu, G. Zhang, N. Chen, Y. Hu, T. Regier, D. Rawach and S. Sun, ACS Energy Lett., 2021, 6, 1153–1161 CrossRef CAS.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- R. Santhosh Kumar, S. Ramakrishnan, S. Prabhakaran, A. R. Kim, D. R. Kumar, D. H. Kim and D. J. Yoo, J. Mater. Chem. A, 2022, 10, 1999–2011 RSC.
- M. B. Poudel, N. Logeshwaran, S. Prabhakaran, A. R. Kim, D. H. Kim and D. J. Yoo, Adv. Mater., 2024, 36, 2305813 CrossRef CAS PubMed.
- Z. Zhang, T. Wang, W. Wang, X. Wang, X. Luo, C. Cheng and X. Liu, Adv. Energy Mater., 2023, 13, 2300325 CrossRef CAS.
- Z. Li, S. Ji, C. Wang, H. Liu, L. Leng, L. Du, J. Gao, M. Qiao, J. H. Horton and Y. Wang, Adv. Mater., 2023, 35, 2300905 CrossRef CAS PubMed.
- R. S. Kumar, S. Prabhakaran, S. Ramakrishnan, S. C. Karthikeyan, A. R. Kim, D. H. Kim and D. J. Yoo, Small, 2023, 19, 2207096 CrossRef CAS PubMed.
- F. Qiang, J. Feng, H. Wang, J. Yu, J. Shi, M. Huang, Z. Shi, S. Liu, P. Li and L. Dong, ACS Catal., 2022, 12, 4002–4015 CrossRef CAS.
- S. Ramakrishnan, D. B. Velusamy, S. Sengodan, G. Nagaraju, D. H. Kim, A. R. Kim and D. J. Yoo, Appl. Catal., B, 2022, 300, 120752 CrossRef CAS.
- S. Ding, L. He, L. Fang, Y. Zhu, T. Li, Z. Lyu, D. Du, Y. Lin and J.-C. Li, Adv. Energy Mater., 2022, 12, 2202984 CrossRef CAS.
- N. Logeshwaran, S. Ramakrishnan, S. S. Chandrasekaran, M. Vinothkannan, A. R. Kim, S. Sengodan, D. B. Velusamy, P. Varadhan, J.-H. He and D. J. Yoo, Appl. Catal., B, 2021, 297, 120405 CrossRef CAS.
- G. Yasin, S. Ali, S. Ibraheem, A. Kumar, M. Tabish, M. A. Mushtaq, S. Ajmal, M. Arif, M. A. Khan, A. Saad, L. Qiao and W. Zhao, ACS Catal., 2023, 13, 2313–2325 CrossRef CAS.
- C. Zhou, X. Chen, S. Liu, Y. Han, H. Meng, Q. Jiang, S. Zhao, F. Wei, J. Sun, T. Tan and R. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704 CrossRef CAS PubMed.
- D. Acharya, I. Pathak, B. Dahal, P. C. Lohani, R. M. Bhattarai, A. Muthurasu, T. Kim, T. H. Ko, K. Chhetri and H. Y. Kim, Carbon, 2023, 201, 12–23 CrossRef CAS.
- Y. Niu, X. Teng, S. Gong and Z. Chen, J. Mater. Chem. A, 2020, 8, 13725–13734 RSC.
- P. Liu, D. Gao, W. Xiao, L. Ma, K. Sun, P. Xi, D. Xue and J. Wang, Adv. Funct. Mater., 2018, 28, 1706928 CrossRef.
- X. Liu, M. Park, M. G. Kim, S. Gupta, G. Wu and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 9654–9658 CrossRef CAS PubMed.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS.
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2022, 34, 2108133 CrossRef CAS PubMed.
- Z. Li, Y. Xie, J. Gao, X. Zhang, J. Zhang, Y. Liu and G. Li, J. Mater. Chem. A, 2023, 11, 26573–26579 RSC.
- Z. Zhang, Y. Qin, M. Dou, J. Ji and F. Wang, Nano Energy, 2016, 30, 426–433 CrossRef CAS.
