
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges and opportunities for characterisation of high-temperature polymer electrolyte membrane fuel cells: a review
Adam
Zucconi
 ab,
Jennifer
Hack
ac,
Richard
Stocker
d,
Theo A. M.
Suter
a,
Alexander J. E.
Rettie
*a and
Dan J. L.
Brett
*a
ab,
Jennifer
Hack
ac,
Richard
Stocker
d,
Theo A. M.
Suter
a,
Alexander J. E.
Rettie
*a and
Dan J. L.
Brett
*a
aElectrochemical Innovation Lab, Department of Chemical Engineering, UCL, London WC1E 7JE, UK. E-mail: a.rettie@ucl.ac.uk
bMobility Innovation Hub, HORIBA MIRA, Nuneaton, Warwickshire CV10 0TU, UK
cDepartment of Materials Science & Engineering, University of Sheffield, Sir Robert Hadfield Building, Mappin Street, Sheffield S1 3JD, UK
dHORIBA Instruments Inc., 2890 John R Rd, Troy, Michigan 48083, USA
First published on 7th March 2024
Abstract
High-temperature (120–200 °C) polymer electrolyte membrane fuel cells (HT-PEMFCs) are promising energy conversion devices that offer multiple advantages over the established low-temperature (LT) PEMFC technology, namely: faster reaction kinetics, improved impurity tolerance, simpler water and thermal management, and increased potential to utilise waste heat. Whilst HT- and LT-PEMFCs share several components, important differences in the membrane materials, transport mechanisms and operating conditions provide new challenges and considerations for characterisation. This review focuses on phosphoric acid-doped HT-PEMFCs and provides a detailed discussion of the similarities and differences compared to LT-PEMFCs, as well as state-of-the-art performance and materials. Commonly used characterisation techniques including electrochemical, imaging, and spectroscopic methods are reviewed with a focus on use in HT-PEMFCs, how experimentation or analyses differ from LT-PEMFCs, and new opportunities for research using these techniques. Particular consideration is given to the presence of phosphoric acid and the absence of liquid water. The importance of accelerated stress tests for effective characterisation and durability estimation for HT-PEMFCs is discussed, and existing protocols are comprehensively reviewed focusing on acid loss, catalyst layer degradation, and start-up/shutdown cycling. The lack of standardisation of these testing protocols in HT-PEMFC research is highlighted as is the need to develop such standards.
 Adam Zucconi | Adam Zucconi is a research scientist at HORIBA, and part-time PhD researcher in the Department of Chemical Engineering at UCL. His main research focuses on in situ and operando characterisation of fuel cells and electrolysers, principally low- and high-temperature polymer electrolyte membrane technologies. He has particular interest in electrochemical and exhaust liquid and gas characterisation. |
 Jennifer Hack | Dr Hack is a Royal Academy of Engineering Research Fellow in the Department of Materials Science and Engineering at the University of Sheffield, UK. She completed her PhD at University College London in 2021 working on fuel cell characterisation. After undertaking an EPSRC Doctoral Prize Fellowship studying zinc–air batteries, followed by a Project Lead role on the Faraday Institution's LiSTAR project, Jennifer joined the University of Sheffield in 2023. Her research focuses on the study of morphology evolution in electrochemical devices, in particular electrolysers and fuel cells, using in situ and operando X-ray and neutron CT. |
 Richard Stocker | Dr. Richard Stocker is a Principle Engineer at HORIBA Instruments Inc. In his 10 years at HORIBA he has focused on Li-ion battery pack projects, including BMS development, cell characterization testing, and simulation modelling. Graduating from the University of Nottingham with a MEng in Mechanical Engineering (2013), they furthered their specialization with a PhD from Coventry University (2020), investigating Li-ion battery cell ageing and developing algorithms to decode ageing mechanisms from electrical cycling data. He has investigated the use of Electrochemical Impedance Spectroscopy (EIS) and Distribution of Relaxation Times (DRT) as applied to batteries and fuel cells. |
 Theo A. M. Suter | Theo A. M. Suter is a postdoctoral researcher in the electrochemical innovation laboratory at UCL focusing on fuel cell fabrication, testing, and characterization. He completed his PhD in nanomaterial chemistry at UCL in 2018 and now specializes in nanoengineering of the fuel cell catalyst layer, particularly via the use of nanomaterials and heterogeneous catalyst layer fabrication. His interests focus on how the fuel cell catalyst layer morphology and microstructure impacts the performance of fuel cells, and how different fabrication techniques can be used as a tool to improve device durability. |
 Alexander J. E. Rettie | Alex Rettie is an Associate Professor in Electrochemical Conversion and Storage in the Department of Chemical Engineering, UCL (UK). His interests are in the experimental discovery and characterisation of electrochemical energy materials and their incorporation into devices, with a focus on electronic and ionic charge transport. He leads a national hydrogen network based at UCL and is the UK's alternate delegate to the IEA's technology collaboration programme on hydrogen. |
 Dan J. L. Brett | Dan Brett specialises in electrochemical power systems. He holds the Royal Academy of Engineering Research Chair in Metrology for Electrochemical Propulsion and is the Director of the UCL Advanced Propulsion Lab. He has co-founded companies including Bramble Energy, Oort Energy, Sention, Element 30, Gaussian and Prosemino. He has been recognised through awards including the De Nora Prize for Applied Electrochemistry (ISE), Baker Medal (ICE) and the Princess Royal Silver Medal (RAEng). He is listed in the Stanford top 2% of scientists in the world and in 2023 the Royal Academy of Engineering and TFL recognised him as an ‘Engineering Icon’. |
1. Introduction
1.1 Emergence of PEMFCs
The global dependency on fossil fuels to provide energy has many disadvantages including high carbon and pollutant emissions (CO2, NOx, SOx, particulate matter, etc.),1–4 evidence of a causal link between economic growth, higher mortality rates and environmental degradation,4–8 fossil fuel price fragility and general increasing price trend,9–11 and higher economic costs due to trends in climate policy favouring non-fossil fuel markets and carbon taxes.12,13 Renewable energy combined with electrified power systems is experiencing rapid growth in light of these issues.14–16Polymer electrolyte membrane (also called proton exchange membrane) fuel cells (PEMFCs) are a promising energy conversion technology. These fuel cells convert the chemical energy of the fuel (hydrogen) and oxidant (oxygen) to electrical energy and heat, with water as the waste product. PEMFCs are favoured for applications that require fast start-up, high power density, zero local carbon emissions, low operating temperature, and the use of air as the oxidant.17,18 Such devices already have an established presence in niche markets such as material handling and remote/back-up power,19,20 and are expected to play a key role in the decarbonisation of heating, power, and transport.19 Over 60% of fuel cell units shipped in 2022 were estimated to be PEMFCs, making up 86% of the total fuel cell power output shipped that year.21
The technology can be separated into two categories: (i) low-temperature (LT-PEMFC), and (ii) high-temperature (HT-PEMFC). The former is typically operated at 60–80 °C, with the latter operating at 120–200 °C.22 PEMFCs that operate at 100–120 °C are sometimes referred to as intermediate-temperature (IT-PEMFCs), and can typically be grouped into two categories: (i) modified LT-PEMFCs where the membrane has been adapted to retain more water at temperatures above 100 °C, often by adding inorganic fillers; and (ii) HT-PEMFCs with phosphoric acid (PA)-doped polybenzimidazole (PBI) membranes.23 Due to the close relation of IT-PEMFCs to LT-PEMFCs and HT-PEMFCs depending on the membrane material and proton conduction method, this technology is not specified independently in the remainder of the review.
1.2 Motivation for HT-PEMFCs
LT-PEMFCs based on conventional perfluorosulphonic acid-based membrane electrolytes suffer from relatively high membrane cost, water management issues, catalyst poisoning, and expensive precious metal catalysts.24 Operating at temperatures above 100 °C offers potential solutions to some of these issues. The primary advantages to higher temperature operation include: increased tolerance of Pt catalysts to fuel impurities, faster reaction kinetics, easier water management, easier thermal management due to greater heat rejection resulting from the higher temperature gradient between the stack and ambient environment, and higher quality waste heat generation.22,24–261.3 Purpose of this review
Many of the review papers on HT-PEMFCs focus on the membrane.24,25,55–63 While there are review papers that discuss the characterisation of materials, components, and cells,38,47 these are limited to subsections of larger reviews. Characterisation techniques for LT-PEMFCs have been reviewed,20,64–66 yet comprehensive assessment of characterisation for HT-PEMFCs is very limited. Due to the differences in materials, and physical and chemical processes, the diagnostic and measurement techniques also require different approaches and analyses. Combining advanced characterisation techniques with accelerated stress tests (ASTs) is critical to studying degradation and durability. The appropriate characterisation technique will also depend on the application. For example, membrane electrode assembly (MEA) research and development typically requires more detailed information across chemical, physical, and electrical domains; whereas, control or system modelling may require more concise characterisation focusing on key parameters and metrics. This review aims to condense the current understanding of how HT-PEMFCs operate with context from the LT-PEMFC field given where appropriate, and how that affects characterisation and ASTs. This is increasingly important as interest and research into HT-PEMFCs grows, and this technology moves towards commercialisation.2. Materials, operation, and differences compared to LT-PEMFCs
The following section provides an overview of HT-PEMFC materials and design, highlighting the differences compared to LT-PEMFCs. This section is not intended to be a comprehensive review of materials and design, as this is well represented in other review articles.24,25,55–632.1 Materials
PFSA membranes have been modified to operate at higher temperatures (up to 120 °C) whilst still using water as the proton conductor.57,81 Inorganic fillers such as graphene oxide (GO), SiO2, TiO2, and ZrO2 can be added to the PFSA membrane to improve water retention and performance at higher temperatures and lower relative humidity.81–88 Sulphonated hydrocarbon polymers are another type of membrane investigated for HT-PEMFCs. While they have advantages in mechanical and thermal properties, as well as increased water uptake, they suffer from catalyst layer delamination and membrane thinning due to dehydration under HT-PEMFC operating conditions.67 Most crucially, both PFSA and sulphonated hydrocarbon membranes still require humidification and liquid water, which retains the complexity of the physical system and control strategy.
The most common membrane for HT-PEMFCs is PA-doped PBI-based. PA is used as the proton conductor due to its high proton conductivity under anhydrous conditions, low gas permeability, and good thermal and chemical stability.47 Proton conductivity can be increased by increasing the acid doping level; however, this decreases mechanical strength due to the strong plasticising effect.89–91 The actual proton conduction mechanisms are complex and depend on the water content, temperature, and amount of acid dopant. These factors affect the amount, and pathway, of conduction via structural diffusion (also referred to as “proton hopping” and “Grotthuss mechanism”), and via vehicular diffusion through H3O+ and H2PO4−.67,92,93 Despite their prevalence, PBI membranes still have unresolved issues. These include: acid leaching, reduced mechanical strength due to doping, and reduction in proton conductivities at high temperatures and low water content due to evaporation and acid condensation.63,67,94–96 Optimisation of acid doping should maximise performance whilst maintaining mechanical strength and minimising acid loss.67 Improvements in these areas have been achieved using PBI-based composite membranes by the use of crosslinking,97–99 and three-layered membranes.100 Advances have also been made with the addition of inorganic fillers,98,101–103 heteropolyacids (HPAs),104–106 carbon nanotubes,107,108 and metal–organic frameworks (MOFs).109–111
A promising alternative to PA-PBI are ion-pair coordinated membranes, specifically quaternary ammonium functionalised polymers (QAPs). The quaternary ammonium species causes complete deprotonation of PA, enabling a strong interaction between ammonium cations and biphosphate anions.55,112 Strong PA retention is possible at lower temperatures (<140 °C), moderate humidification, and higher temperatures (>200 °C).55 Ion-pair coordinated membranes could allow for operation at a much wider temperature range than PA-PBI. Quaternary ammonium-biphosphate (QAPOH) membranes combined with phosphonated ionomers have shown high peak power outputs from 80–240 °C, and excellent durability as low as 40 °C.113,114 Additionally, the use of an intrinsically microporous Tröger's base-derived polymer membrane has been shown to allow operation between −20 °C and 200 °C, whilst displaying exceptional performance retention even after start-up/shutdown cycling at 15 °C and −20 °C.115
The catalyst supports in HT-PEMFCs are usually carbon-based much like LT-PEMFCs. Operating under typical HT-PEMFC conditions poses greater challenges to the catalyst layer due to higher temperatures increasing Pt particle growth rate and carbon corrosion.120,121 Studies using graphene and carbon-walled nanotubes in the catalyst support have shown improved stability and power density; however, the electrochemically active surface area was higher for the conventional carbon supports.120,122,123
Both the catalyst and support materials in HT-PEMFCs are similar to LT-PEMFCs. However, the binder materials that join these components together can be quite different. LT-PEMFCs typically use a similar PFSA ionomer to the electrolyte which requires hydration for proton conduction in the catalyst layer, PTFE may also be added to improve water management.124 PTFE is the most common binder material in HT-PEMFCs due to its hydrophobicity and thermal stability.55 It is an insulating material and therefore requires a proton-conducting material. Ionic conductivity is usually enabled by the presence of PA which originates from the membrane during MEA preparation or cell assembly, or through direct deposition onto the catalyst layer.116 Optimisation of the PA in the catalyst layer is required as too much PA leads to electrode flooding which impedes oxygen transport to catalyst sites, and non-uniform distribution reduces catalyst utilisation.125 The catalyst microstructure should be designed to minimise crack width as this is a pathway for PA to penetrate the GDL, and have a sufficiently porous structure to allow the PA to activate the catalyst layer during activation.126,127 PTFE optimisation is also important as it affects acid uptake and too much will flood the catalyst layers and reduce membrane conductivity.128 The binder can also obstruct reaction pathways and decrease performance.55 A recent study using density functional theory and molecular dynamics simulations determined that an overall binder content of 25 wt% made of equal ratios of Nafion and PTFE yielded the best Pt poisoning protection.129 PA-PBI has also been used as the binder material. Using too much PA-PBI reduces the electrochemically active surface area (ECSA) due to increased blockages of the electron pathway, too little and there is an insufficient ionic conduction and potentially poor binding.130,131 Phosphonated polymers show promise as ionomeric binders. They have the benefits of greatly reduced acid loss in the presence of water, and the low concentration of phosphonic acid mitigates the impact of phosphate poisoning and PA flooding of the electrode.113 Protonation of the phosphonated ionomer has been shown to provide excellent performance and durability compared to non-protonated ionomers and state-of-the-art LT-PEMFCs.114 However, improvements are still required under high voltage conditions, dynamic drive cycles, and partial humidification, along with the practicality of scaling up to a full-size stack (e.g., non-uniform reactant supply and membrane-electrode contact).114
Gaskets must seal the cell, compensate for dimensional changes, be electrically insulating, and provide compression over long periods of time, and they must do this at temperatures up to 200 °C in the presence of PA.47,138 Fluoroelastomers (FKM), ethylene propylene diene monomer (EPDM), silicon, and PTFE are all commonly employed HT-PEMFC gasket materials.52,144
2.2 Operating principles
| 2H2 (g) + O2 (g) → 2H2O (g) | (1) |
 | ||
| Fig. 1 Schematic of HT-PEMFC operation including structural proton conduction and vehicular conductivity mechanisms. Relative component sizes are not to scale. | ||
Due to operating at higher temperatures, the magnitude of the Gibbs free energy is lower than that under LT-PEMFC conditions. This leads to a lower reversible voltage: 1.18 V at 360 K (86.85 °C) and 1.15 V at 440 K (166.85 °C). The voltage decrease is minimal due to the smaller entropy change of forming a gas compared to a liquid.
Operating temperatures typically range from 120–200 °C depending on membrane and proton conduction method (hydrous or anhydrous). HT-PEMFCs relying on water for conduction generally operate up to 120 °C, where PA-based conduction mostly operates in the range of 140–180 °C. The optimum temperature balances a trade-off between cell performance and degradation rate. Operating under humified conditions will also depend on the proton conduction method. Despite typically operating without external humidification, PA-based HT-PEMFCs can achieve conductivity improvements when humidified compared to non-humidified operation due to an increased contribution from the vehicular conduction mechanism (as discussed in detail in the following section) resulting from the higher water content.92,145 However, PA loss increases with higher water content.146,147
(1) Hopping from the N–H sites to PA anions (H2PO4−), most relevant for low doping scenarios i.e., no free acid;
(2) Hopping along PA anions. This occurs in the presence of free acid and leads to conductivity several orders of magnitude higher than (1);
(3) Hopping via water molecules. This mechanism is concurrent with (2);
(4) Direct hopping along nitrogen sites of the PBI chains (only relevant for non-doped PBI).
Mechanisms 1, 2, and 3 are illustrated accordingly in Fig. 1. Proton diffusion involving benzimidazole nitrogen sites can be neglected due to its proton exchange process being nine orders of magnitude slower than proton exchange between phosphate species.93 The high proton conductivity of PA is due to the high degree of hydrogen bond network frustration (a severe imbalance of proton donors to acceptors). This explains the significant contribution of rapid structural diffusion (hopping) to proton conductivity (∼97% in neat PA).92 As temperature increases and water content decreases, PA undergoes condensation reactions and forms higher molecular weight and slower diffusing phosphate species, whilst also reducing the hydrogen bond network frustration, resulting in a slight decrease in conductivity. Increasing water content leads to an increase in conductivity contribution via the vehicular mechanism through the transport of H3O+ and H2PO4−.92 This mechanism is shown in Fig. 1. Fig. 2 shows the peak contribution from structural diffusion in neat PA and an increase in vehicular transport as water content increases. Despite the improvement in total conductivity, operation at high temperatures and water content leads to increased PA loss.149 Although adding PBI to PA decreases the proton conductivity by decreasing the hydrogen bond network frustration, it also reduces the hygroscopicity (water uptake) of PA, which reduces electroosmotic drag and may explain why PA-PBI membranes outperform other PA-based electrolytes.93
 | ||
| Fig. 2 Total ionic conductivity of PA as a function of water content and the contributions from structural diffusion and ionic transport of H3O+ and H2PO4−. Adapted from ref. 92 with permission from the Royal Society of Chemistry. | ||
The activation process occurs after assembly but prior to testing or general operation. The aim of this process is to enable the maximum fuel cell performance to be achieved via a “break-in” period. The activation of LT-PEMFCs is mainly focused on the hydration of the PFSA membrane.166 During HT-PEMFC activation, PA is redistributed from the electrolyte driven by current, capillary force, and hydrophilic and hydrophobic properties of the catalyst layer.155 The optimal interface is achieved when the carbon support, catalyst sites and membrane are connected by a thin film of PA. This optimum is disrupted by the presence of water.167 Sufficient penetration of PA into the catalyst layers is required for catalyst utilisation and limitation of the charge transfer resistance.168 The activation procedure used can greatly influence the uniformity of current distribution and required procedure duration, not just the steady-state voltage.155 The procedure is also required to provide reproducible performance over time.169 Galvanostatic (constant current) activation is reported to yield the best results compared to current cycling, potential cycling, temperature cycling, and increased back pressure activation.155,170,171 Uneven distribution of PA within the MEA can initially lead to severe heterogeneous current density distributions which can cause local hot spots, resulting in local catalyst and membrane degradation.155 Typically, galvanostatic activation is performed at low current densities (0.2 A cm−2) and the duration ranges from 24 to 100 hours.155,169,172–174 This is in contrast to LT-PEMFCs where the activation procedure duration required is typically 6–20 hours, and often utilises potential cycling.175–178
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 ppm),34,200 whereas LT-PEMFCs show significant performance decrease around 0.005% (50 ppm).201,202 Reformate after the water–gas-shift reaction has a typical CO content of 2%.203 Therefore, HT-PEMFCs have the potential to use reformate fuel directly, whereas LT-PEMFCs cannot without additional cleaning, which adds cost and complexity. This is a major advantage over LT-PEMFCs in terms of commercialisation, as methods using steam-methane reforming are currently the cheapest way to produce hydrogen.204 It is even reported that CO can help to mitigate carbon corrosion and ECSA loss during load cycling.193 Engl et al. found CO blocked catalyst sites and inhibited the electrochemical reactions, easing degradation via the reverse current mechanism.205
000–50000 ppm),34,200 whereas LT-PEMFCs show significant performance decrease around 0.005% (50 ppm).201,202 Reformate after the water–gas-shift reaction has a typical CO content of 2%.203 Therefore, HT-PEMFCs have the potential to use reformate fuel directly, whereas LT-PEMFCs cannot without additional cleaning, which adds cost and complexity. This is a major advantage over LT-PEMFCs in terms of commercialisation, as methods using steam-methane reforming are currently the cheapest way to produce hydrogen.204 It is even reported that CO can help to mitigate carbon corrosion and ECSA loss during load cycling.193 Engl et al. found CO blocked catalyst sites and inhibited the electrochemical reactions, easing degradation via the reverse current mechanism.205
Introduction of PA to the system leads to degradation mechanisms that are not present in LT-PEMFCs. One of these mechanisms is acid migration. The number of PA molecules per imidazole group (acid doping level) can be as high as 70.206 This results in significant quantities of free acid with high mobility. As discussed in Section 2.2.2, there are multiple proton conductions mechanisms. One is the transport of H2PO4− from cathode to anode. Anion migration accounts for approximately 2–4% of the total charge transfer under typical HT-PEMFC conditions.207 This migration of H2PO4− increases with current density and doping level, and can lead to sufficient acid pressure at the anode to penetrate the catalyst layer and porous structures, potentially resulting in electrode flooding and acid loss.126,157,208,209 The leaching out of the membrane will cause ohmic resistance to increase, electrode flooding may impede mass transport of reactants, and ultimately reduce cell lifetime.157,208,209 The increase of H2PO4− at the anode leads to a concentration gradient which causes back diffusion towards the cathode, and under steady-state conditions an equilibrium is reached.207 The leaching acid typically travels through overlapping catalyst layer and MPL cracks.157 Designing catalyst layers and MPLs that have smaller cracks, and reducing crack connectivity between the catalyst layer and MPL can significantly reduce acid loss via this mechanism.69,126
Despite the H2PO4− migration mechanism, the majority of PA leaching during operation occurs at the cathode. Phosphoric acid is highly hygroscopic and hydrophilic, and the generation of water at the cathode causes free PA to be washed out.147,149,210 A recent study suggests that it is not the PA–water interaction that leads to leaching, but rather the inability of the polymer to hold both water and PA above a certain level.211 As the membrane is exposed to water, the PA cluster takes up water until it is limited by the polymer chains, the addition of water increases the cluster interaction energy and PA molecules are exchanged for water molecules which results in acid leaching. And as temperature increases, the water vapour pressure exceeds the hydrogen bonding of water to the PA cluster, and water molecules begin to escape; this explains why PA loss has been reported to be negligible at higher temperatures (160–180 °C).186,187,211 This newly proposed PA loss mechanism due to the exposure to water and high temperatures is shown in Fig. 3.
 | ||
| Fig. 3 Schematic of (a) previous PA loss mechanism and (b) newly proposed PA loss mechanism when exposed to water and high temperatures. Reproduced from ref. 211 with permission from the Royal Society of Chemistry. | ||
Another loss mechanism is PA evaporation. Despite the low vapour pressures of PA below 300 °C, acid can still be lost through evaporation, and this loss increases with temperature and reactant gas flow rate.212
Acid can also be lost during MEA assembly. If the hot-pressing conditions are not optimised (particularly over-compression) the diffusion layers can be soaked with acid.22,165 Once this acid enters the electrodes, it will be lost during conditioning or normal operation as the gases remove it from the porous diffusion layers. Therefore, it is important not to over-compress during assembly as this is another mechanism of acid loss. In summary, the acid loss mechanisms are:
(1) Phosphate anion migration from cathode to anode resulting in sufficient acid pressure to cause leaching at the anode;
(2) Significant water content causing free PA to be effectively washed out at the cathode;
(3) Evaporation of PA at high temperatures;
(4) PA penetration into the diffusion layers during hot-pressing.
While not a significant degradation mechanism, it is worth noting that as temperature increases and PA water content decreases, PA condensation reactions occur. The condensation products are polyphosphoric acids of higher molecular weight (e.g., H4P2O7, H5P3O10).92 This results in slower phosphate diffusion and a reduction in hydrogen bond network frustration, leading to a slight decrease in ionic conductivity.92,93 Lower molecular weight species (H3PO4) are regained by increasing the water content and subsequent hydrolysis reactions. A benefit of PBI membranes is the acid–base interaction reduces the concentration of condensation products while increasing the ortho-phosphate species.93
HT-PEMFCs also suffer from phosphate adsorption onto the Pt catalyst which block reaction sites and inhibit ORR and HOR.152,213,214 Adsorption of the phosphate species at the cathode is dependent on temperature and potential. The potential dependence is illustrated in Fig. 4. Hydrogen and oxygen adsorption are dominant below 300 mV and above 800 mV, respectively. Intermediate voltages (300–800 mV) are dominated by phosphate adsorption, and increasing the temperature decreases this window.152 Phosphate adsorption also occurs at the anode but is not expected to cause serious poisoning and may even assist in stabilising hydrogen adsorption. CO in the anode gas feed can increase phosphate adsorption, whilst the presence of water decreases coverage.214
 | ||
| Fig. 4 Dominating species adsorption on Pt catalyst across the polarisation curve. Reprinted with permission from ref. 152, Copyright (2013) American Chemical Society. | ||
HT-PEMFCs that use phosphonated polymers may also suffer from phosphonic acid anhydride formation which decreases proton conductivity; although, polymers can be designed with hydroxyl groups with low reactivity in phosphonic acid to avoid this formation.113,114
2.3 State-of-the-art HT-PEMFC performance
Table 1 gives an overview of HT-PEMFC and LT-PEMFC performance. HT-PEMFCs are clearly able to achieve high power densities and proton conductivities. PA-PBI based HT-PEMFCs show good performance at temperatures of ≈160 °C. Operation at temperatures much higher than this can cause issues with PA evaporation.114,212 A recent study showed good performance and PA retention from −20 to 200 °C using ultramicroporous PA-doped Tröger's base polymers (Fig. 5).115 The improved retention was attributed to the acid–base interactions and syphoning effect of microporosity. Recent work has also shown the promise of ion-pair membranes, specifically QAPOH which have demonstrated excellent PA retention at lower temperature operation.112–114,215 The use of ion-pair and phosphonated polymer ionomeric binders have also been shown to improve HT-PEMFC performance by increasing acid retention and limiting anhydride formation.113,114,215Fig. 6 shows a comparison of polarisation curves for protonated and non-protonated QAPOH-PA, and PBI-PA HT-PEMFCs to a LT-PEMFC and anion exchange membrane (AEM) FC. Use of QAPOH membranes and protonation of the ionomer showed a significant performance improvement over the PBI HT-PEMFC. The exponential voltage decay at high current densities is visible for the LT-PEM and AEM fuel cells, typically due to electrode flooding impeding reactant transport.216 Operating at HT-PEMFC temperatures results in a relatively linear i–V relationship at high current densities, due to the absence of liquid water. The HT-PEMFC using protonated phosphonic acid ionomers can achieve current and power densities comparable to the LT-PEMFC. Although, low current density performance is poorer for HT-PEMFCs due to sluggish ORR rates in concentrated PA.53,213| T (°C) | RH an./ca. (%) | P an./ca. (kPaabs) | Flow rate an./ca. flow rate (mL min−1), (cathode gas) | Membrane/thickness (μm) | Pt loading an./ca. (mg cm−2) | Peak power density (mW cm−2) | Proton conductivity@temperature (mS cm−1@°C) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Value estimated from plot. b Individual electrode loading not specified. | ||||||||
| LT-PEMFCs | ||||||||
| 60 | 100/100 | 101/101 | 35/70 (O2) | Phosphosilicate sol-SPEEK/67 | 0.48/0.48 | 450 | 138@70 | 220 |
| 70 | —/— | 101/101 | 60/60 (O2) | SiO2 SPEEK-Nafion/72 | 0.40/0.40 | 589 | 18@80 | 221 |
| 75 | 100/100 | —/— | 100/100 (O2) | TiSiO4-Nafion/70 | 0.40/0.40 | 803 | 249@70 | 222 |
| 80 | 50/50 | 101/101 | 300/300 (O2) | Phosphotungstic acid-graphene oxide-SPAEK/45 | 0.40/0.40 | 782 | 261@80 | 223 |
| 80 | 100/100 | —/— | 1.4/2.5 stoichiometry (air) | Layered double hydroxide-SPEEK/— | 0.40/0.40 | 560 | 230@80 | 224 |
| 80 | 100/100 | 148/148 | 500/500 (air) | GORE/15 | 0.10/0.40 | 1300a | — | 114 |
| 80 | 30/50 | 250/230 | 1.3/1.5 stoichiometry (air) | —/— | —/— | 1344 | — | 225 |
| 94 | 65/65 | 250/250 | 1.5/2.0 stoichiometry (air) | Nafion/14 | —/0.20 | 1300a | — | 226 |
| 94 | 65/65 | 250/250 | 1.5/2.0 stoichiometry (air) | Nafion/14 | —/0.05 | 960a | — | 226 |
| 100 | 100/100 | 148/148 | 500/500 (air) | Nafion/25 | 0.10/0.10 | 900a | — | 114 |
|
||||||||
| HT-PEMFCs | ||||||||
| 80 | 0/0 | 170/170 | 500/500 (O2) | QAPOH/35 | 0.50/0.70 | 530 | 4a@80 | 114 |
| 120 | 0/0 | 101/101 | 100/200 (air) | Pyridine-PBI/28 | 0.32/0.32 | 779 | 37@120 | 227 |
| 130 | 100/100 | 300/300 | 1.5/2.0 stoichiometry (O2) | Nafion/51 | 0.30/0.30 | 620 | — | 191 |
| 130 | 100/100 | 300/300 | 1.5/2.0 stoichiometry (O2) | Aquivion/30 | 0.30/0.30 | 870 | — | 191 |
| 140 | —/— | —/— | 300/500 (O2) | Phosphonated polystyrene/— | 0.25/0.375 | 184 | 5.8@140 | 228 |
| 160 | —/— | —/— | —/— (O2) | PBI/— | —/— | 638 | 89@160 | 229 |
| 160 | 0/0 | —/— | 80/160 (O2) | PBI/30–50 | —/— | 713 | 150@160 | 230 |
| 160 | 0/0 | —/— | 82/296 (air) | PBI/106 | 1.00/1.00 | 447 | 214@160 | 231 |
| 160 | 0/0 | —/— | —/— (air) | QA-PBI/— | 0.50/1.50 | 412 | 75a@160 | 232 |
| 160 | 0/0 | 101/101 | 200/200 (O2) | Dimethylbiphenyl Tröger's base polymer/160 | 0.50/0.50 | 815 | 140a@160 | 115 |
| 160 | 0/0 | 147/147 | 500/500 (O2) | PBI/50 | 0.50/0.60 | 500a | 100a@160 | 113 |
| 160 | 0/0 | 147/147 | 500/500 (O2) | QAPOH/40 | 0.50/0.60 | 1130 | 110a@160 | 113 |
| 200 | 0/0 | 147/147 | 500/500 (O2) | QAPOH/40 | 0.50/0.60 | 1503 | 110a@200 | 113 |
| 200 | 0/0 | 170/170 | 500/500 (O2) | QAPOH/35 | 0.50/0.70 | 2010 | 10a@160 | 114 |
| 200 | 0/0 | 101/101 | 200/200 (O2) | Dimethylbiphenyl Tröger's base polymer/160 | 0.50/0.50 | 630a | 120a@200 | 115 |
| 240 | 0/0 | 147/147 | 500/500 (O2) | QAPOH/40 | 0.50/0.60 | 1740 | 110a@240 | 113 |
| 240 | 0/0 | 285/285 | 200/500 (air) | SnP2O7-Nafion composite/80 | 0.60b | 410 | 80a@160 | 215 |
 | ||
| Fig. 5 Polarisation curves, power density and high-frequency resistance of HT-PEMFCs based on TB and m-PBI membranes without backpressure or humidification: (a) TB/PA and m-PBI MEAs at 160 °C, (b) PA/dimethylbiphenyl (DMBP)-TB performance 80–200 °C, (c) DMBP-TB MEA performance −20–40 °C, and (d) PA/DMBP-TB and PA/m-PBI MEA performance 40 °C.115 | ||
 | ||
| Fig. 6 H2/air polarisation curves for (a) protonated and non-protonated QAPOH, and PBI HT-PEMFCs at 160 °C and 148 kPa backpressure, and (b) Nafion LT-PEMFC and AEMFC at 80 °C and 148 kPa backpressure, with anode/cathode Pt loadings of 0.5/0.7, 0.5/0.6, 1.0/0.75, 0.1/0.4, and 0.5/0.6 mgPt cm−2, respectively.114 | ||
While high power densities are achieved with HT-PEMFCs, many of the studies use pure oxygen instead of air to achieve this performance. In many applications such as vehicles, air would be supplied instead, and the peak power density will be significantly reduced. The U.S. DoE has set a rated power target of 1800 mW cm−2 to be achieved with air supplied to the cathode by 2025.217 The data suggests commercial LT-PEMFCs are closest to achieving this target, although much of the information about commercial cells and stacks is proprietary. HT-PEMFCs still require performance improvement to achieve power densities >1000 mW cm−2 under H2/air operation. In addition, the total Pt group metal (PGM) content is recommended to be ≤0.10 mgPGM cm−2.217Table 1 and Fig. 6 show that significantly higher Pt loadings are used for HT-PEMFCs to achieve performance similar to LT-PEMFCs. Generally, Pt content needs to be reduced by a factor of 10 to meet the recommended target.
