
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLe Chatelier's principle enables stable and sustainable aqueous sodium/magnesium-ion batteries†
Martin
Karlsmo
 a,
Tomooki
Hosaka
ab and
Patrik
Johansson
*ac
a,
Tomooki
Hosaka
ab and
Patrik
Johansson
*ac
aDepartment of Physics, Chalmers University of Technology, 41296 Göteborg, Sweden. E-mail: patrik.johansson@chalmers.se
bDepartment of Applied Chemistry, Tokyo University of Science, Shinjuku, 1628601 Tokyo, Japan
cALISTORE-European Research Institute, CNRS FR 3104, Hub de I'Energie, Rue Baudelocque, 80039 Amiens, France
First published on 11th January 2024
Abstract
Prussian blue analogue (PBA) based aqueous batteries assembled with organic materials are an up-and-coming and promising technology for less demanding applications. By avoiding scarce, costly, and toxic transition metals (e.g. Ni/Co/Cu), the technology may become low-cost, more environmentally benign, and also safer than today's alternatives. Here we rely on a PBA using the FeII/III redox pair together with aqueous low-to-medium concentrated fluorine/perchlorate-free electrolytes and organic materials to create long-term performant cells. The performance in terms of capacity, coulombic efficiency, cell voltage, and energy density are all comparable with previously reported aqueous PBA-based batteries, while the cycling performance is substantially improved by practically implementing Le Chatelier's principle. Additionally, we investigate the redox process(es) and find no proof for any proton storage, but that both Na+ and Mg2+ likely are active, why we classify it as an aqueous Na/Mg-ion battery.
Introduction
Large scale stationary energy storage and less demanding, non-premium electrical devices, e.g. Internet-of-Things, are applications very suitable for rechargeable batteries as they typically are energy efficient, versatile, and have a simple maintenance.1,2 Unlike smartphones and electric vehicles, however, these applications do not require high-performant lithium-ion batteries (LIBs) since the per-weight stored amount of energy is not necessarily the most crucial factor. Instead, the long-term cycling stability, cost, environmental impact, and total energy throughput are the more sought after metrics, to comply with the transition to a more sustainable energy infrastructure.3,4 In this regard, aqueous batteries based on organic materials and Fe/Mn redox centres are an attractive alternative, as they would provide a safer and more sustainable energy storage chemistry.5 These batteries are made without any scarce, hazardous, and intrinsically costly transition metals, natural graphite anodes, metal current collectors, flammable organic solvents, and fluorinated salts, all of which comprise traditional LIBs. Instead, the energy storage/transfer is facilitated by water-based electrolytes of simple inorganic salts, cellulose-based binders and separators, and carbon-based current collectors.Applying aqueous electrolytes, however, does come with inherent challenges.6 The arguably great solvent water dissolves many electrode active materials, leading to poor cycling stability, and furthermore limits the nominal battery cell voltage to the water splitting reaction. Material dissolution is particularly evident for Prussian blue analogues (PBAs) (AxM1[M2(CN)6]y·nH2O, where A is a mobile metal and M1 and M2 are transition metals), otherwise featuring robust three-dimensional open frameworks and being a most popular cathode active material choice for aqueous batteries.7 One of the more environmentally benign PBAs, NaxMn[Fe(CN)6]y·nH2O (Mn-PBA), which shows higher redox potential and energy density than NaxFe[Fe(CN)6]y·nH2O, suffers from this issue, and it has generally been attributed to Mn dissolution where Jahn–Teller distortion is thought to deform the structure, and the disproportionation reaction of Mn3+ results in Mn2+ dissolution into the electrolyte.8,9 Here, ‘water-in-salt’ electrolytes (WISEs) have been a common remedy, by limiting the concentration of free water molecules available by pushing the electrolytes to their saturation limit.10 It is an interesting path by itself, but these electrolytes will most likely remain academic curiosities as they most often are based on costly, unsafe, and environmentally unfriendly fluorinated/perchlorate salts11,12 – defeating the very purpose of aqueous batteries. There are only a few examples of low-cost and green WISEs or water-in-bisalts, introduced by Han et al. and Lukatskaya et. al.13,14 Still, they are no viable options for Mn-PBA as the alkaline environment caused by the highly soluble Ac− anion induces instability, decomposing Mn-PBA to ferric hydroxide resulting in severe capacity fading. Other possible remedies could potentially be gelifying agents15 or co-solvents,16 although the liquid and aqueous nature of the electrolyte then comes into question.
Herein, active and passive materials aligned with the green ethos are combined to create an aqueous sodium/magnesium-ion battery based on the redox couple perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA)|Mn-PBA. By now tailoring our ‘hybrid electrolyte’, developed by some of us from a Na2SO4(aq) electrolyte, by adding Mg2+ to suppress the PTCDA dissolution and enable stable cycling,17 into a ‘triple electrolyte’, this by now also adding Mn2+ to change the Mn-PBA dissolution equilibrium, we do overcome the main challenge of using Mn-PBA in aqueous media, namely that of loss of Mn from the active material, in a simple and effective manner. We choose PTCDA rather than the more commonly used NaTi2(PO4)3 (NTP),18 due to its commercial availability, cycling stability, and high power performance.19 We start by studying the active charge carrier(s) and the electrode cycling reversibility by basic electrochemical and ex situ material characterization methods, respectively, followed by electrochemical characterization of Mn-PBA in half-cells with the hybrid and triple electrolytes. Finally, we assemble full cells of PTCDA|Mn-PBA and evaluate their functionality.