- H. Liang, Y. Zhang, F. Chen, S. Jing, S. Yin and P. Tsiakaras, Appl. Catal., B, 2019, 244, 559–567 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- A. Muthurasu, I. Pathak, D. Acharya, Y. R. Rosyara and H. Y. Kim, J. Mater. Chem. A, 2024, 12, 1826–1839 RSC.
- Y. Ha, B. Fei, X. Yan, H. Xu, Z. Chen, L. Shi, M. Fu, W. Xu and R. Wu, Adv. Energy Mater., 2020, 10, 2002592 CrossRef CAS.
- Q. Wang, Y. Ji, Y. Lei, Y. Wang, Y. Wang, Y. Li and S. Wang, ACS Energy Lett., 2018, 3, 1183–1191 CrossRef CAS.
- S. Liu, M. Wang, X. Sun, N. Xu, J. Liu, Y. Wang, T. Qian and C. Yan, Adv. Mater., 2018, 30, 1704898 CrossRef PubMed.
- Z. P. Ifkovits, J. M. Evans, P. A. Kempler, M. B. Morla, K. H. Pham, J. A. Dowling, A. I. Carim and N. S. Lewis, ACS Energy Lett., 2022, 7, 4258–4264 CrossRef CAS.
- M. Karuppannan, Y. Kim, S. Gok, E. Lee, J. Y. Hwang, J.-H. Jang, Y.-H. Cho, T. Lim, Y.-E. Sung and O. J. Kwon, Energy Environ. Sci., 2019, 12, 2820–2829 RSC.
- X. Zheng, J. Deng, N. Wang, D. Deng, W.-H. Zhang, X. Bao and C. Li, Angew. Chem., Int. Ed., 2014, 53, 7023–7027 CrossRef CAS PubMed.
- A. Khataee, T. Sadeghi Rad, S. Nikzat, A. Hassani, M. H. Aslan, M. Kobya and E. Demirbaş, Chem. Eng. J., 2019, 375, 122102 CrossRef CAS.
- C. Wu, H. Li, Z. Xia, X. Zhang, R. Deng, S. Wang and G. Sun, ACS Catal., 2020, 10, 11127–11135 CrossRef CAS.
- P. Wei, X. Sun, Q. Liang, X. Li, Z. He, X. Hu, J. Zhang, M. Wang, Q. Li, H. Yang, J. Han and Y. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 31503–31513 CrossRef CAS PubMed.
- Z. Li, X. Wu, X. Jiang, B. Shen, Z. Teng, D. Sun, G. Fu and Y. Tang, Advanced Powder Materials, 2022, 1, 100020 CrossRef.
- S.-Q. Deng, Z. Zhuang, C.-A. Zhou, H. Zheng, S.-R. Zheng, W. Yan and J. Zhang, J. Colloid Interface Sci., 2023, 641, 265–276 CrossRef CAS PubMed.
- T. Li, Y. Hu, K. Liu, J. Yin, Y. Li, G. Fu, Y. Zhang and Y. Tang, Chem. Eng. J., 2022, 427, 131992 CrossRef CAS.
- A. Li, K. Shen, J. Chen, Z. Li and Y. Li, Chem. Eng. Sci., 2017, 166, 66–76 CrossRef CAS.
- A. Kundu, A. Samanta and C. R. Raj, ACS Appl. Mater. Interfaces, 2021, 13, 30486–30496 CrossRef CAS PubMed.
- S. Kim, S. Ji, H. Yang, H. Son, H. Choi, J. Kang and O. L. Li, Appl. Catal., B, 2022, 310, 121361 CrossRef CAS.
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113918 CrossRef CAS PubMed.
- M. A. Ahsan, A. R. Puente Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. T. Sreenivasan and J. C. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- X.-R. Wang, J.-Y. Liu, Z.-W. Liu, W.-C. Wang, J. Luo, X.-P. Han, X.-W. Du, S.-Z. Qiao and J. Yang, Adv. Mater., 2018, 30, 1800005 CrossRef PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07609a |
| This journal is © The Royal Society of Chemistry 2024 |