Power density is not the only important performance criterion. Durability is critical to real-world use of fuel cell technology. LT-PEMFCs have shown their durability in applications, and have met or exceeded durability targets in some cases, but still require improvement.218 The U.S. DoE 2025 targets require dynamic durability exceeding 8000 hours based on its own drive cycle protocol. Tables 2 and 3 show the steady-state and dynamic degradation rates of LT and HT-PEMFCs across a range of tests.
| T (°C) | RH an./ca. (%) | P an./ca. (kPaabs) | Membrane/thickness (μm) | Pt loading an./ca. (mg cm−2) | Voltage (V) | Current density (A cm−2) | Test duration (h) | Degradation rate (μV h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Value estimated from plot. b Average of stack. c Value after initial performance increase. | |||||||||
| LT-PEMFC | |||||||||
| 55 | 20/20 | 101/101 | Gore/— | —/— | 0.58–0.49a,b | 0.50 | 1000 | 90b | 233 |
| 70 | 100/100 | 101/101 | GoreSelect/35 | 0.45/0.60 | 0.65–0.54 | 0.80 | 26300 |
4–6 | 234 |
| 75 | 100/100 | 480/480 | Dow/— | 4.00/4.00 | 0.80–0.79a | 0.86 | 11000 |
1.3 | 235 |
| 75 | 100/100 | 480/480 | Dow/— | 4.00/4.00 | 0.85–0.83a | 0.54 | 11000 |
1.4 | 235 |
| 75 | 81/81 | 101/101 | Nafion/51 | 0.50/0.50 | 0.67c–0.61a | 0.30 | 2700 | 22 | 236 |
| 80 | 100/100 | 150/150 | GoreSelect/25 | 0.40/0.40 | 0.78–0.73a | 0.20 | 2000 | 25 | 237 |
| 80 | 100/100 | 150/150 | GoreSelect/25 | 0.40/0.40 | 0.96–0.93a | 0.00 | 2000 | 20 | 237 |
|
|||||||||
| HT-PEMFC | |||||||||
| 40 | 0/0 | 101/101 | Dimethylbiphenyl Tröger's base polymer/160 | 0.50/0.50 | 0.48–0.41a | 0.30 | 150 | 467 | 115 |
| 80 | 0/0 | 148/148 | QAPOH/40 | 0.50/0.60 | 0.50 | 0.20 | 200 | 0 | 114 |
| 140 | 0/0 | —/— | PBI/50–75 | 1.00/0.75 | 0.62–0.59a | 0.25 | 1600 | 13b | 238 |
| 150 | 0/0 | 101/101 | PBI/— | 1.70 (total) | 0.42–0.38a | 1.00 | 1100 | 41 | 147 |
| 150 | —/— | 101/101 | PBI/— | 0.15/0.35 | 0.43a,b | 0.20 | 1100 | 22b | 239 |
| 150 | 0/0 | 101/101 | PBI/— | 0.90/1.70 | 0.70–0.55a | 0.20 | 4000 | 16–53b | 240 |
| 160 | 0/0 | 148/148 | QAPOH/40 | 0.50/0.60 | 0.60c–0.59 | 0.60 | 2500 | 3.3c | 114 |
| 160 | 0/0 | 147/147 | QAPOH/40 | 0.50/0.60 | 0.55 | 0.60 | 550 | 0.35 | 113 |
| 160 | 0/0 | 101/101 | PBI/40 | 0.30–0.36/0.9 | 0.62–0.55a | 0.20 | 13000 |
4.6c | 241 |
| 160 | 0/0 | 101/101 | PBI/40 | 0.30–0.36/0.9 | 0.59–0.57 | 0.20 | 13000 |
1.4c | 241 |
| 160 | 0/0 | —/— | PBI/45 | 1.00 (total) | 0.67 | 0.20 | 187 | 36 | 110 |
| 160 | 0/0 | —/— | PBI/50–75 | 1.00/0.75 | 0.53–0.39 | 0.40 | 659 | 180b | 242 |
| 167 | 1.5/0 | —/— | PBI/— | —/— | 0.53–0.49a | 0.50 | 810 | 49a,b | 243 |
| 180 | 0/0 | 101/101 | PBI/90 | 0.25/0.78 | 0.65–0.62 | 0.20 | 10400 |
2.3 | 100 |
| 200 | 0/0 | —/— | PBI/70 | 0.10/1.60 | 0.68–0.53 | 0.20 | 2700 | 27c | 244 |
| T (°C) | RH an./ca. (%) | P an./ca. (kPaabs) | Membrane/thickness (μm) | Pt loading an./ca. (mg cm−2) | Test profile | Test duration (h or cycles) | Degradation rate (μV h−1 or μV per cycle) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Value estimated from plot. b Average of stack. c Backpressure. d Three different MEA manufacturers. | ||||||||
| LT-PEMFC | ||||||||
| 40 | 100/100 | —/— | —/20 | 0.40 (total) | Start-up/shutdown | 1200 cycles | 125@0.40 A cm−2 | 245 |
| 65 | 100/100 | —/— | —/20 | 0.40 (total) | Start-up/shutdown | 1200 cycles | 233@0.40 A cm−2 | 245 |
| 80 | 100/100 | —/— | —/20 | 0.40 (total) | Start-up/shutdown | 1200 cycles | 317@0.40 A cm−2 | 245 |
| 55 | 50/50 | 101/101 | Nafion/23 | —/— | Dynamic load cycle (NEDC) | 300 h | 40@0.00 A cm−2a,b | 246 |
| 150@1.00 A cm−2a,b | ||||||||
| 75 | 50/50 | 101/101 | Nafion/23 | —/— | Dynamic load cycle (NEDC) | 300 h | 210@0.00 A cm−2a,b | 246 |
| 560@1.00 A cm−2a,b | ||||||||
| 65 | 95/95 | —/— | Nafion/— | 0.10/0.40 | Start-up/shutdown | 1000 cycles | 70@0.64 A cm−2a | 247 |
| 80@1.00 A cm−2a | ||||||||
| 65 | 50/80 | 101/101 | Nafion/25 | 0.10/0.30 | Current step cycling | 2000 cycles | 19@0.60 A cm−2b | 248 |
| 130@1.00 A cm−2b | ||||||||
| 76 | 50/50 | 100/80c | GORE-Nafion/18 | 0.40 (total) | Dynamic load cycle (NEDC) | 1000 h | 41@1.00 A cm−2a,b | 249 |
| 80 | 90/90 | 101/101 | Nafion/28 | 0.09/0.61 | Start-up only | 750 cycles | 27@0.67 A cm−2 | 250 |
| Shutdown only | 930 cycles | 32@0.67 A cm−2 | ||||||
| 80 | 90/90 | 101/101 | Nafion/28 | 0.09/0.12 | Start-up only | 360 cycles | 472@0.67 A cm−2 | 250 |
| Shutdown only | 660 cycles | 273@0.67 A cm−2 | ||||||
| 80 | 100/100 | 101/101 | —/18 | 0.018/0.10 | Potential cycling | 30000 cycles |
4@1.50 A cm−2 | 251 |
|
||||||||
| HT-PEMFC | ||||||||
| 160 | —/— | 101/101 | PBI/50–75 | 1.00/0.75 | High current cycling | 51 h | 400@0.60 A cm−2 | 189 |
| 500@1.00 A cm−2 | ||||||||
| 80–160 | 1–16/1–16 | 68.9/68.9c | QAPOH/80 | 0.60/0.60 | Thermal cycling | 550 cyclesa | 390@0.15 A cm−2 | 112 |
| 80–160 | 1–16/1–16 | 68.9/68.9c | PBI/50 | 1.00/0.75 | Thermal cycling | 70 cycles | 11143@0.15 A cm−2 (failure) |
112 |
| 160 | 0/1.4 | —/— | PBI/40 | 1.50/1.50 | Potential cycling | 30000 cycles |
9@0.30 A cm−2a | 156 |
| 25–180 | —/— | 101/101 | PBI/— | 1.00/0.75 | Start-up/shutdown | 4000 h | 19@0.20 A cm−2 | 252 |
| 160 | —/— | —/— | PBI/— | 1.00/1.50 | Start-up/shutdown | 600 h | 30@0.20 A cm−2 | 194 |
| 160 | —/— | —/— | PBI/— | 1.00/1.50 | High current cycling | 600 h | 84@0.20 A cm−2 | 194 |
| 160 | —/— | —/— | PBI/— | 1.00/1.50 | Low current cycling | 600 h | 115@0.20 A cm−2 | 194 |
| 160 | 0/0 | 100/100 | PBI/50–75 | 1.00/0.75 | High current cycling | 70 h | 486@1.00 A cm−2 | 146 |
| 160 | 0/0 | 100/100 | PBI/50–75 | 1.0/0.75 | Low current cycling | 70 h | 363@0.30 A cm−2 | 146 |
| 30–160 | —/— | —/— | PBI/— | —/— | Start-up/shutdown | 20 cycles | 4–73@0.30 A cm−2 | 219 |
| 160 | —/— | —/— | PBId/— | —/— | High current cycling | 1100 cycles | 32–62@0.30 A cm−2 | 219 |
| 48–144@0.60 A cm−2 | ||||||||
| 68–314@1.00 A cm−2 | ||||||||
| 80–180 | 0/0 | —/— | PBI/50–75 | 1.00/0.75 | Start-up/shutdown | 1562 cycles | 26@0.25 A cm−2b | 238 |
| 11@0.03 A cm−2b | ||||||||
Differences in durability testing protocols across studies make direct performance comparisons difficult, but overall trends can be inferred. Generally, the reported degradation rates in Tables 2 and 3 are similar for LT and HT-PEMFCs. However, LT-PEMFCs can achieve such rates at higher current densities and voltages and therefore can achieve a higher power density with a similar degradation rate to HT-PEMFCs. LT-PEMFCs are closer to achieving the 2025 durability target of 8000 hours under DoE test protocols, likely due to their maturity and greater testing and characterisation under dynamic conditions. There are far fewer studies of HT-PEMFCs using drive cycles, therefore, it is more difficult to state the current status under these test conditions. However, studies have shown cycling at high current densities typically leads to high degradation rates in HT-PEMFCs due to loss of acid from the membrane and through the diffusion layers, and catalyst layer degradation.146,189,219 Crucially, HT-PEMFCs require significantly higher PGM loadings compared to LT-PEMFCs to achieve these degradation rates as is evident in Tables 2 and 3. Therefore, research efforts should be aimed at improving durability and reducing PGM content, with a key tool in achieving this being effective characterisation.
There are also fewer studies reporting the durability of HT-PEMFC stacks, particularly under dynamic conditions. This may be partly attributable to the increased material cost of testing stacks, as well as the testing hardware available. However, large active area cells assembled into stack hardware is more representative of commercial systems than are small area MEAs. Thus, testing and characterisation at the stack level is an important step in commercialising the technology. The degradation rates in Table 2 for stacks are typically higher than MEAs, suggesting the cell area scale up and stack design may have important effects on durability. These improvements required for HT-PEMFC performance and durability necessitate the development of advanced characterisation techniques and standardised AST protocols. These techniques must allow for comprehensive assessment and comparison across multiple scales from physico-chemical processes, up to overall electrochemical performance.
3. Electrochemical characterisation techniques
The electrochemical performance of the cell must be evaluated as it is one of the most important parameters alongside cost. Electrochemical techniques are used to evaluate the performance, degradation and durability, and are also used to validate and inform modelling efforts.3.1 Polarisation curve
The polarisation curve is a common method of evaluating PEMFC performance. For HT-PEMFCs, the method involves initially stabilising the fuel cell at constant current, typically 0.2 A cm−2 to avoid high potentials which could cause carbon corrosion at lower current densities or PA loss at high current densities.253,254 After stabilisation, the current density is incrementally increased from 0 A cm−2 with a constant step time e.g., 30 s, and the corresponding voltage at each increment is measured, referred to as a galvanostatic sweep. Typically, once the voltage drops below a pre-set value, the current direction is reversed, and the voltage is measured again at each increment. This is particularly important for LT-PEMFCs which can exhibit significant hysteresis behaviour.216,255 The hysteresis effect is attributed to the water dynamics in the membrane due to the water generation at the cathode, and water storage capabilities of the membrane and GDLs.216 The influence of water on proton conduction and electrode flooding means the water dynamics affect the performance, with the backward sweep typically yielding better performance.255 This can be attributed to a greater level of membrane hydration for the backward sweep as water has accumulated at higher current densities during the forward sweep. HT-PEMFCs have also been reported to exhibit hysteresis behaviour, an example of this is shown in Fig. 7.256,257 However, discussion on the phenomenon in HT-PEMFCs is very limited. PA is hygroscopic, and water is known to improve the conductivity of HT-PEMFCs. Therefore, the hysteresis effect may be attributed to water dynamics as in LT-PEMFCs. In this case, it should be expected that the hysteresis effect for HT-PEMFCs is less severe than for LT-PEMFCs due to the greatly diminished influence of water on membrane conductivity characteristics. However, literature reports are inconclusive as both sweep directions have been reported to yield the best performance depending on the study.256,257 Further research is required to confirm the cause of hysteresis and the dependence on operating conditions. The degree of hysteresis may then be useful as a diagnostic technique.216 Due to the length of hold and measurement times, polarisation curves capture the pseudo steady-state performance, which can be useful in steady-state modelling. However, other techniques are required for analysis of dynamic performance. | ||
| Fig. 7 Polarisation curve showing hysteresis behaviour in HT-PEMFC operating at 160 °C with dry input gases. Reprinted from ref. 256, Copyright (2015), with permission from Elsevier. | ||
3.2 Current interrupt (CI)
Current interrupt (CI) is a simple and fast technique for determining high-frequency resistance (HFR). CI involves measuring the voltage response to an abrupt step-change in current. This measurement can be done in parallel with polarisation curve measurements, which is beneficial as it captures the resistance change as a function of current. CI is often used to estimate HFR, and this is predicated on the principle that the voltage response to ohmic resistance is nearly instantaneous, and therefore can be distinguished from other losses in the response.258 CI has been reported to give higher resistance values than electrochemical impedance spectroscopy (EIS) due to the larger perturbation of CI causing non-zero potential distributions within the porous electrodes, yielding an additional ohmic loss contribution.259 Also, if the measurement frequency used to determine HFR during CI is significantly lower than EIS, e.g., 102 Hz compared to >105 kHz, this would result in non-ohmic resistance contributions being measured. Despite this, CI is often used to infer membrane resistance,20 and has the benefit of being less complex than EIS. The CI method does not differ between LT and HT-PEMFC technology.3.3 Electrochemical impedance spectroscopy (EIS)
EIS is another widely-used method for characterising electrochemical systems, and for the theory of the technique and application to PEMFCs in general, the reader is referred to existing review articles.260–262 During an EIS measurement, the fuel cell is held at a constant current (or voltage if using potentiostatic mode), an AC perturbation signal which is typically 5–10% of the DC signal (or approximately 10 mV if using potentiostatic mode) is applied and the voltage (or current) response is measured.260–262 The optimum amplitude requires a trade-off that allows sufficient signal-to-noise without violating linearity or stability requirements; the optimum amplitude may also vary with frequency. Total Harmonic Distortion (THD) has been used to identify the optimum amplitude as a function of frequency, with values ranging from 5–80% of the DC current signal.263 Galvanostatic EIS is generally preferred for fuel cells as it is less likely to result in the cell being overloaded compared to potentiostatic mode whereby small voltage changes result in large current changes.262 However, potentiostatic mode allows for better representation of electrode kinetics which are potential-dependent. The impedance measurement is repeated at intervals across a wide frequency range. The typical frequency range for electrochemical systems is 0.01 Hz to 10 kHz, with 10 points per decade.260 A range of 0.1 Hz to 100 kHz has been recommended specifically for HT-PEMFCs.264 The fuel cell should be held at the EIS measurement conditions of interest prior to recording the spectrum to enable stable measurements; values between 5 min and 30 min are reported for HT-PEMFCs,264–267 and the necessary time may differ with operating conditions. The resulting spectra then only provides a snapshot of the impedance at this condition (e.g. potential, temperature, relative humidity, pressure, etc.). Therefore, many measurements may need to be taken depending on the application of characterisation data.The gases used may also differ depending on the desired analysis. Generally, EIS is run with H2 at the anode, and air (or O2) at the cathode. This is most useful for analysing the fuel cell under typical operation, and allows analysis of ORR and oxygen mass transport impedances to be measured. However, by supplying N2 to the cathode, these impedance mechanisms along with water and electrochemical heat generation are removed. By neglecting the anode by assuming fast HOR and no H2 mass transport limitation, the fuel cell impedance is the sum of the cathode catalyst layer impedance, membrane resistance, and other electronic resistances of the system.268 Therefore, the proton resistance and charge double layer capacitance of the cathode catalyst layer can be determined by fitting a transition line model, where the 45° line at high frequencies is related to the proton resistance.
EIS is very useful for investigating different mechanisms within the fuel cell e.g., charge transfer, double-layer capacitance, mass transport of reactant species, and ohmic resistance.269,270 This is due to its ability to measure the impedance evolution and accumulation over a wide frequency range, which enables separation of mechanisms depending on their characteristic time constants. This capability has been used to determine the effects of operating conditions, degradation, and design optimisation.145,179,271–274 EIS data can be analysed using equivalent circuit models (ECMs). These models can consist of a combination of resistors, capacitors, inductors, and specialised electrochemical components (Warburg diffusion and constant phase elements).270 Analysis using ECMs is not straightforward due to the complexity of fuel cell operation. EIS data can be fit using a variety of ECMs, and the more circuit elements, the closer the fit. However, purely empirical models lack insight into the underlying physical and chemical parameters.20 To combat this, physically-representative models are used, and the simplest and most common is the Randles circuit.270 This ECM consists of a resistor representing the electrolyte resistance connected in series to a parallel combination of a resistor and capacitor representing charge transfer resistance and a double layer capacitance, respectively. However, the simplicity may mean that fidelity is compromised, and impedance processes may be missed. Other more complex models are used to represent a greater number of different impedance mechanisms within fuel cells, these are often developed in conjunction with distribution of relaxation times (DRT) analysis.275,276 Applying these more complex models will require knowledge of what features are expected in the spectrum and an understanding of the frequency dependence.
These models have been used to analyse the effects of operating conditions in more detail as specific resistance and capacitance values are attributed to individual impedance mechanisms.186,275,276 The ECMs can also be used to model fuel cell performance with dynamic capability.276–278
Despite the usefulness and ubiquity of EIS, difficulty remains in obtaining valid spectra, and proof of the data validity is often missing from studies in the literature. To be valid, EIS measurements must fulfil certain criteria:260,270
• Linearity – the response to the input perturbation must be linear- or at least pseudo-linear- for the mathematical analysis to be meaningful. The relationship between voltage and current in a fuel cell is often non-linear. Therefore, sufficiently small perturbations are required to achieve a pseudo-linear response such that the system is under steady-state conditions. However, the signal amplitude must be high enough for a sufficient signal-to-noise ratio.
• Stability – the system response to the perturbation must not cause the system to drift from steady-state. This can be an issue for fuel cells at low frequencies as the system undergoes changes e.g., oxide layer growth, adsorption of impurities, temperature changes, etc.
• Causality – the output of the system must only be caused by the perturbation input.
A validity check can be applied to the EIS data to determine if any corruption occurred. The Kramers–Kronig (KK) transformation is often used to test the validity. The KK relation links the real and imaginary impedance and allows one of these contributions to be uniquely calculated if the other is known. The KK transformations were derived under the same assumptions required for valid EIS: linear, stable, and causal.260 Therefore, if the impedance contribution calculated using the KK transformation does not adequately match the measured spectra, the validity criteria have been violated. The difference between the measured and KK-generated spectra are termed “residuals”, Fig. 8 shows an example of KK and EIS-generated spectra along with KK residuals. The residual limit for determining if the data is valid is not defined mathematically and can vary between studies. Generally, the residual limit is 0.5–1.0%.265,267,279 It is also important to inspect the residuals across the frequency range in order to determine systematic errors that may not yield residuals high residuals but still result in invalid data.280 Maintaining constant conditions during the measurement e.g., temperature, gas flow, pressure, etc., is required to prevent instability or time-variance that can result in invalid data. It is important to note that many EIS studies of PEMFCs in the literature do not include the validity check, and thus interpretation of the EIS data is problematic.
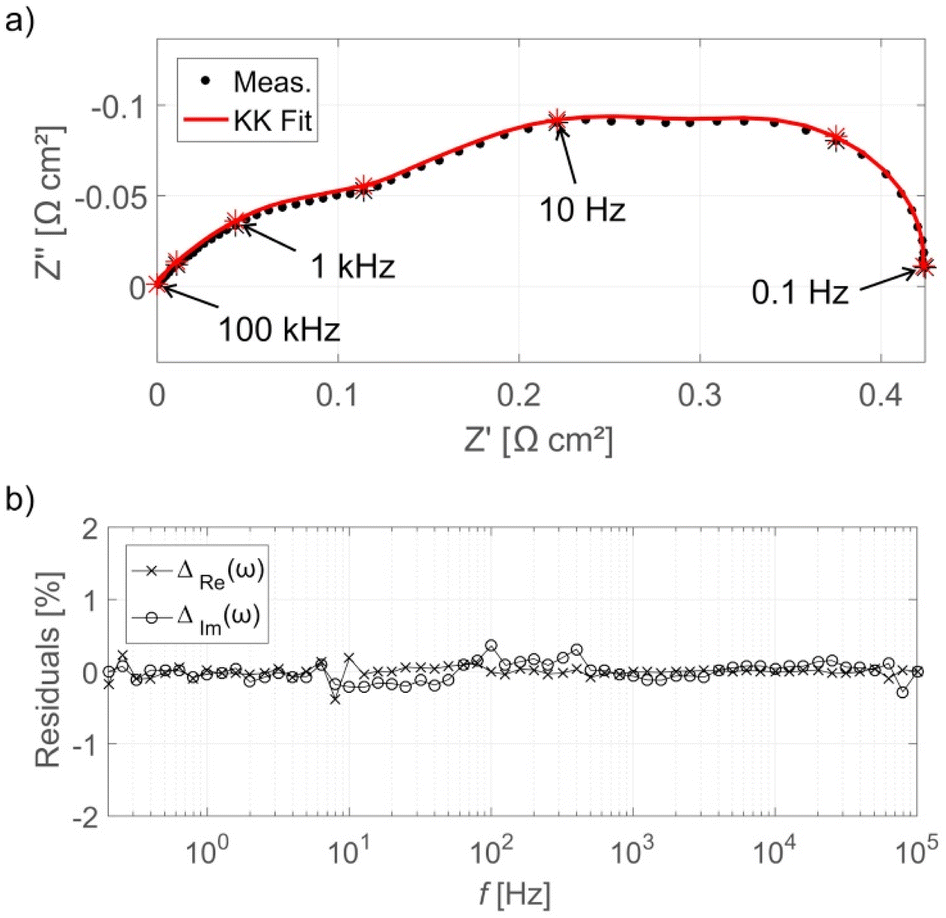 | ||
| Fig. 8 (a) EIS Nyquist plot for HT-PEMFC operating at 160 °C at 0.3 A cm−2 with KK fit, and (b) KK residuals as a function of frequency. The EIS spectra have been normalised by subtracting the high-frequency real axis intercept. Reprinted from ref. 265, Copyright (2017), with permission from Elsevier. | ||
There are no significant differences between conducting EIS measurements for HT-PEMFCs compared to LT-PEMFCs. At the highest frequencies, inductive behaviour is often visible and is caused by the test cables and equipment.269,280 This has been reported in both HT-PEMFCs and LT-PEMFCs.272,279,281 The Zre (also denoted: Z′) axis intercept at high frequencies, often referred to as HFR, is ascribed to ohmic resistance.262 As proton conductivity dominates ohmic resistance, the intercept is used to indicate electrolyte characteristics.267,279,282 This applies to both LT and HT technology and the only difference is the proton conductivity mechanisms. EIS spectra commonly display one or more arcs. These arcs are attributed to different impedance mechanisms. HT-PEMFCs share many of the same impedance mechanisms, and these will show up in the spectra in a similar way to LT-PEMFCs. These include charge transfer and mass transport limitations. These can then be deconvoluted further using DRT analysis which can provide attributions to electrode-specific processes such as proton conduction, HOR, ORR, and H2 and O2 transport impedance.265
The main difference when analysing HT-PEMFC EIS spectra is the behaviour at low frequencies. LT-PEMFCs have been repeatedly reported to exhibit an inductive loop at low frequencies (<0.1 Hz); but often studies do not show this behaviour, and it is unknown if the studies omit it or if it is physically absent.269 An example of the inductive loop is shown in Fig. 9a. The locations of HFR, total polarisation resistance (total R), last measured point (LMP) which is typically limited by equipment or experimentation time, and the DC point which refers to the resistance that would be measured as the slope of the polarisation curve, are indicated.269 The cause of the low-frequency inductive loop is disputed. One explanation for the loop involves side reactions with intermediate species. The multi-step ORR mechanism involves adsorption of species onto the catalyst sites and the formation of intermediates such as peroxide.283–285 One study suggested the inductive loop was caused by an intermediate step and was not dependent on current or potential.286 The inductive loop has also been predicted by models accounting for hydrogen peroxide intermediate formation as part of ORR. It has also been explained by the formation and relaxation of Pt oxide.283,285,287 Another explanation involves water dynamics where the product water transport from cathode to anode at high current densities leads to a membrane resistance reduction.288,289 It has also been suggested that the slow uptake of water by the membrane at high current densities and release at lower current densities causes the inductive loop.290 The slow hydration of the ionomer may also contribute to this inductive effect.291 These explanations may be valid under low relative humidity conditions, but other explanations may be required under fully humidified conditions.269,290,291 CO poisoning is another explanation for the inductive phenomena.292,293 Oxidation of CO and subsequent desorption of CO2 results in an increase in the number of sites for H2 chemisorption onto the Pt surface, reducing the charge transfer resistance.294 Use of pure O2 as the oxidant was reported to prevent the appearance of the inductive loop, this was attributed to the direct chemical oxidation of CO without intermediate steps.294 Overall, the cause of the low-frequency inductive loop is still unclear and may depend on multiple different processes.269
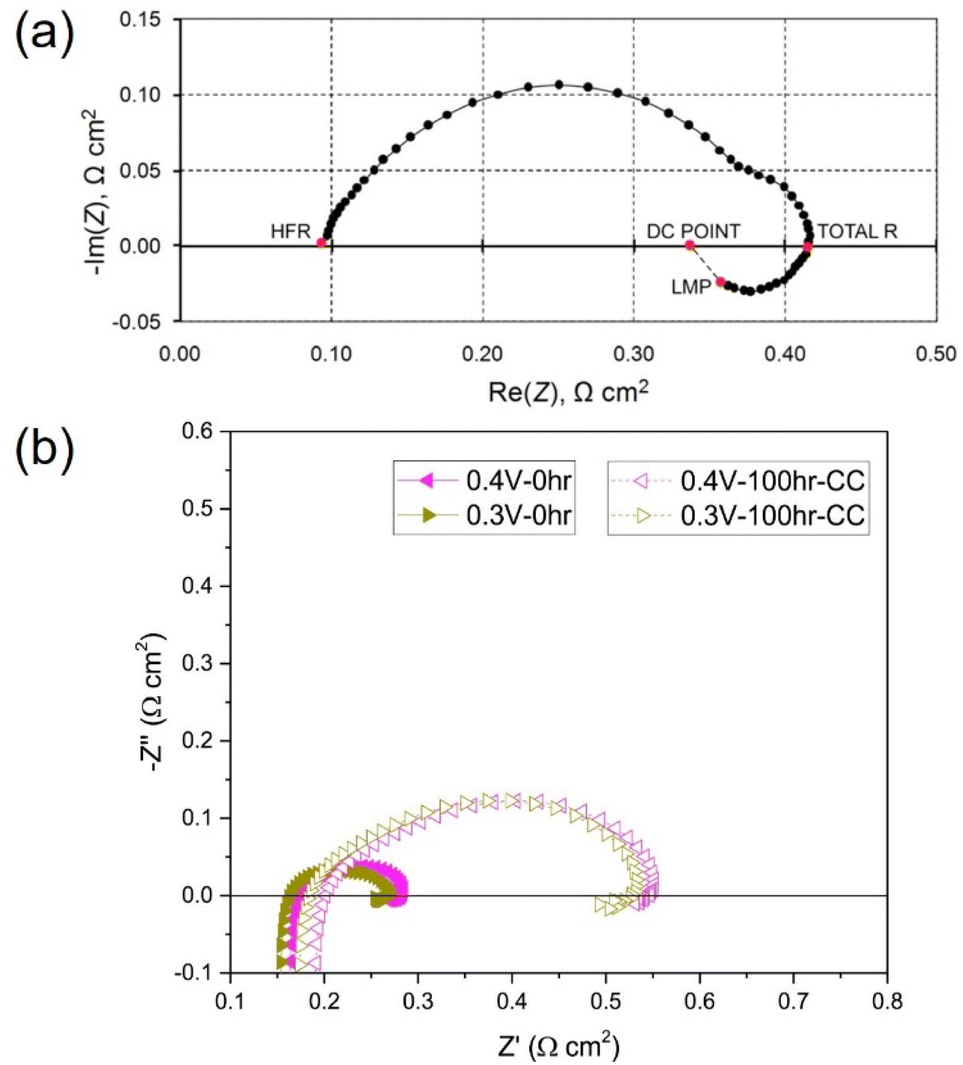 | ||
| Fig. 9 (a) LT-PEMFC EIS Nyquist plot with inductive loop indicating high-frequency resistance (HFR), total R, last measured point (LMP), and DC point locations, reprinted from ref. 269 (open access); (b) HT-PEMFC EIS measurements showing inductive loop at various cell voltages before and after 100 h at 0.2 A cm−2, reprinted from ref. 281, Copyright (2018), with permission from Elsevier. | ||
There are very few studies that report low-frequency inductive behaviour for HT-PEMFCs (an example is shown in Fig. 9b). This may be for several reasons: (i) there are fewer EIS studies of HT-PEMFCs giving the appearance it occurs less frequently; (ii) the behaviour does occur but is not being reported; (iii) the changes in operating conditions i.e., higher temperature and reduced water content mean that the behaviour appears only at lower frequencies than is being tested, or rarely occurs at all. There are studies that do report the inductive phenomena for HT-PEMFCs and solid acid fuel cells.266,281,295 Within these studies, the loop is significantly smaller than in the LT-PEMFC studies. Different conclusions may be drawn from this observation. One is that the change in conditions is reducing the impact of intermediate species adsorption, Pt oxidation, CO poisoning, and/or water dynamics. This is feasible especially as ORR reaction rates and CO tolerance increase with temperature, and HT-PEMFCs do not rely on water for proton conductivity.37 So far, the low-frequency inductive phenomena in HT-PEMFCs has been attributed to phosphate poisoning, although Pt oxide growth and relaxation may also contribute.281,295
EIS has also been used to investigate the effects of different operating conditions on HT-PEMFC impedance,266 as shown in Fig. 10. The effect of increasing temperature on charge transfer is shown in Fig. 10a. The higher temperature was expected to improve the reaction kinetics, reducing charge transfer impedance and activation overpotential. A more complex relationship was visible at higher current densities, as shown in Fig. 10b. There were expected competing impacts of increased temperature on reaction kinetics and changes in humidification due to water vapour pressure changes.266 Whereas, the effect of air stoichiometry was clear at 1.0 A cm−2, decreasing the stoichiometry from 2.0 to 1.5 results in significantly higher mass transport impedance due, and likely caused inhomogeneous current density distributions. The effect of air humidification is shown in Fig. 10c and d. Increased humidification resulted in a decrease in HFR. This is expected as water is known to improve conductivity by increasing vehicular proton transport.92 This improvement is offset by increases in charge transfer and mass transport impedance, expected to be caused by increased phosphate anion adsorption and oxygen diffusion resistance, respectively.266
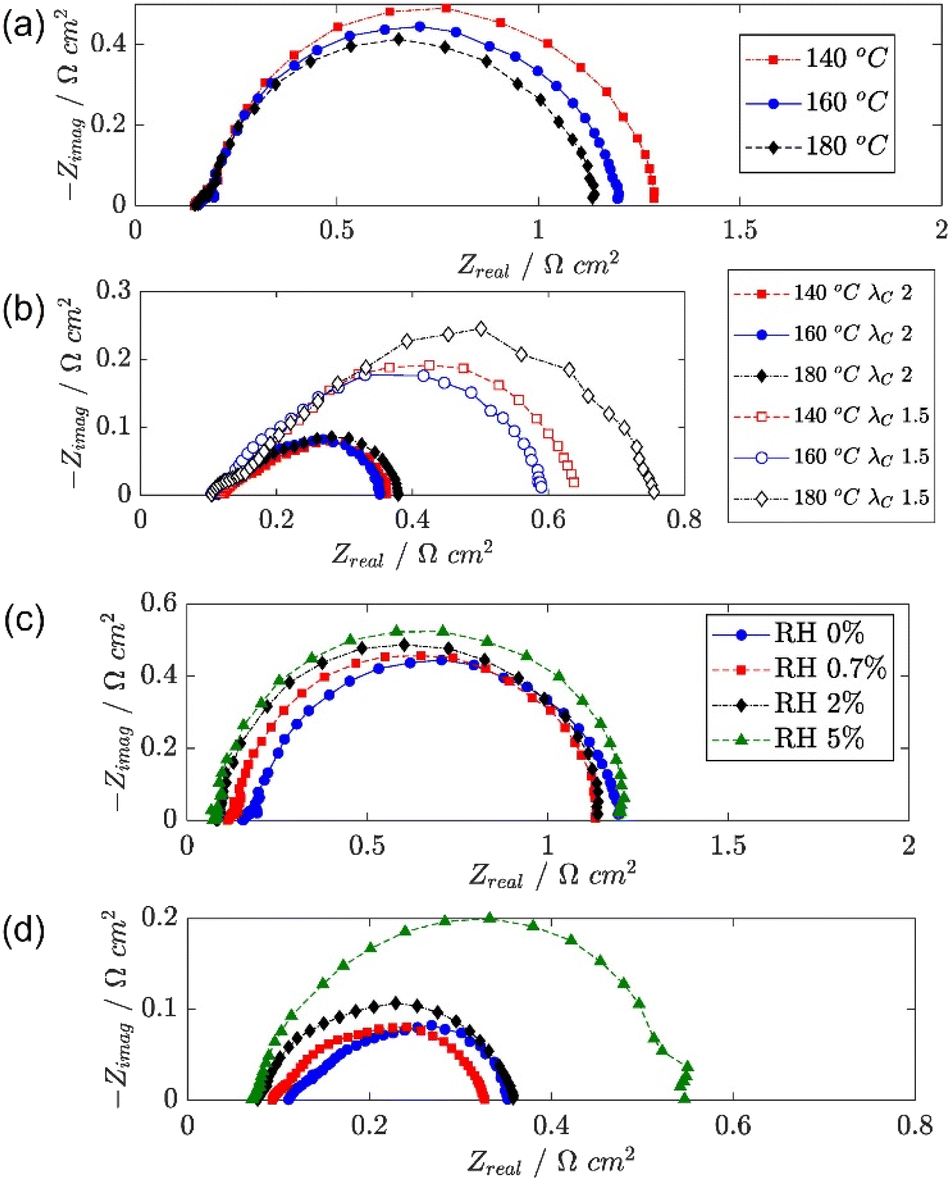 | ||
| Fig. 10 HT-PEMFC EIS Nyquist plots of (a) temperature effect at 0.05 A cm−2, (b) temperature and air stoichiometry effects at 1.0 A cm−2, (c) air relative humidity effect at 0.05 A cm−2 and (d) 1.0 A cm−2. Adapted from ref. 266 (open access). | ||
In summary, EIS is a powerful tool for HT-PEMFC characterisation and modelling. Careful attention must be paid to the validity of the data, this is also true for LT-PEMFCs. And low-frequency inductive phenomena in HT-PEMFCs requires more research to determine the extent to which it affects HT-PEMFCs and the cause(s).