Experimental
Electrolyte preparation
Sodium sulfate (Na2SO4) (anhydrous, Redi-Dri™, free-flowing, ACS reagent, ≥99%), magnesium sulfate (MgSO4) (anhydrous, free-flowing, Redi-Dri™, ReagentPlus®, ≥99.5%), and manganese(II) sulfate monohydrate (MnSO4·H2O) (ReagentPlus®, ≥99%), all purchased from Sigma-Aldrich, were used to create the electrolytes. The hybrid electrolyte (1.9 m Na2SO4 + 2.4 m MgSO4),17,19 and the triple electrolyte (1.9 m Na2SO4 + 2.4 m MgSO4 + 0.3 m MnSO4) were both prepared by first dissolving Na2SO4 to saturation in ultra-pure water (Millipore® Direct-Q® Purification, 18.2 MΩ cm at 25 °C), before adding MgSO4, and for the latter finally also MnSO4, all in magnetically stirred vials at ca. 50 °C.Material synthesis and electrode fabrication
Mn-PBA was synthesized by a chelate-assisted precipitation method according to a previous study.20 The Mn-PBA had a secondary particle size of 5–10 μm (Fig. S1a†), and the composition was estimated to Na1.59Mn[Fe(CN)6]0.86·2.1H2O based on inductively coupled plasma atomic emission spectroscopy (ICP-OES) and thermogravimetric analysis (Fig. S1b†). The PTCDA (Sigma-Aldrich) based electrodes are similar to those used in.17 They were made by first preparing a solution of 3 wt% carboxymethyl cellulose (CMC) (Sigma-Aldrich) in ultra-pure water until complete dissolution by stirring. Subsequently, PTCDA and carbon black (CB) (Super-P, Alfa Aesar) were mixed manually with a mortar before being added to the CMC solution and stirred for 12 h. The slurry was cast onto a graphite foil (SGL Carbon) using a Doctor blade (250 μm wet thickness) followed by vacuum drying at 60 °C for 12 h. The electrode weight ratio PTCDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CB:CMC was 75:15:10 with a final thickness of ca. 40 μm and a loading of 1–2 mg cm−2 active material. Higher active material loading (3–4 mg cm−2) PTCDA electrodes were also made. The same procedure, weight ratio, and mass loading as for PTCDA was used to create the Mn-PBA electrodes.
:CB:CMC was 75:15:10 with a final thickness of ca. 40 μm and a loading of 1–2 mg cm−2 active material. Higher active material loading (3–4 mg cm−2) PTCDA electrodes were also made. The same procedure, weight ratio, and mass loading as for PTCDA was used to create the Mn-PBA electrodes.
Free-standing AC electrodes, used only for the basic electrochemical characterization of the stand-alone electrodes, were made by first stirring AC (Darco G-60, 600 m2 g−1, J. T. Baker) for 12 h in an aqueous 1 M HNO3 solution, before washing with excessive amounts of ultra-pure water in a vacuumed Büchner funnel, followed by vacuum drying at 100 °C for 12 h. The dried AC was mixed with CB and 3–4 ml acetone and put on a stirring hot plate, following the procedure described by Brousse et al.21 While stirring, PTFE (60 wt% in solution, Sigma-Aldrich) was slowly added, and the solution was thereafter left until the solvent had evaporated. The resulting paste was kneaded and spread with a few ml ethanol until resulting in a firm homogeneous film. The free-standing electrodes were dried in an oven at 80 °C for 12 h with a resulting thickness of ca. 400 μm, an active material loading of 10–12 mg cm−2, and a 75:15:10 AC:CB:PTFE weight ratio.
Materials characterization
Mn-PBA electrodes were pre-cycled to charged and discharged states by galvanostatic cycling (GC) between 0.8/0.85 and −0.5 V at 1C using a Biologic VMP3 multichannel potentiostat/galvanostat. Post cycling the electrodes were recovered and washed with 1.0 ml ultra-pure water. After drying in ambient air, ex situ X-ray diffraction (XRD) was performed on the electrodes, using a single crystal sample holder and a Bruker D8 Discover equipment to obtain diffractograms in the range 5–75° 2θ. Cu radiation was used with a Ni filter to cut out the Cu Kβ contributions, with two 2.5° soller slits to improve peak shapes. The incidence slit was set to fixed sample illumination mode with 5 mm illumination, and an anti-scatter shield was placed 2.5 mm above the samples. The samples were measured with 0.02° increments, and 2 s per step, summing up to 7000 s per scan, while rotated at 10 rpm.Fourier-transform infrared (FTIR) vibrational spectra were obtained using a Bruker Alpha attenuated total reflection (ATR-FTIR) spectrometer and a Ge crystal. 512 scans with a resolution of 2 cm−1 were made for each sample. The same electrodes as used for the XRD were used for the ex situ FTIR spectroscopy characterization.
The scanning electron microscopy (SEM) images were taken with a JEOL 7800F Prime at an acceleration voltage of 5.0 kV and 10 mm working distance.
The separator not in direct contact with the Mn-PBA electrode (see below) was recovered after cycling and put in 1 ml 1 M HCl for 24 h. It was thereafter filtered, diluted with 0.1 M HNO3, and then ICP-OES was carried out, using a Thermo Scientific™ – iCAP™ PRO XP ICP-OES in order to quantify the amount of dissolved active material during cycling.
