
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting electrocatalytic hydrogen evolution over a Mo2C–W2C heterostructure by interface-induced electron modulation†
Lijuan
Jiang
,
Ruijing
Wang
,
Huimin
Zhou
,
Guang-Feng
Wei
* and
Xuefeng
Wang
 *
*
Shanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: weigf@tongji.edu.cn; xfwang@tongji.edu.cn
First published on 15th January 2024
Abstract
Carbides have a high d-band electronic density of states at the Fermi level and hence characteristics resembling those of platinum. However, due to the strong adsorption of hydrogen intermediates on the surface of the carbides, the kinetics of the hydrogen evolution reaction (HER) is severely limited. Here, we demonstrate a strategy to improve the HER kinetics of carbides by modulating the surface electronic structure. Mo2C–W2C ultrafine nanoparticles on reduced graphene oxide (Mo2C–W2C/RGO) with strong electron interaction are constructed to improve the HER activity. The interfacial interaction between Mo2C and W2C adjusts the electronic structure of the surface, and the favorable hydrogen desorption on this surface is proved by density functional theory calculations. Benefitting from the adjusted electronic structures on the Mo2C–W2C heterointerface, the hydrogen adsorption free energies on both the Mo and W sites of Mo2C–W2C are close to the ideal values (0 eV). As expected, the Mo2C–W2C/RGO exhibits a superior HER activity with ultra-low overpotentials of 81 mV and 87 mV at a current density of −10 mA cm−2 under acidic and alkaline conditions, respectively, as well as remarkable stability. This work reveals the modulating effect of the electronic structure of carbides on the HER activity, which has a profound guiding effect for the directional design of advanced electrocatalysts.
Introduction
The hydrogen evolution reaction (HER) via water electrolysis enables the conversion of renewable energy sources (such as tidal, solar, wind, and geothermal energy) into green H2 energy, offering a promising route to alleviate the excessive consumption of fossil fuels and related environmental issues.1,2 In order to reduce the reaction energy barrier and overpotential, electrocatalysts play a crucial role in the process of water electrolysis.3–5 The HER is a classic two-electron reduction process with hydrogen intermediates (H*) as the only reaction intermediate (2H+ +2e− → H2 in acidic solution or 2H2O + 2e− → H2 + 2OH− in alkaline solution).6 According to the Sabatier principle,7 an active HER catalyst generally binds hydrogen neither too weakly nor too strongly, regardless of the pH value of the reaction solution. Currently, Pt group noble metals are considered as benchmark HER electrocatalysts showing extremely low overpotential.8–10 Unfortunately, their low abundance and high price hinder their large-scale application; thus, exploiting earth-abundant and high-performance HER electrocatalysts is inevitable.Remarkable advances have been made regarding the use of transition metals and their alloys, carbides, nitrides, chalcogenides, and phosphides, which feature their high activity, outstanding stability, and earth abundance.11,12 Among the candidates, transition-metal carbides, such as molybdenum carbides and tungsten carbides, have attracted increasing interest on account of their noble-metal-like electronic configuration, high electronic conductivity, wide pH applicability and outstanding catalytic activity correlated with tunable phase composition.13,14 In addition, a previous report has proposed that the hybridizing between the d-orbital of metallic tungsten/molybdenum atoms and s- or p-orbital of carbon atoms gave rise to a broadened d-band of molybdenum carbides and tungsten carbides resulting in high similarity to the d-band of Pt.15–18 Despite its unique properties, pure carbides show low activity for the HER.19 On the one hand, density functional theory (DFT) calculations have demonstrated that the strong adsorption of H* on the carbide surface facilitates the reduction of H+ but restricts the hydrogen desorption step.20 On the other hand, the synthesis of carbides usually requires a high-temperature annealing process, resulting in serious agglomeration of particles to form an irregular morphology with low specific surface area, thereby reducing the density of the active site.21,22 Efforts have been devoted to designing carbide nanostructures with large surface area and well-exposed active sites. For instance, Hu et al. reported a scalable and economical method to produce W2C nanodot-decorated CNT networks via a spray-drying approach.23 Zheng et al. presented a strategy to synthesize well-dispersed transition metal carbide nanoparticles.24 These strategies have been successfully used to prepare a variety of carbide nanomaterials with effectively improved activity, but the size of the as-obtained nanoparticles is hard to control and the size distribution range is wide. At the same time, these strategies for adjusting the nanoarchitectures of carbides cannot change their inherent properties of the strong adsorption of hydrogen intermediates, which also leads to the limitation of the activity of carbides. To address this issue, numerous approaches are being pursued to modify carbides by constructing heterophases and doping with heteroatoms to adjust the electronic properties and enhance the HER activity.19,20,25,26 Notably, the strong interaction at the two-phase interface can establish electronic transfer and generate electronic density redistribution between the two adjacently contacted components, adjusting the electronic structure and promoting electrocatalytic performance. Chen et al. identified that a synergy between Mo2N and Mo2C in biomass-derived heterostructures can exert the activity and satisfactory durability.25 From this perspective, constructing small-sized carbide nanoarchitectures and modulating the electronic structure of carbides should be an effective way to branch out for studying catalytic abilities.