DRT analysis has been used in LT-PEMFCs to investigate transport properties of a commercial stack,298 commercial stack fault characterisation,29 analysis and modelling of electrode properties,276,279,299 and investigation of low frequency inductive phenomena.285Fig. 11 shows the DRT plot for a HT-PEMFC, assignment of peaks, and relation to the individual impedance mechanisms within the EIS spectra. In this case, P1 is assigned to mass transport, P2 and P3 to the ORR, and P4–7 to anode-related processes such as charge transfer kinetics of the HOR.265 In HT-PEMFC studies, DRT has already been applied to a variety of studies. The effect of operating conditions including CO concentration in the anode gas steam was investigated using DRT.300 The results suggested the presence of CO increases HOR, ORR, and mass transport impedances. The use of a reference electrode allowed for individual electrode DRT analysis to be conducted. A DRT peak was reported in the anode analysis at around 100 Hz when a CO concentration of 5% was present. This peak was attributed to the influence of CO adsorption on charge transfer. The effect of air stoichiometry on the individual impedance contributions of mass transport, ORR, and HOR has also been investigated and are shown in Fig. 12a.267 Increasing the air stoichiometry significantly reduced the size and increased the frequency of the peak associated with mass transport. Although there were diminishing returns, especially at stoichiometries above 3. A similar, but less prominent trend was observed in the main peak attributed to ORR when increasing the stoichiometry. However, a minor ORR peak and the HOR peak did not show this trend, and an explanation is not provided for this behaviour. These results support a similar stoichiometry investigation from a previous study.265 In the same study, the effect of temperature was investigated, the DRT plot is shown in Fig. 12b. Higher temperatures resulted in a minimal decrease in the peak height associated with mass transport, and a larger decrease in peak height and shift to higher frequencies for the main ORR peak, attributable to faster reaction kinetics.267 An increase in time constants for the minor ORR and HOR/proton transport peaks are not discussed in the study. The effects of activation procedure have been analysed using DRT.155 The mass transport impedance was observed to decrease for all the activation procedures (galvanostatic, temperature cycling, current cycling, and elevating backpressure). The time constant for ORR was seen to decrease for all procedures except temperature cycling, indicating an acceleration in the charge transfer during the activation process. The effects of catalyst layer morphology, composition and manufacturing method have also been investigated using DRT.125,127 The use of a PtCo alloy resulted in a reduction in mass transport peak area, as well as a shift to higher frequencies for the ORR peak, indicating faster reaction kinetics compared to a conventional Pt/C catalyst.127 An Fe–N–C was also included in the same work, and multiple additional mass transport impedance processes were indicated in the DRT analysis that were not present in the other catalyst compositions. This was explained by the Fe–N–C catalyst morphology exacerbating the issue of poor oxygen accessibility to the catalyst sites. Poor impregnation of H3PO4 into the cathode was also expected. The impact of CO and other species such as CO2 have also been studied using DRT.272,301 Generally, the introduction of CO and other species does not improve performance, but synergistic effects may occur when species such as H2O, CO2, and CH3OH are also present. However, improvements appeared to depend on current density and can be minimal. A potential new application of DRT for HT-PEMFCs is the investigation of the inductive phenomena. Typically this behaviour is excluded in the analysis, with an exception for a LT-PEMFC study.285
 | ||
| Fig. 11 (a) EIS spectra of HT-PEMFC operating at 160 °C at 0.3 A cm−2 with individual impedance contributions, (b) DRT plot showing deconvolution and assignment of impedance contributions. Reprinted from ref. 265, Copyright (2017), with permission from Elsevier. | ||
 | ||
| Fig. 12 DRT plots of (a) effect of air stoichiometry and (b) operating temperature on a 45 cm2 HT-PEMFC at 0.2 A cm−2. Adapted from ref. 267, Copyright (2022), with permission from Elsevier. | ||
The depth of DRT analyses varies between studies, and so too does the detail provided about data validation and parameter choice. DRT requires excellent measurement quality due to its sensitivity, and KK relations should be used to indicate validity.265 Despite the lack of a priori assumptions, DRT is not straightforward. The DRT quality is sensitive to the number of measurement points in the impedance data.302 The mathematical calculation of DRT poses challenges, and in order to obtain a stable numerical solution, Tikhonov regularisation is typically used.302,303 This method requires the selection of a regularisation parameter, λ, which greatly influences the output DRT.304 Optimal selection of λ may be obtained through an iterative process combined with user experience, the value should then be applied to all data within that specific dataset.275 The regularisation parameter has a smoothing effect which is used to remove artificial, erroneous peaks. It is advised that λ should be as small as possible for good resolvability and as large as needed to suppress artefacts, and there is no effective “black box” method to determine this value, i.e., λ must be determined by the user through careful experimentation and analysis.304 It should also be noted that DRT approaches may extrapolate to frequencies past those used in obtaining the EIS data, meaning inaccurate conclusions could be drawn if impedance mechanisms did not fully evolve within the EIS measurement frequency range.
As DRT analysis uses EIS data, there are no specific considerations for HT-PEMFCs that do not apply to LT-PEMFCs regarding use of the technique, but knowledge of the differences between these systems is required for interpretation of results. And careful attention should be paid to the information provided on data validity when drawing conclusions.
3.4 Cyclic voltammetry (CV)
Cyclic voltammetry is a widely utilised technique to determine the ECSA in PEMFCs. The ECSA is calculated by dividing the charge density related to H adsorption/desorption (or CO oxidation) obtained in the CV by the product of the charge required to oxidise/reduce a monolayer of the adsorbed species and the catalyst loading.305 The conventional in situ method involves supplying H2 to the electrode of interest and N2 (or another inert gas) to the other electrode. The first acts as the reference and counter electrodes, and the second is the working electrode. This technique is referred to as the “hydrogen adsorption/desorption” method, where the hydrogen underpotential deposition, Hupd, is used for ECSA calculations. The potential is scanned in forward and reverse directions at a rate typically in the range of 10–100 mV s−1.146,171,186,306This technique is frequently used in LT-PEMFC,189,307–310 and HT-PEMFC ECSA measurements,114,130,147,161,186,311 although it has been suggested that this method is unreliable for HT-PEMFC ECSA measurements due to phosphate adsorption suppressing the Hupd peaks, and high faradaic hydrogen evolution reaction currents superimposing pseudocapacitive Hupd currents.116,312 The oxidation of a monolayer of CO or “CO stripping” is another common method for ECSA determination in HT-PEMFCs which makes use of the affinity of Pt to bind with CO, and is only partly affected by the problems affecting the Hupd method.306 However, there are issues with side reactions that influence the ECSA determination at higher temperatures, and the CO adsorption itself is strongly temperature-dependent and a calibration curve needs to be established to measure ECSA based on oxidation of a monolayer of CO at the temperatures of interest.116 There are several CO stripping methods to calculate ECSA. One method is to use the current of a second potential sweep as a baseline to determine the CO-related oxidation peak in the first sweep. Another method is to use a reference CV where no CO adsorption occurs, this can then be used to determine the CO-related oxidation peak when the CO monolayer is present. Both methods attempt to isolate CO oxidation from other current sources.306 A third method involves the detection of CO2 in the working electrode exhaust to infer the oxidation of the adsorbed CO and thus ECSA.313 One study used these methods to investigate the effect of the partial pressure of water on the ESCA.313 It was found that an increase in partial pressure of water above 10 kPa resulted in ECSA reduction when measured with both CV and exhaust CO2 techniques. This was attributed to the uneven distribution of the PA/water mixture on the catalyst and flooding of porous electrode structure. ECSA values determined via CO2 detection have been reported to have similar values to CO stripping using a reference CV across a wide temperature range (40–180 °C).306Fig. 13 shows the trends in ECSA values as a function of temperature using the three methods. The method using the second potential cycle as a baseline was deemed to overestimate ECSA due to the inability to distinguish between CO and other oxidation currents.306 The CO2 method has been shown to exhibit slightly lower ECSA values due to the CV methods being influenced by the formation of water and hydroxide species.313 The CO2 method requires a second cycle to be subtracted from the first to eliminate CO2 that is not produced by CO monolayer oxidation but instead by oxidation of carbonaceous species within the cell.306
 | ||
| Fig. 13 Apparent ECSA trend as a function of temperature using three methods. Reprinted from ref. 306 (open access). | ||
3.5 Linear sweep voltammetry (LSV)
Linear sweep voltammetry is a common technique for detecting electrical shorting and fuel crossover in PEMFCs.314 The technique involves the same gas supply and electrode designation as CV mentioned previously. Hydrogen that crosses from the anode to cathode should be fully oxidised and an instantaneous hydrogen oxidation current density is generated which allows for the estimation of hydrogen crossover rate.315 The potential is typically scanned from approximately 0 V to around 0.5 V with slow scan rates, e.g., 1–5 mV s−1.The measurement method is the same for LT-PEMFCs and HT-PEMFCs and has been used to determine the hydrogen crossover rate for both technologies, and is particularly useful in determining membrane degradation.194,219,274,309,316 The hydrogen crossover was monitored over a 6000 h HT-PEMFC durability test. The crossover current density increased from around 0.5 mA cm−2 at the beginning of test to 13.9 mA cm−2 after 5705 h of operation. This was attributed to membrane thinning and the formation of pinholes and microcracks.187 The impact of single-layer graphene (SLG) on hydrogen crossover has also been investigated for ultrathin (7.7 μm) PBI membranes.274 The addition of SLG was reported to significantly reduce hydrogen crossover, resulting in higher OCV. After 100 h at constant current, the hydrogen crossover current density of ultrathin PBI MEA without SLG increased from 21 to 70 mA cm−2, whereas the use of SLG resulted in a minor increase from 14 to 15 mA cm−2.
3.6 Current distribution mapping
Local variations in the electrochemistry, activity, temperature, and humidity all produce current density inhomogeneities.317 These inhomogeneities affect fuel cell performance and degradation, potentially causing damage to the membrane and catalyst layers.318,319 There are various methods to measure the current distribution within the cell. The measurements are typically obtained by introducing a segmented electronically conductive component. This can be in the form of segmented electrodes, flow fields, or current collectors.318,320 Printed circuit boards (PCBs) are often used to create segmented current collectors.317,321,322 These methods involve physically altering the fuel cell which can affect operation and performance. A non-invasive method that has been used involves the calculation of current density from the magnetic field surrounding the fuel cell.323,324The current distribution mapping technique for HT-PEMFCs follows the same principles as for LT-PEMFCs. The materials used in the devices may differ due to the different operational environments. Components in HT-PEMFCs must be able to withstand the higher temperatures and presence of PA.
Current distribution mapping in LT-PEMFCs has been used to investigate reactant starvation,318,320 start-up/shut-down,250,317,325 effects of operating conditions on single cells,319,321,326–328 effects of operating conditions on a stack,329 and localised EIS.327 One application of current distribution mapping used in HT-PEMFC research is the investigation of flow field geometry effects.154,330Fig. 14 and 15 show how this mapping technique has been used to investigate the impact on current density distribution of four different flow field patterns. This is important as current density distribution homogeneity is important to reduce hotspots. The serpentine and pin-type flow fields resulted in lower variation and more even distribution of current density compared to the parallel and interdigitated flow fields. Although, performance when using interdigitated flow channels was comparable to serpentine when using air at high flow rates. Interdigitated geometries have also been reported to achieve higher current and power densities than other geometries.331,332 Recent work suggested the improvement when using interdigitated geometries is likely due to the enhanced convective mass transport in the GDL, rather than from the pressure and subsequent oxygen concentration increase resulting from the dead-end geometry.332 The benefit of this is the reduced requirement of pressure drop compared to conventional designs such as serpentine, thus reducing parasitic power losses from the compressor.
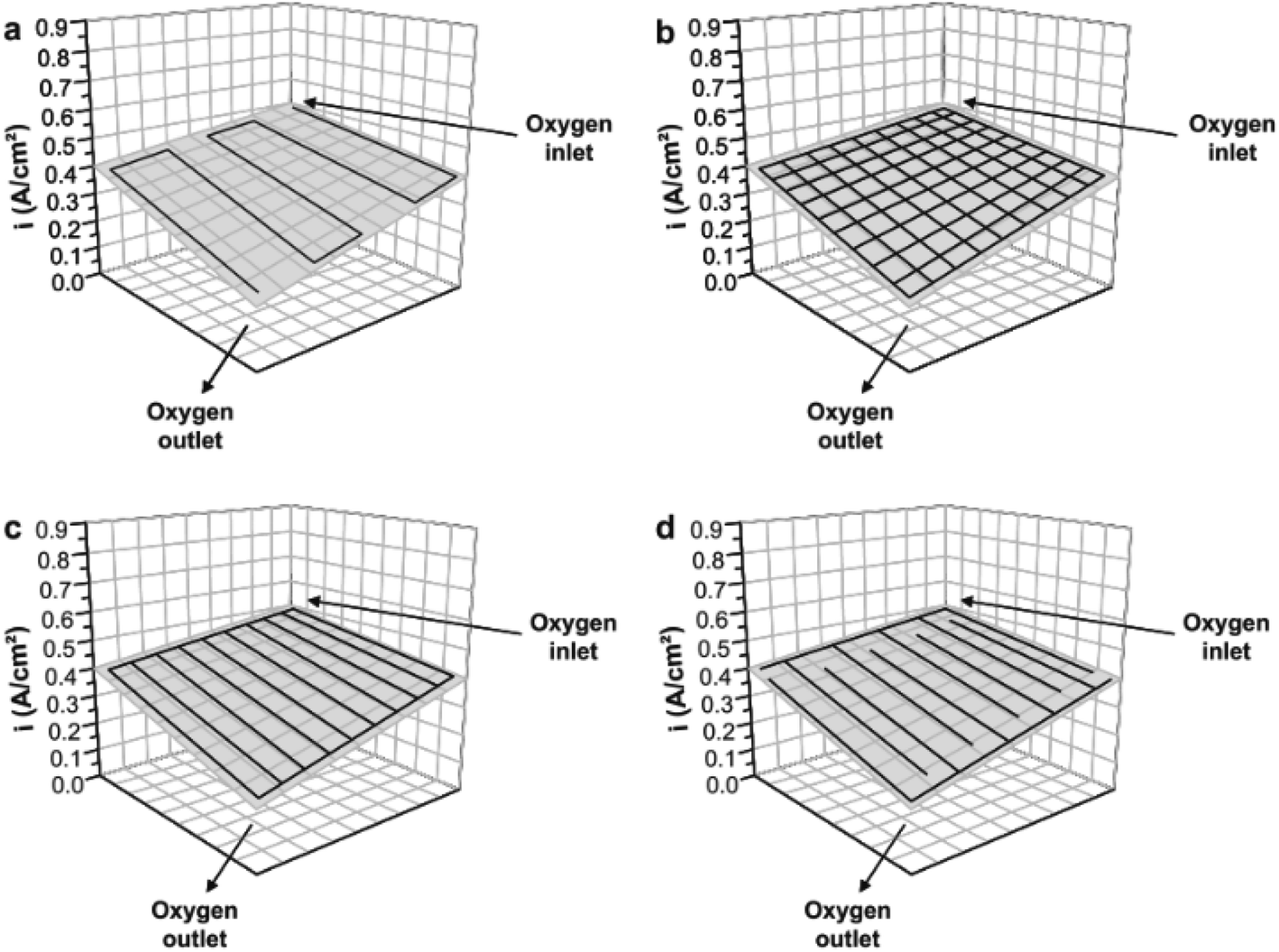 | ||
| Fig. 14 Flow channel pattern overlayed onto current distribution map with oxygen inlet and outlet indicated. (a) Serpentine, (b) pin-type, (c) parallel, and (d) interdigitated. Reprinted from ref. 154, Copyright (2011), with permission from Elsevier. | ||
 | ||
| Fig. 15 Current distribution maps at an average current density of 0.38 A cm−2 (H2/O2 228/76 ml min−1). Flow geometries correspond with those in Fig. 11. Reprinted from ref. 154, Copyright (2011), with permission from Elsevier. | ||
The influence of CO concentration in the anode fuel supply from 0% to 3% has also been investigated using current density distribution mapping.30 The results showed inhomogeneities in current distribution increased with increasing CO content, and this effect was worse at higher current densities. Current values were typically lower at the inlet and highest at the outlet, as increased poisoning effect was suspected at the inlet. Increasing the temperature from 160 °C to 180 °C reduced the relative difference between inlet and outlet current densities. It was also noted that load operation in potentiostatic mode resulted in more even current distribution than galvanostatic mode. Although the explanation for this is unclear.203 In another study, the current density distribution within a HT-PEMFC stack operating under both hydrogen and reformate conditions was investigated.51 Under hydrogen/air conditions with co-flow, the local current density was highest at the air inlet and lowest at the air outlet, which was explained by reactant consumption along the flow path. Under reformate/air co-flow conditions with an anode stoichiometry of 2, the depletion of both oxygen and hydrogen along their respective flow paths results in a greater degree of homogeneity. Although current is still higher closer to the anode and cathode inlets compared to outlets. Reducing the anode stoichiometry to 1.2 led to a significant increase in inhomogeneity which was attributed to CO poisoning. However, in this case local current density was highest at the inlet and lowest at the outlet. Operating in counter-flow mode was found to reduce the current density inhomogeneity compared to co-flow mode. Studies of the effects of reformate on LT-PEMFC current density distribution have also been conducted, but these use CO concentrations 3 orders of magnitude lower than HT-PEMFC. Once again, highlighting the significant improvement in CO tolerance of HT-PEMFCs. Other applications of this technique include durability testing,151,153 effect of catalyst layer deposition,333 effects of reactant stoichiometry using local EIS,334 and fuel starvation,190,335
Potential future opportunities for this technique to be used in HT-PEMFC include the effects of start-up and shutdown processes, particularly under sub-zero conditions. As mentioned in Section 2.2.4 there are several start-up strategies that are typically analysed regarding time taken to heat up rather than impact on cell performance, and using current distribution mapping to assess these different methods may provide useful insight. As noted previously, sub-zero operation of HT-PEMFCs is likely to be easier than LT-PEMFCs due to the reduced sensitivity to liquid water.115,185 Greater understanding of degradation and inhomogeneities at these conditions could be provided by current distribution mapping, as long as the device is capable of operation under these conditions.
4. X-ray techniques
Techniques such as those using X-rays and neutrons provide detailed material information that is not accessible using electrochemical techniques. These techniques provide useful information about the internal structure and processes occurring within the MEA spanning nm to mm length scales, which can be used in conjunction with electrochemical characterisation to progress development of fuel cell technology.4.1 X-ray radiography and CT
X-ray computed tomography (X-ray CT) is now a widely used technique for analysing the morphology of PEMFCs across multiple length- and time-scales. The technique works by passing a beam of X-rays through a sample, with the X-rays that pass through the sample being collected by a detector. The attenuation of X-rays is proportional to the atomic number, as well as the density of the material. Radiographs are collected at angles through 360°, termed “projections”, and these projections are then reconstructed using a computer algorithm to create a 3D dataset comprising of voxels (volume pixels).X-ray CT is a particularly adaptable technique, owing to the fact that multiple lenses and optics can be added to the detector to match the spatial resolution to the feature of interest, allowing for features from catalyst layer pores to flow fields to be resolved.336,337 Furthermore, as well as ex situ studies of PEMFC morphology, in situ/operando studies are now becoming more common for time-resolved studies of PEMFCs.
Since HT-PEMFC research is not as mature as for LT-PEMFCs, most of the studies carried out using X-ray CT have been ex situ, and the majority have focussed on the GDE. Diedrichs et al.271 and later Hoppe et al.342 used in situ compression rigs to correlate morphology changes in the GDL to fuel cell performance, arising from different compression levels and different flow-field misalignments, respectively. Start-stop conditions have also been investigated in HT-PEMFCs using X-ray CT, including studies into the idling temperature, showing that degradation leads to catalyst deactivation and pore collapse in the GDE,343 as well as a decrease in tortuosity of the GDL.219
As well as understanding the relationship between morphology and performance, ex situ X-ray CT has been used to investigate PA behaviour across the MEA. These have included visualisation of PA migration away from the membrane into the GDL fibres as a result of operation,69,344 and demonstration that the presence of an MPL can enhance PA redistribution in the catalyst layer, but prevent PA leaching into the flow channel, a common challenge for HT-PEMFC operation.68 PA has also been injected into different GDL materials and the 3D invasion pathways were investigated, as well as the effect of intrusion through an MPL (Fig. 16).69 Because of the poor contrast between PA, membrane, MPL and GDL, synchrotron X-ray sources have been widely used in these studies because of the higher flux and enhanced contrast that can be achieved.68,69 However, recent work by Bailey et al. has proven that lab-based X-ray CT can be used for imaging the PA distribution, when combined with a machine learning (ML) approach to segmentation.344 This work was extended to show that the addition of SLG can somewhat block the release of PA into the GDL/MPL.274 However, this work still relies on the method of subtracting a “dry” scan containing no PA from a “wet” scan where PA has been introduced to the same component e.g., GDL (Fig. 16). This is not ideal as it increases imaging and analysis complexity and limits in situ and operando studies where morphology may change. Future work should continue to find methods for improving contrast/segmentation of phases in HT-PEMFCs; for example, by applying deep-learning algorithms for segmentation.345
 | ||
| Fig. 16 PA breakthrough in various GDLs, with and without an MPL (top), reprinted from ref. 69, Copyright (2017), with permission from Elsevier; lab-based X-ray CT of dry and wet GDEs, with the differences between dry and wet GDL samples, (a), highlighted by the histogram, (b), (bottom), reprinted from ref. 344, Copyright (2021), with permission from Elsevier. | ||
As well as microscale studies of MEA morphology, X-ray nano-CT has been used to study the pore-structure of LT-PEMFC catalyst layers.338,346,347 Whilst unable to resolve individual particles of the catalyst layer or MPL, X-ray nano-CT can be used to investigate the morphology of these layers,348 as well as the pore size distribution, with the comparison of results to mercury intrusion porosimetry (MIP) showing that nano-CT can capture information about pores greater than 50 nm in diameter.347 X-ray nano-CT has also been used to study changes in the nano-scale morphology of the catalyst layers before and after AST,349 with results showing an increase in the average pore size from the beginning to end-of-life (EOL) sample with a decrease in the porosity. There is some opportunity for X-ray nano-CT of HT-PEMFC catalyst layers, although since the properties of low- and high-temperature catalyst layers tend to be similar, the scope for novel nanoscale investigations is potentially limited.
In situ/operando X-ray CT studies of HT-PEMFCs are emerging, with a focus primarily on PA distribution in the MEA. Halter et al. used operando X-ray CT to study the effect of current on the re-distribution of PA in the MEA of a HT-PEMFC. Results showed that PA breakthrough from the membrane to the catalyst layer happened only in the anode, and only in regions where the catalyst layer and MPL cracks overlapped.157 This highlights the presence of a potential degradation mechanism for HT-PEMFCs that could be studied in more detail over a longer period of time in future studies. Further work by the group also highlighted that by designing catalyst layers with controlled crack widths, connectivity and accessibility, PA migration from the catalyst layer can be mitigated.126
Remaining challenges for in situ/operando studies of HT-PEMFCs is the development of cells suitable for imaging that can also achieve elevated temperatures of 160 °C. Whilst this is more feasible for beamline-based experiments, owing to the larger spaces and cell sizes that can be used because of the higher beam flux, achieving this in the small cavity of a lab-based system is a challenge. Difficulties remain when separating the PA from other components of the fuel cell during the segmentation as part of the analysis, this is typically overcome using a subtraction method whereby the GDL is first imaged without PA and this dataset is subtracted from the main dataset of interest;69,157,344,363 however, this is time-consuming and limits in situ/operando studies. The authors recently reported the use of a contrast enhancement agent to overcome this issue in HT-PEMFC X-ray imaging.165 The agent used was a Cs compound which acted as a phosphoric acid tracer. The higher atomic number of Cs significantly improved the contrast between the PA and surrounding materials. This technique was demonstrated using an in situ study of the hot-press process. Radiographs before and after hot-pressing, with and without the tracer, are shown in Fig. 17a–d. The tracer allowed visualisation of the PA in the electrode (appearing dark due to X-ray attenuation), which was imaged in real-time (videos available in the supplementary data of the source publication).165 Orthoslices from the reconstructed X-ray CT data are shown in Fig. 17e–h. The PA appears bright with high contrast, making it easily distinguishable from the other materials, allowing for simpler image analysis without the need for the subtraction of a reference dataset. Nonetheless, innovations in fixture design, cell operating environments, and post-scan analysis techniques will bring about significant advancements for studying PA dynamics, as well as HT-PEMFC MEA degradation.
 | ||
| Fig. 17 A comparison of the effect of using a contrast enhancement tracer on PA visualisation during HT-PEMFC hot-pressing. Radiographs: (a) hot-press start – no tracer (b) hot-press start – with tracer, (c) hot-press end – no tracer, and (d) hot-press end – with tracer. Orthoslices of 3D reconstructed datasets: (e) no tracer and (f) with tracer. Cathode cracks and PA visualisation: (g) before hot-press and (h) after hot-press. All scale bars are 500 μm. Reprinted from ref. 165 (open access). | ||
4.2 X-ray absorption spectroscopy (XAS)
X-ray absorption spectroscopy (XAS) is a technique that can be used to study the local environment of PEMFC catalysts, which includes information about the electronic structure and redox state, as well as the coordination environment and bond lengths around atoms. XAS works by delivering X-rays to a sample, upon which they are absorbed, resulting in the release of a photoelectron of a specified energy, and a corresponding sharp increase in the absorption intensity, or ‘edge’, in the XAS spectrum. There are two main components of XAS spectra that can be investigated: extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge spectroscopy (XANES). The differences are the energy at which the features occur. In EXAFS, higher energies are probed, where absorptions give information about inter-atomic lengths and coordination numbers of atoms. XANES probes the absorption edge of an element, which allows the oxidation state and symmetry of elements in the sample to be understood.Whilst XAS has been widely used for ex situ characterisation of novel PEMFC catalysts for both LT-PEMFCs and HT-PEMFCs.364–367In situ and operando studies are the ideal use of XAS, and are thought to be powerful tools for developing new catalysts with higher activity and stability.368 The ability to study and identify changes occurring within the catalysts during operation will ultimately inform the development of novel, durable catalysts. This is especially important when considering the need to reduce the amount of Pt in the catalyst layer, or removal altogether. In situ/operando XAS studies have been carried out on both three-electrode and two-electrode cells.369 The difference between these is that three-electrode cells operate in an aqueous environment, which do not fully represent the operating conditions of PEMFCs; studies are typically done at room temperature, with no gas flow to/from the electrodes. In contrast, two-electrode cells operate under standard fuel cell conditions, with elevated operating temperatures, gas flow on the anode and cathode and water formation/removal through the flow channels. Thus, with the change from three- to two-electrode cells, the behaviour of electrocatalysts under operando conditions can be observed.369
For HT-PEMFCs, XAS has been used in a number of studies for investigating catalyst poisoning.152,214,370,371Operando XAS has been used to track the changing adsorption of species, from H, to PO4 to O, with increasing voltage, and PO4 coverage at different temperatures (Fig. 18).152,372In situ half-cells, with and without electrolyte flow under ambient conditions, have also been used to study these adsorbed intermediate species in more detail.371 While these studies shed light on the catalyst poisoning mechanisms in HT-PEMFCs, there is clear opportunity to extend such studies to operando systems operating at standard and elevated temperatures, to further elucidate the poisoning mechanism and allow for improved catalyst layer design. Given that HT-PEMFCs are tolerant to less pure hydrogen, adsorbates on the anode were studied using in situ XAS, using an anode gas feed containing CO and water to represent reformate hydrogen.214 Results showed that CO concentrations above 2% significantly decreased the HOR activity on the anode.