Electrochemical and battery assessments
The GC and cyclic voltammetry (CV) studies were carried out using a Biologic VMP3 multichannel potentiostat/galvanostat and a Scribner Associates Incorporated 580 Battery Test System at room temperature (ca. 22 °C). For the CV, 3-electrode Swagelok cells were assembled with a Mn-PBA working electrode (WE) and a free-standing AC or a high loading PTCDA counter electrode (CE), all 10 mm Ø, and a Ag/AgCl pseudo reference electrode (Ag/AgCl from here on) (RE) (5 mm Ø, 0.127 mm silver foil, Alfa Aesar/AgCl ink, ALS Japan) ca. 0.34 V vs. Ag/AgCl (3.4 M KCl, Leakless Ag/AgCl Reference Electrode, eDAQ), together with two Whatman cellulose filter separators (Grade 44, Sigma-Aldrich). Scan rates of 0.2–5 mV s−1 and potential ranges between −0.2/0 and 0.7 V vs. Ag/AgCl were used for the CV. Coin-cells (CR2032) for GC were assembled with 10 mm Ø electrodes (PTCDA|Mn-PBA full cell) or with a 10 mm Ø WE combined with an AC or high loading PTCDA CE (13 mm Ø) (half-cell), using the same sort of separators. The GC tests were performed between 0.2 and 1.3–1.6 V (full cell), between −0.5/−0.7 and 0.8/0.85 V (half-cell with AC), and between −0.7 and 1.1/1.2 V (half-cell with high loading PTCDA) at current densities from 0.2C (0.02 A g−1) to 50C (5 A g−1). The balanced PTCDA|Mn-PBA cells were put together with a PTCDA/Mn-PBA weight ratio of ca. 1.1, and 40 μl electrolyte was used in for all cells. 100 mA h g−1 was used to calculate the C-rate, thus 1C = 0.1 A g−1.The specific capacity Q (mAh g−1) was calculated as:
 | (1) |
The active material level specific energy density E (W h kg−1) and ditto power density P (W kg−1) were then calculated as:
 | (2) |
 | (3) |
The logarithm of the peak current (i) was plotted vs. the sweep rate (v), and the b parameter from:
| logi = blogv + loga | (4) |
The relationship between v and i can also be written as
| i = k1v + k2v1/2 | (5) |
Results and discussion
To start off, the redox reactions of PTCDA and Mn-PBA are investigated using CV by studying the effect of electrolyte salt and charge species concentration. Ex situ FTIR spectroscopy and ex situ XRD are thereafter used to assess the cycling reversibility of the reactions of Mn-PBA, followed by half-cell characterization of Mn-PBA by CV and GC using the hybrid and triple electrolytes. We end by relating the PTCDA|Mn-PBA full cells' performance to the state-of-the-art.Active charge carrier and cycling reversibility
Unlike several other PBAs,24 the particular Mn-PBA used herein provides compelling evidence of not having the H+/H3O+ as active charge species, neither for mild nor acidic conditions, as the redox potential remains constant over several orders of magnitude of [H+/H3O+] (Fig. 1a). The CV studies rather suggest that both Na+ and Mg2+ ions contribute to the redox reactions of MnPBA, having redox potentials roughly following what is expected by the Nernst equation (Fig. 1b and c) (+59/+29 mV per decade of increasing concentration of salt/mono/di-valent charge species). A slight divergence from the theoretical approximation is to be expected since: (i) the concentration exceeds the dilute regime, yielding activity coefficients ≠ 1, and (ii) the electrode net current flow ≠ 0, also affecting the activity and moreover introducing additional overpotential and resistive loss terms. The ascending current density for the three electrolytes follows as: 1.0 m Na2SO4 < 1.0 m MgSO4 < hybrid electrolyte (Fig. 1a). Mn-PBA thus seems to require a higher concentration of charge carriers and/or chaotropic Mg2+ to facilitate a more pronounced charge transfer reaction. The higher salt concentration of the hybrid electrolyte (1.9 m Na2SO4 + 2.4 m MgSO4), as compared to the 0.1 m and 1.0 m electrolytes, also up-shifts the CV redox peaks (Fig. 1a–c). | ||
| Fig. 1 Mn-PBA electrode voltammograms (cycle 1, 0.2 mV s−1) at different (a) pH, (b) [Na+], and (c) [Mg2+]. | ||
The voltammograms of PTCDA are also rather unaffected by pH (Fig. S2a†); even though pH 4 stands out, there is no clear trend and there was also a notable peak shape variance for identical PTCDA cells. In stark contrast, Wang et al. reported PTCDA to be electrochemically active with a 1 M H2SO4(aq) electrolyte (pH < 1),25 and therefore H+/H3O+ co-intercalation should be possible, but our analysis points to the opposite at milder conditions. It seems like, however, unlike our previous belief,17 that both Na+ and Mg2+ are active charge species for PTCDA, displaying an unambiguous Nernst trend for increasing electrolyte concentrations of both salts (Fig. S2b and c†). Therefore, if the materials discussed are used to create full cells, PTCDA|Mn-PBA is an aqueous Na/Mg-ion battery.
The ex situ FTIR spectra suggest a FeII/FeIII redox reaction for MnPBA as the characteristic FeII–CN–MnII vibration band at 2070 cm−1 decreases while the ν(FeIII–CN–MnII) band at 2145 cm−1 simultaneous appears when oxidizing (charging) the material (Fig. 2a and S3†).26 Moreover, a very reversible cycling behavior is indicated as both bands return to their original shapes and intensities when the material is discharged. A similar behavior has been reported before for K2Ni[Fe(CN)6] using aqueous K-ion and Mg-ion electrolytes,24 and this material furthermore incorporated a fair amount of water in the open framework structure after cation deintercalation. This is, however, not observed here, with the OH-stretching vibration bands at ca. 3000–3600 cm−1 remaining unpronounced in the charged state (Fig. 2a).
 | ||
| Fig. 2 Ex situ (a) FTIR and (b) XRD of Mn-PBA electrodes cycled with the hybrid electrolyte. The noisy peaks in (a) originate from the ambient air gases. | ||
The diffractogram of Mn-PBA (Fig. S1c†) agrees well with the hydrated monoclinic phase previously reported, belonging to the P21/n space group.27 Furthermore, the ex situ XRD investigation reveals a transition from monoclinic to cubic phase, and also a slightly changed crystal structure between the pristine electrode and the subsequent discharged states, as a new peak emerges at ca. 22° after charging with the intensity decreasing/increasing during the following discharge/charge (Fig. 2b). This was not observed for Mn-PBA when using a (9 m NaOTf + 22 m TEAOTf)(aq) electrolyte11 and could possibly be explained by SO42− intercalation into graphite.28 To test this, an ‘empty’ graphite CC was oxidized and the resulting diffractogram indeed exhibits such a peak, although the capacity from this reaction is by all means negligible (Fig. S4†). Altogether, the cation intercalation/deintercalation process is highly reversible, with the highlighted peaks changing accordingly (Fig. 2b).
Electrochemical characterization
The voltammograms display two overlapping reversible redox peaks both during oxidation, at ca. 0.35 and 0.43 V vs. Ag/AgCl, and reduction, at ca. 0.37 and 0.31 V during the first cycle with the hybrid electrolyte (Fig. S5a†), similar to what has been reported before for Mn–PBAs,11 and will from now on be referred to as the first redox reaction. This feature has previously been associated with the FeII/FeIII redox,29 in accordance with our FTIR spectroscopy results. There is also a second peak at higher potential involving MnII/MnIII, but in our systems this is unfortunately situated right at the edge of the electrochemical stability window (ESW) (Fig. S5b†). To utilize this reaction would require precise potential control to avoid oxygen evolution reaction (OER), or further modification of the electrolyte configuration to expand the ESW. Moreover, the triple electrolyte based cell renders a redox response almost identical during the first cycle, and only a slight shift is observed by cycle 10, making us believe Mn2+(aq) is not involved in the redox process (Fig. S5a†), which is plausible as the standard redox potentials of Mn2+(aq) + 2e− → Mn(s) and Mn3+(aq) + e− → Mn2+(aq) are −1.18 V and 1.51 V vs. SHE, respectively.From the analysis of the logarithm of the peak current (ip) using the linear relations (eqn (4)) we find a mix of faradaic and non-faradaic oxidation and reduction reactions (box,red ≈ 0.75) (Fig. 3a and b), thus in the same range as NiCo-PBA using a 2 M NaNO3(aq) electrolyte,30 and moreover as the scan rate increases a larger fraction of the current originates from non-faradaic (capacitive), or pseudocapacitive behaviour (Fig. 3b).31
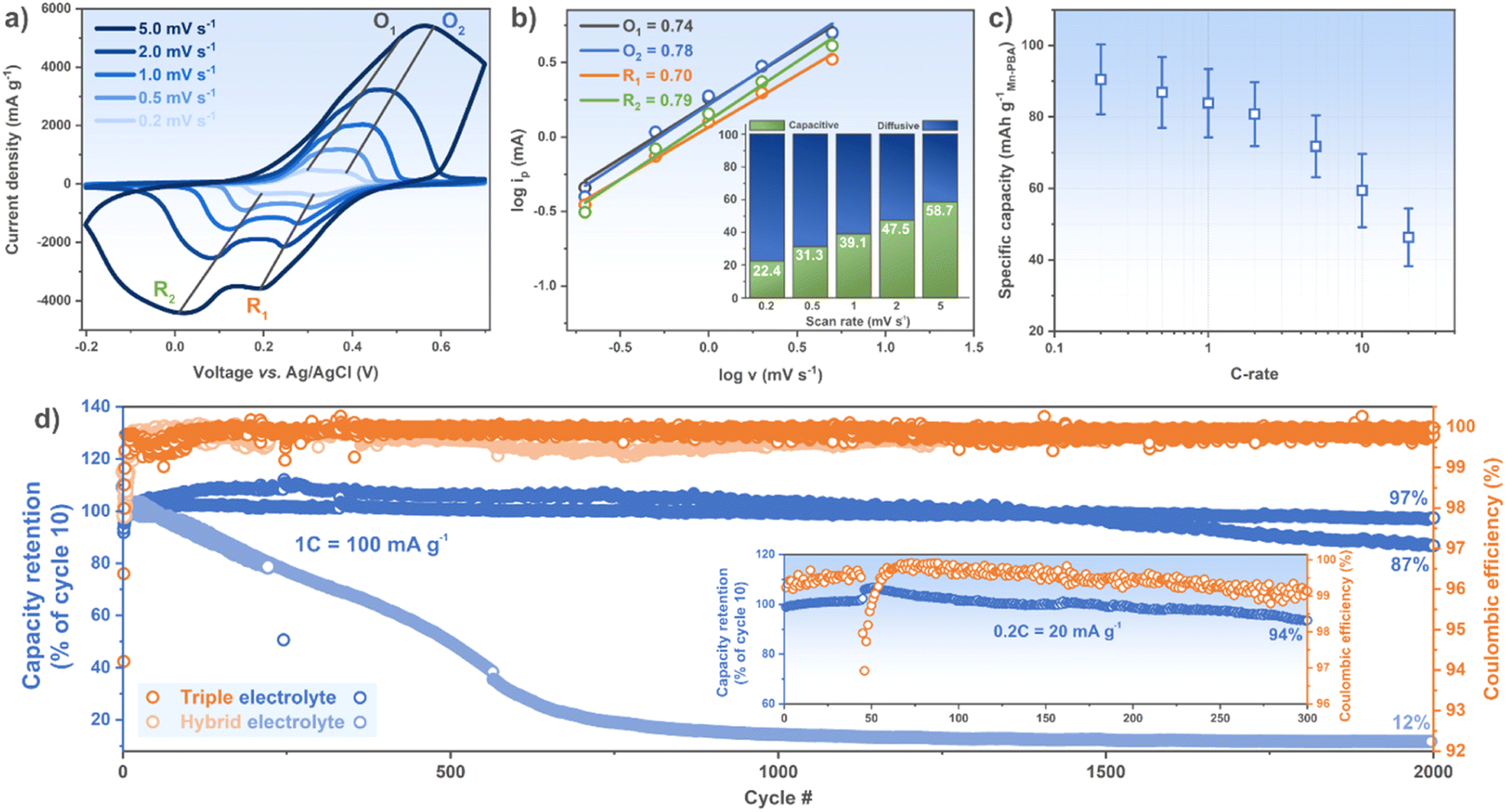 | ||
| Fig. 3 Mn-PBA electrode (a) voltammogram at different scan rates, (b) the corresponding logipvs. logv, (c) rate capability (mean and standard deviation of 10 cells), and (d) long-term cycling tests with (a–d) the triple and (d) the hybrid electrolytes. (b and d) Insets show the current contributions at the different scan rates and the long-term cycling at 0.2C, respectively. | ||
By restricting the voltage range to the first redox reaction(s), to avoid the OER (Fig. S5b†), the Mn-PBA electrode delivers a stable specific capacity of ca. 90 mA h g−1 at 0.2C and retains a substantial amount up to 20C, at coulombic efficiencies ≥99.5% for all C-rates (Fig. 3c and S7a†). This is slightly higher capacity than what has been reported for other Mn–PBAs (first reaction(s) only), whereas the rate performance in previous works differ vastly; from similar to much better.11–13,32–34 The large standard deviation is a result of our graphite current collector having a substantial areal weight variance.
The cycling stability of Mn-PBA using the hybrid and the triple electrolytes show the cells with the former to experience severe capacity decay right from the start and continuing throughout, whereas the latter performs remarkably well – both in terms of capacity retention (Table 1 and Fig. 3d) and coulombic efficiency (≥99.75% at 1C & ≥99% at 0.2C). For a fairer comparison, we use the retention relative to cycle 10 since the capacity initially increases during the first cycles for most cells. The hybrid electrolyte half-cell(s) on the other hand performs more in line with previous studies using Mn-PBA and 17 m NaClO4(aq) (Table 1), with almost no capacity left at cycle 2000 (Fig. 3d).
| Battery chemistry | Mn-PBA half-cell | Full cell (capacity retention) | Full cell (rate retention) | Ref. |
|---|---|---|---|---|
| Na3Fe2(PO4)3|17 M NaClO4|Mn-PBA | 66%@1000 at 10C | 75%@700 at 5C | — | 32 |
| CrCr-PBA|17 M NaClO4|Mn-PBA | 92.5%@100 at 0.5 A g−1 | 90%@100 at 1C | — | 33 |
| KMn[Cr(CN)6]|17 M NaClO4|Mn-PBA | — | 50%@100 at 5C | 70% from 5–20C | 12 |
| NaTi2(PO4)3|17 M NaClO4|Mn-PBA | 74%@100 at 2.0 mA cm−1 | 81%@50 at 2.0 mA cm−2 | 48% from 2–20 mA cm−2 | 34 |
| NaTi2(PO4)3|32 M KAc + 8 M NaAc|Mn-PBA | — | 36%@85 at 0.1 A g−1 | <50% from 0.1–0.5 A g−1 | 13 |
| NaTi2(PO4)3|80 M NaTFSi0.375EMIMTFSi0.625|Mn-PBA | — | 70%/79%@300 at 1C | — | 35 |
| NaTiOPO4|9 M NaOTf + 22 M TEAOTF|Mn-PBA | 98%@50 at 1C | 76%@800 at 1C | 60% from 0.25–10C | 11 |
| PTCDA|4.6 M triple electrolyte|Mn-PBA | 92%@2000 at 1C (0.1 A g− 1 ) | 86%@1000 at 1C | 73% from 0.2–10C | This work |
| 94%@300 at 0.2C | 89%@800 at 0.2C | 85% from 5–20C |
We attribute the enhanced cycling stability to a suppressed Mn dissolution, in accordance with Le Chatelier's principle.36 To investigate this further, separators were recovered after 200 cycles and there are no signs of side-reactions for the cell with the triple electrolyte, unlike for the cell with the hybrid electrolyte (Fig. S6a and b†). The dissolved amount was furthermore quantified using ICP-OES, and after 1000 cycles the cell's separator contained 112 mg Mn with the hybrid electrolyte. The elemental analysis also reveals the presence of Fe (110 mg), as did Jiang et al.,11 leading us to speculate that a quadruple electrolyte could possibly enable even better cycling stability. The decreased coulombic efficiency for the slower cycling could indicate some irreversible phenomena at play (inset Fig. 3d), but the disassembled cell does not point to any severe side-reactions, whereas only a little bit of corrosion (brown) is present on the edges of the part of the separator that was in contact with the stainless steel casing (Fig. S6c†). We attribute the lower coulombic efficiency to possibly be due to corrosion of stainless steel and some water decomposition, considering the catalyzing high surface area AC CE37 and the longer time spent close to the edge of the ESW when cycling at 0.2C as compared to at 1C.
Next, the electrochemical performance of balanced PTCDA|Mn-PBA cells were assessed and they manage to retain a large amount of capacity even up to 20 and 50C (Fig. 4a and S7b), and slightly better than other Mn-PBA batteries (Table 1). This could be due to either: (i) the use of a high power performant anode, not acting as a bottle neck, and/or (ii) the lower electrolyte concentration (<5 m vs. ≥17 m), generating lower viscosity and higher ionic conductivity, and therefore allowing for faster charging/discharging rates. Furthermore, the active material level energy density (35 W h kg−1PTCDA+Mn-PBA at 0.2C) is more than twice that of our previous PTCDA|AC hybrid supercapacitor device (17 W h kg−1PTCDA+AC),19 and similar to previously reported PBA-based ASIBs (Fig. 4b),11,12,32,33,35,38–40 while simultaneously retaining a very good power density (1700 W kg−1PTCDA+Mn-PBA at 50C) – highlighting Mn-PBA's cathode material capability for both energy and power, and its fitting electrochemical traits with PTCDA.
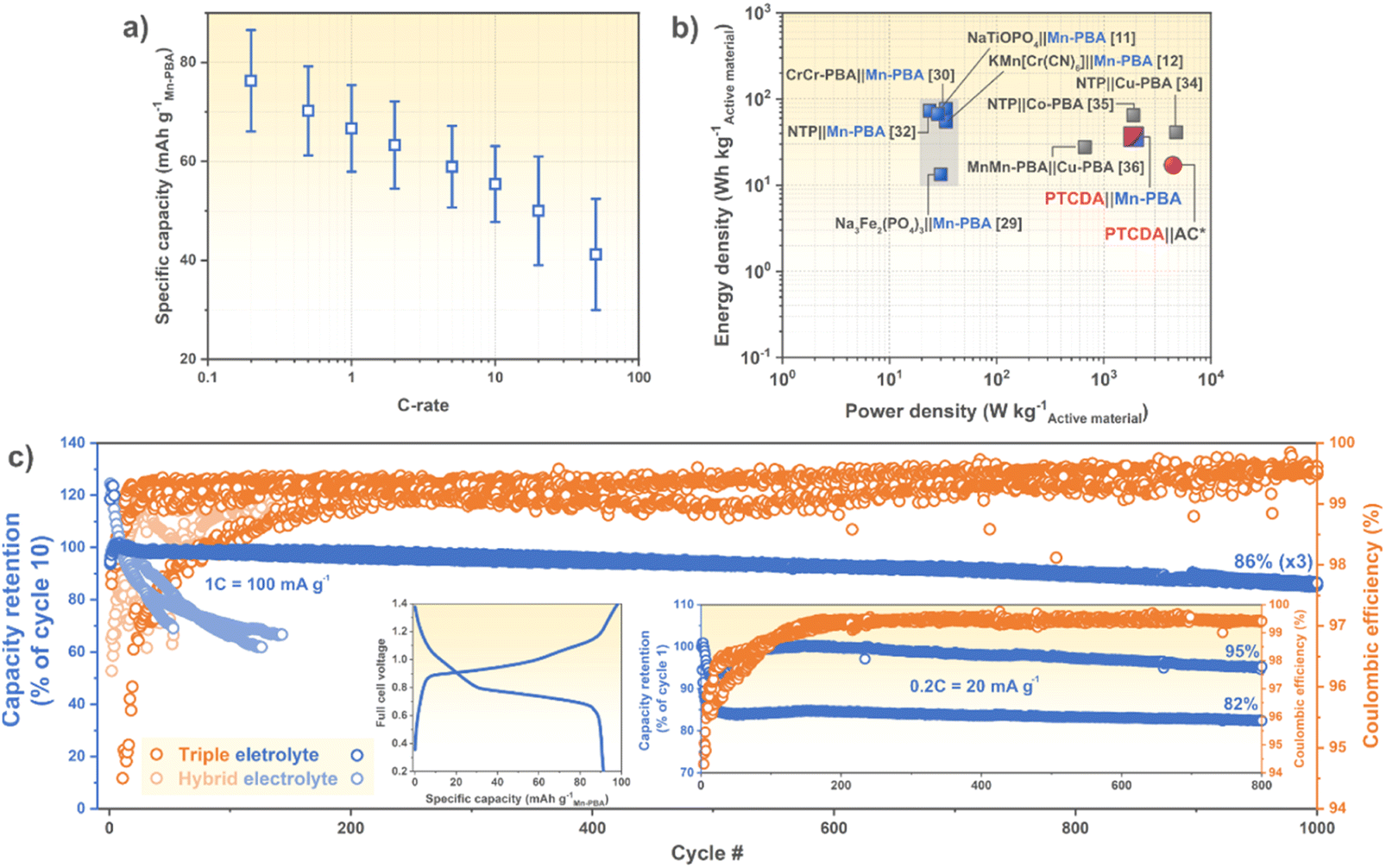 | ||
| Fig. 4 (a) Rate capability (mean and standard deviation of 8 cells), (b) Ragone plot, and (c) long-term cycling of PTCDA|Mn-PBA cells with (a–c) the triple and (c) hybrid electrolytes. Left and right (c) insets show typical charge/discharge curves at 0.2C and the long-term cycling performance at 0.2C, respectively. The grey area papers in (b) did not report a powder density. *Our previous work.19 | ||
In order to exclude the 2nd redox reaction the full cell voltage was cut at 1.3–1.5 V, yielding an average voltage of roughly half (Fig. 4c), in the same range as the NaTi2(PO4)3|32 m KAc + 8 m NaAc|Mn-PBA and the Na3Fe2(PO4)3|17 m NaClO4|Mn-PBA cells (Table 1). By including the 2nd redox reaction (or not) the cell voltage can readily be tuned for application specific needs; by increasing the cut-off voltage, the FeII/III redox reaction gradually decreases, and by cycle 20 the material exchanges electrons mainly via the MnII/III redox pair (Fig. S8a†). This substantially increases the full cell nominal voltage, at the cost of lower coulombic efficiency (Fig. S8b†).
The long-term cycling performance was also evaluated for the PTCDA|Mn-PBA chemistry, displaying an outstanding capacity retention with the triple electrolyte at both 1C (86%@1000, n = 3) and at 0.2C (89%@800, n = 2), with coulombic efficiencies ≥99.5% (Fig. 4c and Table 1). The hourly (dis)charge time of 1C is not only required by many practical applications, but as compared to the much faster cycling (5–10C) often applied (Table 1), the time spent at the electrolyte electrochemical stability window edges is longer and thus any impaired coulombic efficiency and cycling stability would be revealed. Here neither are much affected at the even slower cycling of 0.2C (Fig. 4c), which we attribute to only using the FeII/III redox reaction and minimized Mn dissolution.
Concluding remarks
The herein presented novel battery chemistry PTCDA|Mn-PBA shuttles Na+ and Mg2+ ions during operation using low-to-medium concentrated electrolytes in a very reversible manner. We can exclude any proton storage mechanism for both electrodes and thereby we can categorize the cells made as aqueous Na/Mg-ion batteries. Moreover, a solution is proposed to the previously chronic transition metal dissolution of Mn–PBAs, and this without having to resort to any extreme electrolyte or electrode engineering. We see the here presented tailored electrolyte as a true alternative to the more popular 10–100× price tagged17 [TFSI]− and ClO4− based WISEs. The assembled full cells also highlight the possibility to build sustainable and cost-effective batteries that in principle can supply energy for less demanding applications and in tandem with more performant LIBs help to fulfil the ever-increasing electricity needs of our modern society.Author contributions
M. K. designed the study and did all the measurements, formal analysis, validation, visualization, writing of the original draft, and together with the co-authors reviewed & edited the manuscript drafts throughout the writing process. T. H synthesized the Mn-PBA and participated in validating the results. P. J. was responsible for the general conceptualization, project administration, supervision of M. K., and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our sincere appreciation to the Swedish Research Council for Sustainable Development (FORMAS) (grant #2018-01450) and the Swedish Research Council (VR) (grant #2021-00613) for supporting this work.Notes and references
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- H. Pan, Y. S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 RSC.
- H. S. Hirsh, Y. Li, D. H. S Tan, M. Zhang, E. Zhao and Y. Shirley Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, J. Mater. Chem. A, 2014, 3, 22–42 RSC.
- R. Arvidsson, M. Chordia and A. Nordelöf, Int. J. Life Cycle Assess., 2022, 27, 1106–1118 CrossRef CAS.
- Y. Liang and Y. Yao, Nat. Rev. Mater., 2022, 82(8), 109–122 CrossRef.
- S. Qiu, Y. Xu, X. Wu and X. Ji, Electrochem. Energy Rev., 2021, 1, 3 Search PubMed.
- H. Li, W. Zhang, K. Sun, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, Z. Zhang, Y. Lai, H. Sun, H. Li, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, H. Sun, W. Zhang, K. Sun, Z. Zhang and Y. Lai, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- W. Li, C. Han, W. Wang, Q. Xia, S. Chou, Q. Gu, B. Johannessen, H. Liu, S. Dou, W. Li, C. Han, W. Wang, Q. Xia, S. Chou, H. Liu, S. Dou, Q. Gu and B. Johannessen, Adv. Energy Mater., 2020, 10, 1903006 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- L. Jiang, L. Liu, J. Yue, Q. Zhang, A. Zhou, O. Borodin, L. Suo, H. Li, L. Chen, K. Xu and Y. S. Hu, Adv. Mater., 2020, 32, 1904427 CrossRef CAS PubMed.
- K. Nakamoto, R. Sakamoto, Y. Sawada, M. Ito, S. Okada, K. Nakamoto, M. Ito, S. Okada, R. Sakamoto and Y. Sawada, Small Methods, 2019, 3, 1800220 CrossRef CAS.
- J. Han, H. Zhang, A. Varzi and S. Passerini, ChemSusChem, 2018, 11, 3704–3707 CrossRef CAS PubMed.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- H. Ao, C. Chen, Z. Hou, W. Cai, M. Liu, Y. Jin, X. Zhang, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2020, 8, 14190–14197 RSC.
- M. Karlsmo, R. Bouchal and P. Johansson, Angew. Chem., Int. Ed., 2021, 60, 24709–24715 CrossRef CAS PubMed.
- D. Bin, F. Wang, A. G. Tamirat, L. Suo, Y. Wang, C. Wang and Y. Xia, Adv. Energy Mater., 2018, 8, 1703008 CrossRef.
- M. Karlsmo and P. Johansson, Batteries Supercaps, 2022, 5, e202200306 CrossRef CAS.
- T. Hosaka, T. Fukabori, H. Kojima, K. Kubota and S. Komaba, ChemSusChem, 2021, 14, 1166–1175 CrossRef CAS PubMed.
- T. Brousse, P. L. Taberna, O. Crosnier, R. Dugas, P. Guillemet, Y. Scudeller, Y. Zhou, F. Favier, D. Bélanger and P. Simon, J. Power Sources, 2007, 173, 633–641 CrossRef CAS.
- Z. Zhang, Y. Zhu, M. Yu, Y. Jiao and Y. Huang, Nat. Commun., 2022, 131(13), 1–11 Search PubMed.
- H. Zhang, M. Hu, Q. Lv, Z. H. Huang, F. Kang and R. Lv, Small, 2020, 16, 1902843 CrossRef CAS PubMed.
- A. I. Komayko, S. V. Ryazantsev, I. A. Trussov, N. A. Arkharova, D. E. Presnov, E. E. Levin and V. A. Nikitina, ChemSusChem, 2021, 14, 1574–1585 CrossRef CAS PubMed.
- X. Wang, C. Bommier, Z. Jian, Z. Li, R. S. Chandrabose, I. A. Rodríguez-Pérez, P. A. Greaney and X. Ji, Angew. Chem., Int. Ed., 2017, 56, 2909–2913 CrossRef CAS PubMed.
- H. Tokoro, S. Miyashita, K. Hashimoto and S.-I. Ohkoshi, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 172415 CrossRef.
- J. Song, L. Wang, Y. Lu, J. Liu, B. Guo, P. Xiao, J. J. Lee, X. Q. Yang, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2658–2664 CrossRef CAS PubMed.
- F. M. Alcorn, K. L. Kuntz, D. L. Druffel and S. C. Warren, ACS Appl. Energy Mater., 2018, 1, 5062–5067 CrossRef CAS.
- M. Pasta, R. Y. Wang, R. Ruffo, R. Qiao, H. W. Lee, B. Shyam, M. Guo, Y. Wang, L. A. Wray, W. Yang, M. F. Toney and Y. Cui, J. Mater. Chem. A, 2016, 4, 4211–4223 RSC.
- Y. Zeng, Y. Wang, Z. Huang, H. Luo, H. Tang, S. Dong and P. Luo, Nanotechnology, 2023, 34, 475403 CrossRef PubMed.
- A. González, E. Goikolea, J. A. Barrena and R. Mysyk, Renewable Sustainable Energy Rev., 2016, 58, 1189–1206 CrossRef.
- S. Qiu, X. Wu, M. Wang, M. Lucero, Y. Wang, J. Wang, Z. Yang, W. Xu, Q. Wang, M. Gu, J. Wen, Y. Huang, Z. J. Xu and Z. Feng, Nano Energy, 2019, 64, 103941 CrossRef CAS.
- J. Chen, C. Liu, Z. Yu, J. Qu, C. Wang, L. Lai, L. Wei and Y. Chen, Chem. Eng. J., 2021, 415, 129003 CrossRef CAS.
- K. Nakamoto, R. Sakamoto, M. Ito, A. Kitajou and S. Okada, Electrochem., 2017, 85, 179–185 CrossRef CAS.
- D. Reber, R. Grissa, M. Becker, R.-S. Kühnel, C. Battaglia, D. Reber, R. Grissa, M. Becker, R. Kühnel and C. Battaglia, Adv. Energy Mater., 2021, 11, 2002913 CrossRef CAS.
- H. Le Chatelier, Comptes rendus, 1884, 99, 786–789 Search PubMed.
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- X. Y. Wu, M. Y. Sun, Y. F. Shen, J. F. Qian, Y. L. Cao, X. P. Ai and H. X. Yang, ChemSusChem, 2014, 7, 407–411 CrossRef CAS PubMed.
- X. Wu, M. Sun, S. Guo, J. Qian, Y. Liu, Y. Cao, X. Ai and H. Yang, ChemNanoMat, 2015, 1, 188–193 CrossRef CAS.
- M. Pasta, C. D. Wessells, N. Liu, J. Nelson, M. T. McDowell, R. A. Huggins, M. F. Toney and Y. Cui, Nat. Commun., 2014, 51(5), 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06826a |
| This journal is © The Royal Society of Chemistry 2024 |