Herein, a heterostructure composed of Mo2C and W2C anchored to reduced graphene oxide (denoted as Mo2C–W2C/RGO) is constructed via a sequential two-step approach. The Mo2C–W2C heterostructure with ultrafine size (about 2–3 nm) and abundant interfaces maximizes the exposure of active sites and facilitates the mass transfer process, while the interfacial interaction optimizes the electronic configuration of the catalyst. DFT calculations reveal the modulating effect of the electronic structure of the Mo2C–W2C heterostructure on the HER activity. The Mo2C–W2C heterostructure shows a favourable hydrogen desorption step, resulting in the optimized free energy for hydrogen adsorption (ΔGH*). As expected, the Mo2C–W2C/RGO affords an ultra-low overpotential (η10 = 81 and 87 mV for reaching a current density of −10 mA cm−2) under acidic and alkaline conditions.
Materials and methods
Chemicals
The graphene oxide (GO) powder was obtained from Qitaihe Baotailong Graphene New Material Co., Ltd. Ammonium metatungstate hydrate ((NH4)6H2W12O40·xH2O), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O), melamine and potassium hydroxide (KOH) were purchased from Aladdin. Concentrated sulfuric acid (H2SO4) and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. Nafion solution (5%) was purchased from Sigma-Aldrich.Synthesis of electrocatalysts
Take Mo2C–W2C/RGO-0.56 as an example: GO (0.20 g), (NH4)6Mo7O24·4H2O (0.36 g), (NH4)6H2W12O40·xH2O (0.50 g) and melamine (2.0 g) in double-distilled water (40 mL) were sonicated for 60 min. Then, after drying at 80 °C for a whole night, the obtained well-mixed precursor powder was pyrolyzed at 800 °C under Ar flow for 3 h, and the Mo2C–W2C/RGO catalyst was produced. According to the varied Mo/W molar ratio, a series of Mo2C–W2C/RGO were obtained, the products were denoted as Mo2C–W2C/RGO-n, in which n refers to the measured nMo/(Mo+W). According to a similar procedure, Mo2C/RGO and W2C/RGO were obtained in the absence of (NH4)6H2W12O40·xH2O and (NH4)6Mo7O24·4H2O, respectively. Graphene oxide (GO) was directly annealed in an Ar atmosphere at 800 °C for 3 h to obtain reduced graphene oxide (RGO).Material characterization
Scanning electron microscopy (SEM) was performed on a Hitachi S-4800 instrument. Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), energy dispersive X-ray spectroscopy (EDS), and elemental mapping investigations were performed with a JEOL JEM 2100 instrument. X-ray diffraction (XRD) analysis was performed on a Bruker D8 diffractometer using Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS) was conducted on a Thermo Scientific™ K-Alpha™+ spectrometer equipped with a monochromatic Al Kα X-ray source (1486.6 eV) operating at 100 W. All peaks were calibrated to the C 1s peak binding energy at 284.8 eV for adventitious carbon. The Raman investigation was performed on a laser confocal Raman microspectrometer (XploRA, Horiba Jobin Yvon, Ltd.)Electrochemical measurements
All electrochemical measurements were performed using an electrochemical workstation (CHI 660, CH Instruments, Inc., Shanghai, China) in a three-electrode system. As for the HER test, the graphite rod and saturated calomel electrode (SCE, Hg/Hg2Cl2 in saturated KCl) were used as the counter electrode and reference electrode, respectively. The synthesized samples (5 mg) were first dispersed in 1.0 mL of ethanol mixture and 30 μL of 5 wt% Nafion solution prior to a ≥ 30 min sonication to form an electrocatalyst ink and then loaded onto a glassy carbon electrode (GCE, 4 mm in diameter) as a working electrode. The potential values used in this study were converted to E(RHE) from E(SCE) according to the formula E(RHE) = E(SCE) + 0.241 + 0.059 × pH. For comparison, commercial 20 wt% Pt/C with the same mass loading was also tested. Linear sweep voltammetry (LSV) tests for the HER were conducted with a scan rate of 5 mV s−1 in electrolytes (0.5 M H2SO4 or 1.0 M KOH). The double-layer capacitances (Cdl) were calculated through cyclic voltammograms (CV) at various sweep rates from 20 to 200 mV s−1. Electrochemical impedance spectroscopy (EIS) was carried out at an overpotential of 200 mV in a frequency range from 10−2 Hz to 105 Hz and an amplitude of 5 mV.Computation details