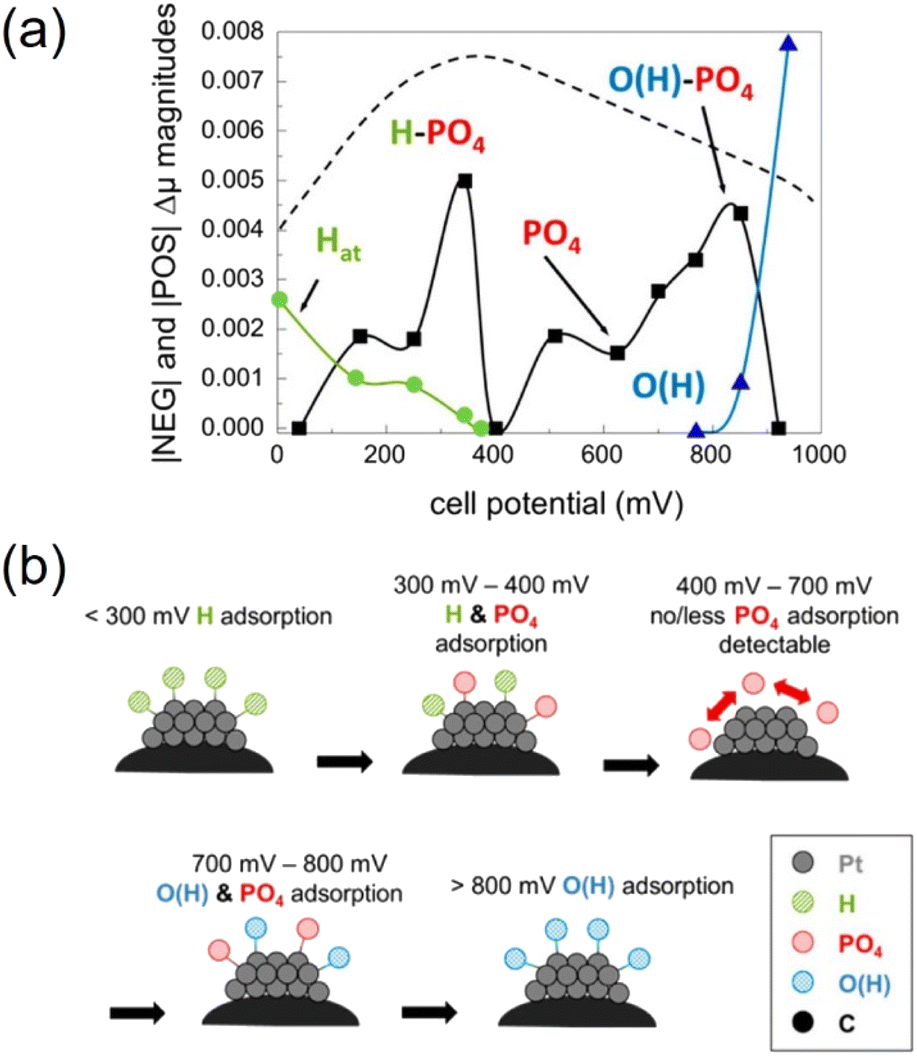 | ||
| Fig. 18 (a) Coverage of different adsorbates at across cell potential range, dashed line indicates expected PO4 coverage according to He et al.,372 (b) diagram illustrating adsorbate condition at different cell potentials. Figures reprinted with permission from ref. 152, Copyright (2013) American Chemical Society. | ||
Recent work has shown it is possible to spatially resolve the XAS spectra across a catalyst layer using XAS-CT, which collects XAS information at each projection during a CT scan.373 This has allowed for a range of studies of catalysts, including observation of heterogeneous degradation and ceria-migration occurring in LT-PEMFC MEAs during stress-testing.374,375 Applying similar methods to HT-PEMFCs represents a new opportunity for XAS-CT. Furthermore, the emergence of dedicated beamlines for in situ/operando XAS combined with a range of other X-ray techniques (XRD, X-ray CT, X-ray emission spectroscopy (XES)), such as the BL36XU beamline at Spring-8, will allow the realisation of such advanced experiments in the coming years.376
4.3 X-ray diffraction and XRD-CT
X-ray diffraction (XRD) is used to study the crystalline structure of samples. Incident X-rays focussed on the sample are scattered by the atomic planes in the sample and if they are coherent, the diffracted X-rays are collected at the detector. Information about the crystal lattice can be obtained, as well as changes to the lattice parameters if carrying out in situ/operando studies. Information about phase changes can also be obtained since different compounds will have different crystal structures.XRD has been widely used for PEMFCs, especially for analysing new materials. Since each phase has a unique set of diffraction lines and intensities, various features of novel HT-PEMFC components have been characterised. This includes the study of novel catalysts, like PtNiCu,377 confirming the successful distribution of GO in PBI/GO membranes,378 or analysing the coating of Ni onto electrospun fibres for novel GDEs.379 XRD has also been used for studying degradation in HT-PEMFCs. Batet et al. conducted XRD over the course of a durability study of a HT-PEMFC stack, by extracting one MEA from the stack at each interval. XRD was then done on the anode and cathode catalyst layers, with results showing an increase in average crystallite size over the durability test.380 XRD was also used to confirm the successful impregnation of silica nanoparticles into the PBI membrane, with results showing that an intermediate silica content leads to improvement in performance and a lower degradation rate.381
Regarding in situ/operando studies, a few studies have been conducted on LT-PEMFCs to monitor the evolution of Pt surface oxides in an operating fuel cell,382 as well as in combination with small angle scattering (SAXS) to measure crystallite size and decouple different Pt degradation modes (like aggregation, coalescence, Ostwald ripening).383 The combination of XRD with spatially resolved CT (XRD-CT) has further advanced the ability to study fuel cell catalysts in five dimensions, i.e., three spatial dimensions plus time and spatially-resolved diffraction data. XRD-CT involves the collection of XRD spectra for each voxel in a sample across a slice in a tomogram. Thus, as well as 3D information about the evolution of catalyst layer morphology that can be gathered from CT, the chemical composition of a phase can be spatially resolved, as shown by Martens et al.384 In the same study, the group combined XRD-CT with SAXS-CT, to simultaneously resolve the local nanostructure of the PEMFC components. Whilst such studies hold significant potential for elucidating information about PEMFCs across length-scales, they are practically very challenging to carry out, which may limit their use in practice. Nonetheless, there is significant scope for application to HT-PEMFCs, for example, to study phenomena such as the phase change of PA during heating, and degradation of the various MEA components during ASTs.
5. Neutron techniques
The neutron interaction with matter occurs at the atomic nucleus, where incoming neutrons interact with the nucleus, resulting in both absorption and scattering. Unlike X-rays, where the strength of the interaction scales with the elemental number, Z, the neutron interaction is not related to the atomic number. Thus, elements across the periodic table have characteristic cross-sections, with the added advantage that some isotopes can be distinguished using neutrons. The advantage of this for PEMFCs is that common materials found inside fuel cells have widely different cross-sections, where the neutron cross-section is a measure of the interaction between the neutron and the sample, measured in barn (10−28 m2). For example, hydrogen has a large cross-section, so appears bright in a neutron image, whereas carbon has a small cross-section and is less visible in neutron images. Thus, the visualisation of water is particularly possible, which cannot be as easily achieved using X-rays, given the low atomic number of hydrogen. Furthermore, given the isotope effect, deuterium (i.e., hydrogen with a proton and a neutron) has a very low neutron cross-section and is hardly visible in neutron images, meaning that deuterated water can be used for humidification to further differentiate between water formed from the electrochemical reaction and water from humidification.5.1 Neutron imaging
The use of neutrons for imaging is one of the most widely used applications of neutrons for PEMFCs. Because of the relatively low flux of neutron sources, the trade-off is that spatial resolutions (i.e., the size of the smallest resolvable features) are typically on the order of 10–100 μm.385,386 Furthermore, exposure times for each radiograph are on the order of milliseconds (for high-speed imaging) to minutes (for high-resolution imaging),387 with longer exposure times reducing the signal-to-noise ratio (with a resulting loss of temporal resolution). The result of these two conditions is that the feature of interest, especially for operando studies, tends to be water formation in the flow channels. Most studies have been radiography studies, where a 2D radiograph, which is an averaged image through the through- or in-plane direction, is collected. These studies are normally time-resolved, meaning that images are gathered continuously throughout an experiment. Through-plane measurements are performed to understand the distribution of water along the length of the flow channel, with many studies on LT-PEMFCs observing the tendency for water to pool at the bends of serpentine flow channels,388 or studying the effect of introducing multiple serpentine channels into the design.389 In-plane measurements are done with the cell in line with the beam direction and can be used to observe the back-diffusion of water into the anode flow channels,390,391 as well as studying the preferential formation of water under “land” regions (rather than under channel regions) during operation.392 Advances in beamline technologies mean that the spatial resolution achievable using neutron sources continues to improve. Pixel sizes as low as 2.5 μm have been reported,393 which is a particular advantage for in-plane studies, since it allows for water in each layer of the MEA to be resolved. Finally, LT-PEMFC start-up has been investigated using neutron radiography to observe the formation of ice during cold-start,394 the phase transition between ice and liquid water,395 and the effect of residual water in the flow channels on the start-up profile of LT-PEMFCs.396 Such start-up behaviour is also of interest for HT-PEMFCs, as will be discussed later in the section.As well as using in- or through-plane imaging, isotope exchange has been applied in neutron studies of LT-PEMFCs. Since the cross section of deuterium is much smaller than that of hydrogen, switching the inlet anode gas between H2 and D2 has allowed for distinction of the ion exchange in the membrane,397 as well as water exchange between the GDL and the flow channels.398
For HT-PEMFCs, the effect of water on fuel cell performance is less of a concern during operation, since operating temperatures are above 100 °C.22,47 This means that liquid water is less likely to exist in a transient form in the GDL or flow channels during normal operation. Thus, the majority of studies using neutrons carried out on HT-PEMFCs have taken advantage of H/D isotope contrast to monitor acid distribution and exchange of protons/deuterons through the membrane during operation. Work by Arlt et al. used H/D contrast to monitor the distribution of hydrogen in a PBI membrane.399 By switching the anode feed gas between hydrogen and deuterium, the exchange between protons and deuterons could be observed. It was found that exchange occurred first at the gas inlet, with exchange at the gas outlet occurring later (Fig. 19a). Later work by Lin et al. built on this to monitor the rate of exchange at different current densities, with increasing current density resulting in a faster exchange through the anode flow channels (Fig. 19b).400 The movement of PA through the cell has also been studied, with the amount of PA measured from radiographs being in reasonable agreement with the expected amount of acid inside the cell.401 Deuteration was also used in this study to observe isotope exchange at different dewpoints of 30 and 70 °C.
 | ||
| Fig. 19 (a) Neutron radiographs showing the H-to-D and D-to-H exchange in a HT-PEMFC operating at 160 °C, reprinted from ref. 399, Copyright (2015), with permission from Elsevier; and (b) the effect of current density (200, 400 and 600 mA cm−2) on the speed of H-to-D exchange, with faster exchange at higher current density, reprinted from ref. 400 (open access). | ||
A handful of neutron tomography studies have been carried out so far on LT-PEMFCs. Because the flux of neutron sources are relatively low, exposure times for imaging have typically been on the order of multiple seconds. Thus, early studies carried out a single tomography on the order of hours.402,403 High-resolution neutron tomography has been used in recent years for observing fine structure, such as work by Manzi-Orezzoli et al. to study coating on GDLs, combined with X-ray CT, with spatial resolution down to 10 μm.404 Alrwashdeh et al. reported a voxel size of 6.5 μm for the study of water distribution in different GDL materials, although the exposure time for a single radiograph, in this case, was still 15 s.405 Most recently, work by Ziesche et al. showed that it was possible to collect a single tomogram in 40 s, which allowed for operando monitoring of water distribution in the flow channels.406 Owing to the third spatial dimension, the volume of water was quantified, allowing for comparison of the actual volume of water residing in the flow channels to the theoretical volume calculated according to Faraday's law.
With these recent advancements in 3- and 4D neutron imaging, there is clear scope for similar studies to be conducted on HT-PEMFCs. Furthermore, whilst the radiography studies of HT-PEMFCs described here show some initial understanding of ion-exchange and PA migration, there is considerable opportunity for studying the role of water during start-up and shutdown of HT-PEMFCs in four dimensions, as well as investigating freeze–thaw dynamics.
5.2 Neutron scattering
During neutron scattering, neutrons scattered by the sample are collected and analysed and neutrons transmitted through the sample in the plane of the incident beam are ignored. In comparison to neutron imaging, which has a relatively low spatial resolution (on the order of microns), neutron scattering experiments can resolve changes at the angstrom level,407 meaning it is a powerful technique for probing properties of electrocatalysts, catalyst layers, and diffusion in PEMFCs. Furthermore, as with imaging, H/D isotope distinction is used during scattering experiments to allow for distinction between the “mobile” protons contributing to structural diffusion and those which are bound to the polymer chain.408 As with imaging, neutron scattering experiments must be done at national facilities, since neutron sources are not available in the lab. There are different types of neutron scattering experiments, with two of the most common being quasi-elastic neutron scattering (QENS) and small-angle neutron scattering (SANS). They are distinct in that QENS experiments are spectroscopic and are used to study dynamics by measuring changes in neutron velocity. In contrast, SANS uses diffraction to study the structure of samples by measuring the change in neutron direction.409,410In situ SANS has been used to study water dynamics in the catalyst layer of LT-PEMFCs, in combination with neutron radiography (NR) for capturing macroscale information about water distribution.411 Furthermore, information about local hydration, as well as catalyst layer microstructure, was elucidated using the technique, including the use of H2O/D2O.412 Contrast variation (CV)-SANS allows for the investigation of partial scattering functions, i.e. the scattering function of each component in a sample, such as the constituent materials of a catalyst layer ink.413 Thus, the local structure of the catalyst layer components, Pt, carbon and ionomer, can be deconvoluted. For LT-PEMFCs, CV-SANS studies have included investigations into the effect of Pt-loading on ionomer adsorption, where the ionomer layer decreased as Pt-loading increased.414 Further work by Harada et al. used CV-SANS to distinguish between adsorbed ionomer (forming a thin film on the Pt-surface) and deposited ionomer (found at throats in aggregated carbon particles).415 Given the importance of electrolyte/Pt interfaces in HT-PEMFCs, CV-SANS is also expected to be a useful technique for studying novel catalyst layer designs or compositions with optimised performance as research into HT-PEMFC catalyst layers continues to increase.
For HT-PEMFCs, SANS has been applied to the study of catalyst layers, where the structure of a range of catalyst layers was studied across a range of length scales, by including wider angles.416 SANS was also combined with SAXS to highlight the structure of different components in HT-PEMFC electrodes, using H3PO4/D3PO4 contrast.417
QENS studies have been used to probe water dynamics in LT-PEMFCs, including investigations into water distribution in the catalyst layers.418 QENS data allowed for the quantification of water molecules per sulphonic acid group of different types (immobile, slow and fast), as well as identifying the effect of temperature on the type of water molecules observed. Although there is minimal liquid water in operating HT-PEMFCs, QENS could still be a useful technique for understanding water dynamics, particularly during start-up/shutdown. Furthermore, QENS has been applied to the study of PA at different Pt-loadings of the catalyst layer of HT-PEMFCs, with results suggesting stronger trapping of PA at higher Pt-loadings.419 QENS has also been applied to the study of proton motion in the membrane. Early QENS application to HT-PEMFC membranes compared the proton dynamics measured with QENS to conductivity studies, which showed diffusion processes occurring at a rate between PA doped in a PBI membrane and pure PA.420 QENS has also been used to study proton diffusion in the membrane, with the extraction of fractal diffusion dimensions allowing for study and verification of diffusion processes across a broad range, from ps to ns.408 The proton dynamics of PA from the solid to the molten state has been studied using QENS.421 This application may be useful in studying the performance of HT-PEMFCs at sub-zero conditions during cold start-ups – an area which has received little attention to date.
6. Raman and infrared spectroscopy
6.1 Raman spectroscopy
Raman spectroscopy uses the inelastic scattering of light at a given wavelength to differentiate between chemical structures in samples. The peaks in the spectra are used to identify specific local bonding arrangements, allowing for detailed structure determination. Raman spectroscopy is a flexible, non-invasive analytical technique with micron spatial resolutions.422 Raman spectroscopy in LT-PEMFC research has been used to investigate the chemical structure of catalyst and support materials,423–425 analyse membrane degradation,426 including the distinction between loss of sulphonic end groups and fluorinated backbone,427 and determine membrane water content.428 One benefit of Raman over infrared spectroscopy is that the spectra are not overwhelmed by the presence of liquid water. This has allowed for in situ studies of water transport in LT-PEMFCs.429 A particular type of Raman spectroscopy, coherent anti-Stokes Raman scattering (CARS), can be used to investigate dynamic studies of water transport due to the higher intensity signal and faster time resolution.430 One study used CARS spectroscopy and a bespoke cell design with a quartz window to investigate the hydration states of the membrane as a function of relative humidity and current density.430Research in HT-PEMFCs share similarities due to similar components in the catalyst layer; for example, the technique has been used to investigate the structure of catalyst and carbon supports.379,431,432 The structure of composite membranes has also been analysed in HT-PEMFC research.274,433 Raman spectroscopy was used to study the effects of acid doping on PBI membranes as it is highly sensitive to the structural changes that occur during the acid–base proton exchange.38 Also, Raman mapping has been used to determine the acid distribution as a function of the doping method.434In situ/operando studies using operating cells are not reported in the current literature, and there are only a small number of these studies for LT-PEMFCs. Studying the chemical structure in the membrane during operation at HT-PEMFCs conditions is a potentially useful yet challenging application of Raman spectroscopy that is yet to be demonstrated.
6.2 Infrared spectroscopy
Fourier Transform Infrared (FTIR) Spectroscopy uses the absorption and transmission of incident IR light to obtain information about the sample. The frequencies of light absorbed are related to the physical characteristics of the material. IR spectra can be obtained using a monochromatic beam which changes in wavelength over time, or now more commonly, by using a Fourier transform instrument to measure a wide range of wavelengths at the same time. Software combined with the FTIR spectroscopy method can search through a large number of reference spectra to determine the identity of the substance under measurement.435 FTIR spectroscopy has been used to investigate the acid-doping process similar to Raman spectroscopy.436 The FTIR spectra showed evidence of an acid–base proton exchange reaction resulting in the formation of H2PO4− and protonated imidazolium cations. Fig. 20 shows the presence of H2PO4− (942 cm−1) was clear at lower doping levels but became partially masked by the large amount of free H3PO4 (998 cm−1) at high acid concentrations.436 Other applications of FTIR spectroscopy in HT-PEMFC research include characterisation of acid groups in membranes,99,437,438 characterisation of electrocatalysts,438 and investigation of adsorption of PA onto Pt.439 A cell design allowing for in situ FTIR measurements of an operating HT-PEMFC was used to measure the spectroscopic signature of adsorbed CO on the Pt surface as a function of temperature.440 An FTIR gas spectrometer has been used to measure the CO2 content in the cathode exhaust caused by carbon corrosion induced by simulated start-up/shutdown in a LT-PEMFC.441 A similar study was conducted for an HT-PEMFC whereby the cell was subjected to 30 minutes at potentials of 0.9, 1.0, 1.1 and 1.2 V, and the CO2 in the cathode exhaust gas was measured using an IR spectrometer.442 The higher the potential, the greater the carbon corrosion and the more CO2 was emitted. Operating at 160 °C and 1.2 V resulted in a peak CO2 content of approximately 1600 ppm for a 100 cm−2 cell. The measurement of CO2 in the exhaust using IR spectroscopy has also been utilised in determining the ECSA of an HT-PEMFC electrode and is deemed to be a reliable method.306Fig. 21 shows the IR spectra of PA–water mixtures. The broad intense absorption above 3000 cm−1 corresponds to the stretching vibration of OH involved in a hydrogen bond with the oxygen of another water molecule. This feature decreases with increasing PA content and disappears at a PA–water ratio of 1:1 (λ = 5). This was explained by water molecules being separated and only able to form hydrogen bonds with phosphate species.92
 | ||
| Fig. 20 FTIR spectra of pristine ABPBI, acid-doped ABPBI and phosphoric acid. Spectra numbered (1)–(4) correspond to ABPBI membranes with increase acid doping levels. Reprinted from ref. 436, Copyright (2014), with permission from Elsevier. | ||
 | ||
| Fig. 21 IR spectra of water and PA–water mixtures as a function of water content, λ. Reprinted from ref. 92 with permission from the Royal Society of Chemistry. | ||
7. Optical and infrared imaging
7.1 Optical imaging
Transparent cells have been designed to investigate water flooding in LT-PEMFCs through direct visualisation of the water behaviour using charge-coupled device (CCD) digital cameras.443–448 The transparent cells are comprised of the same components as a conventional PEMFC except that a window made from materials such as polycarbonate, acrylic, or plexiglass allows for optical observation inside the cell.448 Careful consideration and design are required to minimise the difference in the performance of a typical PEMFC and a transparent PEMFC, important aspects include size and shape of the active area, uniform heating, and validation against typical PEMFC.448 The technique is also limited due to the difficultly in quantifying water transport inside the cell which is not visible to the camera. This is where X-ray and neutron techniques provide advantages with greater penetration into the sample and visualisation of the cell interior. However, the advantages of transparent cells and optical imaging are that they provide high temporal and spatial resolutions, allow for operando experiments whilst being lower cost and are simpler and less hazardous.447,449 Due to the high operating temperatures, visualisation of liquid water for HT-PEMFCs in this way is not as useful, other than during start-up or any operation below 100 °C. A possible application may be visualisation of PA since it is in liquid form at typical operating temperatures and leaches out to the GDL and bipolar plates. However, the quantities may be insufficient to make detection using this method practical or possible.Optical fibres have gained interest and application as a sensor for PEMFCs. Optical fibre sensors have been used to detect temperature,450 relative humidity,450,451 membrane water content,452 and species related to membrane degradation.453,454 The application of optical fibres in HT-PEMFC is less common. One use has the fibres included as part of micro-electro-mechanical systems (MEMS) technology for detecting internal conditions such as temperature.455 Optical fibres have been used as pH sensors,456,457 which could be useful to infer acid distribution in HT-PEMFCs, but there are no studies applying optical fibres to sense PA in HT-PEMFCs to the best of our knowledge.
7.2 Infrared imaging
Infrared (IR) imaging, also known as infrared thermography, detects the infrared radiation emitted by an object and creates an image from which the temperature distribution can be inferred. IR thermography can be used in single-frame mode to take a snapshot of the system, or transient imaging can be used to image the system as a function of time.458 Exothermic chemical reactions and ohmic heating in the fuel cell release heat energy and can be used for multiple types of analysis. One such type is to investigate fuel crossover, which can in turn be used to investigate membrane degradation. If the membrane is truly impermeable to the reactants, hydrogen and oxygen cannot react directly (as opposed to the electrochemical route required for fuel cell operation). However, low levels of fuel crossover do occur and this is increased by perforations forming in the membrane.27 The direct reaction leads to local temperature hotspots where higher crossover is present, this enables the identification of pinhole size and location.459 IR transparent windows have been used in LT-PEMFCs to investigate cathode and flow channel temperature distributions.460,461 The presence of liquid water interferes with the IR radiation and temperature estimation becomes difficult.462 However, water transport visualisation has been achieved using optical and IR imaging.463 To the authors' knowledge, there are no reported studies using IR thermography combined with IR transparent windows in HT-PEMFC research. However, IR thermography has been used to detect the external stack temperature distribution.4648. Gas and ion chromatography
8.1 Gas chromatography
Gas chromatography (GC) involves a sample being injected into the system and carried to a column by a carrier gas known as the mobile phase. Once in the column, the sample mixture interacts with the stationary phase (typically a liquid film) and is separated into its various components based on the time taken for the compound to progress through the column to the detector (retention time). GC is typically combined with a mass spectrometer (MS) to allow for the separation and analysis of trace elements of chemical species in the exhaust gas at the anode and cathode outlets.20 GC has been used in LT-PEMFC research to investigate catalyst poisoning effects,465–467 degradation products,468 water distribution,469 and gas crossover.470–472GC requires a column to separate the gases prior to analysis. However, some columns may be suitable for certain gases e.g., H2 but not others e.g., CO2.473 In some studies, gas is directed to the mass spectrometer without separation, and this is referred to as direct gas mass spectrometry (DGMS). This technique has been used to analyse degradation products such as HF, H2O2, CO2, SO, SO2, H2SO2, and H2SO3.473,474
There are only a couple of studies in the literature that use GC or DGMS to analyse HT-PEMFC exhaust gas.190,475 Attention has focused on the CO2 released at the anode during hydrogen starvation,190,476 or ECSA calculation by monitoring CO2 during CO stripping.313 Another use of GC is to identify the concentration of gas species exiting a reformer and entering the HT-PEMFC.477,478 One of the disadvantages of GC techniques is the relatively slow sampling time (min); therefore, real-time gas analysers using techniques such as DGMS which can provide faster sampling may be useful for more dynamic studies.65
8.2 Ion chromatography
Ion chromatography (IC) shares a similar process to GC whereby the sample is injected into a mobile phase (eluent) and enters the analytical column, here the ions in the sample adsorb onto a stationary phase (either anion or cation exchange resins). Ions with a higher affinity to the stationary phase will take longer to pass through the column and be detected and thus different ions are separated. IC is utilised more in HT-PEMFCs than GC. IC can be used to detect fluoride, sulphate, nitrate, and phosphate anions, as well as sodium and calcium cations.479 IC has been used to determine the main PA degradation products in the fuel cell exhaust: PO43− along with HPO42−, H2PO4−, and H3PO4.147 Detection and quantification of phosphate ions are used to investigate PA content and loss in HT-PEMFCs.147,149,208,212,363,479 Inductively coupled plasma mass spectrometry (ICP-MS) has also been used to quantify acid loss; here exhaust water is analysed and Pt loss can also be detected.146,480 Several studies use the detection of HPO42− to determine the loss rate of PA (H3PO4) by assuming all forms of PA are converted to HPO42− due to the equilibrium associated with this tri-protic acid.149,212 Across the studies it was generally found that increasing operating temperature, reactant flow rate, and current density led to faster PA loss and reduced durability, with the cathode loss typically higher than anode loss.147,149,208,212 IC has also been used to investigate the effects of contaminated air on HT-PEMFC operation.481,482 Sampling frequency in these studies is typically in the tens of hours or several days, meaning that highly dynamic studies of PA loss are unlikely to use IC.IC has likewise been used in LT-PEMFC research to study degradation. Most commonly IC is used to detect fluoride and sulphate content of water samples which is indicative of membrane degradation.483–485 Online measurements of fluoride ions from a PEM water electrolyser have also been made using IC; however, dynamic capability is limited by the sampling time, which in turn, was limited by the rinsing and recording time to 24 min.486
9. Electron microscopy
9.1 Scanning electron microscopy
Scanning electron microscopy (SEM) is used for imaging structures and materials from the nm–μm range. This technique utilises a beam of electrons that is focused onto the surface of a sample, the interaction with the electron beam and the sample create several different types of electrons and X-rays, including secondary electrons and backscattered electrons. This allows for direct surface imaging across a wide variety of length scales depending on the exact magnification chosen. Most samples for LT- and HT-PEMFCs are either individual components (membranes, catalysts, support materials) or full MEAs (top surfaces, cross-section). Samples are typically deposited onto carbon tape and then coated with a thin layer of conductive materials (either gold or more typically carbon), to minimise sample charging. However, cross-sectional SEM samples typically require encasement in a resin followed by polishing, microtoming or freeze fracturing to ensure a flat sample.487Focussed ion beam (FIB)-SEM is the most advanced preparation technique, in which an ion beam is used to mill the surface of the sample, creating a flat surface to image.70,127,186,488,489 The FIB-SEM can be used to alternate between period of milling followed by imaging, thereby building a 3D image of the sample, which can in turn be used as a basis for 3D image-based modelling. The high resolution of SEM allows for the particle and pore structure of the MPL and catalyst layer to be imaged, which are beyond the typical resolution limit of other 3D imaging techniques like X-ray CT. Since the catalyst layers in LT- and HT-PEMFCs are similar, studies using FIB-SEM are largely similar for the two different technologies. For HT-PEMFCs, Prokop et al. used FIB-SEM to evaluate the 3D microstructure of catalyst layers,490 and used the microstructures to model transport properties, with models showing good agreement with experimental studies.491 Further modelling work has also used PEMFC catalyst layer datasets to generate deep-learning algorithms for accurate segmentation of FIB-SEM datasets, i.e., ensuring the correct assignment of a label to each phase in the catalyst layer.492 Finally, the morphology of different HT-PEMFC catalysts, including Pt/C and Pt3Co/C, was studied using FIB-SEM, with analysis of the porosity showing that larger average pore size results in better acid distribution.127
SEM is also typically combined with energy-dispersive X-ray spectroscopy (EDX or EDS), which can measure and quantify the amounts of each element within the sample by collecting the characteristic X-rays of the element. EDX has been used in HT-PEMFC samples in conjunction with SEM imaging,159,480,493 most interestingly it has been utilised to map the PA distribution within the structure of the catalyst layer (shown in Fig. 22).168 SEM has also been widely used for qualitatively assessing the surface of the deposited material or layers; such as catalyst layer or membranes.493–499 Cross-sectional SEM is a useful technique to assess membrane and electrode thickness, pore size distribution, catalyst location, catalyst layer structure and defects.70,103,127,161,378,380,488,489,493–495,499–501 For HT-PEMFCs this has been shown to be useful for tracking acid loss out of the membrane via thickness changes.380 Cross-sectional SEM has also been shown to significantly improve understanding of structural degradation during ASTs by observing the direct impact of the MEA thickness, pore structure, catalyst migration and support degradation.122,124,125,127,186,488,489,496 While the experimental methods and analysis are very similar between LT- and HT-PEMFCs the differences in electrode composition, in particular the importance of PA in the catalyst layer, gives SEM an increased importance for HT-PEMFC compared to LT-PEMFC.
 | ||
| Fig. 22 Cross-sectional SEM images of HT-PEMFC MEAs with EDX mapping of phosphorous. The MEAs feature various anode/cathode GDL combinations imaged at the end of testing: (a) H23C2/H23C2, (b) H23C2/H23C4, (c) H23C2/H24C3, and (d) H23C2/H24C5. The distinct band in the centre is the membrane, with the anode and cathode on the left and right sides respectively. Reprinted from ref. 168, Copyright (2018), with permission from Wiley. | ||
9.2 Transmission electron microscopy
Transmission electron microscopy (TEM) is a sample analysis technique that focuses a beam of electrons onto a sample, the electrons pass through the sample, interacting with it before reaching the detector behind the sample. TEM has high resolution; however, given the electron beam must pass through the sample, the materials have to be exceptionally thin. Samples are typically prepared via deposition onto specialised TEM grids, leaving clusters of materials no more than 10–100 nm thick. As a result of this preparation method this technique is only useful for imaging a small part of fuel cell materials, typically support materials, ionomer and most commonly catalysts.493,498,502 TEM is often used to investigate catalyst characteristics due to the nano-scale resolution of this technique. Examples include catalyst particle size before and after cell reversal caused by air and fuel starvation,503,504 catalyst aggregation and surface area,505 catalyst particle size and distribution.506 TEM has also been applied to HT-PEMFCs to determine Pt particle size and size distribution.120,162,192 There is little to no difference in sample preparation reported between LT and HT-PEMFCs, although, sample preparation typically involves sonication in water which is likely to significantly disrupt the PA distribution in HT-PEMFCs.10. Magnetic resonance
10.1 Nuclear magnetic resonance
Nuclear Magnetic Resonance (NMR) is the absorption and reemission of radiofrequency (RF) pulses by nuclear spins in a magnetic field, or the responses of spins to external RF excitations.507 NMR techniques can only probe nuclei with non-zero spins, essentially nuclei with odd number of protons and/or neutrons e.g., 1H, 2H, 13C, 19F, 31P, 195Pt. There are many different techniques that use NMR such as solution-state, solid-state, pulsed-field gradient (PFG) and static-field gradient (SFG), relaxometry, and others. For a more in-depth review of different NMR techniques and applications to PEMFCs, the reader is referred to dedicated review articles.507,508 NMR techniques are used in LT-PEMFCs to characterise structural properties, and proton and water transport properties. Membrane structural characterisation studies typically use NMR to target 1H, 13C, 19F and 31P nuclei, and can be used to identify structural changes caused by degradation.507–509 Analysis of exhaust water following membrane degradation has utilised 19F NMR.510 Water and proton transport properties such as the water diffusion and electroosmotic drag coefficients have been investigated using NMR methods.511–513 NMR has been used similarly in HT-PEMFCs, although focusing on proton transport in acid-doped membranes rather than water-saturated PFSA membranes. Proton conduction mechanisms in PA–water and PA–benzimidazole systems have been investigated using 1H, 17O and 31P NMR spectroscopy.92,93 The studies identified the changes in proton transport mechanism as a function of water content, as well as identifying the importance of hydrogen bond frustration and why PA-PBI are suited to HT-PEMFCs. 31P NMR has also been used to examine the impact of water on the energetics of PA interaction with the membrane and suggest a new acid loss mechanism based on the change in PA cluster energy.211Fig. 23 shows the chemical peaks of 31P NMR for hydrous and anhydrous PA-PBI and biphosphate-TMA systems. The chemical shift downfield indicates a higher cluster interaction energy. Adding water to PA-PBI significantly increases the interaction energy; whereas, only a minor increase occurs when adding water to the biphosphate-TMA system. The lower interaction energy of the PA-PBI system is used to explain the poorer PA retention when water is present in contrast to the QAP-based systems.211 | ||
| Fig. 23 31P NMR spectra comparison between hydrous and anhydrous PA-PBI and biphosphate-TMA. Hydrous conditions have water to PA ratio of 10. Reproduced from ref. 211 with permission from the Royal Society of Chemistry. | ||
10.2 Magnetic resonance imaging
Magnetic resonance imaging (MRI) is based on the principles of NMR, and is commonly used in medical applications, whereby magnetic field gradients enable for spatial reconstruction to form the image. MRI has been used for in situ water visualisation in LT-PEMFCs.514–516 Local current density has also been measured using the shift in obtained NMR signal caused by the magnetic field of the local current density.517 While MRI has benefits such as being non-invasive and compatible with operating fuel cells, it does face challenges regarding the fuel cell material,518 being incompatible with ferromagnetic and paramagnetic materials. This means that the typical materials within the electrode are not suitable for MRI, and MRI-specific component designs are required to enable successful imaging.507 Currently, there are no studies in the literature using MRI to study HT-PEMFCs. Other than requiring specialist equipment and cell componentry, the main reason is likely that the lack of liquid water to be visualised means there is little interest in using this technique. However, 31P MRI is utilised in medical applications to noninvasively detect phosphorous-containing biological tissues.519,520 It may therefore be theoretically possible to use 31P MRI as a method to investigate PA distribution in HT-PEMFCs.11. Accelerated stress tests
Accelerated stress tests (ASTs) are tests designed to replicate long-term operation, allowing characterisation of degradation in a controlled manner and estimation of durability. ASTs may also be referred to as accelerated durability/degradation tests (ADTs), and ageing tests.11.1 Background on LT-PEMFC ASTs
Given the importance of durability testing and the relative maturity of LT-PEMFCs, multiple organisations have developed AST standards, including the U.S. DoE and those developed as part of the ID-FAST project funded by H2020.521–523Chemical failure modes arise due to chemical attack on various components in the MEA and are most relevant for the membrane and the GDL. The main reason for the chemical degradation of the membrane is a result of the formation of hydrogen peroxide (H2O2) and subsequent breakdown to free-radicals (e.g., ·OH, ·OOH and ·H);524,525 these react with both side-chains and the polymer backbone in the PFSA membrane,526via a complex number of reactions that are discussed in detail by Coms.525 It is thought that the presence of impurities, like Fe2+ or Cu2+, accelerates the formation of free radicals in the membrane,525 as shown by an OCV durability test.527
Hydrogen peroxide can also result in degradation of the GDL via oxidation.528 To emulate this form of degradation, GDLs can be soaked in H2O2 for a number of hours, followed by performance testing and comparison of MEAs prepared with aged GDLs to those prepared with pristine GDLs. A range of conditions for GDL soaking are found in the literature: 35 wt% H2O2 for 12 h at 90 °C;529 30 wt% for 16 h or 24 h at 90 °C;339,530 30 wt% H2O2 for 15 h at 95 °C;531 H2O at 80 °C for 1000 h.532 Despite differences in the degradation protocol used, the result of the immersion in most cases was found to be reduced fuel cell performance, along with increased water accumulation in the GDL.
Mechanical failure modes mostly affect the membrane (though carbon corrosion could be considered a mechanical failure mode of the catalyst layer due to the formation of cracks). Humidity cycling during MEA operation can cause the membrane to crack and pinholes can be formed.362,533 This results in performance loss by way of gas crossover or significant inhomogeneity in current density distribution.534,535 Furthermore, pinhole formation, in particular, is thought to be accelerated by way of combined chemical and mechanical degradation,536 where gas crossover, growth of pinholes and side/main-chain degradation are all observed.536,537
Carbon corrosion is most likely to occur during start-up/shutdown of the fuel cell, where significant overpotentials arising as a result of an air/hydrogen boundary lead to oxidation of the carbon and gas formation in the form of carbon monoxide and carbon dioxide.538,539 By cycling in a triangular-sweep profile between 1.0 and 1.5 V for 5000 cycles, the carbon support can be degraded. As well as the formation of gases, the morphology of the catalyst layer is significantly impacted, with the formation of cracks through the catalyst layer,349,360 as well as the collapse of the electrode leading to reduced porosity.540 Both effects increase the mass transport losses in the cell, since gas diffusion and water transport are inhibited in the CL and electron transport pathways are minimised due to the cracks.541
Electrocatalyst degradation occurs during operation over many thousands of hours, and the AST used to resemble this involves square-wave cycling between 0.6 and 0.95 V for 30000 cycles. The voltage window relates to the activation region of the polarisation curve, where the performance of the fuel cell is limited most by electrocatalyst kinetics. Over the course of cycling, the Pt undergoes degradation via several key mechanisms, including agglomeration, Ostwald ripening and dissolution, as discussed by Meier et al.542 Cyclic voltammetry is one of the key techniques for characterising and understanding the progress of degradation in the cycle, since CV allows for quantification of the ECSA and as the catalyst nanoparticles coalesce/agglomerate, their active surface area available for reaction decreases.543 The electrocatalyst AST is particularly useful in the search for low- to non-Pt-containing electrocatalysts,251 since the long-term stability of these can be measured using the AST. Other effects, like the particle size effect,544 the use of alternative supports like carbon nanofibres,545 tantalum-modified titanium oxide,546 have been investigated using the electrocatalyst AST.
11.2 HT-PEMFC ASTs
A comprehensive list of ASTs for HT-PEMFCs is given in Table 4 and will be discussed in detail in this section. ASTs commonly focus on acid loss, catalyst degradation, and the effects of start-up/shutdown procedures.| Type of stress | Cycle description | Temperature range | Gases supplied (anode//cathode) | Duration | Ref. |
|---|---|---|---|---|---|
| Acid loss | |||||
| Current cycling | 0.60 A cm−2 (4 min) | 160 °C | H2//air (dry) | 70–230 h | 146 |
| 1.00 A cm−2 (16 min) | |||||
| 0 A cm−2 (10 min every 6 h) | |||||
| Current cycling | 0.60 A cm−2 (4 min) | 150 °C | H2//O2 (dry) | 70 h | 274 and 548 |
| 1.00 A cm−2 (16 min) | |||||
| 0.00 A cm−2 (10 min every 6 h) | |||||
| Current cycling | 0.60 A cm−2 (4 min) | 160 °C | H2//air (unspecified) | 500 h | 219 and 549 |
| 1.00 A cm−2 (16 min) | |||||
| Current cycling | 0.20 A cm−2 | 160 °C | H2//air (unspecified) | 600 h | 194 |
| 0.80 A cm−2 (20 min per cycle) | |||||
| Current cycling | 0.20 A cm−2 (15–120 s hold times) | 160 °C | H2//air (dry) | 2000 h | 547 |
| 0.80 A cm−2 (21–168 s hold times) | |||||
| Current cycling | 0.20–0.60 A cm−2 | 160 °C | H2//air (unspecified) | 100 h | 186 |
| (0.05 A cm−2 step every 5 min) | |||||
| Constant current | 0.20 A cm−2 | 190 °C | H2//air (dry) | 2830 h | 212 |
| Constant potential | 0.20 V | 150 °C | H2//air (dry) | 100 h | 564 |
| Thermal cycling | 0.20 A cm−2 | 140 °C (1 h) | H2//air (unspecified) | 100 h | 186 |
| 160 °C (1 h) | |||||
| 180 °C (1 h) | |||||
| Thermal cycling | 0.15 A cm−2 | 80–160 °C | H2//O2 (pH2O = 9.7 kPa) | 550 cycles | 112 |
| (10 °C min−1 ramp rate) | |||||
|
|||||
| Catalyst layer degradation | |||||
| Potential cycling | 0.60–1.00 V (0.05 mV s−1 scan rate) | 160 °C | H2//N2 (dry) | 10000 cycles |
123 |
| Potential cycling | 0.60 V (8 s) | 160 °C | H2//N2 (dry and wet N2 tests) | 30000 cycles |
125 and 156 |
| 1.00 V (8 s) | |||||
| Potential cycling | OCV (3 min) | 160 °C | H2//air (unspecified) | 100 h | 281 |
| 0.50 V (30 s) | |||||
| Potential cycling | 0.90 V (3 min) | 160 °C | H2//air (dry) | 5000 cycles | 551 |
| 0.50 V (3 min) | |||||
| Potential cycling | 0.60–1.20 V (0.05 mV s−1 scan rate) | 160 °C | H2//N2 (dry) | 5000 cycles | 122 |
| Current cycling | 0.00 A cm−2 (20 min) | 160 °C | H2//air (dry) | 100 h | 480 |
| 0.20 A cm−2 (20 min) | |||||
| Current cycling | 0.0–0.2 A cm−2 | 160 °C | H2//air (unspecified) | 100 h | 186 |
| (0.05 A cm−2 step every 5 min) | |||||
| Current cycling | 0.04 A cm−2 | 160 °C | H2//air (unspecified) | 600 h | 194 |
| 0.20 A cm−2 (20 min per cycle) | |||||
| Current cycling | 0.00 A cm−2 (4 min) | 160 °C | H2//air (unspecified) | 1000 h | 550 |
| 0.30 A cm−2 (16 min) | |||||
|
|||||
| Bipolar plate degradation | |||||
| Potential cycling | 0.10 V (15 min) | 120 °C | H2//O2 (unspecified) | 83 h | 142 |
| 0.97 V (15 min) | |||||
|
|||||
| Membrane degradation | |||||
| Temperature cycling | 0.00 A cm−2 | 30 °C (30 min) | Unspecified | 150 h | 557 |
| 180 °C (30 min) | |||||
| Temperature cycling | 0.6 V | 40 °C (2 h) | Unspecified | 2500 h | 559 |
| 160 °C (2 h) | |||||
|
|||||
| Start-up/shutdown | |||||
| Current and thermal cycling | 0.00 A cm−2 (25 → 200 °C OCV stabilise) | 25–200 °C | H2/N2//air/N2 (dry) | 100 cycles | 555 |
| 0.60 A cm−2 (1 h at 200 °C) | |||||
| 0.00 A cm−2 (200 → 25 °C) | |||||
| Potential cycling at constant temperature | 1.0 V (5 min, fuel valves open) | −20 °C | H2//O2 (dry) | 150 cycles | 115 |
| 0.10 V (5 min, cell off, valves closed) | 15 °C | ||||
| (0.025 mV min−1 scan rate) | 40 °C | ||||
| Thermal cycling at constant voltage | 0.50 V | 40–160 °C (15 °C min−1 ramp rate) | H2//air (dry) | 700 cycles | 114 |
| Current and thermal cycling | 0.30 A cm−2 (approx. 12 h, 160 °C) | 30–160 °C | H2/N2//air/N2 (unspecified) | 500 h | 219 |
| 0.00 A cm−2 (approx. 12 h, ≤120 °C) | |||||
| Current and thermal cycling | 0.20 A cm−2 (16 h, 180 °C) | 25–180 °C | H2//air (dry) | 157 cycles | 252 |
| 0.00 A cm−2 (approx. 8 h, 25 °C) | |||||
| Current and thermal cycling | 0.00 A cm−2 (30 s, 115 °C) | 80–165 °C | H2/reformate/N2//air (dry) | 1562 cycles | 238 |
| 0.25 A cm−2 (990 s, 165 °C) | (2.5 °C min−1 ramp rate) | ||||
| 0.03 A cm−2 (600 s, 25 °C) | |||||
| Fuel cycling | No load applied | 180 °C | H2/air//air | 150 cycles | 252 |
| Anode gas switched between H2 and air in 90 s intervals (air as cathode gas) | |||||
| Potential cycling | 1.5 V (30 min) | 160 °C | H2//N2 (100% RH) | 2.5 h | 565 |
|
|||||
| Duty cycle | |||||
| Current cycling | Duty cycle based on new European drive cycle (NEDC) | 160 °C | H2//air | 500 cycles | 566 |
| Current cycling | Annual CHP unit profile | 160 °C | H2/CO//air | 970 h | 567 |
| 0.40 A cm−2 (250 h) | |||||
| 0.20–0.40 A cm−2 (120 h) | |||||
| 0.20 A cm−2 (120 h) | |||||
| 0.00–0.20 A cm−2 (240 h) | |||||
| 0.20–0.40 A cm−2 (120 h) | |||||
| 0.20 A cm−2 (120 h) | |||||
 | ||
| Fig. 24 AST profiles for inducing PA loss using (a) high (LC1a) and (b) low (LC2) current density cycling, and (c) mass of phosphorous in the fuel cell exhaust water determined by ICP-MS. Adapted from ref. 146, Copyright (2016), with permission from Wiley. | ||
The length of time the cell is held at the current density set points also has an effect. Longer hold times lead to lower degradation rates; this was explained by more time at low current densities allowing for a greater amount of acid which had migrated into the electrodes to re-enter the membrane.547 Multiple studies have reported using the same or very similar current cycling profiles; however, the test duration, gas supply parameters, and temperature are not consistent across all studies.146,219,274,548,549 This makes it difficult to reliably compare MEA performance between different studies. Constant current or potential testing is reported in the literature with the former being more common for acid loss testing due to the dependence on water generation and therefore current rather than potential, but current cycling is used most and induces greater degradation rates.146,550 Thermal cycling is also used to accelerate acid loss due to the impact of temperature on evaporation and the impact of water partial pressure changes occurring when cycling with set points above and below phase change temperatures. Thermal cycling has been reported to cause more significant PA leaching than low or high load current cycling.186 It is clear that ASTs, including high temperatures >180 °C and/or current density cycling with upper set points ≥0.6 A cm−2, are effective and common methods to study acid loss in HT-PEMFCs. However, a standardised approach is lacking and is required to allow reliable comparisons across studies.
000 cycles with subsequent 24 hours of operation for both humidified and dry H2/N2 conditions can be seen.
 | ||
| Fig. 25 Polarisation curves at beginning of life (BoL), after specified number of cycles, and end of test (EoT) with 24 hours of operation at 0.2 A cm−2. (a) Dry and (b) humidified H2/N2 cycling between 0.6 and 1.0 V (graph modified from original).156 | ||
Triangular and square wave cycling is frequently used in catalyst layer degradation protocols. Other than the potential limits, triangular and square wave cycles are defined by their scan rate and hold times respectively. Generally, square wave ASTs lead to faster degradation rates due to rapid potential changes and longer hold times at high potential.553,554 However, which type of wave used during AST cycling will accelerate degradation more depends on the conditions of the individual cycles.552 Current cycling is also used to investigate catalyst degradation in HT-PEMFCs. These cycles are typically from 0.0 A cm−2 to 0.2 A cm−2 and are generally square waves with up to 20 min hold times. This type of cycling is more representative of real fuel cell operation as OCV and non-zero currents are used under hydrogen and air conditions. To the authors' knowledge there are currently no standards for ASTs targeting catalyst degradation in HT-PEMFCs. Studies do use established cycles used for LT-PEMFCs, and this is reasonable as the catalyst layer materials and structure shares many similarities with LT-PEMFCs. However, higher temperature operation, water in the vapour phase, the presence of PA, use of reformate gas, and different binder materials mean that catalyst degradation in HT-PEMFCs ultimately occurs under different conditions. Therefore, specific catalyst degradation AST protocols should be defined for HT-PEMFCs. It is also worth noting that AST studies of catalyst degradation in the literature typically run a predetermined number of cycles or duration, whereas acid loss studies may do this or run the cell until failure.
Research focusing on bipolar plate degradation and durability in HT-PEMFCs is relatively low.563 ASTs for HT-PEMFC bipolar plates are even rarer. Only one AST was found in the literature. The study itself did not solely focus on bipolar plate degradation, but instead assessed the effects of potential cycling on both the MEA and bipolar plates.142 Therefore, ASTs for HT-PEMFC bipolar plates are still lacking.
11.3 Summary of recommendations
Due to the importance of ASTs in allowing for controlled inducement of degradation mechanisms and estimation of lifetime, focus must be given to the development and standardisation of ASTs for HT-PEMFCs. Recommendations from this review include:• Standardisation of acid loss ASTs. These should use high current density cycling with upper set points ≥0.6 A cm−2, higher temperatures (>180 °C) may also be combined with the current density cycling to induce acid loss. However, the expected operating temperature of the fuel cell should be considered when estimating lifetime, with many PBI-based MEAs operating at 160 °C.
• ASTs targeting catalyst layer degradation (e.g., carbon corrosion, catalyst surface area loss) can be adapted from standardised LT-PEMFC ASTs such as those generated by the U.S. DoE. However, the difference in operating conditions must be factored in when making adaptions to existing LT-PEMFC ASTs (e.g., higher temperature, and dry reactant gases). The impact of the presence of water during potential cycling using N2 on degradation and lifetime estimation should also be considered.
• Start-up/shutdown ASTs are the most complex with significant variation, and also require standardisation. Adaption of existing protocols such as the U.S. DoE AST for LT-PEMFC start-up/shutdown may be a relatively simple pathway to achieving improved standardisation, as only the operating conditions would need to be altered. However, targeting specific HT-PEMFC degradation mechanisms during start-up/shutdown such as acid loss may require additional protocol adaption.
• ASTs for other components and degradation mechanisms have received less attention and require more development before standardisation can be achieved.
• Whilst adaptions to existing LT-PEMFC AST protocols may hasten standardisation of HT-PEMFC ASTs, it is not sufficient to cover all degradation mechanisms that are unique to HT-PEMFCs. Therefore, efforts should be made to build on the work of the DENMEA (grant agreement ID: 245156) and CISTEM projects with similar efforts to those of LT-PEMFC AST development (e.g., H2020 European ID-FAST Project, U.S. DoE) to develop standardised ASTs for HT-PEMFCs.
12. Summary and concluding remarks
HT-PEMFCs offer multiple potential benefits over conventional LT-PEMFCs including higher impurity tolerance, simplified water and thermal management as well as potentially faster reaction kinetics. The recent development of PA-based ion-pair membranes for high-temperature operation offers significant improvements in performance across a wide temperature range, and improved PA retention in the presence of water. However, state-of-the-art performance and durability of HT-PEMFCs do not match that of commercial LT-PEMFCs. Additionally, the Pt loading is significantly higher for HT-PEMFCs. Thus, further work is required to address these issues.This review introduced HT-PEMFC technology, the applications and important considerations of common characterisation techniques, how this differed from LT-PEMFCs, and highlighted areas required to advance characterisation and progress HT-PEMFC technology to commercialisation. Key conclusions and future directions are summarised below:
(1) Operando and in situ characterisation techniques are required to provide information about the fuel cell under realistic operating conditions. Methods that are non-destructive and allow the fuel cell to operate as it would in real-world applications require fewer assumptions and caveats when analysing the results, and therefore are the most useful. While there are operando and in situ techniques applied currently, the existing mature application of techniques to LT-PEMFCs may provide useful insight for further development and opportunities for HT-PEMFCs.
(2) Electrochemical characterisation techniques are essential for operando characterisation of HT-PEMFC performance. These characterisation methods share many similarities in their application with LT-PEMFCs, and the differences mainly occur in the results and their interpretation. The greatly reduced sensitivity to proton conduction via water molecules appears to lead to a reduction in hysteresis behaviour during polarisation curves, and the low-frequency inductance loop commonly found in LT-PEMFC EIS is scarcely reported. Further research is required to determine the extent to which these phenomena occur across operating conditions and to conclusively determine the impact of low-frequency inductive behaviour for HT-PEMFCs, and thus inform more accurate ECMs. The importance of linear EIS validity is also highlighted, especially when using DRT analysis which requires high-quality, valid data to allow meaningful interpretation. CV methods using the hydrogen underpotential method to determine ECSA may not be accurate for HT-PEMFCs, and CO stripping may be a better alternative.
(3) Imaging techniques such as X-ray and neutron radiography and CT, and optical and MRI imaging are commonly used in LT-PEMFCs to visualise water content and some offer similar applicability in characterising PA distribution in HT-PEMFCs. X-ray and neutron tomography techniques permit 3D and 4D in situ and operando studies providing useful information on the fuel cell morphology and dynamic transport of water and PA. Challenges arise with these techniques due to limited access to synchrotron facilities. Although X-ray radiography and CT are possible using lab-based systems, lab-based studies typically require a subtraction method to allow segmentation of the PA which necessitates time-consuming additional experimental and image processing steps. This ultimately limits the scope for in situ or operando studies where morphology changes prevent subtraction or preliminary “dry” scans are not possible. Therefore, new methods to improve PA segmentation by enhancing contrast would simplify existing PA visualisation methodologies.
(4) Non-invasive operando measurement of exhaust gases or analysis of effluent water can provide information on degradation. Gas and ion chromatography techniques are typically used for this purpose, with the latter most common for determining PA loss in HT-PEMFCs. These techniques offer the potential to monitor various degradation mechanisms such as carbon corrosion, membrane degradation, and PA loss; ICP-MS can also be used to identify trace elements such as Pt in effluent water. Current methods require long sampling times or for the sample to be collected and analysed ex situ. Development of techniques or methodologies with short sampling times (seconds to a couple of minutes) would allow for non-invasive dynamic studies of degradation as a function of operating conditions.
(5) Raman and IR spectroscopy are used to characterise the structure of catalyst layers and membranes in HT-PEMFCs. Raman spectroscopy has the advantage of high sensitivity even in the presence of liquid water, and this has allowed for dynamic studies of water transport in LT-PEMFCs. Operando studies of PA transport in HT-PEMFCs could offer insight into the chemical states of the acid-doped membrane. IR spectroscopy allows for in situ and operando studies of HT-PEMFC exhaust gases, often focusing on CO2 caused by corrosion. NMR spectroscopy has been useful in probing the water and proton transport properties in LT-PEMFCs and has been applied similarly in PA-based systems in HT-PEMFCs; this has allowed a new PA loss mechanism to be suggested and insights into the success of PA-PBI systems and the improved PA retention of ion-pair membranes.
(6) ASTs are crucial for estimating real-world durability in a controlled lab-based setting. When combined with advanced characterisation techniques, ASTs are valuable in targeting specific degradation mechanisms, which ultimately assists the progress towards increased durability. Standardised ASTs for HT-PEMFCs are lacking, making meaningful durability comparisons between different studies impossible. Modifying existing standardised ASTs for LT-PEMFCs may be a potential remedy for this issue, an example being the adjustment of existing ASTs (e.g., U.S. DoE) targeting catalyst degradation as the technologies share similar catalyst and support componentry. Careful consideration must also be given to the humidification state of the inlet gas in common catalyst layer ASTs. This parameter varies across tests and has been shown to have a significant impact on the degradation and representation of real-world operation. Unique mechanisms such as PA loss require new protocols. The most effective existing ASTs for inducing PA loss use high current density cycling (upper set point ≥0.6 A cm−2), yet standardisation across studies is missing. The presence of PA must also be considered when designing new ASTs for components such as bipolar plates. Start-up/shutdown ASTs show considerable variation between studies, likely due to the complex nature of the process and the number of parameters involved. The initial and final temperatures, ramp rate, gases supplied, current and voltage cycling all vary significantly and have a critical impact on degradation, making it difficult to draw comparisons across studies and highlighting the need for standardisation.
The progression of HT-PEMFC technology to commercialisation relies on the utilisation of existing characterisation techniques as well as the development of new methodologies. A combination of electrochemical, multi-scale imaging, and spectroscopic techniques under fuel cell operating conditions is required for a deeper understanding of underlying physical and chemical mechanisms as well as the characterisation of performance, degradation and durability of cells and stacks. Another key takeaway is the identification of standardised ASTs as an essential next step to allow comparisons of technologies across studies.
Author contributions
Adam Zucconi: conceptualisation, investigation, project administration, visualisation, writing – original draft, writing – review & editing. Jennifer Hack: investigation, writing – original draft, writing – review & editing. Richard Stocker: writing – review & editing. Theo A. M. Suter: investigation, writing – original draft. Alexander J. E. Rettie: writing – review & editing. Dan J. L. Brett: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AZ acknowledges funding support from HORIBA MIRA. JH acknowledges The Royal Academy of Engineering under the Research Fellowship programme and funding from EPSRC (EP/T517793/1) and the Faraday Institution (FIRG014 and FIRG058). DB acknowledges The Royal Academy of Engineering, the National Physical Laboratory and HORIBA MIRA for sponsoring his Research Chair in Metrology for Electrochemical Propulsion, and the authors are grateful for to EPSRC for sponsoring hydrogen research in the Electrochemical Innovation Lab (EP/X023656/1, EP/W03395X/1, EP/W033321/1, EP/S018204/2).References
- R. Turconi, A. Boldrin and T. Astrup, Renewable Sustainable Energy Rev., 2013, 28, 555–565 CrossRef CAS.
- M. A. Mac Kinnon, J. Brouwer and S. Samuelsen, Prog. Energy Combust. Sci., 2018, 64, 62–92 CrossRef.
- G. Wang, J. Deng, Y. Zhang, Q. Zhang, L. Duan, J. Hao and J. Jiang, Sci. Total Environ., 2020, 741, 140326 CrossRef CAS PubMed.
- I. Hanif, S. M. Faraz Raza, P. Gago-de-Santos and Q. Abbas, Energy, 2019, 171, 493–501 CrossRef.
- L. Jamel and A. Derbali, Cogent Econ. Finance, 2016, 4, 1170653 CrossRef.
- E. Rasoulinezhad, F. Taghizadeh-Hesary and F. Taghizadeh-Hesary, Energies, 2020, 13, 2255 CrossRef CAS.
- A. Rehman, A. Rauf, M. Ahmad, A. A. Chandio and Z. Deyuan, Environ. Sci. Pollut. Res., 2019, 26, 21760–21773 CrossRef CAS PubMed.
- F. Perera, Int. J. Environ. Res. Public Health, 2017, 15, 16 CrossRef PubMed.
- S. Shafiee and E. Topal, Appl. Energy, 2010, 87, 988–1000 CrossRef.
- C. Day and G. Day, Econ. Model., 2017, 63, 153–160 CrossRef.
- IEA, World Energy Outlook 2022, 2022 Search PubMed.
- M. D. Delis, K. de Greiff and S. R. G. Ongena, EBRD Working Paper No. 231, 2019, DOI:10.2139/ssrn.3451335, https://ssrn.com/abstract=3451335.
- World Bank, State and Trends of Carbon Pricing 2021, The World Bank, Washington, DC, 2021 Search PubMed.
- IEA, Renewables Information 2022https://www.iea.org/data-and-statistics/data-tools/energy-statistics-data-browser?country=WORLD%26fuel=Energysupply%26indicator=RenewGenBySource, accessed 7 June 2023.
- R. Zhang and S. Fujimori, Environ. Res. Lett., 2020, 15, 034019 CrossRef.
- N. Rietmann, B. Hügler and T. Lieven, J. Cleaner Prod., 2020, 261, 121038 CrossRef CAS.
- T. Wilberforce, Z. El-Hassan, F. N. Khatib, A. Al Makky, A. Baroutaji, J. G. Carton and A. G. Olabi, Int. J. Hydrogen Energy, 2017, 42, 25695–25734 CrossRef CAS.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Review of the Proton Exchange Membranes for Fuel Cell Applications, Elsevier Ltd, 2010, vol. 35 Search PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- G. Hinds, Curr. Opin. Electrochem., 2017, 5, 11–19 CrossRef CAS.
- ERM, The Fuel Cell Industry Review 2022, 2024 Search PubMed.
- R. E. Rosli, A. B. Sulong, W. R. W. Daud, M. A. Zulkifley, T. Husaini, M. I. Rosli, E. H. Majlan and M. A. Haque, Int. J. Hydrogen Energy, 2017, 42, 9293–9314 CrossRef CAS.
- T. Xiao, R. Wang, Z. Chang, Z. Fang, Z. Zhu and C. Xu, Prog. Nat. Sci.: Mater. Int., 2020, 30, 743–750 CrossRef CAS.
- G. Li, W. Kujawski and E. Rynkowska, Rev. Chem. Eng., 2020, 87–100 CAS.
- T. Myles, L. Bonville and R. Maric, Catalysts, 2017, 7, 16 CrossRef.
- J. O. Jensen, H. A. Hjuler, D. Aili and Q. Li, in High Temperature Polymer Electrolyte Membrane Fuel Cells, ed. Q. Li, D. Aili, H. A. Hjuler and J. O. Jensen, Springer International Publishing, Cham, 2016, pp. 1–4 Search PubMed.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- J. Tjønnås, F. Zenith, I. J. Halvorsen, M. Klages and J. Scholta, IFAC-PapersOnLine, 2016, vol. 49, pp. 302–307 Search PubMed.
- S. Simon Araya, F. Zhou, S. Lennart Sahlin, S. Thomas, C. Jeppesen and S. Knudsen Kær, Energies, 2019, 12, 152 CrossRef.
- M. Boaventura, H. Sander, K. A. Friedrich and A. Mendes, Electrochim. Acta, 2011, 56, 9467–9475 CrossRef CAS.
- B. Shabani, M. Hafttananian, S. Khamani, A. Ramiar and A. A. Ranjbar, J. Power Sources, 2019, 427, 21–48 CrossRef CAS.
- H. P. Dhar, L. G. Christner and A. K. Kush, J. Electrochem. Soc., 1987, 134, 3021–3026 CrossRef CAS.
- Q. Li, R. He, J.-A. Gao, J. O. Jensen and N. J. Bjerrum, J. Electrochem. Soc., 2003, 150, A1599 CrossRef CAS.
- S. K. Das, A. Reis and K. J. Berry, J. Power Sources, 2009, 193, 691–698 CrossRef CAS.
- V. B. Venkatesh, G. Varghese, T. V. Joseph and P. Chippar, Int. J. Hydrogen Energy, 2021, 46, 8179–8196 CrossRef.
- C. Song, Y. Tang, J. L. Zhang, J. Zhang, H. Wang, J. Shen, S. McDermid, J. Li and P. Kozak, Electrochim. Acta, 2007, 52, 2552–2561 CrossRef CAS.
- J. Zhang, Y. Tang, C. Song and J. Zhang, J. Power Sources, 2007, 172, 163–171 CrossRef CAS.