The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional27 and a 450 eV cutoff for the plane-wave basis set28 were employed to perform all the density functional theory (DFT) computations of the materials within the framework of VASP packages29 with projector augmented wave (PAW) pseudopotentials.30 The Monkhorst–Pack k-point mesh with a maximum spacing of 0.05 Å was utilized for the first Brillouin zone integrations.31 The DFT-D3(BJ) method of Grimme was employed to describe long-range van der Waals interactions.32,33 To correct the zero-point energy for the reaction barrier, the vibrational frequency calculations were performed via the finite-difference approach. (2 × 2) four-layer slab models were utilized to simulate the exposed (001) surfaces of Mo2C and W2C. The top two layers of the slab models were fully relaxed and the other layers were fixed to the bulk structure. Due to the small lattice mismatch (<1%), the junction between W2C (001) and Mo2C (001) with two layers at each side was used to build the theoretical model of the Mo2C–W2C interface. The convergence criterion for an electronic self-consistent iteration was set at 1.0 × 10−5 eV. The Quasi-Newton lBFGS method was used for geometry relaxation until the maximal force became less than 0.01 eV Å−1.The Gibbs free energy changes (ΔG) for hydrogen adsorption were expressed as:
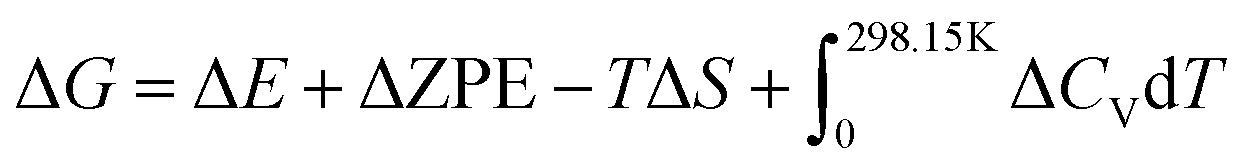
Results and discussion
The synthesis of Mo2C–W2C/RGO is schematically depicted in Fig. 1. According to the varied Mo/W molar ratio, the products were denoted as Mo2C–W2C/RGO-n, in which n refers to the measured nMo/(Mo+W) (Table S1†). Mo2C/RGO and W2C/RGO were prepared by similar synthesis methods for comparison. The morphologies of the samples were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM image of RGO in Fig. 2a shows the morphology of nanosheets with ripples. We further examined the nanoscale structure of Mo2C–W2C/RGO. Taking Mo2C–W2C/RGO-0.56 as a model sample, numerous well-distributed and ultrafine Mo2C–W2C nanoparticles (about 2.46 nm in average size, inset of Fig. 2c) are anchored to the graphene sheets (Fig. 2b and c). The smaller particle size and uniform nature of Mo2C–W2C would effectively reduce the loss of active sites and promote fast mass transfer. Similarly, Mo2C/RGO, Mo2C–W2C/RGO-0.75, Mo2C–W2C/RGO-0.24, and W2C/RGO also displayed ultrafine nanoparticles (Fig. S1†). The lattice fringes with spacings of 2.36 Å and 2.29 Å nm observed in Fig. 2d are associated with the (002) crystal facet and the (101) crystal facet of Mo2C or W2C; the high-resolution TEM (HR-TEM) image also demonstrates that abundant interfaces exist in the structure owing to the alternative arrangement of Mo2C and W2C nanocrystals. In addition, the energy-dispersive X-ray spectra (EDS) manifest that the Mo2C–W2C/RGO composites were composed of Mo, W, C, N and O elements (Fig. S2†), with a small amount of N doped from melamine. The O element results from the inevitable oxidation caused by exposure to air. Furthermore, the elemental mapping of W, Mo, and C from the SEM image (Fig. 2e) indicates a uniform spatial distribution throughout the whole scanning region, confirming the homogeneous growth of Mo2C–W2C. | ||
| Fig. 1 Schematic illustration for fabricating Mo2C–W2C/RGO. | ||
 | ||
| Fig. 2 SEM images of (a) RGO and (b) Mo2C–W2C/RGO-0.56. (c) TEM image and (d) HR-TEM image of Mo2C–W2C/RGO-0.56. (e) SEM image and corresponding element mappings of Mo2C–W2C/RGO-0.56. The inset in panel (c) is the size distribution diagram. | ||
The XRD investigation confirmed the formation of Mo2C–W2C/RGO heterostructures in the final products (Fig. 3a), and the results of Mo2C/RGO, W2C/RGO and Mo2C–W2C/RGO show the characteristic pattern of hexagonal W2C (JCPDS no. 35-0776) and Mo2C (JCPDS no. 35-0787). The diffraction peaks of all samples are relatively weak and broad, consistent with the ultrafine nanoparticles shown by SEM and TEM results. Accordingly, these samples present two distinct characteristic peaks of the carbon-based networks, located in the D (1350 cm−1) and G (1580 cm−1) bands in Raman spectra (Fig. 3b).35,36 The IG/ID value for GO sheets (1.25) is significantly higher than that for RGO nanosheets (0.98) because of the decrease in oxygenic groups during the thermal treatment (Fig. S3†). In contrast, the IG/ID values of Mo2C–W2C/RGO, W2C/RGO, and Mo2C/RGO are markedly increased compared with RGO, which could be due to the coupling between carbide nanoparticles and RGO resulting in a more disordered carbon structure.