- R. Zeis, Beilstein J. Nanotechnol., 2015, 6, 68–83 CrossRef PubMed.
- R. Sur, T. J. Boucher, M. W. Renfro and B. M. Cetegen, J. Electrochem. Soc., 2010, 157, B45 CrossRef CAS.
- J. P. Owejan, J. E. Owejan, W. Gu, T. A. Trabold, T. W. Tighe and M. F. Mathias, J. Electrochem. Soc., 2010, 157, B1456 CrossRef CAS.
- J. M. LaManna and S. G. Kandlikar, Int. J. Hydrogen Energy, 2011, 36, 5021–5029 CrossRef CAS.
- Y. Wu, J. I. S. Cho, M. Whiteley, L. Rasha, T. P. Neville, R. Ziesche, R. Xu, R. Owen, N. Kulkarni, J. Hack, M. Maier, N. Kardjilov, H. Markötter, I. Manke, F. R. Wang, P. R. Shearing and D. J. L. Brett, Int. J. Hydrogen Energy, 2020, 45, 2195–2205 CrossRef CAS.
- L. Liu, W. Chen and Y. Li, J. Membr. Sci., 2016, 504, 1–9 CrossRef CAS.
- S. Deabate, G. Gebel, P. Huguet, A. Morin and G. Pourcelly, Energy Environ. Sci., 2012, 5, 8824 RSC.
- I. A. Schneider, D. Kramer, A. Wokaun and G. G. Scherer, Electrochem. Commun., 2005, 7, 1393–1397 CrossRef CAS.
- X. D. Wang, Y. Y. Duan, W. M. Yan and F. B. Weng, J. Power Sources, 2008, 176, 247–258 CrossRef CAS.
- S. S. Araya, F. Zhou, V. Liso, S. L. Sahlin, J. R. Vang, S. Thomas, X. Gao, C. Jeppesen and S. K. Kær, Int. J. Hydrogen Energy, 2016, 41, 21310–21344 CrossRef CAS.
- P. Xiao, J. Li, H. Tang, Z. Wang and M. Pan, J. Membr. Sci., 2013, 442, 65–71 CrossRef CAS.
- S. H. Woo, A. Taguet, B. Otazaghine, A. Mosdale, A. Rigacci and C. Beauger, Electrochim. Acta, 2019, 319, 933–946 CrossRef CAS.
- A. Chandan, M. Hattenberger, A. El-Kharouf, S. Du, A. Dhir, V. Self, B. G. Pollet, A. Ingram and W. Bujalski, J. Power Sources, 2013, 231, 264–278 CrossRef CAS.
- L. Lüke, H. Janßen, M. Kvesić, W. Lehnert and D. Stolten, Int. J. Hydrogen Energy, 2012, 37, 9171–9181 CrossRef.
- J. Zhang, Z. Xie, J. Zhang, Y. Tang, C. Song, T. Navessin, Z. Shi, D. Song, H. Wang, D. P. Wilkinson, Z. S. Liu and S. Holdcroft, J. Power Sources, 2006, 160, 872–891 CrossRef CAS.
- D. Aili, D. Henkensmeier, S. Martin, B. Singh, Y. Hu, J. O. Jensen, L. N. Cleemann and Q. Li, Electrochem. Energy Rev., 2020, 3, 793–845 CrossRef CAS.
- J. Zhang, D. Aili, S. Lu, Q. Li and S. P. Jiang, Research, 2020, 2020, 1–15 CAS.
- J. Jung, J. Ku, Y. S. Park, C.-H. Ahn, J.-H. Lee, S. S. Hwang and A. S. Lee, Polym. Rev., 2022, 1–37 Search PubMed.
- E. Quartarone, S. Angioni and P. Mustarelli, Materials, 2017, 10, 687 CrossRef PubMed.
- X. Sun, S. Simonsen, T. Norby and A. Chatzitakis, Membranes, 2019, 9, 83 CrossRef CAS PubMed.
- M. Ebrahimi, W. Kujawski, K. Fatyeyeva and J. Kujawa, Int. J. Mol. Sci., 2021, 22, 5430 CrossRef CAS PubMed.
- A. C. Dupuis, Prog. Mater. Sci., 2011, 56, 289–327 CrossRef CAS.
- N. P. Chikhaliya, Y. J. Rathwa and T. Likhariya, High Perform. Polym., 2021, 33, 998–1011 CrossRef CAS.
- S. Subianto, Polym. Int., 2014, 63, 1134–1144 CrossRef CAS.
- S. Bose, T. Kuila, T. X. H. Nguyen, N. H. Kim, K. Lau and J. H. Lee, Prog. Polym. Sci., 2011, 36, 813–843 CrossRef CAS.
- Z. Guo, M. Perez-Page, J. Chen, Z. Ji and S. M. Holmes, J. Energy Chem., 2021, 63, 393–429 CrossRef CAS.
- A. E. Russell and A. Rose, Chem. Rev., 2004, 104, 4613–4635 CrossRef CAS PubMed.
- J. Wu, X. Zi Yuan, H. Wang, M. Blanco, J. J. Martin and J. Zhang, Int. J. Hydrogen Energy, 2008, 33, 1747–1757 CrossRef CAS.
- Q. Meyer, Y. Zeng and C. Zhao, Adv. Mater., 2019, 31, 1–25 CrossRef PubMed.
- R. Haider, Y. Wen, Z.-F. Ma, D. P. Wilkinson, L. Zhang, X. Yuan, S. Song and J. Zhang, Chem. Soc. Rev., 2021, 50, 1138–1187 RSC.
- S. Chevalier, M. Fazeli, F. Mack, S. Galbiati, I. Manke, A. Bazylak and R. Zeis, Electrochim. Acta, 2016, 212, 187–194 CrossRef CAS.
- N. Bevilacqua, M. G. George, S. Galbiati, A. Bazylak and R. Zeis, Electrochim. Acta, 2017, 257, 89–98 CrossRef CAS.
- F. Mack, M. Klages, J. Scholta, L. Jörissen, T. Morawietz, R. Hiesgen, D. Kramer and R. Zeis, J. Power Sources, 2014, 255, 431–438 CrossRef CAS.
- L. Xia, M. Ni, Q. He, Q. Xu and C. Cheng, Appl. Energy, 2021, 300, 117357 CrossRef.
- P. O. Olapade, J. P. Meyers, R. L. Borup and R. Mukundan, J. Electrochem. Soc., 2011, 158, B639 CrossRef CAS.
- E. Carcadea, M. Varlam, M. Ismail, D. B. Ingham, A. Marinoiu, M. Raceanu, C. Jianu, L. Patularu and D. Ion-Ebrasu, Int. J. Hydrogen Energy, 2020, 45, 7968–7980 CrossRef CAS.
- L. Xing, Y. Wang, P. K. Das, K. Scott and W. Shi, Chem. Eng. Sci., 2018, 192, 699–713 CrossRef CAS.
- L. Karpenko-Jereb, C. Sternig, C. Fink, V. Hacker, A. Theiler and R. Tatschl, J. Power Sources, 2015, 297, 329–343 CrossRef CAS.
- A. Ozden, S. Shahgaldi, X. Li and F. Hamdullahpur, Prog. Energy Combust. Sci., 2019, 74, 50–102 CrossRef.
- G. Lin and T. Van Nguyen, J. Electrochem. Soc., 2005, 152, A1942 CrossRef CAS.
- J. H. Chun, K. T. Park, D. H. Jo, S. G. Kim and S. H. Kim, Int. J. Hydrogen Energy, 2011, 36, 1837–1845 CrossRef CAS.
- Y. Wang, B. Seo, B. Wang, N. Zamel, K. Jiao and X. C. Adroher, Energy AI, 2020, 1, 100014 CrossRef.
- Y. Chang, Y. Qin, Y. Yin, J. Zhang and X. Li, Appl. Energy, 2018, 230, 643–662 CrossRef CAS.
- A. Ibrahim, O. Hossain, J. Chaggar, R. Steinberger-Wilckens and A. El-Kharouf, Int. J. Hydrogen Energy, 2020, 45, 5526–5534 CrossRef CAS.
- Z. Jie, T. Haolin and P. Mu, J. Membr. Sci., 2008, 312, 41–47 CrossRef.
- D. C. Lee, H. N. Yang, S. H. Park and W. J. Kim, J. Membr. Sci., 2014, 452, 20–28 CrossRef CAS.
- M. Amjadi, S. Rowshanzamir, S. J. Peighambardoust, M. G. Hosseini and M. H. Eikani, Int. J. Hydrogen Energy, 2010, 35, 9252–9260 CrossRef CAS.
- E. Chalkova, M. B. Pague, M. V. Fedkin, D. J. Wesolowski and S. N. Lvov, J. Electrochem. Soc., 2005, 152, A1035 CrossRef CAS.
- K. T. Park, U. H. Jung, D. W. Choi, K. Chun, H. M. Lee and S. H. Kim, J. Power Sources, 2008, 177, 247–253 CrossRef CAS.
- Y. Zhai, H. Zhang, J. Hu and B. Yi, J. Membr. Sci., 2006, 280, 148–155 CrossRef CAS.
- K. T. Adjemian, R. Dominey, L. Krishnan, H. Ota, P. Majsztrik, T. Zhang, J. Mann, B. Kirby, L. Gatto, M. Velo-Simpson, J. Leahy, S. Srinivasan, J. B. Benziger and A. B. Bocarsly, Chem. Mater., 2006, 18, 2238–2248 CrossRef CAS.
- J. Yang, D. Aili, Q. Li, Y. Xu, P. Liu, Q. Che, J. O. Jensen, N. J. Bjerrum and R. He, Polym. Chem., 2013, 4, 4768 RSC.
- J. Yang, J. Wang, C. Liu, L. Gao, Y. Xu, Q. Che and R. He, J. Membr. Sci., 2015, 493, 80–87 CrossRef CAS.
- Y. Oono, A. Sounai and M. Hori, J. Power Sources, 2009, 189, 943–949 CrossRef CAS.
- J.-P. Melchior, K.-D. Kreuer and J. Maier, Phys. Chem. Chem. Phys., 2017, 19, 587–600 RSC.
- J. P. Melchior, G. Majer and K. D. Kreuer, Phys. Chem. Chem. Phys., 2017, 19, 601–612 RSC.
- N. Üregen, K. Pehlivanoğlu, Y. Özdemir and Y. Devrim, Int. J. Hydrogen Energy, 2017, 42, 2636–2647 CrossRef.
- B. Heggen, S. Roy and F. Müller-Plathe, J. Phys. Chem. C, 2008, 112, 14209–14215 CrossRef CAS.
- R. A. Krueger, L. Vilčiauskas, J.-P. Melchior, G. Bester and K.-D. Kreuer, J. Phys. Chem. B, 2015, 119, 15866–15875 CrossRef CAS PubMed.
- Y. Özdemir, N. Özkan and Y. Devrim, Electrochim. Acta, 2017, 245, 1–13 CrossRef.
- N. N. Krishnan, S. Lee, R. V. Ghorpade, A. Konovalova, J. H. Jang, H.-J. Kim, J. Han, D. Henkensmeier and H. Han, J. Membr. Sci., 2018, 560, 11–20 CrossRef CAS.
- S. Bhadra, N. H. Kim and J. H. Lee, J. Membr. Sci., 2010, 349, 304–311 CrossRef CAS.
- A. Kannan, D. Aili, L. N. Cleemann, Q. Li and J. O. Jensen, Int. J. Hydrogen Energy, 2020, 45, 1008–1017 CrossRef CAS.
- E. Quartarone, P. Mustarelli, A. Carollo, S. Grandi, A. Magistris and C. Gerbaldi, Fuel Cells, 2009, 9, 231–236 CrossRef CAS.
- P. Mustarelli, E. Quartarone, S. Grandi, A. Carollo and A. Magistris, Adv. Mater., 2008, 20, 1339–1343 CrossRef CAS.
- Y. Devrim, H. Devrim and I. Eroglu, Int. J. Hydrogen Energy, 2016, 41, 10044–10052 CrossRef CAS.
- D. Aili, J. Zhang, M. T. Dalsgaard Jakobsen, H. Zhu, T. Yang, J. Liu, M. Forsyth, C. Pan, J. O. Jensen, L. N. Cleemann, S. P. Jiang and Q. Li, J. Mater. Chem. A, 2016, 4, 4019–4024 RSC.
- A. Verma and K. Scott, J. Solid State Electrochem., 2010, 14, 213–219 CrossRef CAS.
- C. Xu, X. Wu, X. Wang, M. Mamlouk and K. Scott, J. Mater. Chem., 2011, 21, 6014 RSC.
- R. Kannan, H. N. Kagalwala, H. D. Chaudhari, U. K. Kharul, S. Kurungot and V. K. Pillai, J. Mater. Chem., 2011, 21, 7223 RSC.
- C.-M. C. Suryani, Y.-L. Liu and Y. M. Lee, J. Mater. Chem., 2011, 21, 7480 RSC.
- J. Escorihuela, Ó. Sahuquillo, A. García-Bernabé, E. Giménez and V. Compañ, Nanomaterials, 2018, 8, 775 CrossRef PubMed.
- Y. Wu, X. Liu, F. Yang, L. Lee Zhou, B. Yin, P. Wang and L. Wang, J. Membr. Sci., 2021, 630, 119288 CrossRef CAS.
- J. Chen, L. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 41350–41358 CrossRef CAS PubMed.
- K. S. Lee, J. S. Spendelow, Y. K. Choe, C. Fujimoto and Y. S. Kim, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- V. Atanasov, A. S. Lee, E. J. Park, S. Maurya, E. D. Baca, C. Fujimoto, M. Hibbs, I. Matanovic, J. Kerres and Y. S. Kim, Nat. Mater., 2021, 20, 370–377 CrossRef CAS PubMed.
- K. H. Lim, A. S. Lee, V. Atanasov, J. Kerres, E. J. Park, S. Adhikari, S. Maurya, L. D. Manriquez, J. Jung, C. Fujimoto, I. Matanovic, J. Jankovic, Z. Hu, H. Jia and Y. S. Kim, Nat. Energy, 2022, 7, 248–259 CrossRef CAS.
- H. Tang, K. Geng, L. Wu, J. Liu, Z. Chen, W. You, F. Yan, M. D. Guiver and N. Li, Nat. Energy, 2022, 7, 153–162 CrossRef CAS.
- T. Engl, L. Gubler and T. J. Schmidt, in High Temperature Polymer Electrolyte Membrane Fuel Cells, Springer International Publishing, Cham, 2016, pp. 297–313 Search PubMed.
- N. Bevilacqua, R. R. Gokhale, A. Serov, R. Banerjee, M. A. Schmid, P. Atanassov and R. Zeis, ECS Trans., 2018, 86, 221–229 CrossRef CAS.
- N. M. Zagudaeva and M. R. Tarasevich, Russ. J. Electrochem., 2010, 46, 530–536 CrossRef CAS.
- A. Schenk, C. Grimmer, M. Perchthaler, S. Weinberger, B. Pichler, C. Heinzl, C. Scheu, F. A. Mautner, B. Bitschnau and V. Hacker, J. Power Sources, 2014, 266, 313–322 CrossRef CAS.
- Y. Devrim, E. D. Arıca and A. Albostan, Int. J. Hydrogen Energy, 2018, 43, 11820–11829 CrossRef CAS.
- H.-S. Oh, J.-H. Lee and H. Kim, Int. J. Hydrogen Energy, 2012, 37, 10844–10849 CrossRef CAS.
- Y. Devrim and E. D. Arıca, Int. J. Hydrogen Energy, 2019, 44, 18951–18966 CrossRef CAS.
- Y. Devrim and E. D. Arıca, Int. J. Hydrogen Energy, 2020, 45, 3609–3617 CrossRef CAS.
- T. A. M. Suter, K. Smith, J. Hack, L. Rasha, Z. Rana, G. M. A. Angel, P. R. Shearing, T. S. Miller and D. J. L. Brett, Adv. Energy Mater., 2021, 2101025, 2101025 CrossRef.
- J. Zhang, H. Wang, W. Li, J. Zhang, D. Lu, W. Yan, Y. Xiang and S. Lu, J. Power Sources, 2021, 505, 230059 CrossRef CAS.
- J. Halter, N. Bevilacqua, R. Zeis, T. J. Schmidt and F. N. Büchi, J. Electroanal. Chem., 2020, 859, 113832 CrossRef CAS.
- N. Bevilacqua, T. Asset, M. A. Schmid, H. Markötter, I. Manke, P. Atanassov and R. Zeis, J. Power Sources Adv., 2021, 7, 100042 CrossRef CAS.
- F. Mack, T. Morawietz, R. Hiesgen, D. Kramer, V. Gogel and R. Zeis, Int. J. Hydrogen Energy, 2016, 41, 7475–7483 CrossRef CAS.
- S. H. Kwon, S. Y. Lee, H.-J. Kim, S.-D. Yim, Y.-J. Sohn and S. G. Lee, Sci. Rep., 2022, 12, 3810 CrossRef CAS PubMed.
- J. Lobato, P. Cañizares, M. A. Rodrigo, J. J. Linares and F. J. Pinar, Int. J. Hydrogen Energy, 2010, 35, 1347–1355 CrossRef CAS.
- J.-H. Kim, H.-J. Kim, T.-H. Lim and H.-I. Lee, J. Power Sources, 2007, 170, 275–280 CrossRef CAS.
- D. Lee, J. W. Lim and D. G. Lee, Compos. Struct., 2017, 167, 144–151 CrossRef.
- D. Lee, J. W. Lim, S. Nam, I. Choi and D. G. Lee, Compos. Struct., 2015, 128, 284–290 CrossRef.
- C. Hartnig and T. J. Schmidt, Electrochim. Acta, 2011, 56, 4237–4242 CrossRef CAS.
- M. Kim, J. W. Lim and D. G. Lee, Compos. Struct., 2015, 119, 630–637 CrossRef.
- A. Ghosh and A. Verma, Fuel Cells, 2014, 14, 259–265 CrossRef CAS.
- D. Lee and D. G. Lee, J. Power Sources, 2016, 327, 119–126 CrossRef CAS.
- I. Kundler and T. Hickmann, in High Temperature Polymer Electrolyte Membrane Fuel Cells, Springer International Publishing, Cham, 2016, pp. 425–440 Search PubMed.
- C. J. Tseng, Y. J. Heush, C. J. Chiang, Y. H. Lee and K. R. Lee, Int. J. Hydrogen Energy, 2016, 41, 16196–16204 CrossRef CAS.
- C.-Y. Chen and S.-C. Su, Int. J. Hydrogen Energy, 2018, 43, 13430–13439 CrossRef CAS.
- W.-M. Yan, C.-Y. Chen and C.-H. Liang, Energy, 2019, 186, 115836 CrossRef CAS.
- C. Alegre, L. Álvarez-Manuel, R. Mustata, L. Valiño, A. Lozano and F. Barreras, Int. J. Hydrogen Energy, 2019, 44, 12748–12759 CrossRef CAS.
- A. Tang, L. Crisci, L. Bonville and J. Jankovic, J. Renewable Sustainable Energy, 2021, 13, 022701 CrossRef CAS.
- S. Galbiati, A. Baricci, A. Casalegno and R. Marchesi, Int. J. Hydrogen Energy, 2012, 37, 2462–2469 CrossRef CAS.
- F. Zhou, D. Singdeo and S. K. Kær, Fuel Cells, 2019, 19, 2–9 CrossRef CAS.
- D. Schonvogel, M. Rastedt, P. Wagner, M. Wark and A. Dyck, Fuel Cells, 2016, 16, 480–489 CrossRef CAS.
- Y. H. Jeong, K. Oh, S. Ahn, N. Y. Kim, A. Byeon, H. Y. Park, S. Y. Lee, H. S. Park, S. J. Yoo, J. H. Jang, H. J. Kim, H. Ju and J. Y. Kim, J. Power Sources, 2017, 363, 365–374 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Energy Environ. Sci., 2012, 5, 6436 RSC.
- S. Yu, L. Xiao and B. C. Benicewicz, Fuel Cells, 2008, 8, 165–174 CrossRef CAS.
- Y. Özdemir, N. Üregen and Y. Devrim, Int. J. Hydrogen Energy, 2017, 42, 2648–2657 CrossRef.
- D. Úbeda, F. J. Pinar, D. C. Orozco, P. Cañizares, M. A. Rodrigo and J. Lobato, J. Appl. Electrochem., 2012, 42, 711–718 CrossRef.
- S. Kaserer, K. M. Caldwell, D. E. Ramaker and C. Roth, J. Phys. Chem. C, 2013, 117, 6210–6217 CrossRef CAS.
- D. Úbeda, P. Cañizares, M. A. Rodrigo, F. J. Pinar and J. Lobato, Int. J. Hydrogen Energy, 2014, 39, 21678–21687 CrossRef.
- J. Lobato, P. Cañizares, M. A. Rodrigo, F. J. Pinar and D. Úbeda, J. Power Sources, 2011, 196, 4209–4217 CrossRef CAS.
- J. Li, L. Yang, H. Sun and G. Sun, Energy Technol., 2022, 2100809, 2100809 CrossRef.
- T. Søndergaard, L. N. Cleemann, L. Zhong, H. Becker, T. Steenberg, H. A. Hjuler, L. Seerup, Q. Li and J. O. Jensen, Electrocatalysis, 2018, 9, 302–313 CrossRef.
- J. Halter, F. Marone, T. J. Schmidt and F. N. Büchi, J. Electrochem. Soc., 2018, 165, F1176–F1183 CrossRef CAS.
- L. N. Cleemann, F. Buazar, Q. Li, J. O. Jensen, C. Pan, T. Steenberg, S. Dai and N. J. Bjerrum, Fuel Cells, 2013, 822–831 CrossRef CAS.
- D. E. Hussin, Y. Budak and Y. Devrim, Int. J. Energy Res., 2022, 46, 4174–4186 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Energy Environ. Sci., 2012, 5, 6436–6444 RSC.
- E. Lee, D. H. Kim and C. Pak, Appl. Surf. Sci., 2020, 510, 145461 CrossRef CAS.
- G. Liu, H. Zhang, J. Hu, Y. Zhai, D. Xu and Z. gang Shao, J. Power Sources, 2006, 162, 547–552 CrossRef CAS.
- J.-C. Lin, C.-M. Lai, F.-P. Ting, S.-D. Chyou and K.-L. Hsueh, J. Appl. Electrochem., 2009, 39, 1067–1073 CrossRef CAS.
- Q. Meyer, N. Mansor, F. Iacoviello, P. L. Cullen, R. Jervis, D. Finegan, C. Tan, J. Bailey, P. R. Shearing and D. J. L. Brett, Electrochim. Acta, 2017, 242, 125–136 CrossRef CAS.
- A. Zucconi, J. Hack, T. A. M. Suter, M. Braglia, P. R. Shearing, D. J. L. Brett and A. J. E. Rettie, J. Power Sources, 2023, 584, 233574 CrossRef CAS.
- P. Choi, N. H. Jalani and R. Datta, J. Electrochem. Soc., 2005, 152, E123 CrossRef CAS.
- M. Geormezi, F. Paloukis, A. Orfanidi, N. Shroti, M. K. Daletou and S. G. Neophytides, J. Power Sources, 2015, 285, 499–509 CrossRef CAS.
- A. Kannan, Q. Li, L. N. Cleemann and J. O. Jensen, Fuel Cells, 2018, 18, 103–112 CrossRef CAS.
- S. Thomas, S. S. Araya, J. R. Vang and S. K. Kær, Int. J. Hydrogen Energy, 2018, 43, 14691–14700 CrossRef CAS.
- T. Tingelöf and J. K. Ihonen, Int. J. Hydrogen Energy, 2009, 34, 6452–6456 CrossRef.
- M. Boaventura and A. Mendes, Int. J. Hydrogen Energy, 2010, 35, 11649–11660 CrossRef CAS.
- S. Galbiati, A. Baricci, A. Casalegno, G. Carcassola and R. Marchesi, Int. J. Hydrogen Energy, 2012, 37, 14475–14481 CrossRef CAS.
- J. R. Vang, S. J. Andreasen, S. S. Araya and S. K. Kær, Int. J. Hydrogen Energy, 2014, 39, 14959–14968 CrossRef CAS.
- G. Qian and B. C. Benicewicz, ECS Trans., 2011, 41, 1441–1448 CrossRef CAS.
- M. Zhiani, S. Majidi and M. M. Taghiabadi, Fuel Cells, 2013, 13(5), 946–955 CrossRef CAS.
- M. Zhiani and S. Majidi, Int. J. Hydrogen Energy, 2013, 38, 9819–9825 CrossRef CAS.
- X. Z. Yuan, S. Zhang, J. C. Sun and H. Wang, J. Power Sources, 2011, 196, 9097–9106 CrossRef CAS.
- G. Tsotridis, A. Pilenga, G. De Marco and T. Malkow, EU Harmonised Test Protocols for PEMFC MEA Testing in Single Cell Configuration for Automotive Applications, JRC Science for Policy report, 2015 Search PubMed.
- Y. Wang, D. U. Sauer, S. Koehne and A. Ersöz, Int. J. Hydrogen Energy, 2014, 39, 19067–19078 CrossRef CAS.
- R. K. Abdul Rasheed, S. M. M. Ehteshami and S. H. Chan, Int. J. Hydrogen Energy, 2014, 39, 2246–2260 CrossRef CAS.
- H. Huang, Y. Zhou, H. Deng, X. Xie, Q. Du, Y. Yin and K. Jiao, Int. J. Hydrogen Energy, 2016, 41, 3113–3127 CrossRef CAS.
- M. Choi, M. Kim, Y.-J. Sohn and S.-G. Kim, Int. J. Hydrogen Energy, 2021, 46, 36982–36994 CrossRef CAS.
- J. Zhang, C. Zhang, D. Hao, M. Ni, S. Huang, D. Liu and Y. Zheng, Int. J. Hydrogen Energy, 2021, 46, 2577–2593 CrossRef CAS.
- R. E. Rosli, A. B. Sulong, W. R. W. Daud, M. A. Zulkifley, M. I. Rosli, E. H. Majlan, M. A. Haque and N. A. M. Radzuan, Int. J. Hydrogen Energy, 2019, 44, 30763–30771 CrossRef CAS.
- J. Zhao, D. Song, J. Jia, N. Wang, K. Liu, T. Zuo and Q. Che, Appl. Surf. Sci., 2022, 585, 152579 CrossRef CAS.
- S. Liu, M. Rasinski, Y. Rahim, S. Zhang, K. Wippermann, U. Reimer and W. Lehnert, J. Power Sources, 2019, 439, 227090 CrossRef CAS.
- S. Galbiati, A. Baricci, A. Casalegno and R. Marchesi, Int. J. Hydrogen Energy, 2013, 38, 6469–6480 CrossRef CAS.
- Y. Oono, T. Fukuda, A. Sounai and M. Hori, J. Power Sources, 2010, 195, 1007–1014 CrossRef CAS.
- M. Rastedt, D. Schonvogel and P. Wagner, ECS Trans., 2014, 64, 741–753 CrossRef CAS.
- F. Zhou, S. J. Andreasen and S. K. Kær, Int. J. Hydrogen Energy, 2015, 40, 6672–6680 CrossRef CAS.
- A. Stassi, I. Gatto, E. Passalacqua, V. Antonucci, A. S. Arico, L. Merlo, C. Oldani and E. Pagano, J. Power Sources, 2011, 196, 8925–8930 CrossRef CAS.
- A. Suzuki, Y. Oono, M. C. Williams, R. Miura, K. Inaba, N. Hatakeyama, H. Takaba, M. Hori and A. Miyamoto, Int. J. Hydrogen Energy, 2012, 37, 18272–18289 CrossRef CAS.
- T. Engl, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2015, 162, F291–F297 CrossRef CAS.
- J. Li, L. Yang, Z. Wang, H. Sun and G. Sun, Int. J. Hydrogen Energy, 2021, 46, 24353–24365 CrossRef CAS.
- Z. Chang, H. Pu, D. Wan, L. Liu, J. Yuan and Z. Yang, Polym. Degrad. Stab., 2009, 94, 1206–1212 CrossRef CAS.
- J. Liao, J. Yang, Q. Li, L. N. Cleemann, J. O. Jensen, N. J. Bjerrum, R. He and W. Xing, J. Power Sources, 2013, 238, 516–522 CrossRef CAS.
- H. Schmies, D. Schonvogel, J. Büsselmann, P. Wagner and A. Dyck, ECS Meet. Abstr., 2020, MA2020-02, p. 2161 Search PubMed.
- J. Büsselmann, M. Rastedt, T. Klicpera, K. Reinwald, H. Schmies, A. Dyck and P. Wagner, Energies, 2020, 13, 567 CrossRef.
- F. Zhou, S. J. Andreasen, S. K. Kær and J. O. Park, Int. J. Hydrogen Energy, 2015, 40, 14932–14941 CrossRef CAS.
- Y. Rahim, H. Janßen and W. Lehnert, Int. J. Hydrogen Energy, 2017, 42, 1189–1202 CrossRef CAS.
- C.-Y. Chen, C.-C. Chen, S.-W. Hsu, M.-P. Lai, W.-H. Lai and W.-M. Yang, Energy Procedia, 2012, 29, 64–71 CrossRef CAS.
- H. Lu, L. Rihko-Struckmann, R. Hanke-Rauschenbach and K. Sundmacher, Top. Catal., 2008, 51, 89–97 CrossRef CAS.
- V. F. Valdés-López, T. Mason, P. R. Shearing and D. J. L. Brett, Prog. Energy Combust. Sci., 2020, 79, 100842 CrossRef.
- A. Al-Qahtani, B. Parkinson, K. Hellgardt, N. Shah and G. Guillen-Gosalbez, Appl. Energy, 2021, 281, 115958 CrossRef CAS.
- T. Engl, J. Kase, L. Gubler and T. J. Schmidt, ECS Electrochem. Lett., 2014, 3, F47–F49 CrossRef CAS.
- T. J. Schmidt and J. Baurmeister, J. Power Sources, 2008, 176, 428–434 CrossRef CAS.
- H. Becker, U. Reimer, D. Aili, L. N. Cleemann, J. O. Jensen, W. Lehnert and Q. Li, J. Electrochem. Soc., 2018, 165, F863–F869 CrossRef CAS.
- S. H. Eberhardt, F. Marone, M. Stampanoni, F. N. Büchi and T. J. Schmidt, J. Electrochem. Soc., 2016, 163, F842–F847 CrossRef CAS.
- S. H. Eberhardt, M. Toulec, F. Marone, M. Stampanoni, F. N. Büchi and T. J. Schmidt, J. Electrochem. Soc., 2015, 162, F310–F316 CrossRef CAS.
- Q. Li, R. He, R. W. Berg, H. A. Hjuler and N. J. Bjerrum, Solid State Ionics, 2004, 168, 177–185 CrossRef CAS.
- A. S. Lee, Y. K. Choe, I. Matanovic and Y. S. Kim, J. Mater. Chem. A, 2019, 7, 9867–9876 RSC.
- S. H. Eberhardt, T. Lochner, F. N. Büchi and T. J. Schmidt, J. Electrochem. Soc., 2015, 162, F1367–F1372 CrossRef CAS.
- Q. He, X. Yang, W. Chen, S. Mukerjee, B. Koel and S. Chen, Phys. Chem. Chem. Phys., 2010, 12, 12544–12555 RSC.
- K. M. Caldwell, S. Kaserer, C. Roth and D. E. Ramaker, ChemElectroChem, 2015, 2, 1502–1509 CrossRef CAS.
- K. S. Lee, S. Maurya, Y. S. Kim, C. R. Kreller, M. S. Wilson, D. Larsen, S. E. Elangovan and R. Mukundan, Energy Environ. Sci., 2018, 11, 979–987 RSC.
- J. Hou, Int. J. Hydrogen Energy, 2011, 36, 7199–7206 CrossRef CAS.
- U.S. Drive, Fuel Cell Technical Team, Fuel Cell Technical Team Roadmap, https://www.energy.gov/sites/prod/files/2017/11/f46/FCTT_Roadmap_Nov_2017_FINAL.pdf, accessed 17 February 2022.
- Y. Wang, D. F. Ruiz Diaz, K. S. Chen, Z. Wang and X. C. Adroher, Mater. Today, 2020, 32, 178–203 CrossRef CAS.
- J. Büsselmann, M. Rastedt, V. Tullius, K. Yezerska, A. Dyck and P. Wagner, Int. J. Hydrogen Energy, 2019, 4, 19384–19394 CrossRef.
- Q. Xie, Y. Li, X. Chen, J. Hu, L. Li and H. Li, J. Power Sources, 2015, 282, 489–497 CrossRef CAS.
- Z. Di, Q. Xie, H. Li, D. Mao, M. Li, D. Zhou and L. Li, J. Power Sources, 2015, 273, 688–696 CrossRef CAS.
- Y. Devrim, S. Erkan, N. Baç and I. Eroglu, Int. J. Hydrogen Energy, 2012, 37, 16748–16758 CrossRef CAS.
- K. Oh, B. Son, J. Sanetuntikul and S. Shanmugam, J. Membr. Sci., 2017, 541, 386–392 CrossRef CAS.
- N. H. Kim, A. K. Mishra, D.-Y. Kim and J. H. Lee, Chem. Eng. J., 2015, 272, 119–127 CrossRef CAS.
- European Commission, Volume Manufacturing of PEM FC Stacks for Transportation and In-line Quality Assurance, https://cordis.europa.eu/project/id/671465/reporting, accessed 17 February 2022.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- M. R. Berber and N. Nakashima, J. Membr. Sci., 2019, 591, 117354 CrossRef CAS.
- V. Elumalai, R. Annapooranan, M. Ganapathikrishnan and D. Sangeetha, J. Appl. Polym. Sci., 2018, 135, 45954 CrossRef.
- C. Gao, M. Hu, L. Wang and L. Wang, Polymers, 2020, 12, 515 CrossRef CAS PubMed.
- L. Wang, Y. Wu, M. Fang, J. Chen, X. Liu, B. Yin and L. Wang, J. Membr. Sci., 2020, 602, 117981 CrossRef CAS.
- N. Nambi Krishnan, A. Konovalova, D. Aili, Q. Li, H. S. Park, J. H. Jang, H.-J. Kim and D. Henkensmeier, J. Membr. Sci., 2019, 588, 117218 CrossRef CAS.
- H. Chen, S. Wang, F. Liu, D. Wang, J. Li, T. Mao, G. Liu, X. Wang, J. Xu and Z. Wang, J. Membr. Sci., 2020, 596, 117722 CrossRef CAS.
- B. WAHDAME, D. CANDUSSO, X. FRANCOIS, F. HAREL, M. PERA, D. HISSEL and J. KAUFFMANN, Int. J. Hydrogen Energy, 2007, 32, 4523–4536 CrossRef CAS.
- S. J. C. Cleghorn, D. K. Mayfield, D. A. Moore, J. C. Moore, G. Rusch, T. W. Sherman, N. T. Sisofo and U. Beuscher, J. Power Sources, 2006, 158, 446–454 CrossRef CAS.
- J. St-Pierre and N. Jia, J. New Mater. Electrochem. Syst., 2002, 5, 263–271 CAS.
- J. Yu, T. Matsuura, Y. Yoshikawa, M. N. Islam and M. Hori, Electrochem. Solid-State Lett., 2005, 8, A156 CrossRef CAS.
- P. J. Ferreira, G. J. la O’, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256 CrossRef.
- A. Kannan, A. Kabza and J. Scholta, J. Power Sources, 2015, 277, 312–316 CrossRef CAS.
- F. J. Pinar, P. Cañizares, M. A. Rodrigo, D. Úbeda and J. Lobato, J. Power Sources, 2015, 274, 177–185 CrossRef CAS.
- J. R. Kim, J. S. Yi and T. W. Song, J. Power Sources, 2012, 220, 54–64 CrossRef CAS.
- T. Søndergaard, L. N. Cleemann, H. Becker, D. Aili, T. Steenberg, H. A. Hjuler, L. Seerup, Q. Li and J. O. Jensen, J. Power Sources, 2017, 342, 570–578 CrossRef.
- P. Moçotéguy, B. Ludwig, J. Scholta, R. Barrera and S. Ginocchio, Fuel Cells, 2009, 9, 325–348 CrossRef.
- C. Jeppesen, S. S. Araya, S. L. Sahlin, S. Thomas, S. J. Andreasen and S. K. Kær, J. Power Sources, 2017, 359, 37–47 CrossRef CAS.
- J. Zhang, D. Aili, J. Bradley, H. Kuang, C. Pan, R. De Marco, Q. Li and S. P. Jiang, J. Electrochem. Soc., 2017, 164, F1615–F1625 CrossRef CAS.
- Y. Y. Jo, E. Cho, J. H. Kim, T.-H. Lim, I.-H. Oh, S.-K. Kim, H.-J. Kim and J. H. Jang, J. Power Sources, 2011, 196, 9906–9915 CrossRef CAS.
- J. Han, J. Han and S. Yu, Int. J. Hydrogen Energy, 2020, 45, 13045–13054 CrossRef CAS.
- K. Eom, G. Kim, E. Cho, J. H. Jang, H.-J. Kim, S. J. Yoo, S.-K. Kim and B. K. Hong, Int. J. Hydrogen Energy, 2012, 37, 18455–18462 CrossRef CAS.
- K. Meng, H. Zhou, B. Chen and Z. Tu, Energy, 2021, 224, 120168 CrossRef CAS.
- T. Chu, M. Xie, Y. Yu, B. Wang, D. Yang, B. Li, P. Ming and C. Zhang, Energy, 2022, 239, 122356 CrossRef CAS.
- S. Komini Babu, D. Spernjak, J. Dillet, A. Lamibrac, G. Maranzana, S. Didierjean, O. Lottin, R. L. Borup and R. Mukundan, Appl. Energy, 2019, 254, 113659 CrossRef CAS.
- R. K. Ahluwalia, X. Wang, J.-K. Peng, N. N. Kariuki, D. J. Myers, S. Rasouli, P. J. Ferreira, Z. Yang, A. Martinez-Bonastre, D. Fongalland and J. Sharman, J. Electrochem. Soc., 2018, 165, F3316–F3327 CrossRef CAS.
- C. Hartnig and T. J. Schmidt, J. Power Sources, 2011, 196, 5564–5572 CrossRef CAS.
- S. Rigal, C. Turpin, A. Jaafar, T. Hordé, J. -B. Jollys and N. Chadourne, Fuel Cells, 2020, 20, 272–284 CrossRef CAS.
- M. Baudy, A. Jaafar, C. Turpin, S. Abbou and S. Rigal, Int. J. Hydrogen Energy, 2023, 48, 20945–20956 CrossRef CAS.
- S. S. Araya, S. J. Andreasen and S. K. Kær, J. Fuel Cell Sci. Technol., 2014, 11, 1–7 Search PubMed.
- C. Zhang, Z. Liu, W. Zhou, S. H. Chan and Y. Wang, Energy, 2015, 90, 1949–1955 CrossRef CAS.
- U. Reimer, B. Schumacher and W. Lehnert, J. Electrochem. Soc., 2015, 162, F153–F164 CrossRef CAS.
- M. A. Rubio, A. Urquia and S. Dormido, J. Power Sources, 2007, 171, 670–677 CrossRef CAS.
- K. R. Cooper and M. Smith, J. Power Sources, 2006, 160, 1088–1095 CrossRef CAS.
- S. Wang, J. Zhang, O. Gharbi, V. Vivier, M. Gao and M. E. Orazem, Nat. Rev. Methods Primers, 2021, 1, 41 CrossRef CAS.
- S. M. Rezaei Niya and M. Hoorfar, J. Power Sources, 2013, 240, 281–293 CrossRef CAS.
- X. Yuan, H. Wang, J. Colin Sun and J. Zhang, Int. J. Hydrogen Energy, 2007, 32, 4365–4380 CrossRef CAS.
- J. J. Giner-Sanz, E. M. Ortega and V. Pérez-Herranz, Fuel Cells, 2016, 16, 469–479 CrossRef CAS.
- F. J. Pinar, M. Rastedt, N. Pilinski and P. Wagner, in High Temperature Polymer Electrolyte Membrane Fuel Cells, Springer International Publishing, Cham, 2016, pp. 353–386 Search PubMed.
- A. Weiß, S. Schindler, S. Galbiati, M. A. Danzer and R. Zeis, Electrochim. Acta, 2017, 230, 391–398 CrossRef.
- A. Baricci and A. Casalegno, Energies, 2023, 16, 4671 CrossRef CAS.
- K. Ruan, L. Yang, H. Sun and G. Sun, J. Power Sources, 2022, 523, 231000 CrossRef CAS.
- T. Gaumont, G. Maranzana, O. Lottin, J. Dillet, S. Didierjean, J. Pauchet and L. Guétaz, Int. J. Hydrogen Energy, 2017, 42, 1800–1812 CrossRef CAS.
- I. Pivac and F. Barbir, J. Power Sources, 2016, 326, 112–119 CrossRef CAS.
- X.-Z. Yuan, C. Song, H. Wang and J. Zhang, Electrochemical Impedance Spectroscopy in PEM Fuel Cells, Springer London, London, 2010 Search PubMed.
- A. Diedrichs, M. Rastedt, F. J. Pinar and P. Wagner, J. Appl. Electrochem., 2013, 43, 1079–1099 CrossRef CAS.
- S. Simon Araya, S. Thomas, A. Lotrič, S. Lennart Sahlin, V. Liso and S. J. Andreasen, Energies, 2021, 14, 2994 CrossRef.
- Q. Meyer, I. Pivac, F. Barbir and C. Zhao, J. Power Sources, 2020, 470, 228285 CrossRef CAS.
- J. Chen, J. J. Bailey, L. Britnell, M. Perez-Page, M. Sahoo, Z. Zhang, A. Strudwick, J. Hack, Z. Guo, Z. Ji, P. Martin, D. J. L. Brett, P. R. Shearing and S. M. Holmes, Nano Energy, 2022, 93, 106829 CrossRef CAS.
- S. Dierickx, A. Weber and E. Ivers-Tiffée, Electrochim. Acta, 2020, 355, 136764 CrossRef CAS.
- M. Heinzmann, A. Weber and E. Ivers-Tiffée, J. Power Sources, 2019, 444, 227279 CrossRef CAS.
- M. Hinaje, S. Raël, P. Noiying, D. A. Nguyen and B. Davat, Energies, 2012, 5, 2724–2744 CrossRef CAS.
- F. Tiss, R. Chouikh and A. Guizani, Int. J. Hydrogen Energy, 2013, 38, 8532–8541 CrossRef CAS.
- M. Heinzmann, A. Weber and E. Ivers-Tiffée, J. Power Sources, 2018, 402, 24–33 CrossRef CAS.
- D. Klotz, A. Weber and E. Ivers-Tiffée, Electrochim. Acta, 2017, 227, 110–126 CrossRef CAS.
- D. Yao, T. C. Jao, W. Zhang, L. Xu, L. Xing, Q. Ma, Q. Xu, H. Li, S. Pasupathi and H. Su, Int. J. Hydrogen Energy, 2018, 43, 21006–21016 CrossRef CAS.
- X. Zhang, T. Zhang, H. Chen and Y. Cao, Appl. Energy, 2021, 286, 116481 CrossRef CAS.
- S. K. Roy, M. E. Orazem and B. Tribollet, J. Electrochem. Soc., 2007, 154, B1378–B1388 CrossRef CAS.
- O. Antoine, Y. Bultel and R. Durand, J. Electroanal. Chem., 2001, 499, 85–94 CrossRef CAS.
- A. Schiefer, M. Heinzmann and A. Weber, Fuel Cells, 2020, 20, 499–506 CrossRef CAS.
- H. Kuhn, A. Wokaun and G. G. Scherer, Electrochim. Acta, 2007, 52, 2322–2327 CrossRef CAS.
- B. P. Setzler and T. F. Fuller, J. Electrochem. Soc., 2015, 162, F519–F530 CrossRef CAS.
- K. Wiezell, P. Gode and G. Lindbergh, J. Electrochem. Soc., 2006, 153, A749–A758 CrossRef CAS.
- N. Holmström, K. Wiezell and G. Lindbergh, J. Electrochem. Soc., 2012, 159, F369–F378 CrossRef.
- I. A. Schneider, M. H. Bayer, A. Wokaun and G. G. Scherer, J. Electrochem. Soc., 2008, 155, B783 CrossRef CAS.
- C. Gerling, M. Hanauer, U. Berner and K. A. Friedrich, J. Electrochem. Soc., 2023, 170, 014504 CrossRef CAS.
- T. V. Reshetenko, K. Bethune and R. Rocheleau, J. Power Sources, 2012, 218, 412–423 CrossRef CAS.
- N. Wagner and M. Schulze, Electrochim. Acta, 2003, 48, 3899–3907 CrossRef CAS.
- T. V. Reshetenko, K. Bethune, M. A. Rubio and R. Rocheleau, J. Power Sources, 2014, 269, 344–362 CrossRef CAS.
- M. Wagner, O. Lorenz, F. P. Lohmann-Richters, Á. Varga and B. Abel, J. Mater. Chem. A, 2021, 9, 11347–11358 RSC.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- F. Ciucci and C. Chen, Electrochim. Acta, 2015, 167, 439–454 CrossRef CAS.
- A. Kulikovsky, J. Phys. Chem. C, 2022, 126, 2424–2429 CrossRef CAS.
- T. Reshetenko and A. Kulikovsky, J. Electrochem. Soc., 2020, 167, 144505 CrossRef CAS.
- N. Bevilacqua, M. A. Schmid and R. Zeis, J. Power Sources, 2020, 471, 228469 CrossRef CAS.
- J. Xu, Y. Wu, S. Xiao, Y. Wang and X. Xu, Renewable Energy, 2023, 211, 669–680 CrossRef CAS.
- Y. Zhang, Y. Chen, M. Yan and F. Chen, J. Power Sources, 2015, 283, 464–477 CrossRef CAS.
- J. Weese, Comput. Phys. Commun., 1992, 69, 99–111 CrossRef.
- E. Ivers-Tiffée and A. Weber, J. Ceram. Assoc. Jpn., 2017, 125, 193–201 CrossRef.
- E. Brightman, G. Hinds and R. O'Malley, J. Power Sources, 2013, 242, 244–254 CrossRef CAS.
- T. Engl, K. E. Waltar, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2014, 161, F500–F505 CrossRef CAS.
- F. Wang, D. Yang, B. Li, H. Zhang, C. Hao, F. Chang and J. Ma, Int. J. Hydrogen Energy, 2014, 39, 14441–14447 CrossRef CAS.
- K. H. Kim, K. Y. Lee, S. Y. Lee, E. Cho, T. H. Lim, H. J. Kim, S. P. Yoon, S. H. Kim, T. W. Lim and J. H. Jang, Int. J. Hydrogen Energy, 2010, 35, 13104–13110 CrossRef CAS.
- H. Niu, C. Ji, S. Wang and C. Liang, Int. J. Hydrogen Energy, 2022, 47, 2662–2672 CrossRef CAS.
- J. Xie, D. L. Wood, D. M. Wayne, T. A. Zawodzinski, P. Atanassov and R. L. Borup, J. Electrochem. Soc., 2005, 152, A104 CrossRef CAS.
- J. Parrondo, C. Venkateswara Rao, S. L. Ghatty and B. Rambabu, Int. J. Electrochem., 2011, 2011, 1–8 CrossRef.
- P. N. Ross and P. C. Andricacos, J. Electroanal. Chem. Interfacial Electrochem., 1983, 154, 205–215 CrossRef CAS.
- E. Zagoraiou, F. Paloukis, S. G. Neophytides and M. K. Daletou, Electrochim. Acta, 2020, 356, 136778 CrossRef CAS.
- S. S. Kocha, J. Deliang Yang and J. S. Yi, AIChE J., 2006, 52, 1916–1925 CrossRef CAS.
- S. Zhang, X.-Z. Yuan and H. Wang, in PEM Fuel Cell Diagnostic Tools, CRC Press, 2011, pp. 87–100 Search PubMed.
- S. R. Choi, D. Y. Kim, W. Y. An, S. Choi, K. Park, S. D. Yim and J. Y. Park, Mater. Sci. Energy Technol., 2022, 5, 66–73 CAS.
- M. Geske, M. Heuer, G. Heideck and Z. A. Styczynski, Energies, 2010, 3, 770–783 CrossRef CAS.
- G. Zhang, S. Shen, L. Guo and H. Liu, Int. J. Hydrogen Energy, 2012, 37, 1884–1892 CrossRef CAS.
- D. Gerteisen, N. Zamel, C. Sadeler, F. Geiger, V. Ludwig and C. Hebling, Int. J. Hydrogen Energy, 2012, 37, 7736–7744 CrossRef CAS.
- Y. Yu, X. Z. Yuan, H. Li, E. Gu, H. Wang, G. Wang and M. Pan, Int. J. Hydrogen Energy, 2012, 37, 15288–15300 CrossRef CAS.
- D. J. L. Brett, S. Atkins, N. P. Brandon, N. Vasileiadis, V. Vesovic and A. R. Kucernak, J. Power Sources, 2007, 172, 2–13 CrossRef CAS.
- D. J. Brett, S. Atkins, N. P. Brandon, V. Vesovic, N. Vasileiadis and A. R. Kucernak, Electrochem. Commun., 2001, 3, 628–632 CrossRef CAS.
- T. Nasu, Y. Matsushita, J. Okano and K. Okajima, J. Int. Counc. Electr. Eng., 2012, 2, 391–396 CrossRef.
- K.-H. Hauer, R. Potthast, T. Wüster and D. Stolten, J. Power Sources, 2005, 143, 67–74 CrossRef CAS.
- J. Dillet, D. Spernjak, A. Lamibrac, G. Maranzana, R. Mukundan, J. Fairweather, S. Didierjean, R. L. Borup and O. Lottin, J. Power Sources, 2014, 250, 68–79 CrossRef CAS.
- V. Lilavivat, S. Shimpalee, J. W. Van Zee, H. Xu and C. K. Mittelsteadt, Electrochim. Acta, 2015, 174, 1253–1260 CrossRef CAS.
- D. Gerteisen, W. Mérida, T. Kurz, P. Lupotto, M. Schwager and C. Hebling, Fuel Cells, 2011, 11, 339–349 CrossRef CAS.
- Q. Zhang, R. Lin, L. Técher and X. Cui, Energy, 2016, 115, 550–560 CrossRef CAS.
- Q. Meyer, K. Ronaszegi, J. B. Robinson, M. Noorkami, O. Curnick, S. Ashton, A. Danelyan, T. Reisch, P. Adcock, R. Kraume, P. R. Shearing and D. J. L. Brett, J. Power Sources, 2015, 297, 315–322 CrossRef CAS.
- C. Siegel, G. Bandlamudi, F. Filusch and A. Heinzel, Fuel Cells, 2011, 11, 489–500 CrossRef CAS.
- T. Sousa, M. Mamlouk, K. Scott and C. M. Rangel, Fuel Cells, 2012, 12, 566–576 CrossRef CAS.
- H. Agarwal, A. U. Thosar, S. D. Bhat and A. K. Lele, J. Power Sources, 2022, 532, 231319 CrossRef CAS.
- D. Úbeda, P. Cañizares, P. Ferreira-Aparicio, A. M. Chaparro, J. Lobato and M. A. Rodrigo, Int. J. Hydrogen Energy, 2016, 41, 20294–20304 CrossRef.
- A. Bergmann, T. Kurz, D. Gerteisen and C. Hebling, in 18th World Hydrogen Energy Conference 2010 - WHEC 2010 Parallel Sessions Book 1: Fuel Cell Basics/Fuel Infrastructures, 2010 Search PubMed.
- K. Yezerska, A. Dushina, F. Liu, M. Rastedt, P. Wagner, A. Dyck and M. Wark, Int. J. Hydrogen Energy, 2019, 44, 18330–18339 CrossRef CAS.
- S. Litster, W. K. Epting, E. A. Wargo, S. R. Kalidindi and E. C. Kumbur, Fuel Cells, 2013, 935–945 CrossRef CAS.
- V. S. Bethapudi, J. Hack, P. Trogadas, J. I. S. Cho, L. Rasha, G. Hinds, P. R. Shearing, D. J. L. Brett and M.-O. Coppens, Energy Convers. Manage., 2019, 202, 112198 CrossRef.
- Q. Meyer, J. Hack, N. Mansor, F. Iacoviello, J. J. Bailey, P. R. Shearing and D. J. L. Brett, Fuel Cells, 2019, 19, 35–42 CrossRef CAS.
- J. Park, H. Oh, H. Park, J. W. Moon, S. J. Lee and S. Y. Jung, Int. J. Energy Res., 2022, 46, 9058–9070 CrossRef CAS.
- J. Hack, T. M. M. Heenan, F. Iacoviello, N. Mansor, Q. Meyer, P. Shearing, N. Brandon and D. J. L. Brett, J. Electrochem. Soc., 2018, 165, F3045–F3052 CrossRef CAS.
- T. M. M. Heenan, C. Tan, J. Hack, D. J. L. Brett and P. R. Shearing, Mater. Today, 2019, 31, 69–85 CrossRef CAS.
- E. Hoppe, H. Janßen, M. Müller and W. Lehnert, J. Power Sources, 2021, 501, 230036 CrossRef CAS.
- F. Javier Pinar, M. Rastedt, N. Pilinski and P. Wagner, Int. J. Hydrogen Energy, 2016, 41, 19463–19474 CrossRef CAS.
- J. J. Bailey, J. Chen, J. Hack, M. Perez-Page, S. M. Holmes, D. J. L. Brett and P. R. Shearing, J. Power Sources, 2021, 509, 230347 CrossRef CAS.
- K. Tang, Q. Meyer, R. White, R. T. Armstrong, P. Mostaghimi, Y. Da Wang, S. Liu, C. Zhao, K. Regenauer-Lieb and P. K. M. Tung, Comput. Chem. Eng., 2022, 161, 107768 CrossRef CAS.
- O. Sekizawa, T. Uruga, N. Ishiguro, H. Matsui, K. Higashi, T. Sakata, Y. Iwasawa and M. Tada, J. Phys.: Conf. Ser., 2017, 849, 012022 CrossRef.
- W. K. Epting, J. Gelb and S. Litster, Adv. Funct. Mater., 2012, 22, 555–560 CrossRef CAS.
- S. Komini Babu, H. T. Chung, P. Zelenay and S. Litster, ACS Appl. Mater. Interfaces, 2016, 8, 32764–32777 CrossRef CAS PubMed.
- A. Pokhrel, M. El Hannach, F. P. Orfino, M. Dutta and E. Kjeang, J. Power Sources, 2016, 329, 330–338 CrossRef CAS.
- A. Mularczyk, Q. Lin, D. Niblett, A. Vasile, M. J. Blunt, V. Niasar, F. Marone, T. J. Schmidt, F. N. Büchi and J. Eller, ACS Appl. Mater. Interfaces, 2021, 13, 34003–34011 CrossRef CAS PubMed.
- M. Sepe, P. Satjaritanun, I. V. Zenyuk, N. Tippayawong and S. Shimpalee, J. Electrochem. Soc., 2021, 168, 074507 CrossRef CAS.
- A. D. Shum, C. P. Liu, W. H. Lim, D. Y. Parkinson and I. V. Zenyuk, Transp. Porous Media, 2022, 144, 715–737 CrossRef CAS.
- Y.-C. Chen, A. Berger, S. De Angelis, T. Schuler, M. Bozzetti, J. Eller, V. Tileli, T. J. Schmidt and F. N. Büchi, ACS Appl. Mater. Interfaces, 2021, 13, 16227–16237 CrossRef CAS PubMed.
- S. Chevalier, N. Ge, J. Lee, M. G. George, H. Liu, P. Shrestha, D. Muirhead, N. Lavielle, B. D. Hatton and A. Bazylak, J. Power Sources, 2017, 352, 281–290 CrossRef CAS.
- S. S. Alrwashdeh, A. M. Al-Falahat, H. Markötter and I. Manke, Case Stud. Chem. Environ. Eng., 2022, 6, 100260 CrossRef CAS.
- A. Kato, S. Kato, S. Yamaguchi, T. Suzuki and Y. Nagai, J. Power Sources, 2022, 521, 230951 CrossRef CAS.
- D. Kulkarni, S. J. Normile, L. G. Connolly and I. V Zenyuk, JPhys Energy, 2020, 2, 044005 CrossRef CAS.
- R. T. White, A. Wu, M. Najm, F. P. Orfino, M. Dutta and E. Kjeang, J. Power Sources, 2017, 350, 94–102 CrossRef CAS.
- R. T. White, S. H. Eberhardt, Y. Singh, T. Haddow, M. Dutta, F. P. Orfino and E. Kjeang, Sci. Rep., 2019, 9, 1–12 CrossRef CAS PubMed.
- J. Hack, L. Rasha, P. L. Cullen, J. J. Bailey, T. P. Neville, P. R. Shearing, N. P. Brandon and D. J. L. Brett, Electrochim. Acta, 2020, 352, 136464 CrossRef CAS.
- D. Ramani, Y. Singh, R. T. White, M. Wegener, F. P. Orfino, M. Dutta and E. Kjeang, Int. J. Hydrogen Energy, 2020, 45, 10089–10103 CrossRef CAS.
- Y. Singh, R. T. White, M. Najm, A. Boswell, F. P. Orfino, M. Dutta and E. Kjeang, J. Electrochem. Soc., 2021, 168, 034521 CrossRef CAS.
- S. H. Eberhardt, F. Marone, M. Stampanoni, F. N. Büchi and T. J. Schmidt, J. Synchrotron Radiat., 2014, 21, 1319–1326 CrossRef CAS PubMed.
- S. Pollastri, M. Bogar, R. Fiala, H. Amenitsch, Y. Yakovlev, A. Lavacchi, G. Aquilanti and V. Matolin, Int. J. Hydrogen Energy, 2022, 47, 8799–8810 CrossRef CAS.
- Y. Cheng, J. Zhang, X. Wu, C. Tang, S. Yang, P. Su, L. Thomsen, F. Zhao, S. Lu, J. Liu and S. P. Jiang, Nano Energy, 2021, 80, 105534 CrossRef CAS.
- Y. Cheng, M. Wang, S. Lu, C. Tang, X. Wu, J.-P. Veder, B. Johannessen, L. Thomsen, J. Zhang, S. Yang, S. Wang and S. P. Jiang, Appl. Catal., B, 2021, 284, 119717 CrossRef CAS.
- Y. Ji, Y. il Cho, Y. Jeon, C. Lee, D.-H. Park and Y.-G. Shul, Appl. Catal., B, 2017, 204, 421–429 CrossRef CAS.
- C. Roth, S. Kaserer, C. Brieger, K. M. Caldwell, J. Melke and D. E. Ramaker, ECS Trans., 2015, 69, 147–156 CrossRef CAS.
- A. S. Leach, J. Hack, M. Amboage, S. Diaz-Moreno, H. Huang, P. L. Cullen, M. Wilding, E. Magliocca, T. S. Miller, C. A. Howard, D. J. L. Brett, P. R. Shearing, P. F. McMillan, A. E. Russell and R. Jervis, J. Phys.: Condens.Matter, 2021, 33, 314002 CrossRef CAS PubMed.
- H.-Y. Park, I. Y. Cha, Y.-H. Chung, M. K. Cho, S. J. Yoo, H.-J. Kim, D. Henkensmeier, J. Y. Kim, S. W. Nam and J. H. Jang, ECS Meet. Abstr., 2015, MA2015-01, p.1855 Search PubMed.
- B. F. Gomes, M. Prokop, T. Bystron, R. Loukrakpam, J. Melke, C. M. S. Lobo, M. Fink, M. Zhu, E. Voloshina, M. Kutter, H. Hoffmann, K. V. Yusenko, A. G. Buzanich, B. Röder, K. Bouzek, B. Paulus and C. Roth, ACS Catal., 2022, 12, 11472–11484 CrossRef CAS.
- Q. He, B. Shyam, M. Nishijima, D. Ramaker and S. Mukerjee, J. Phys. Chem. C, 2013, 117, 4877–4887 CrossRef CAS.
- K. Higashi, S. Takao, G. Samjeské, H. Matsui, M. Tada, T. Uruga and Y. Iwasawa, Phys. Chem. Chem. Phys., 2020, 22, 18919–18931 RSC.
- H. Matsui, N. Ishiguro, Y. Tan, N. Maejima, Y. Muramoto, T. Uruga, K. Higashi, D. Nguyen, H. Dam, G. Samjeské and M. Tada, ChemNanoMat, 2022, 8(4), e202200008 CrossRef CAS.
- H. Matsui, S. Takao, K. Higashi, T. Kaneko, G. Samjeské, T. Uruga, M. Tada and Y. Iwasawa, ACS Appl. Mater. Interfaces, 2022, 14, 6762–6776 CrossRef CAS PubMed.
- T. Uruga, M. Tada, O. Sekizawa, Y. Takagi, T. Yokoyama and Y. Iwasawa, Chem. Rec., 2019, 19, 1444–1456 CrossRef CAS PubMed.
- H. Park, D.-K. Kim, H. Kim, S. Oh, W. S. Jung and S.-K. Kim, Appl. Surf. Sci., 2020, 510, 145444 CrossRef CAS.
- N. Üregen, K. Pehlivanoğlu, Y. Özdemir and Y. Devrim, Int. J. Hydrogen Energy, 2017, 42, 2636–2647 CrossRef.
- I. I. Ponomarev, K. M. Skupov, A. V. Naumkin, V. G. Basu, O. M. Zhigalina, D. Y. Razorenov, I. I. Ponomarev and Y. A. Volkova, RSC Adv., 2019, 9, 257–267 RSC.
- D. Batet, F. T. Zohra, S. B. Kristensen, S. J. Andreasen and L. Diekhöner, Appl. Energy, 2020, 277, 115588 CrossRef CAS.
- T. Ossiander, C. Heinzl, S. Gleich, F. Schönberger, P. Völk, M. Welsch and C. Scheu, J. Membr. Sci., 2014, 454, 12–19 CrossRef CAS.
- I. Martens, R. Chattot, M. Rasola, M. V. Blanco, V. Honkimäki, D. Bizzotto, D. P. Wilkinson and J. Drnec, ACS Appl. Energy Mater., 2019, 2, 7772–7780 CrossRef CAS.
- I. Martens, R. Chattot and J. Drnec, J. Power Sources, 2022, 521, 230851 CrossRef CAS.
- I. Martens, A. Vamvakeros, N. Martinez, R. Chattot, J. Pusa, M. V. Blanco, E. A. Fisher, T. Asset, S. Escribano, F. Micoud, T. Starr, A. Coelho, V. Honkimäki, D. Bizzotto, D. P. Wilkinson, S. D. M. Jacques, F. Maillard, L. Dubau, S. Lyonnard, A. Morin and J. Drnec, ACS Energy Lett., 2021, 6, 2742–2749 CrossRef CAS.
- P. Trtik and E. H. Lehmann, J. Phys.: Conf. Ser., 2016, 746, 012004 CrossRef.
- M. Grosse and N. Kardjilov, Phys. Procedia, 2017, 88, 266–274 CrossRef CAS.
- N. Kardjilov, I. Manke, R. Woracek, A. Hilger and J. Banhart, Mater. Today, 2018, 21, 652–672 CrossRef CAS.
- T. A. Trabold, J. P. Owejan, D. L. Jacobson, M. Arif and P. R. Huffman, Int. J. Heat Mass Transfer, 2006, 49, 4712–4720 CrossRef CAS.
- Y. Wu, J. I. S. Cho, T. P. Neville, Q. Meyer, R. Ziesche, P. Boillat, M. Cochet, P. R. Shearing and D. J. L. Brett, J. Power Sources, 2018, 399, 254–263 CrossRef CAS.
- A. Iranzo and P. Boillat, Int. J. Hydrogen Energy, 2014, 39, 17240–17245 CrossRef CAS.
- N. Kulkarni, J. I. S. Cho, L. Rasha, R. E. Owen, Y. Wu, R. Ziesche, J. Hack, T. Neville, M. Whiteley, N. Kardjilov, H. Markötter, I. Manke, P. R. Shearing and D. J. L. Brett, J. Power Sources, 2019, 439, 227074 CrossRef CAS.