 | ||
| Fig. 3 (a) XRD patterns and (b) Raman spectra of the Mo2C/RGO, W2C/RGO and all Mo2C–W2C/RGO samples. XPS profiles of (c) Mo 3d and (d) W 4f in the Mo2C/RGO, W2C/RGO and all Mo2C–W2C/RGO samples. | ||
To analyze the surface chemical composition and electronic structure of the samples, X-ray photoelectron spectroscopy (XPS) was performed. As depicted in Fig. S4a,† an XPS survey was used to identify the composition of the Mo, W, and C elements in these samples. The profiles of Mo 3d in Mo2C–W2C/RGO and Mo2C/RGO can be deconvoluted into six peaks (Fig. 3c), suggesting that there are three species (Mo–C, Mo4+, and Mo6+) for Mo on the surface.37,38 For the Mo2C/RGO sample, the couple at 228.5 and 231.5 eV should be ascribed to the Mo 3d5/2 and 3d3/2 of Mo–C species. The peaks located at 229.2 and 232.9 eV are attributed to Mo4+ species, and the two at 232.2 and 235.5 eV are associated with Mo6+ species. They are commonly observed when carbides are exposed to air.39 The W 4f spectrum of W2C/RGO (Fig. 3d) can be fitted into four peaks with binding energies at 35.5 and 37.7 eV corresponding to W 4f7/2 and 4f5/2 of surface oxidized W6+, while the two peaks at 31.8 and 33.8 eV can be attributed to W 4f7/2 and 4f5/2 of W–C species.40 Similarly, Mo 3d and W 4f for all Mo2C–W2C/RGO samples can also be fitted into corresponding peaks. Regarding the consistent binding energy of the oxidation state affected by air (Mo6+, Mo4+ and W6+) in the peak fitting of all the samples, we focus on the binding energy of Mo–C and W–C species affected by the heterostructure, which are regarded as the active centers for electrocatalytic HER.40,41 The Mo2C/RGO sample presents Mo 3d5/2 and 3d3/2 at 228.5 and 231.5 eV for Mo–C species, respectively. By contrast, these peaks for Mo–C species slightly shift toward lower binding energies in all Mo2C–W2C/RGO samples, indicating that a change in the electronic structure occurs due to introduction of the W2C. On the other hand, the binding energy of the W–C species of Mo2C–W2C/RGO is positively shifted from that of W2C/RGO samples. Given the higher electronegativity of Mo (2.16) compared to W (1.7),42 the negative shift of Mo–C and positive shift of W–C species in all Mo2C–W2C/RGO samples can reasonably be attributed to electron transfer from W to Mo. Notably, the binding energies of Mo shifted negatively with increasing W composition in Mo2C–W2C/RGO, suggesting that the electronic structure of the surface of the samples can be effectively modified by varying the Mo/W ratio (Fig. 3c and d). The above high-resolution XPS analysis shows that the construction of the Mo2C–W2C/RGO heterostructure modulates the electronic structure of the original carbide, which is expected to regulate the adsorption/desorption strength between active sites and adsorbates. Fig. S4b† shows the high-resolution C 1s XPS spectrum for the all samples, which can be deconvoluted into five peaks (C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, CO, OC–O, and C–Mo/C–W).43,44 Moreover, the N 1s XPS profile (Fig. S4c†) can be fitted into four peaks, assigned to Mo 3p, pyridinic-N, pyrrolic-N, and quaternary-N, respectively.45–47 In general, the planar structures of pyrrolic-N and pyridinic-N favor electron transport in π–π-conjugated graphite layers.40 Regarding the similarity in N type and content, the contribution to conductivity by N-doping into the carbon matrix can be considered to be equivalent for W2C/RGO, Mo2C/RGO, and all Mo2C–W2C/RGO samples.
C, C–N, CO, OC–O, and C–Mo/C–W).43,44 Moreover, the N 1s XPS profile (Fig. S4c†) can be fitted into four peaks, assigned to Mo 3p, pyridinic-N, pyrrolic-N, and quaternary-N, respectively.45–47 In general, the planar structures of pyrrolic-N and pyridinic-N favor electron transport in π–π-conjugated graphite layers.40 Regarding the similarity in N type and content, the contribution to conductivity by N-doping into the carbon matrix can be considered to be equivalent for W2C/RGO, Mo2C/RGO, and all Mo2C–W2C/RGO samples.
The electrocatalytic HER performances of the samples were first evaluated in the standard three electrode system under 0.5 M H2SO4 conditions at 5 mV s−1. All the samples are supported on glassy carbon electrodes with the same mass loading of 0.24 mg cm−2. The synthesis conditions of different molar ratios of W and Mo are at first screened (Fig. S5†), and all the Mo2C–W2C/RGO materials are superior to the W2C/RGO and Mo2C/RGO (Fig. S5 and Table S2†). At the same time, it is suggested that the optimal electrocatalyst with an appropriate Mo/W molar ratio is Mo2C–W2C/RGO-0.56. Fig. 3a displays the polarization curves of the W2C/RGO, Mo2C–W2C/RGO-0.56 and Mo2C/RGO, along with bare RGO and the commercial Pt/C (20 wt%) benchmark for reference. It is obvious that RGO has a negligible HER activity. The W2C/RGO and Mo2C/RGO afford overpotentials (η10) of 158 mV and 110 mV to achieve a current density (j) of −10 mA cm−2, and Mo2C–W2C/RGO-0.56 exhibits an ultralow η10 of only 81 mV. This result reveals that the unique two-phase heterostructure under an optimal Mo/W ratio can dramatically boost the catalytic activity. In addition, the catalytic performance of Mo2C–W2C/RGO-0.56 is much better than those of molybdenum- and/or tungsten-based multi-phase heterostructures at −10 mA cm−2, such as twinned WCN (128 mV),48 W2C/WP@NC (116.37 mV),49 Mo2N–Mo2C/HGr (157 mV),50 and WC-W2C/HCDs (96 mV),51 and is the best among the non-noble catalysts reported recently (Table S3†).
To further investigate the HER mechanism, the Tafel plots (Fig. 4b) were derived by plotting the overpotential against log(j) according to the Tafel equation (η = b × log(j) + a).52 Mo2C–W2C/RGO-0.56 delivers a low onset overpotential (ηonset) of 30 mV and a low Tafel slope (b) of 56 mV dec−1, superior to those of Mo2C/RGO (ηonset = 43 mV, b = 73 mV dec−1) and W2C/RGO (ηonset = 67 mV, b = 94 mV dec−1). Generally, the HER process includes two steps, namely the Volmer, Heyrovsky or Tafel steps,1,53 corresponding to Tafel slopes of 120, 40, and 30 mV dec−1, respectively. The above results suggest that the HER kinetics for Mo2C–W2C/RGO-0.56, Mo2C/RGO and W2C/RGO should follow the Volmer–Heyrovsky mechanism and Mo2C–W2C/RGO-0.56 suggests a more rapid HER kinetics.54,55 Extrapolating the above Tafel plots, the higher value of the calculated exchange current density (j0) also indicates the high HER activity of Mo2C–W2C/RGO-0.56 (Table S4†).
 | ||
| Fig. 4 Electrochemical measurements of Mo2C–W2C/RGO-0.56 for the HER in 0.5 M H2SO4. (a) Polarization curve and (b) Tafel plot of Mo2C–W2C/RGO-0.56 in comparison with those of 20 wt% Pt/C, Mo2C/RGO, W2C/RGO and RGO alone. (c) Electrochemical impedance spectrum of Mo2C–W2C/RGO-0.56 at η = 200 mV in comparison with those of Mo2C/RGO, W2C/RGO and RGO. The inset of panel c shows the equivalent circuit diagram. (d) Long-term durability test of Mo2C–W2C/RGO-0.56. The inset shows the LSV curves before and after the durability test. | ||
The electrochemically active surface area (ECSA) of the samples was estimated by the double layer capacitance (Cdl).56,57 In accordance with the obtained Cdl values (Fig. S6†), Mo2C–W2C/RGO-0.56 (38.5 mF cm−2) has the highest ECSA of all samples, thereby further confirming the higher performance of Mo2C–W2C/RGO-0.56. In addition, the electrochemical impedance spectroscopy (EIS) measurements (Fig. 4c) show that Mo2C–W2C/RGO-0.56 has the smallest electron transfer resistance and the EIS trend is in good agreement with the HER activity trend. The HER activity of the above samples along with that of the commercial Pt/C catalyst is summarized in Table S4.†
As another important assessment of the electrocatalyst, a long-term electrochemical stability test of Mo2C–W2C/RGO-0.56 in acidic media was also performed (Fig. 4d). The activity of the working electrode remained almost completely stable in the continuous test of 30 h, and the polarization curve of the catalyst after the stability test showed no measurable decline of the cathodic current (inset of Fig. 4d). Accordingly, SEM investigation confirmed the well-retained nanostructures after the stability test, as shown in Fig. S7.† These results demonstrate that Mo2C–W2C/RGO-0.56 possesses outstanding durability in acidic electrolyte.
Additionally, Mo2C–W2C/RGO-0.56 also exhibited good HER performance in the alkaline medium (Fig. 5 and Table S5†). Apparently, the performance improvement after the formation of the Mo2C–W2C heterostructure can be obviously observed from Fig. S8 and Table S6.† By contrast, Mo2C–W2C/RGO-0.56 displays a relatively small overpotential of 87 mV at −10 mA cm−2, lower than those of Mo2C/RGO (134 mV), W2C/RGO (159 mV), and RGO (491 mV). Besides, commercial Pt/C still presents the best HER performance, requiring an overpotential of merely 27 mV to deliver a current density of −10 mA cm−2. The faster HER kinetics of Mo2C–W2C/RGO-0.56 can be reflected by its small Tafel slope of 59 mV dec−1 and low onset overpotential of 33 mV, lower than the values of Mo2C/RGO (ηonset = 66 mV, b = 64 mV dec−1) and W2C/RGO (ηonset = 76 mV, b = 93 mV dec−1) (Fig. 5b and Table S5†). Besides, the Tafel slope of Mo2C–W2C/RGO-0.56 within the theoretical value indicates that the reaction kinetics for hydrogen generation is the Volmer–Heyrovsky mechanism.54,55 Furthermore, capacitance values (Fig. S9 and Table S5†), Nyquist diagram (Fig. 5d), and j0 (Table S5†) confirm that the Mo2C–W2C/RGO-0.56 electrode has the largest electrochemical surface area, the smallest charge transfer impedance, and higher intrinsic activity, which is consistent with its HER activity. At the same time, Mo2C–W2C/RGO-0.56 also has satisfactory long-term stability in 1.0 M KOH, delivering a current density of about −10 mA cm−2 for 30 h. In addition, SEM analysis results (Fig. S10†) prove that the structure of Mo2C–W2C/RGO is well maintained after a long-term HER process, which further confirms the remarkable stability of Mo2C–W2C/RGO. Remarkably, the Mo2C–W2C/RGO delivers prominent HER activity in 1.0 M KOH, representing one of the current best carbide-based HER catalysts (Table S3†). All of these results indicate that Mo2C–W2C/RGO shows excellent hydrogen production performance over the entire pH range, providing a promising HER electrocatalyst for practical application.
 | ||
| Fig. 5 Electrochemical measurements of Mo2C–W2C/RGO-0.56 for the HER in 1.0 M KOH. (a) Polarization curve and (b) Tafel plot of Mo2C–W2C/RGO-0.56 in comparison with those of 20 wt% Pt/C, Mo2C/RGO, W2C/RGO and RGO alone. (c) Electrochemical impedance spectrum of Mo2C–W2C/RGO-0.56 at η = 200 mV in comparison with those of Mo2C/RGO, W2C/RGO and RGO. The inset of panel c shows the equivalent circuit diagram. (d) Long-term durability test of Mo2C–W2C/RGO-0.56. The inset shows the LSV curves before and after the durability test. | ||
The aforementioned results indicate the formation of the Mo2C–W2C heterostructure coupled with RGO, which shows superior acidic and alkaline HER performance. The origin of the efficient HER activity of Mo2C–W2C/RGO is worthy of investigation. Therefore, a series of control samples, including W2C, Mo2C, and Mo2C–W2C (Fig. S11a, c and e†), were constructed in the absence of GO, in addition to the RGO, W2C/RGO and Mo2C/RGO already mentioned. Electrochemical tests were performed to compare the activity of the HER under acidic and alkaline conditions. The poor performance of the RGO indicates that RGO is not an active component (Fig. S12†). In addition, W2C/RGO and Mo2C/RGO show superior HER activity over W2C and Mo2C, respectively, and the HER activity of Mo2C–W2C/RGO is better than that of Mo2C–W2C. The results indicate the positive role of RGO in the improvement of the HER performance. This is related to the good electrical conductivity and large surface area of RGO. In the case of the catalysts without a graphene substrate, W2C, Mo2C, and Mo2C–W2C catalysts exhibit severe agglomeration and accumulation (Fig. S11b, d and f†). The good coupling between graphene and nanoparticles inhibits the agglomeration of nanosheets and plays a good dispersion role, which helps to improve the active surface area of the catalyst and expose the active site. Furthermore, graphene as a good conductor can effectively reduce the interfacial barrier of electron transfer, which is helpful for the enhancement of reaction kinetics. The improved activity of Mo2C–W2C/RGO over Mo2C/RGO and W2C/RGO implies the important role of the heterostructure for the HER due to its strong electronic interaction. Thus, we propose that the Mo2C–W2C heterostructure is the main active component, and RGO is also an important factor because of its role in exposing the active site, as well as in mass/charge transfer.
To gain a fundamental understanding of the high catalytic activity of the Mo2C–W2C heterostructure for the HER, density functional theory (DFT) calculations were performed. Here, the free energy profiles of H adsorption on the (001) surface of pure W2C (Fig. 6a) and Mo2C (Fig. 6b), along with the corresponding surfaces of Mo2C–W2C (Fig. 6c) were computed. Regarding the overall HER rate, the hydrogen adsorption free energy (ΔGH*) is regarded as a good descriptor to evaluate the activity of the material, and optimal HER catalysts should have ΔGH* close to zero.58–62 As illustrated in Fig. 6d, on the W site of W2C, the ΔGH* is calculated to be −0.32 eV, and the ΔGH* on the Mo site of Mo2C is −0.10 eV. The relatively negative ΔGH* on the W site shows that the strong adsorption of H on the surface of W2C restricts the Heyrovsky process, resulting in a low HER efficiency, which is consistent with our experimental results of electrochemical tests. For Mo2C–W2C, the ΔGH* values on the Mo and W sites of Mo2C–W2C are −0.03 eV and −0.25 eV, respectively, both of which are closer to the ideal ΔGH* (0 eV) than the corresponding Mo and W sites on pure Mo2C and pure W2C. The calculations demonstrate that the interface structure of Mo2C–W2C plays a crucial role in its excellent HER activity.
 | ||
| Fig. 6 Schematic model of (a) W2C (001), (b) Mo2C (001), and (c) Mo2C–W2C. (d) Calculated free energy diagram of H adsorption. (e) PA-CDD and CDD of the Mo2C–W2C. The yellow and cyan areas represent an increase and decrease in charge density, respectively. The purple, grey, brown, and pink spheres denote Mo, W, C, and H atoms, respectively. | ||
Additionally, we have calculated the charge-density differences (CDD) as well as the planar-average charge-density difference (PA-CDD) along the Z direction of the heterostructure to evaluate interfacial charge-transfer processes. As shown in Fig. 6e, the electron accumulation (yellow) and depletion (cyan) are concentrated at the interfaces formed between Mo2C and W2C, indicating the strong charge redistribution at the interface, consistent with the electron modulation observed by XPS (Fig. 3c and d). Fig. S13† displays the total density of states (DOS) of the Mo2C, W2C and Mo2C–W2C. The DOSs of all three materials cross their Fermi level (corresponding to the energy of the highest electron occupied orbital of the material), demonstrating that all of them can possess good conductivity during the HER. The high density of states of Mo2C–W2C adjacent to the negative side of the Fermi level further proves its excellent ability to donate electrons during the electrocatalytic HER process. The better HER activity of Mo2C–W2C may also originate from the interface charge distribution. This charge redistribution adjusts the surface electronic structure of the material, which weakens the H adsorption and facilitates H2 desorption resulting in enhanced HER kinetics and reduced overpotential.
Based on all the above experiments and theoretical calculation results, the remarkable electrocatalytic activity and stability of Mo2C–W2C for the HER can be attributed to the following reasons: (i) the material is supported by graphene sheets with good electrical conductivity and large specific surface area, showing ultra-fine and evenly distributed nanoparticle morphology, high ECSA and good electron transport capacity, which is conducive to the transport of reactants and products, thus accelerating the occurrence of the HER. (ii) The optimized electronic structure at the Mo2C–W2C interfaces improves the kinetics of the hydrogen evolution process, which endows the Mo2C–W2C with enhanced kinetics and high activity.
Conclusions
In summary, an ultrafine and interface-rich Mo2C–W2C heterostructure anchored to reduced graphene oxide is successfully prepared and used as a highly efficient electrocatalyst for pH-universal HER. The ultrafine size and abundant interfaces of the Mo2C–W2C heterostructure maximize the exposure of active sites and facilitate the mass transfer process, while the interfacial interaction optimizes the electronic configuration of the catalyst. As a result, the Mo2C–W2C heterostructure afforded ultra-low overpotentials (η10 = 81 and 87 mV for reaching a current density of −10 mA cm−2) under acidic and alkaline conditions. DFT calculations further indicate that the constructed Mo2C–W2C heterostructure can achieve almost ideal ΔGH* for the HER. In addition, the charge density difference analysis also reveals the modulating effect of the electronic structure of the heterostructures. The adjusted electronic structures of the Mo2C–W2C effectively promotes the desorption step of H*, which is responsible for the enhanced kinetic metrics and high HER activity of the Mo2C–W2C heterostructure. Both experiments and theoretical calculations have confirmed that the electrocatalytic HER performance of materials can be effectively modulated by adjusting the nanoarchitectures and electronic structure of carbides. This work provides good guidance for the future design of advanced materials with superior HER activity.Author contributions
All authors have contributed to the preparation of the manuscript and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22273066, 22173069), Shanghai Municipal Natural Science Foundation (Grant No. 19DZ2271500, 21ZR1467800) and the Fundamental Research Funds for the Central Universities.References
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- T. Kou, S. Wang and Y. Li, ACS Mater. Lett., 2021, 3, 224–234 CrossRef CAS.
- T. Ouyang, Y.-Q. Ye, C.-Y. Wu, K. Xiao and Z.-Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923 CrossRef CAS PubMed.
- L. Jiang, R. Wang, Z. Xiang and X. Wang, Int. J. Hydrogen Energy, 2024, 11, 898–906 CrossRef.
- R. Mohili, N. R. Hemanth, H. Jin, K. Lee and N. Chaudhari, J. Mater. Chem. A, 2023, 11, 10463–10472 RSC.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846 CrossRef.
- F. Li, G.-F. Han, H.-J. Noh, Y. Lu, J. Xu, Y. Bu, Z. Fu and J.-B. Baek, Angew. Chem., Int. Ed., 2018, 57, 14139–14143 CrossRef CAS PubMed.
- W. Li, Z.-Y. Hu, Z. Zhang, P. Wei, J. Zhang, Z. Pu, J. Zhu, D. He, S. Mu and G. V. Tendeloo, J. Catal., 2019, 375, 164–170 CrossRef CAS.
- Q. Xue, X. Y. Bai, Y. Zhao, Y. N. Li, T. J. Wang, H. Y. Sun, F. M. Li, P. Chen, P. Jin, S. B. Yin and Y. Chen, J. Energy Chem., 2022, 65, 94–102 CrossRef CAS.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- R. Wang, L. Jiang, Q. Wang, G. F. Wei and X. Wang, Adv. Compos. Hybrid Mater., 2023, 6, 157 CrossRef CAS.
- S. Hussain, D. Vikraman, A. Feroze, W. Song, K.-S. An, H.-S. Kim, S.-H. Chun and J. Jung, Front. Chem., 2019, 7, 716 CrossRef CAS.
- A. Nadar, A. M. Banerjee, M. R. Pai, R. P. Antony, A. K. Patra, P. U. Sastry, H. Donthula, R. Tewari and A. K. Tripathi, Int. J. Hydrogen Energy, 2020, 45, 12691–12701 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS.
- X. F. Lu, L. Yu, J. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900699 CrossRef.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef.
- B. Ren, D. Li, Q. Jin, H. Cui and C. Wang, J. Mater. Chem. A, 2017, 5, 13196–13203 RSC.
- H. Jin, J. Chen, S. Mao and Y. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 22094–22101 CrossRef CAS.
- N. Han, K. R. Yang, Z. Lu, Y. Li, W. Xu, T. Gao, Z. Cai, Y. Zhang, V. S. Batista, W. Liu and X. Sun, Nat. Commun., 2018, 9, 924 CrossRef PubMed.
- W.-F. Chen, J. T. Muckermana and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC.
- X. Tao, Y. Li, J. Du, Y. Xia, Y. Yang, H. Huang, Y. Gan, W. Zhang and X. Li, J. Mater. Chem., 2011, 21, 9095–9102 RSC.
- Y. Hu, B. Yu, W. Li, M. Ramadoss and Y. Chen, Nanoscale, 2019, 11, 4876–4884 RSC.
- W. Zheng, L. Wang, F. Deng, S. A. Giles, A. K. Prasad, S. G. Advani, Y. Yan and D. G. Vlachos, Nat. Commun., 2017, 8, 418 CrossRef.
- W.-F. Chen, S. Iyer, S. Iyer, K. Sasaki, C.-H. Wang, Y. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818 RSC.
- P. Yan, Y. Wu, X. Wei, X. Zhu and W. Su, Nanomaterials, 2020, 10, 1621–1711 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimm, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- F. Malara, S. Carallo, E. Rotunno, L. Lazzarini, E. Piperopoulos, C. Milone and A. Naldoni, ACS Catal., 2017, 7, 4786–4795 CrossRef CAS.
- Z. Zheng, S. Hu, W. Yin, J. Peng, R. Wang, J. Jin, B. He, Y. Gong, H. Wang and H. J. Fan, Adv. Energy Mater., 2023, 2303064 CrossRef.
- D. Guo, M. Yang, S. Xu, S. Zhu, G. Liu, N. Wu, A. Cao, H. Mi and X. Liu, Nanoscale, 2022, 14, 14575–14584 RSC.
- Y. Shen, Y. Jiang, Z. Yang, J. Dong, W. Yang, Q. An and L. Mai, Adv. Sci., 2022, 9, 2104504 CrossRef CAS.
- Y. Sun, X. Hu, J. C. Yu, Q. Li, W. Luo, L. Yuan, W. Zhang and Y. Huang, Energy Environ. Sci., 2011, 4, 2870–2877 RSC.
- Q. Gong, Y. Wang, Q. Hu, J. Zhou, R. Feng, P. N. Duchesne, P. Zhang, F. Chen, N. Han, Y. Li, C. Jin, Y. Li and S.-T. Lee, Nat. Commun., 2016, 7, 13216 CrossRef CAS PubMed.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Am. Chem. Soc., 2015, 137, 110 CrossRef CAS PubMed.
- J. G. Speight, Lange's Handbook of Chemistry, McGraw-Hill, Professional, New York, NY, USA, 16 edn, 2005 Search PubMed.
- H. Lin, W. Zhang, Z. Shi, M. Che, X. Yu, Y. Tang and Q. Gao, ChemSusChem, 2017, 10, 2597–2604 CrossRef CAS PubMed.
- Y. Hu, B. Yu, W. Li, M. Ramadoss and Y. Chen, Nanoscale, 2019, 11, 4876–4884 RSC.
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755 CrossRef CAS PubMed.
- M. Shi, W. Li, J. Fang, Z. Jiang, J. Gao, Z. Chen, F. Sun and Y. Xu, Electrochim. Acta, 2018, 283, 834–841 CrossRef CAS.
- Y. Ding, K. W. Cao, J. W. He, F. M. Li, H. Huang, P. Chen and Y. Chen, Chin. J. Catal., 2022, 43, 1535–1543 CrossRef CAS.
- Z. Kou, T. Wang, H. Wu, L. Zheng, S. Mu, Z. Pan, Z. Lyu, W. Zang, S. J. Pennycook and J. Wang, Small, 2019, 15, 1900248 CrossRef PubMed.
- P. Wei, X. Sun, M. Wang, J. Xu, Z. He, X. Li, F. Cheng, Y. Xu, Q. Li, J. Han, H. Yang and Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 53955–53964 CrossRef CAS.
- H. Yan, Y. Xie, Y. Jiao, A. Wu, C. Tian, X. Zhang, L. Wang and H. Fu, Adv. Mater., 2018, 30, 1704156 CrossRef PubMed.
- S.-C. Sun, F.-X. Ma, H. Jiang, M.-X. Chen, P. Xu, L. Zhen, B. Song and C.-Y. Xu, Chem. Eng. J., 2023, 462, 142132 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- S. Xu, F. Wang, L. Mai, L. Wang and P. K. Chu, Electrochim. Acta, 2013, 90, 344–349 CrossRef CAS.
- J. Yu, W.-J. Li, H. Zhang, F. Zhou, R. Li, C.-Y. Xu, L. Zhou, H. Zhong and J. Wang, Nano Energy, 2019, 57, 222–229 CrossRef CAS.
- J. Zhao, N. Liao and J. Luo, J. Mater. Chem. A, 2023, 11, 9682–9690 RSC.
- Z. Li, M. Hu, P. Wang, J. Liu, J. Yao and C. Li, Coord. Chem. Rev., 2021, 439, 213953 CrossRef CAS.
- Z. Pu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, S. Mu, W. Zhao, F. Su, G. Zhang and S. Liao, Nano-Micro Lett., 2020, 12, 1–29 CrossRef.
- J. K. Nørskov, T. Bligaard and A. Logadottir, J. Electrochem. Soc., 2005, 152, 23–26 CrossRef.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS.
- H. J. Yan, Y. Xie, A. P. Wu, Z. C. Cai, L. Wang, C. G. Tian, X. M. Zhang and H. G. Fu, Adv. Mater., 2019, 31, 1901174 CrossRef PubMed.
- L. Wang, Y. Hao, L. Deng, F. Hu, S. Zhao, L. Li and S. Peng, Nat. Commun., 2022, 13, 5785 CrossRef CAS.
- E. Yu and Y. Pan, J. Mater. Chem. A, 2022, 10, 24866–24876 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07373d |
| This journal is © The Royal Society of Chemistry 2024 |