- Q. Meyer, S. Ashton, P. Boillat, M. Cochet, E. Engebretsen, D. P. Finegan, X. Lu, J. J. Bailey, N. Mansor, R. Abdulaziz, O. O. Taiwo, R. Jervis, S. Torija, P. Benson, S. Foster, P. Adcock, P. R. Shearing and D. J. L. Brett, Electrochim. Acta, 2016, 211, 478–487 CrossRef CAS.
- P. Boillat, D. Kramer, B. C. Seyfang, G. Frei, E. Lehmann, G. G. Scherer, A. Wokaun, Y. Ichikawa, Y. Tasaki and K. Shinohara, Electrochem. Commun., 2008, 10, 546–550 CrossRef CAS.
- M. Siegwart, F. Huang, M. Cochet, T. J. Schmidt, J. Zhang and P. Boillat, J. Electrochem. Soc., 2020, 167, 064510 CrossRef CAS.
- J. Biesdorf, P. Oberholzer, F. Bernauer, A. Kaestner, P. Vontobel, E. H. Lehmann, T. J. Schmidt and P. Boillat, Phys. Rev. Lett., 2014, 112, 248301 CrossRef CAS PubMed.
- P. Stahl, J. Biesdorf, P. Boillat and K. A. Friedrich, Fuel Cells, 2017, 17, 778–785 CrossRef CAS.
- P. Boillat, G. G. Scherer, A. Wokaun, G. Frei and E. H. Lehmann, Electrochem. Commun., 2008, 10, 1311–1314 CrossRef CAS.
- I. Manke, C. Hartnig, N. Kardjilov, M. Messerschmidt, A. Hilger, M. Strobl, W. Lehnert and J. Banhart, Appl. Phys. Lett., 2008, 92, 244101 CrossRef.
- T. Arlt, W. Lüke, N. Kardjilov, J. Banhart, W. Lehnert and I. Manke, J. Power Sources, 2015, 299, 125–129 CrossRef CAS.
- Y. Lin, T. Arlt, N. Kardjilov, I. Manke and W. Lehnert, Energies, 2018, 11, 2214 CrossRef.
- P. Boillat, J. Biesdorf, P. Oberholzer, A. Kaestner and T. J. Schmidt, J. Electrochem. Soc., 2014, 161, F192–F198 CrossRef CAS.
- A. Santamaria, H.-Y. Tang, J. W. Park, G.-G. Park and Y.-J. Sohn, Int. J. Hydrogen Energy, 2012, 37, 10836–10843 CrossRef CAS.
- H.-Y. Tang, A. Santamaria, J. Kurniawan, J. W. Park, T.-H. Yang and Y.-J. Sohn, J. Power Sources, 2010, 195, 6774–6781 CrossRef CAS.
- V. Manzi-Orezzoli, A. Mularczyk, P. Trtik, J. Halter, J. Eller, T. J. Schmidt and P. Boillat, ACS Omega, 2019, 4, 17236–17243 CrossRef CAS PubMed.
- S. S. Alrwashdeh, F. M. Alsaraireh, M. A. Saraireh, H. Markötter, N. Kardjilov, M. Klages, J. Scholta and I. Manke, AIMS Energy, 2018, 6, 607–614 CAS.
- R. F. Ziesche, J. Hack, L. Rasha, M. Maier, C. Tan, T. M. M. Heenan, H. Markötter, N. Kardjilov, I. Manke, W. Kockelmann, D. J. L. Brett and P. R. Shearing, Nat. Commun., 2022, 13, 1616 CrossRef CAS PubMed.
- N. Martinez, A. Morin, Q. Berrod, B. Frick, J. Ollivier, L. Porcar, G. Gebel and S. Lyonnard, J. Phys. Chem. C, 2018, 122, 1103–1108 CrossRef CAS.
- B. Hopfenmüller, R. Zorn, O. Holderer, O. Ivanova, W. Lehnert, W. Lüke, G. Ehlers, N. Jalarvo, G. J. Schneider, M. Monkenbusch and D. Richter, J. Chem. Phys., 2018, 148(20), 204906 CrossRef PubMed.
- J. Malugani, M. Tachez, R. Mercier, A. Dianoux and P. Chieux, Solid State Ionics, 1987, 23, 189–196 CrossRef CAS.
- M. Kruteva, Adsorption, 2021, 27, 875–889 CrossRef CAS.
- A. Putra, H. Iwase, D. Yamaguchi, S. Koizumi, Y. Maekawa, M. Matsubayashi and T. Hashimoto, J. Phys.: Conf. Ser., 2010, 247, 012044 CrossRef.
- S. Koizumi, S. Ueda, T. Inada, Y. Noda and R. A. Robinson, J. Appl. Crystallogr., 2019, 52, 791–799 CrossRef CAS.
- T. Kusano, T. Hiroi, K. Amemiya, M. Ando, T. Takahashi and M. Shibayama, Polym. J., 2015, 47, 546–555 CrossRef CAS.
- W. Yoshimune and M. Harada, Chem. Lett., 2019, 48, 487–490 CrossRef CAS.
- M. Harada, S. Takata, H. Iwase, S. Kajiya, H. Kadoura and T. Kanaya, ACS Omega, 2021, 6, 15257–15263 CrossRef CAS PubMed.
- O. Holderer, M. Carmo, M. Shviro, W. Lehnert, Y. Noda, S. Koizumi, M.-S. Appavou, M. Appel and H. Frielinghaus, Materials, 2020, 13, 1474 CrossRef CAS PubMed.
- O. Holderer, M. Khaneft, Y. Lin, S. Liu, A. Feoktystov, M. Kruteva, R. Zorn and W. Lehnert, ECS Trans., 2017, 80, 19–25 CrossRef CAS.
- K. Ito, T. Yamada, A. Shinohara, S. Takata and Y. Kawakita, J. Phys. Chem. C, 2021, 125, 21645–21652 CrossRef CAS.
- M. Khaneft, O. Holderer, O. Ivanova, W. Lüke, E. Kentzinger, M. S. Appavou, R. Zorn and W. Lehnert, Fuel Cells, 2016, 16, 406–413 CrossRef CAS.
- O. Holderer, O. Ivanova, B. Hopfenmüller, M. Zamponi, W. Maier, A. Majerus, W. Lehnert, M. Monkenbusch and R. Zorn, Int. J. Hydrogen Energy, 2014, 39, 21657–21662 CrossRef CAS.
- B. Frick, L. Vilčiauskas, P. P. Deen and S. Lyonnard, Solid State Ionics, 2013, 252, 26–33 CrossRef CAS.
- D. J. L. Brett, A. R. Kucernak, P. Aguiar, S. C. Atkins, N. P. Brandon, R. Clague, L. F. Cohen, G. Hinds, C. Kalyvas, G. J. Offer, B. Ladewig, R. Maher, A. Marquis, P. Shearing, N. Vasileiadis and V. Vesovic, ChemPhysChem, 2010, 11, 2714–2731 CrossRef CAS PubMed.
- E. Öner, A. Öztürk and A. B. Yurtcan, Int. J. Hydrogen Energy, 2020, 45, 34818–34836 CrossRef.
- W. Zhang, J. Chen, G. F. Swiegers, Z.-F. Ma and G. G. Wallace, Nanoscale, 2010, 2, 282–286 RSC.
- F. Charreteur, F. Jaouen, S. Ruggeri and J.-P. Dodelet, Electrochim. Acta, 2008, 53, 2925–2938 CrossRef CAS.
- P. Gode, J. Ihonen, A. Strandroth, H. Ericson, G. Lindbergh, M. Paronen, F. Sundholm, G. Sundholm and N. Walsby, Fuel Cells, 2003, 3, 21–27 CrossRef CAS.
- M. Holber, P. Johansson and P. Jacobsson, Fuel Cells, 2011, 11, 459–464 CrossRef CAS.
- Z. Peng, P. Huguet, S. Deabate, A. Morin and A. K. Sutor, J. Raman Spectrosc., 2013, 44, 321–328 CrossRef CAS.
- S. Deabate, P. Huguet, A. Morin, G. Gebel, Y. Lanteri, Z. Peng and A.-K. Sutor, Fuel Cells, 2014, 14, 677–693 CrossRef CAS.
- H. Nishiyama, S. Takamuku, K. Oshikawa, S. Lacher, A. Iiyama and J. Inukai, J. Phys. Chem. C, 2020, 124, 9703–9711 CrossRef CAS.
- V. G. Zhigalina, O. M. Zhigalina, I. I. Ponomarev, K. M. Skupov, D. Y. Razorenov, I. I. Ponomarev, N. A. Kiselev and G. Leitinger, CrystEngComm, 2017, 19, 3792–3800 RSC.
- R. Wang, Z. Chang, Z. Fang, T. Xiao, Z. Zhu, B. Ye, C. Xu and J. Cheng, Int. J. Hydrogen Energy, 2020, 45, 28190–28195 CrossRef CAS.
- P. Ghosh, S. Mandal, S. Majumdar, A. Sarkar, S. Ganguly and K. Kargupta, Int. J. Hydrogen Energy, 2020, 45, 16708–16723 CrossRef CAS.
- F. Mack, S. Heissler, R. Laukenmann and R. Zeis, J. Power Sources, 2014, 270, 627–633 CrossRef CAS.
- A. Haug, H. Renate, M. Schulze, G. Schiller and K. A. Friedrich, in PEM Fuel Cell Diagnostic Tools, CRC Press, 2011, pp. 389–400 Search PubMed.
- G. A. Giffin, F. Conti, S. Lavina, A. Majerus, G. Pace, C. Korte, W. Lehnert and V. Di Noto, Int. J. Hydrogen Energy, 2014, 39, 2776–2784 CrossRef CAS.
- M. Wilhelm, M. Jeske, R. Marschall, W. L. Cavalcanti, P. Tölle, C. Köhler, D. Koch, T. Frauenheim, G. Grathwohl, J. Caro and M. Wark, J. Membr. Sci., 2008, 316, 164–175 CrossRef CAS.
- M. S. M. Yusof, A. A. Jalil, A. Ahmad, S. Triwahyono, M. H. D. Othman, T. A. T. Abdullah, M. L. Firmansyah, H. D. Setiabudi, A. Johari and W. Nabgan, Int. J. Hydrogen Energy, 2019, 44, 20760–20769 CrossRef CAS.
- F. C. Nart and T. Iwasita, Electrochim. Acta, 1992, 37, 385–391 CrossRef CAS.
- G. Neophytides, L. Quaroni, F. N. Büchi, A. Orfanidi, S. G. Neophytides and T. J. Schmidt, Electrochem. Commun., 2013, 34, 200–203 CrossRef CAS.
- E. Yli-Rantala, A. Pasanen, P. Kauranen, V. Ruiz, M. Borghei, E. Kauppinen, A. Oyarce, G. Lindbergh, C. Lagergren, M. Darab, S. Sunde, M. Thomassen, S. Ma-Andersen and E. Skou, Fuel Cells, 2011, 11, 715–725 CrossRef CAS.
- V. Bandlamudi, P. Bujlo, C. Sita and S. Pasupathi, Mater. Today: Proc., 2018, 5, 10602–10610 CAS.
- K. Tüber, D. Pócza and C. Hebling, J. Power Sources, 2003, 124, 403–414 CrossRef.
- A. Hakenjos, H. Muenter, U. Wittstadt and C. Hebling, J. Power Sources, 2004, 131, 213–216 CrossRef CAS.
- X. G. Yang, F. Y. Zhang, A. L. Lubawy and C. Y. Wang, Electrochem. Solid-State Lett., 2004, 7, A408 CrossRef CAS.
- X. Liu, H. Guo and C. Ma, J. Power Sources, 2006, 156, 267–280 CrossRef CAS.
- M. Rahimi-Esbo, A. Ramiar, A. A. Ranjbar and E. Alizadeh, Int. J. Hydrogen Energy, 2017, 42, 11673–11688 CrossRef CAS.
- M. I. Rosli, D. J. Borman, D. B. Ingham, M. S. Ismail, L. Ma and M. Pourkashanian, J. Fuel Cell Sci. Technol., 2010, 7, 1–7 CrossRef.
- C.-Y. Hsu and F.-B. Weng, in PEM Fuel Cell Diagnostic Tools, CRC Press, 2011, pp. 209–227 Search PubMed.
- N. A. David, P. M. Wild, J. Jensen, T. Navessin and N. Djilali, J. Electrochem. Soc., 2010, 157, B1173 CrossRef CAS.
- N. David, K. Von Schilling, P. M. Wild and N. Djilali, Int. J. Hydrogen Energy, 2014, 39, 17638–17644 CrossRef CAS.
- Y. P. Patil, T. A. P. Seery, M. T. Shaw and R. S. Parnas, Ind. Eng. Chem. Res., 2005, 44, 6141–6147 CrossRef CAS.
- V. Prabhakaran, C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1029–1034 CrossRef CAS PubMed.
- J. F. Botero-Cadavid, P. Wild and N. Djilali, Electrochim. Acta, 2014, 129, 416–424 CrossRef CAS.
- C. Y. Lee, Y. C. Chiang, F. B. Weng, S. C. Li, P. H. Wu and H. I. Yueh, Renewable Energy, 2017, 108, 126–131 CrossRef.
- S. Wu, W. Cheng, Y. Qiu, Z. Li, S. Shuang and C. Dong, Sens. Actuators, B, 2010, 144, 255–259 CrossRef CAS.
- O. Belhadj Miled, D. Grosso, C. Sanchez and J. Livage, J. Phys. Chem. Solids, 2004, 65, 1751–1755 CrossRef CAS.
- J. Robinson, P. Shearing and D. Brett, J. Imaging, 2016, 2, 2 CrossRef.
- L. Dubau, L. Castanheira, M. Chatenet, F. Maillard, J. Dillet, G. Maranzana, S. Abbou, O. Lottin, G. De Moor, A. El Kaddouri, C. Bas, L. Flandin, E. Rossinot and N. Caqué, Int. J. Hydrogen Energy, 2014, 39, 21902–21914 CrossRef CAS.
- R. Shimoi, M. Masuda, K. Fushinobu, Y. Kozawa and K. Okazaki, J. Energy Resour. Technol., 2004, 126, 258–261 CrossRef CAS.
- A. Hakenjos and C. Hebling, J. Power Sources, 2005, 145, 307–311 CrossRef CAS.
- H. Guo, M. H. Wang, J. X. Liu, Z. H. Nie, F. Ye and C. F. Ma, J. Power Sources, 2015, 273, 775–783 CrossRef CAS.
- M. M. Daino, Z. Lu, J. M. LaManna, J. P. Owejan, T. A. Trabold and S. G. Kandlikar, Electrochem. Solid-State Lett., 2011, 14, B51 CrossRef CAS.
- J. Scholta, M. Messerschmidt, L. Jörissen and C. Hartnig, J. Power Sources, 2009, 190, 83–85 CrossRef CAS.
- H. Becker, T. Bacquart, M. Perkins, N. Moore, J. Ihonen, G. Hinds and G. Smith, J. Power Sources Adv., 2020, 6, 100036 CrossRef.
- L. C. Pérez, P. Koski, J. Ihonen, J. M. Sousa and A. Mendes, J. Power Sources, 2014, 258, 122–128 CrossRef.
- J. St-Pierre, J. Ge, Y. Zhai, T. V. Reshetenko and M. Angelo, ECS Trans., 2013, 58, 519–528 CrossRef.
- D. Liang, Q. Shen, M. Hou, Z. Shao and B. Yi, J. Power Sources, 2009, 194, 847–853 CrossRef CAS.
- M. M. Mench, C. Y. Wang and M. Ishikawa, J. Electrochem. Soc., 2003, 150, A1052 CrossRef CAS.
- J. Zhang, H. Zhang, J. Wu and J. Zhang, Pem Fuel Cell Test Diagnosis, 2013, pp. 171–185s Search PubMed.
- F. Liu, B. Yi, D. Xing, J. Yu and H. Zhang, J. Membr. Sci., 2003, 212, 213–223 CrossRef CAS.
- E. Endoh, S. Terazono, H. Widjaja and Y. Takimoto, Electrochem. Solid-State Lett., 2004, 7, A209 CrossRef CAS.
- K. Teranishi, K. Kawata, S. Tsushima and S. Hirai, Electrochem. Solid-State Lett., 2006, 9, A475 CrossRef CAS.
- J. Wu, X.-Z. Yuan, J. J. Martin, H. Wang, D. Yang, J. Qiao and J. Ma, J. Power Sources, 2010, 195, 1171–1176 CrossRef CAS.
- C. Noack, J. Kallo and A. K. Friedrich, J. Appl. Electrochem., 2012, 42, 833–841 CrossRef CAS.
- F. Zhou, S. J. Andreasen, S. K. Kær and D. Yu, Int. J. Hydrogen Energy, 2015, 40, 2833–2839 CrossRef CAS.
- E. Jannelli, M. Minutillo and A. Perna, Appl. Energy, 2013, 108, 82–91 CrossRef CAS.
- A. Jo, K. Oh, J. Lee, D. Han, D. Kim, J. Kim, B. Kim, J. Kim, D. Park, M. Kim, Y.-J. Sohn, D. Kim, H. Kim and H. Ju, Int. J. Hydrogen Energy, 2017, 42, 1698–1714 CrossRef CAS.
- N. Pilinski and P. Wagner, in 4th PEFC and H2 Forum B1108, 2013 Search PubMed.
- S. Liu, K. Wippermann and W. Lehnert, Int. J. Hydrogen Energy, 2021, 46, 14687–14698 CrossRef CAS.
- D. Schonvogel, J. Büsselmann, H. Schmies, H. Langnickel, P. Wagner and A. Dyck, Int. J. Hydrogen Energy, 2021, 46, 33934–33947 CrossRef CAS.
- S. T. Ali, Q. Li, C. Pan, J. O. Jensen, L. P. Nielsen and P. Møller, Int. J. Hydrogen Energy, 2011, 36, 1628–1636 CrossRef CAS.
- X. Z. Yuan, S. Zhang, S. Ban, C. Huang, H. Wang, V. Singara, M. Fowler, M. Schulze, A. Haug, K. Andreas Friedrich and R. Hiesgen, J. Power Sources, 2012, 205, 324–334 CrossRef CAS.
- L. Luo, B. Huang, Z. Cheng and Q. Jian, Int. J. Energy Res., 2020, 44, 4784–4799 CrossRef.
- Y. Zhai, K. Bethune, G. Bender and R. Rocheleau, J. Electrochem. Soc., 2012, 159, B524–B530 CrossRef CAS.
- P. Marocco, K. Sundseth, T. Aarhaug, A. Lanzini, M. Santarelli, A. O. Barnett and M. Thomassen, J. Power Sources, 2021, 483, 1–38 CrossRef.
- H. Yu, J. M. Roller, S. Kim, Y. Wang, D. Kwak and R. Maric, J. Electrochem. Soc., 2014, 161, F622–F627 CrossRef CAS.
- S. Liu, M. Rasinski, Y. Lin, K. Wippermann, A. Everwand and W. Lehnert, Electrochim. Acta, 2018, 289, 354–362 CrossRef CAS.
- K. Hengge, C. Heinzl, M. Perchthaler, D. Varley, T. Lochner and C. Scheu, J. Power Sources, 2017, 364, 437–448 CrossRef CAS.
- M. Prokop, M. Vesely, P. Capek, M. Paidar and K. Bouzek, Electrochim. Acta, 2022, 413, 140133 CrossRef CAS.
- M. Prokop, P. Capek, M. Vesely, M. Paidar and K. Bouzek, Electrochim. Acta, 2022, 413, 140121 CrossRef CAS.
- X. Liu, K. Park, M. So, S. Ishikawa, T. Terao, K. Shinohara, C. Komori, N. Kimura, G. Inoue and Y. Tsuge, J. Power Sources Adv., 2022, 14, 100084 CrossRef CAS.
- K. Sasiwimonrit and W.-C. Chang, Int. J. Hydrogen Energy, 2020, 45, 14491–14499 CrossRef CAS.
- F. Seland, T. Berning, B. Børresen and R. Tunold, J. Power Sources, 2006, 160, 27–36 CrossRef CAS.
- H. Su, C. Felix, O. Barron, P. Bujlo, B. J. Bladergroen, B. G. Pollet and S. Pasupathi, Electrocatalysis, 2014, 5, 361–371 CrossRef CAS.
- H. Su, Q. Xu, J. Chong, H. Li, C. Sita and S. Pasupathi, J. Power Sources, 2017, 341, 302–308 CrossRef CAS.
- D. Yao, W. Zhang, Q. Ma, Q. Xu, S. Pasupathi and H. Su, J. Power Sources, 2019, 426, 124–133 CrossRef CAS.
- M.-J. Choo, K.-H. Oh, H.-T. Kim and J.-K. Park, ChemSusChem, 2014, 7, 2335–2341 CrossRef CAS PubMed.
- W. Xiao, Z. Xia, H. Li, R. Sun, S. Wang and G. Sun, Energy Technol., 2020, 8, 2000085 CrossRef CAS.
- Z. Jabbari, B. Nassernejad, N. Fallah, M. Javanbakht and N. Afsham, Surf. Eng., 2019, 35, 1013–1020 CrossRef CAS.
- Y. Özdemir, N. Özkan and Y. Devrim, Electrochim. Acta, 2017, 245, 1–13 CrossRef.
- A. Orfanidi, M. K. Daletou and S. G. Neophytides, Appl. Catal., B, 2011, 106, 379–389 CrossRef CAS.
- A. Taniguchi, T. Akita, K. Yasuda and Y. Miyazaki, Int. J. Hydrogen Energy, 2008, 33, 2323–2329 CrossRef CAS.
- A. Taniguchi, T. Akita, K. Yasuda and Y. Miyazaki, J. Power Sources, 2004, 130, 42–49 CrossRef CAS.
- Y. Kiros, Int. J. Electrochem. Sci., 2007, 2, 285–300 CrossRef CAS.
- K. Wikander, H. Ekström, A. E. C. Palmqvist and G. Lindbergh, Electrochim. Acta, 2007, 52, 6848–6855 CrossRef CAS.
- L. Yan, Y. Hu, X. Zhang and B. Yue, Annu. Rep. NMR Spectrosc., 2016, 88, 149–213 CrossRef CAS.
- S. Suarez and S. Greenbaum, Chem. Rec., 2010, 10, 377–393 CrossRef CAS PubMed.
- M. Robert, A. El Kaddouri, J.-C. Perrin, S. Leclerc and O. Lottin, J. Electrochem. Soc., 2018, 165, F3209–F3216 CrossRef CAS.
- J. Healy, C. Hayden, T. Xie, K. Olson, R. Waldo, M. Brundage, H. Gasteiger and J. Abbott, Fuel Cells, 2005, 5, 302–308 CrossRef CAS.
- C. Simari, G. Potsi, A. Policicchio, I. Perrotta and I. Nicotera, J. Phys. Chem. C, 2016, 120, 2574–2584 CrossRef CAS.
- F. Xu, S. Leclerc, D. Stemmelen, J.-C. Perrin, A. Retournard and D. Canet, J. Membr. Sci., 2017, 536, 116–122 CrossRef CAS.
- E. Galitskaya, A. F. Privalov, M. Weigler, M. Vogel, A. Kashin, M. Ryzhkin and V. Sinitsyn, J. Membr. Sci., 2020, 596, 117691 CrossRef CAS.
- S. Tsushima, T. Ikeda, T. Koido and S. Hirai, J. Electrochem. Soc., 2010, 157, B1814 CrossRef CAS.
- T. Kotaka, S. Tsushima and S. Hirai, ECS Trans., 2007, 11, 445–450 CrossRef CAS.
- J. Bedet, G. Maranzana, S. Leclerc, O. Lottin, C. Moyne, D. Stemmelen, P. Mutzenhardt and D. Canet, Int. J. Hydrogen Energy, 2008, 33, 3146–3149 CrossRef CAS.
- K. Ogawa, Y. Yokouchi, T. Haishi and K. Ito, J. Magn. Reson., 2013, 234, 147–153 CrossRef CAS PubMed.
- Z. Zhang and B. Balcom, in PEM Fuel Cell Diagnostic Tools, CRC Press, 2011, pp. 229–254 Search PubMed.
- Y. Liu, Y. Gu and X. Yu, Quant. Imaging Med. Surg., 2017, 7, 707–716 CrossRef PubMed.
- R. Brown, O. Khegai and P. Parasoglou, Sci. Rep., 2016, 6, 30568 CrossRef CAS PubMed.
- U.S. Department of Energy, 3.4 Fuel Cells - Multi-Year Research, Development, and Demonstration Plan, 2016 Search PubMed.
- The European Commission, 2021.
- A. Bisello, E. Colombo, A. Baricci, C. Rabissi, L. Guetaz, P. Gazdzicki and A. Casalegno, J. Electrochem. Soc., 2021, 168, 054501 CrossRef CAS.
- Y. Nosaka, K. Ohtaka, N. Ohguri and A. Y. Nosaka, J. Electrochem. Soc., 2011, 158, B430 CrossRef CAS.
- F. D. Coms, ECS Trans., 2008, 16, 235–255 CrossRef CAS.
- K. H. Wong and E. Kjeang, J. Electrochem. Soc., 2014, 161, F823–F832 CrossRef CAS.
- M. Inaba, T. Kinumoto, M. Kiriake, R. Umebayashi, A. Tasaka and Z. Ogumi, Electrochim. Acta, 2006, 51, 5746–5753 CrossRef CAS.
- J. W. Frisk, M. T. Hicks, R. T. Atanasoski, W. M. Boand, A. K. Schmoeckel and M. J. Kurkowski, in 2004 Fuel Cell Seminar, San Antonio, TX, November, 2004 Search PubMed.
- H. Liu, M. G. George, R. Banerjee, N. Ge, J. Lee, D. Muirhead, P. Shrestha, S. Chevalier, J. Hinebaugh, R. Zeis, M. Messerschmidt, J. Scholta and A. Bazylak, J. Electrochem. Soc., 2017, 164, F704–F713 CrossRef CAS.
- T. Arlt, M. Klages, M. Messerschmidt, J. Scholta and I. Manke, Energy, 2017, 118, 502–511 CrossRef CAS.
- R. Mukundan, J. R. Davey, K. Rau, D. A. Langlois, D. Spernjak, J. D. Fairweather, K. Artyushkova, R. Schweiss and R. L. Borup, ECS Trans., 2013, 58, 919–926 CrossRef.
- G. Athanasaki, N. Chauhan, R. Ahmad and A. M. Kannan, Int. J. Hydrogen Energy, 2021, 46, 31754–31763 CrossRef CAS.
- V. Stanic, ECS Proc, 2004, 21, 391–401 Search PubMed.
- A. Jung, I. M. Kong, C. Y. Yun and M. S. Kim, J. Membr. Sci., 2017, 523, 138–143 CrossRef CAS.
- R. Lin, E. Gülzow, M. Schulze and K. A. Friedrich, J. Electrochem. Soc., 2011, 158, B11 CrossRef CAS.
- C. Lim, L. Ghassemzadeh, F. Van Hove, M. Lauritzen, J. Kolodziej, G. G. Wang, S. Holdcroft and E. Kjeang, J. Power Sources, 2014, 257, 102–110 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, J. Electrochem. Soc., 2014, 161, E3311–E3322 CrossRef CAS.
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS.
- A. Pandy, Z. Yang, M. Gummalla, V. V. Atrazhev, N. Y. Kuzminyh, V. I. Sultanov and S. Burlatsky, J. Electrochem. Soc., 2013, 160, F972–F979 CrossRef CAS.
- H. Schulenburg, B. Schwanitz, N. Linse, G. G. Scherer, A. Wokaun, J. Krbanjevic, R. Grothausmann and I. Manke, J. Phys. Chem. C, 2011, 115, 14236–14243 CrossRef CAS.
- N. Macauley, D. D. Papadias, J. Fairweather, D. Spernjak, D. Langlois, R. Ahluwalia, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3148–F3160 CrossRef CAS.
- J. C. Meier, C. Galeano, I. Katsounaros, J. Witte, H. J. Bongard, A. A. Topalov, C. Baldizzone, S. Mezzavilla, F. Schüth and K. J. J. Mayrhofer, Beilstein J. Nanotechnol., 2014, 5, 44–67 CrossRef PubMed.
- Q. Meyer, Y. Zeng and C. Zhao, J. Power Sources, 2019, 437, 226922 CrossRef CAS.
- D. J. S. Sandbeck, N. M. Secher, F. D. Speck, J. E. Sørensen, J. Kibsgaard, I. Chorkendorff and S. Cherevko, ACS Catal., 2020, 10, 6281–6290 CrossRef CAS.
- P. Podleschny, U. Rost, R. Muntean, G. Marginean, A. Heinzel, V. Peinecke, I. Radev, M. Muhler and M. Brodmann, Fuel Cells, 2018, 18, 586–593 CrossRef CAS.
- A. Kumar and V. Ramani, ACS Catal., 2014, 4, 1516–1525 CrossRef CAS.
- S. Thomas, C. Jeppesen, T. Steenberg, S. S. Araya, J. R. Vang and S. K. Kær, Int. J. Hydrogen Energy, 2017, 42, 27230–27240 CrossRef CAS.
- Z. Guo, J. Chen, J. J. Byun, M. Perez-Page, Z. Ji, Z. Zhao and S. M. Holmes, J. Membr. Sci., 2022, 641, 119868 CrossRef CAS.
- N. Pilinski, M. Rastedt and P. Wagner, ECS Trans., 2015, 69, 323–335 CrossRef CAS.
- F. J. Pinar, M. Rastedt, N. Bruns and P. Wagner, EFC 2013 - Proc. 5th Eur. Fuel Cell Piero Lunghi Conf., 2013, pp. 19–20 Search PubMed.
- V. Bandlamudi, P. Bujlo, V. Linkov and S. Pasupathi, Fuel Cells, 2019, 19, 231–243 CrossRef CAS.
- S. Stariha, N. Macauley, B. T. Sneed, D. Langlois, K. L. More, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018, 165, F492–F501 CrossRef CAS.
- C. H. Paik, G. S. Saloka and G. W. Graham, Electrochem. Solid-State Lett., 2007, 10, B39 CrossRef CAS.
- G. S. Harzer, J. N. Schwämmlein, A. M. Damjanović, S. Ghosh and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F3118–F3131 CrossRef CAS.
- J. O. Leader, Y. Yue, M. R. Walluk and T. A. Trabold, Int. J. Hydrogen Energy, 2022, 47, 18820–18830 CrossRef CAS.
- E. Colombo, A. Bisello, A. Casalegno and A. Baricci, J. Electrochem. Soc., 2021, 168, 054508 CrossRef CAS.
- J. Park, L. Wang, S. G. Advani and A. K. Prasad, Electrochim. Acta, 2014, 120, 30–38 CrossRef CAS.
- P. Ren, P. Pei, Y. Li, Z. Wu, D. Chen and S. Huang, Prog. Energy Combust. Sci., 2020, 80, 100859 CrossRef.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef CAS.
- N. Garland, T. Benjamin and J. Kopasz, ECS Trans., 2007, 11, 923–931 CrossRef CAS.
- R. Mukundan, A. M. Baker, A. Kusoglu, P. Beattie, S. Knights, A. Z. Weber and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3085–F3093 CrossRef CAS.
- J. Kerres and V. Atanasov, Int. J. Hydrogen Energy, 2015, 40, 14723–14735 CrossRef CAS.
- V. Weissbecker, K. Wippermann and W. Lehnert, J. Electrochem. Soc., 2014, 161, F1437–F1447 CrossRef CAS.
- L. Tian, W. Zhang, Z. Xie, K. Peng, Q. Ma, Q. Xu, S. Pasupathi and H. Su, Acta Phys.-Chim. Sin., 2020, 37, 2009049 Search PubMed.
- G. Bin Jung, H. H. Chen and W. M. Yan, J. Power Sources, 2014, 247, 354–359 CrossRef.
- M. Tomas, P. Novotny, F. Gholami, O. Tucek and F. Marsik, Environ. Clim. Technol., 2020, 24, 529–544 CrossRef CAS.
- P. Moçotéguy, B. Ludwig, J. Scholta, Y. Nedellec, D. J. Jones and J. Rozière, Fuel Cells, 2010, 10, 299–311 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |