
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA MOF-derived pyrrolic N-stabilized Ni single atom catalyst for selective electrochemical reduction of CO2 to CO at high current density†
Jin Wook
Lim‡
a,
Dong Heon
Choo‡
b,
Jin Hyuk
Cho‡
c,
Jaehyun
Kim
d,
Won Seok
Cho
a,
Odongo Francis
Ngome Okello
a,
Kisoo
Kim
 a,
Sungwon
Lee
a,
Junwoo
Son
a,
Si-Young
Choi
a,
Jong Kyu
Kim
a,
Ho Won
Jang
*d,
Soo Young
Kim
*c and
Jong-Lam
Lee
*ab
a,
Sungwon
Lee
a,
Junwoo
Son
a,
Si-Young
Choi
a,
Jong Kyu
Kim
a,
Ho Won
Jang
*d,
Soo Young
Kim
*c and
Jong-Lam
Lee
*ab
aDepartment of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang 790-784, Korea. E-mail: jllee@postech.ac.kr
bDivision of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), Pohang 790-784, Korea
cDepartment of Materials Science and Engineering, Korea University, Seoul, Korea. E-mail: sooyoungkim@korea.ac.kr
dDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Korea. E-mail: hwjang@snu.ac.kr
First published on 27th February 2024
Abstract
Electrochemical reduction of CO2 to chemical fuels with a transition metal-based single atom catalyst (SAC) offers a promising strategy to reduce CO2 with high catalytic selectivity. To date, the study of atomically dispersed SACs has been mainly conducted by using a conventional H-type cell system with limited solubility of CO2 in aqueous electrolytes, resulting in large overpotentials and low current density. Here, we reported a pyrrolic N-stabilized Ni SAC with low-coordinated Ni–Nx sites by thermal activation of Ni ZIF-8, which was tested in a 3-compartment microfluidic flow cell system at the industrial level. When the pyrolysis temperature increased from 800 °C (Ni SAC-800) to 1000 °C (Ni SAC-1000), the content ratio of pyrrolic N/pyridinic N increased from 0.37 to 1.01 as well as the coordination number of Ni in Ni–Nx sites decreased from 3.14 to 2.63. Theoretical calculations revealed that the synergistic effect between the high content ratio of pyrrolic N and low-coordinated Ni can decrease the energy barrier for the desorption of *CO during the CO2RR. Therefore, Ni SAC-1000 exhibited superior catalytic performances with high CO selectivity (FECO = 98.24% at −0.8 VRHE) compared to that of Ni SAC-800 (FECO = 40.76% at −0.8 VRHE). Moreover, Ni SAC-1000 based on the flow cell system showed a higher current density (∼200 mA cm−2) compared to that of the H-type cell system (∼20 mA cm−2). As a result, this study experimentally demonstrated that the pyrrolic N-stabilized and low-coordinated Ni SAC-1000 in the microfluidic flow cell reactor provides great chances for scaling up the productivity of the CO2RR at the industrial level.
Introduction
The conversion of CO2 to chemical fuels by the electrochemical CO2 reduction reaction (CO2RR) with renewable electricity is an effective way to solve global climate changes and achieve carbon recycling in human society.1–5 Among the possible chemical fuels of the CO2RR, carbon monoxide (CO) is one of the most desired products because it was generated by low electron consumption of CO2 and could be further converted to methanol, hydrocarbons, and petroleum through Fischer–Tropsch synthesis.6 To date, nanostructure-based metal catalysts such as Ag nanowires,7,8 3D hierarchical Cu,9,10 and Au nanoneedles11 have been demonstrated to improve the catalytic performances of CO production because they have a large catalytic surface area for the CO2RR.7–16 But, it still suffered from large overpotentials and highly competitive hydrogen evolution reaction (HER) in an aqueous system.17,18Recently, single atom catalysts (SACs) have emerged as a promising candidate for the CO2RR because they have atomically dispersed active sites and can easily change the local coordination environments.19–22 Especially, a non-noble and earth-abundant Ni-based SAC coordinated in a carbon matrix with neighboring N configurations (Ni–Nx–C structure) has been demonstrated to be highly selective in CO production.23–27 Because the Ni–Nx configurations play a role in active sites for the CO2RR, there were several studies to modulate the coordination structure and local environment of the Ni species.25 For instance, Yan et al. suggested that unsaturated Ni–Nx sites effectively strengthen the CO2 adsorption, leading to an increase in the catalytic selectivity of CO (FECO = 92% at −1.03 VRHE).28 In addition, low-valent Ni+ species anchored on a N-doped graphene matrix showed high selectivity toward CO production (FECO = 94% at −0.9 VRHE).29 Pranav et al. reported that various nitrogen species bonded with metal, such as graphitic N, pyridinic N, and pyrrolic N, acted as the active sites for the CO2RR based on density functional theory (DFT) calculations.30 Despite several kinds of research on Ni-based SACs for the CO2RR, the reason for the enhanced catalytic activities remains unclear due to the complexity of various factors, and more importantly, the role of different types of N configurations in the CO2RR still remains elusive.
The most effective and easy way to control the N configurations in Ni-based SACs could be pyrolysis treatment at high temperatures. During the pyrolysis process, the Ni and N species react with each other to form various active Ni–Nx sites and make different N configurations depending on the process conditions. Thus, understanding the role of types of N configurations fabricated under different pyrolysis conditions can provide insights into the relationship between the local Ni–Nx structure and catalytic performances, enabling the making of efficient catalysts.
So far, CO current density for previously reported Ni SACs with a Ni–Nx–C structure has been limited because most of them were evaluated in conventional H-type electrochemical cell systems.31–35 In a typical H-type cell system, the CO2 gas was supplied to the catalytic surface through aqueous electrolytes. The current density of CO2 conversion could be restricted due to the low CO2 solubility and slow diffusion of CO2 in aqueous solution, resulting in hardly satisfying the current density of the industrial level (>100 mA cm−2).36 To scale up the productivity of the CO2RR at the practical level, a gas diffusion electrode (GDE)-based flow cell system could be employed, where the CO2 gas was directly supplied to the catalytic surface without containing the aqueous solution. The flow cell system can produce high current density because of the continuous contact between CO2 and the catalyst as well as the fast diffusion of CO2 gas. Thus, the microfluidic flow cell system is a promising candidate for enhancing the productivity of the CO2RR in the Ni-based SAC.
In this study, we demonstrated that a simple pyrolysis process can modulate the local structure of the Ni–Nx site and types of N configurations in Ni SACs, which affects the catalytic performances of the CO2RR. To manipulate the atomic environments of Ni SACs, we fabricated three types of samples with Ni ZIF-8 (not heat-treated), Ni SAC-800, and Ni SAC-1000 (after pyrolysis at 800 and 1000 °C). As the pyrolysis temperature increased from 800 to 1000 °C, the coordination number of Ni–N decreased from 3.14 to 2.63 as well as the relative ratio of pyrrolic N/pyridinic N increased from 0.37 to 1.01, indicating that the pyrrolic N can be thermally activated and stabilized in low-coordinated Ni SACs. Ni SAC-1000 showed relatively higher catalytic performances (FECO = 98.24% at −0.8 VRHE and jCO = 148.25 mA cm−2 at −1.4 VRHE) compared to that of Ni SAC-800 (FECO = 40.76% at −0.8 VRHE and jCO = 4.41 mA cm−2 at −1.4 VRHE) in a microfluidic flow cell system due to the pyrrolic N-stabilized Ni SAC with low coordinated Ni–Nx sites. Because of the continuous contact between CO2 and the catalyst as well as the fast diffusion of CO2 gas in the flow cell system, Ni SAC-1000 maintained FECO > 95% at potentials from −0.8 to −1.4 VRHE and showed significantly higher current density (∼200 mA cm−2) compared to that of the H-type cell system (∼20 mA cm−2). DFT calculations showed that the synergistic effect between the high content ratio of pyrrolic N and low-coordinated Ni can decrease the energy barrier for the desorption of *CO during the CO2RR, leading to high production of CO. As a result, the high performance of Ni SAC-1000 in the microfluidic flow cell system presents great chances for scaling up the productivity of CO2 reduction products toward the industrial level.
Results and discussion
Morphology and microstructure
The Ni SAC was fabricated by solution-based synthesis and a subsequent pyrolysis process, which are schematically outlined in Fig. 1a (synthesis procedures are detailed in the Experimental section). ZIF-8 was fabricated through solution-based synthesis with Ni and Zn precursors, and 2-MeIm. ZIF-8 with a porous structure serves as a molecular cage by trapping the Ni and Zn ions with the organic ligands of 2-MeIm. After the pyrolysis process, a metal (M)–Nx–C structure supported on an N-rich carbon matrix was clearly obtained because the ordered assembly of Ni and Zn was placed in the ZIF-8 structure. To investigate the effect of pyrolysis temperature, we conducted heat treatment at 800 and 1000 °C at a rate of 25 °C min−1 and maintained the temperature for 4 h under an Ar atmosphere in a tube furnace (denoted as Ni SAC-800 and Ni SAC-1000). The morphology of Ni ZIF-8 (before pyrolysis), Ni SAC-800, and Ni SAC-1000 was investigated using a FE-SEM. The SEM images showed that the as-prepared samples exhibited similar cubic-like morphology even after the pyrolysis process (Fig. 1b).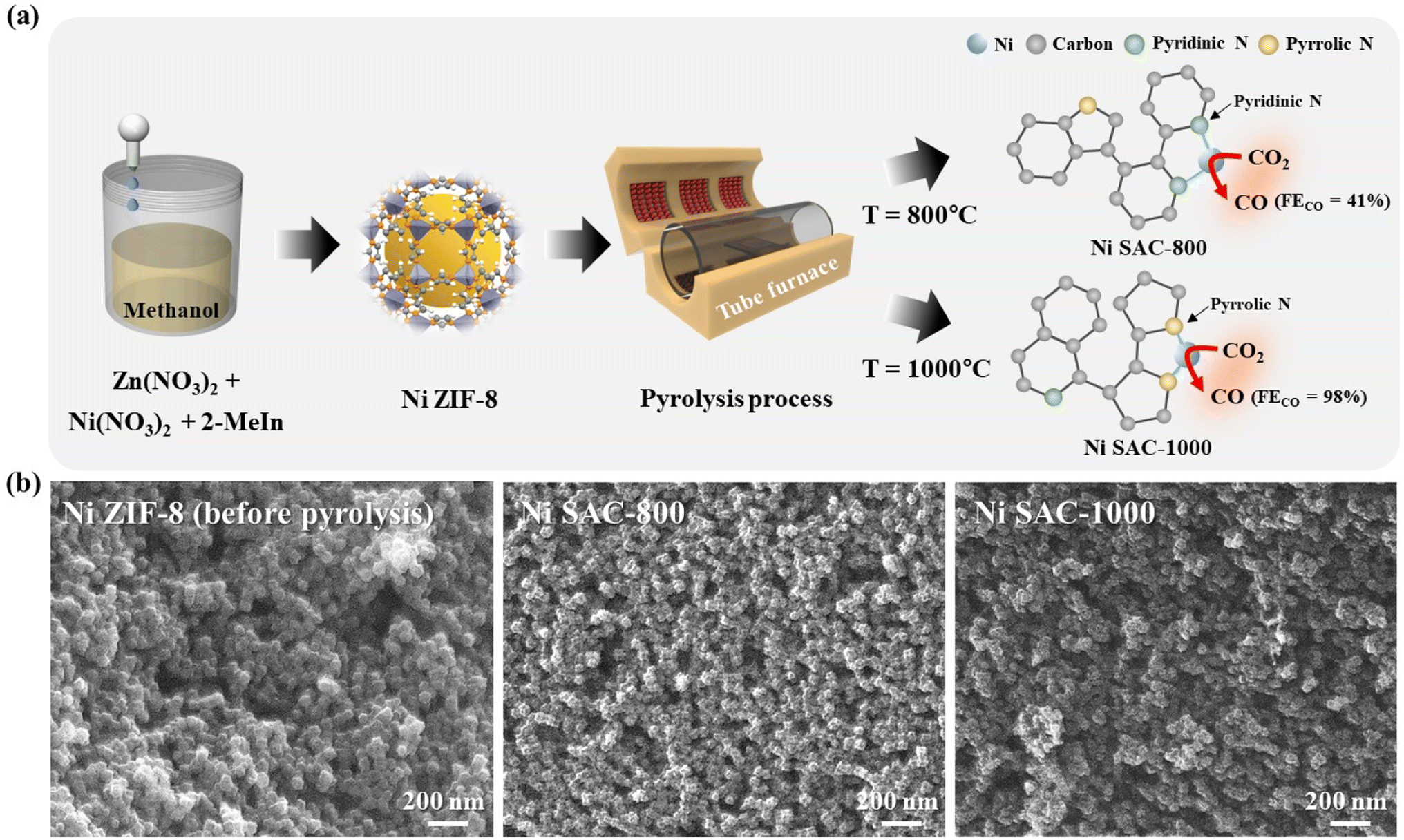 | ||
| Fig. 1 (a) Schematic illustration of the fabrication process of the pyrrolic N-stabilized low-coordinated Ni SAC and proposed Ni–Nx active site structure. (b) SEM images of Ni ZIF-8 (before pyrolysis) and the Ni SAC (after pyrolysis at 800 and 1000 °C). | ||
The microstructure of the as-prepared samples was further investigated with HR-TEM. It showed a uniform rhombic dodecahedron structure with an average size of approximately 40–50 nm in all samples (Fig. 2a and S1†). However, the morphology started to aggregate because the MOF structure in ZIF-8 changed to an M–Nx–C structure under the pyrolysis process. The inseted SAED patterns revealed an amorphous-like carbon structure, which means that there are not any metal-containing nanoparticles in the carbon matrix. HAADF-STEM analysis was conducted to investigate the atomic distribution of Ni atoms in the as-prepared samples (Fig. 2b). In the Ni ZIF-8 skeleton structure, no bright dots representing isolated single Ni atoms are observed. In contrast, isolated single Ni atoms were clearly distributed in the entire N-rich carbon matrix in both Ni SAC-800 and -1000. Thus, it is important to thermally activate the Ni ZiF-8 structure to achieve highly dispersed Ni single atoms in the M–Nx–C structure. TEM-EDS images showed that the elements of C, N, Zn, and Ni were distributed in the as-prepared samples (Fig. S2†). The elemental content of Ni increased with increasing pyrolysis temperature, which was determined by ICP-AES. It showed that the Ni content was 0.63 wt%, 1.41 wt%, and 2.01 wt% for Ni ZIF-8, Ni SAC-800, and Ni SAC-1000, respectively (Fig. S3 and Table S1†). Notably, most of the Zn species were evaporated during the pyrolysis process, but residual Zn species still remained and could be anchored in the carbon matrix. The residual Zn can prevent the aggregation of Ni species and plays a role in the construction of the porous structure.28
 | ||
| Fig. 2 (a and b) TEM and HAADF-STEM images of Ni ZIF-8 (before pyrolysis) and the Ni SAC (after pyrolysis at 800 and 1000 °C) with the SAED patterns in the inset. | ||
Crystal structure, chemical compositions, and electronic configuration
The crystal structure was detected by XRD and Raman spectroscopy (Fig. S4†). Both Ni SAC-800 and -1000 samples exhibited no diffraction patterns from crystalline Ni and Zn-based nanoparticles. It showed a broad graphitic carbon peak of the (002) plane at 21.5°, which was totally different from the sharp peaks of the ZIF-8 structure. So, there are no peaks of the Ni and Zn metallic phases in Ni SAC-800 and -1000, which were consistent with the results of HR-TEM and HAADF-STEM. The Raman spectra showed a higher integrated area ratio of ID/IG for Ni SAC-1000 (1.49) compared to those of Ni SAC-800 (1.13), leading to a higher disordered structure related to the structural defects. As a result, we confirmed that the Ni single atoms are highly distributed on the defect-rich carbon matrix, and they are detected by only HAADF-STEM analysis.XPS was conducted to elucidate the surface compositions and related valence states of the as-prepared samples (Fig. 3). The Zn 2p spectra of the as-prepared samples contained two peaks centered at 1044.30 and 1021.40 eV, which can be assigned to Zn2+ in both Zn 2p1/2 and Zn 2p3/2 (Fig. S5a†).22,37 As the pyrolysis temperature increased, the intensity of the Zn 2p spectra gradually decreased but it still remained as also confirmed by TEM-EDS, and the residual Zn plays role in the prevention of aggregation of Ni species.28 The Ni 2p3/2 spectra of the as-prepared samples were deconvoluted into the peaks of Ni2+ (856.22 eV), Ni+ (855.54 eV), and metallic Ni0 (854.28 eV) states (Fig. 3a).29,37,38 Ni ZIF-8 (before pyrolysis) showed only the Ni2+ peak, which was consistent with the valence state of the MOF structure in reference NiPc. On the other hand, there are three distinct peaks of Ni2+, Ni+, and metallic Ni0 for both Ni SAC-800 and -1000 samples, indicating that the valence state of Ni atoms is likely to be between 0 and +2. Notably, as the pyrolysis temperature increased, the integrated area ratio of Ni+/Ni2+ increased in the as-prepared samples (Fig. 3c). So, it means that the valence state of Ni atoms in Ni SAC-1000 was relatively lower than that of Ni SAC-800. The N 1 s spectra were deconvoluted into seven N-related configurations, originating from graphitic N (403.69 eV), N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (imidazole) (402.12 eV), pyrrolic N (401.48 eV), N–Ni (400.59 eV), N–Zn (399.13 eV), pyridinic N (398.55 eV), and N–H (imidazole) (398.24 eV), respectively (Fig. 3b).25,28,37,39 Ni ZIF-8 showed distinct MOF structure-related peaks of NC (imidazole) and N–H (imidazole), and they vanished after the pyrolysis because the MOF structure could be changed to a M–Nx–C structure. It is worth noting that the relative ratio of pyrrolic N/pyridinic N was higher in Ni SAC-1000 compared to that of Ni SAC-800, indicating that some of the pyridinic N species were damaged during the pyrolysis process; meanwhile most of the pyrrolic N species remained in Ni SAC samples (Fig. 3d, S5b and Table S1†). When the pyridinic N species were removed from the carbon matrix, it will leave several defects such as vacancies and also can be confirmed by Raman spectroscopy results of the ID/IG value (Table S2†). Thus, it can be concluded that the N configuration coordinated in Ni species is strongly affected by thermal activation energies.
C (imidazole) (402.12 eV), pyrrolic N (401.48 eV), N–Ni (400.59 eV), N–Zn (399.13 eV), pyridinic N (398.55 eV), and N–H (imidazole) (398.24 eV), respectively (Fig. 3b).25,28,37,39 Ni ZIF-8 showed distinct MOF structure-related peaks of NC (imidazole) and N–H (imidazole), and they vanished after the pyrolysis because the MOF structure could be changed to a M–Nx–C structure. It is worth noting that the relative ratio of pyrrolic N/pyridinic N was higher in Ni SAC-1000 compared to that of Ni SAC-800, indicating that some of the pyridinic N species were damaged during the pyrolysis process; meanwhile most of the pyrrolic N species remained in Ni SAC samples (Fig. 3d, S5b and Table S1†). When the pyridinic N species were removed from the carbon matrix, it will leave several defects such as vacancies and also can be confirmed by Raman spectroscopy results of the ID/IG value (Table S2†). Thus, it can be concluded that the N configuration coordinated in Ni species is strongly affected by thermal activation energies.
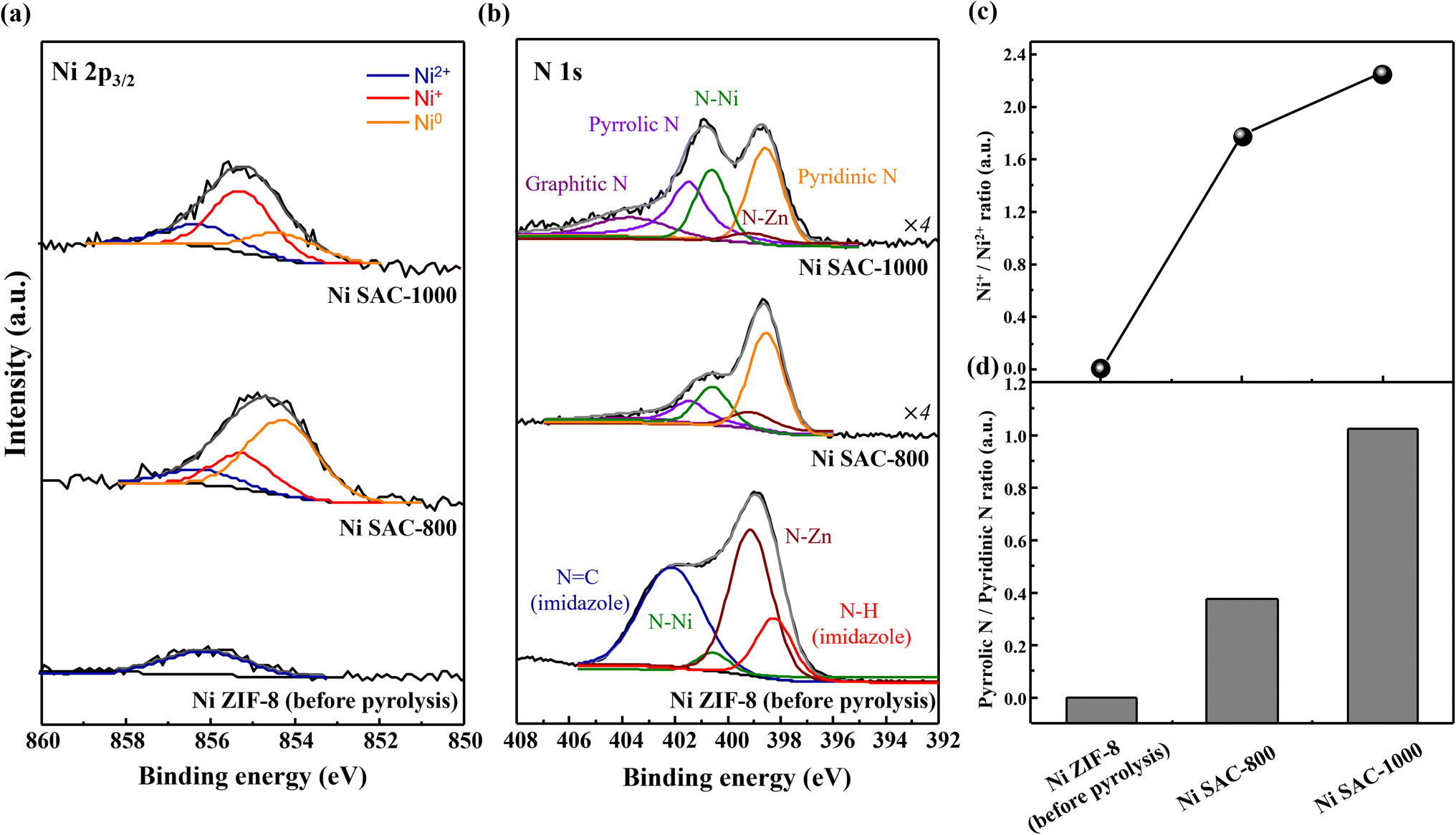 | ||
| Fig. 3 XPS spectra of (a) Ni 2p3/2 and (b) N 1s for Ni ZIF-8 (before pyrolysis) and the Ni SAC (after pyrolysis at 800 and 1000 °C). Relative integrated peak area ratio of (c) Ni+/Ni2+ content and (d) pyrrolic N/pyridinic N content in Ni-based samples. | ||
To identify the local electronic structure of the as-prepared samples, we further conducted XANES and EXAFS (Fig. 4). In the Ni K-edge XANES spectra, the near-edge absorption energy of Ni ZIF-8, Ni SAC-800, and Ni SAC-1000 is placed between those of reference Ni foil (Ni0) and NiPc (Ni2+), indicating a valence state between 0 and +2 (Fig. 4a). Also, the valence state of the as-prepared samples negatively shifted as the pyrolysis temperature increased, suggesting that Ni atoms in Ni SAC-1000 are in a relatively low-valent state compared to the other samples. The coordination environment of Ni atoms in the as-prepared samples was investigated by the corresponding FT-EXAFS. It showed the presence of Ni–N peaks and the absence of Ni–Ni peaks in the Ni SAC samples, indicating that Ni species are distributed at an atomic level in the carbon matrix (Fig. 4b and S6†). The average Ni–N coordination number in Ni ZIF-8, Ni SAC-800, and Ni SAC-1000 is 4.0, 3.14, and 2.63, respectively, suggesting that the Ni atoms were unsaturated with N species after the pyrolysis process (Table S3†). So, it indicated that Ni SAC samples have the configurations of Ni–Nx (x = 2 ∼ 4) and showed more unsaturated coordination environments in Ni SAC-1000 compared to that of Ni SAC-800. Moreover, after the pyrolysis, Ni–N interatomic distances gradually decreased compared to that of the MOF structure in NiPc (Ni–N4), suggesting that the pyrolysis process leads to the destruction of the Ni–N4 structures and forms structural defects in the carbon matrix. Furthermore, through the electron paramagnetic resonance (EPR) measurement technique, the g-value is calculated to be 2.14, indicating abundant nitrogen vacancies in Ni SAC-1000 (Fig. S7†). From these results, we conclude that the highly distributed Ni atoms in Ni SAC-1000 have a relatively low-coordinated and low-valent Ni state (Ni+–Nx) with a high content of pyrrolic N species compared to that of Ni SAC-800.
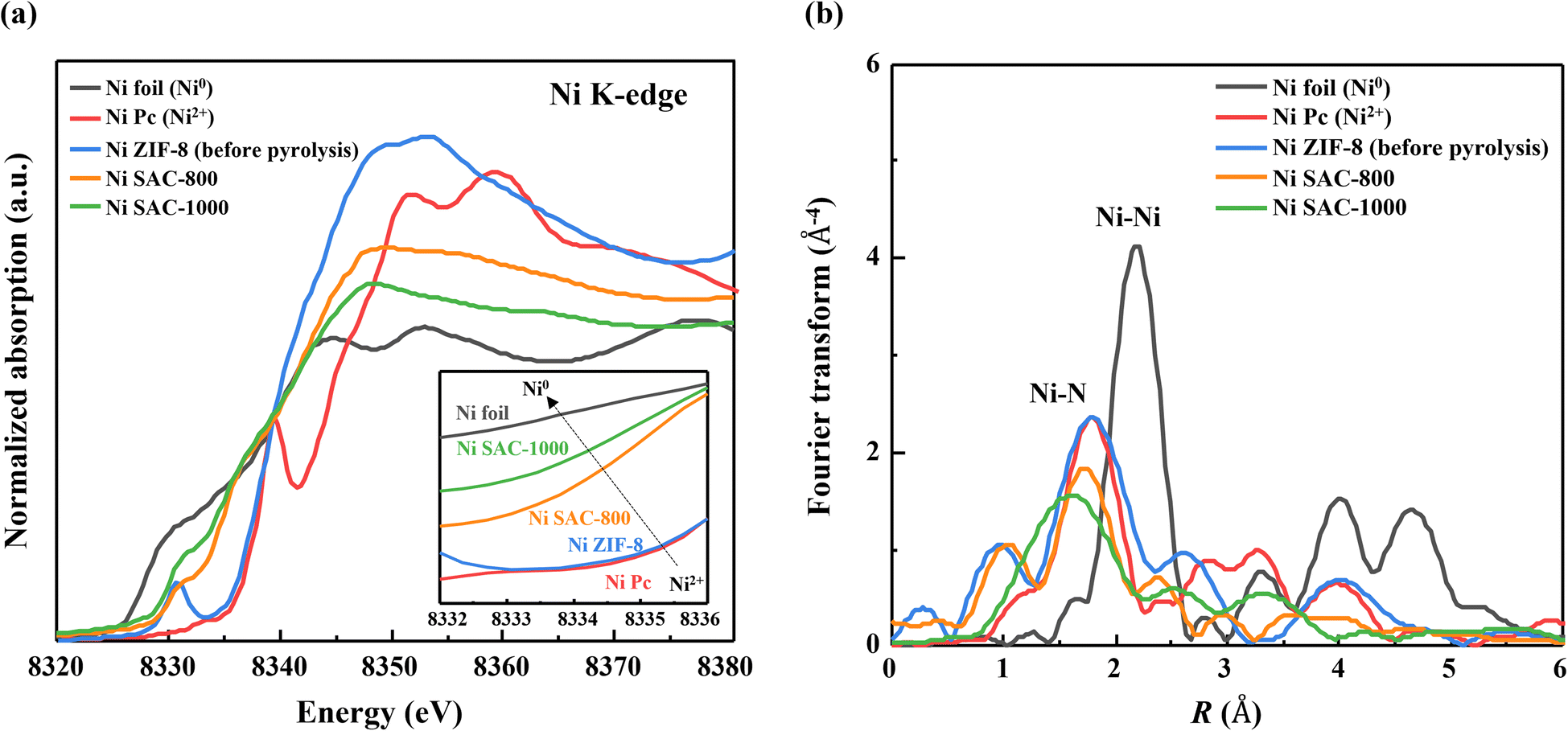 | ||
| Fig. 4 (a) Ni K-edge XANES spectra of Ni ZIF-8 (before pyrolysis), the Ni SAC (after pyrolysis at 800 and 1000 °C), and reference Ni foil and NiPc samples. (b) FT-EXAFS spectra of the Ni K-edge for Ni-based samples in the R space. | ||
Electrochemical CO2 reduction in a flow cell system
A conventional H-type cell system has a critical issue with restricted current densities by the limited solubility of CO2 in an aqueous electrolyte. Thus, we employed a GDE-based flow cell system and evaluated the Ni ZIF-8 (before pyrolysis), Ni SAC-800, and Ni SAC-1000 samples to identify the effect of pyrolysis temperature on the catalytic properties of the CO2RR in a CO2-purged electrolyte (Fig. 5a). LSV was conducted to investigate the current-related performance of the as-prepared samples in CO2 and N2-purged 0.5 M KHCO3 electrolyte at a scan rate of 100 mV s−1 (Fig. 5b). It showed more negative current densities under the CO2-purged electrolyte than that under the N2-purged one. This is because the reduction of CO2 and H2O can occur simultaneously under the former conditions but only H2O could be reduced under the latter one, indicating that there is an activity for CO2 reduction in all samples. Especially, Ni SAC-1000 showed the highest magnitude of current density among the samples (∼200 mA cm−2) and revealed a relatively higher value compared to that of the H-type cell system. The kinetics of the electron-transfer process were investigated by EIS measurements (Fig. 5c). The first semi-circle in the high-frequency region indicates a capacitive/resistive behavior between the solution, reference electrode, and working electrode constituted by the samples. The second semi-circle in the low-frequency region is the resistance from the charge transfer during an electrocatalytic reaction.40,41 The result that the radius of the second semi-circle is the smallest for Ni SAC-1000 means that the charge-transfer activity could be enhanced by increasing the pyrolysis temperature in as-prepared samples. ECSA measurement was conducted to identify the catalytically active surface area for the CO2RR (Fig. 5d and S8†). The double-layer capacitance (Cdl) of Ni SAC-1000 was 13.08 μF cm−2, which was the highest value among the samples. So, the larger active surface area of Ni SAC-1000 is one of the reasons for enhanced catalytic activity. The faradaic efficiencies (FEs) of the as-prepared samples were characterized at different applied potentials from −0.4 to −1.4 VRHE in CO2-purged 0.5 M KHCO3 electrolyte (Fig. 6a). Before pyrolysis, Ni ZIF-8 primarily produces H2 (FEH2 > 40%) with only a trace amount of CO (FECO < 10%). After pyrolysis at 800 °C, the FECO of Ni SAC-800 improved as high as 40.76% at −0.8 VRHE. As the pyrolysis temperature further increased to 1000 °C, the FECO of Ni SAC-1000 drastically increased to a maximum value of 98.24% at −0.8 VRHE, due to the increased content of catalytically active pyrrolic N species and abundant structural defects in Ni SAC-1000. Also, Ni SAC-1000 showed the highest partial current density of CO among the samples (jCO = 148.25 mA cm−2 at −1.4 VRHE) (Fig. 6b). The high selectivity and current density are due to the significantly suppressed H2 evolution in Ni SAC-1000 (FEH2 < 5% at all applied potentials) (Fig. 6c and Table S4†). For practical applications, the stability of catalysts is important for the CO2RR in CO2-purged electrolytes. The stability of Ni SAC-1000 was evaluated for 24 h at a constant potential of −0.8 VRHE in CO2-purged 0.5 M KHCO3 (Fig. 6d). To maintain the pH value of the electrolyte, CO2 gas was continuously provided to the electrolyte during the reaction. The total current density was almost constant for 24 h (∼50 mA cm−2 at −0.8 VRHE) and a high FECO of ∼95% was consistently obtained in long-term reactions. Moreover, there was negligible change in the morphology, crystal structure, and atomic environment of Ni SAC-1000 before and after the reaction for 24 h, indicating that Ni SAC-1000 maintained its catalytic activities and chemical stability for a long-term reaction in the microfluidic flow cell system (Fig. S9†). Thus, the thermal activation in the as-prepared samples was found to be beneficial not only for the catalytic activity but also for excellent stability. Furthermore, we investigated the electrochemical stability of Ni-SAC-1000 through 1000 cycles in cyclic voltammetry (CV), confirming its ability to impede nickel aggregation (Fig. S10†).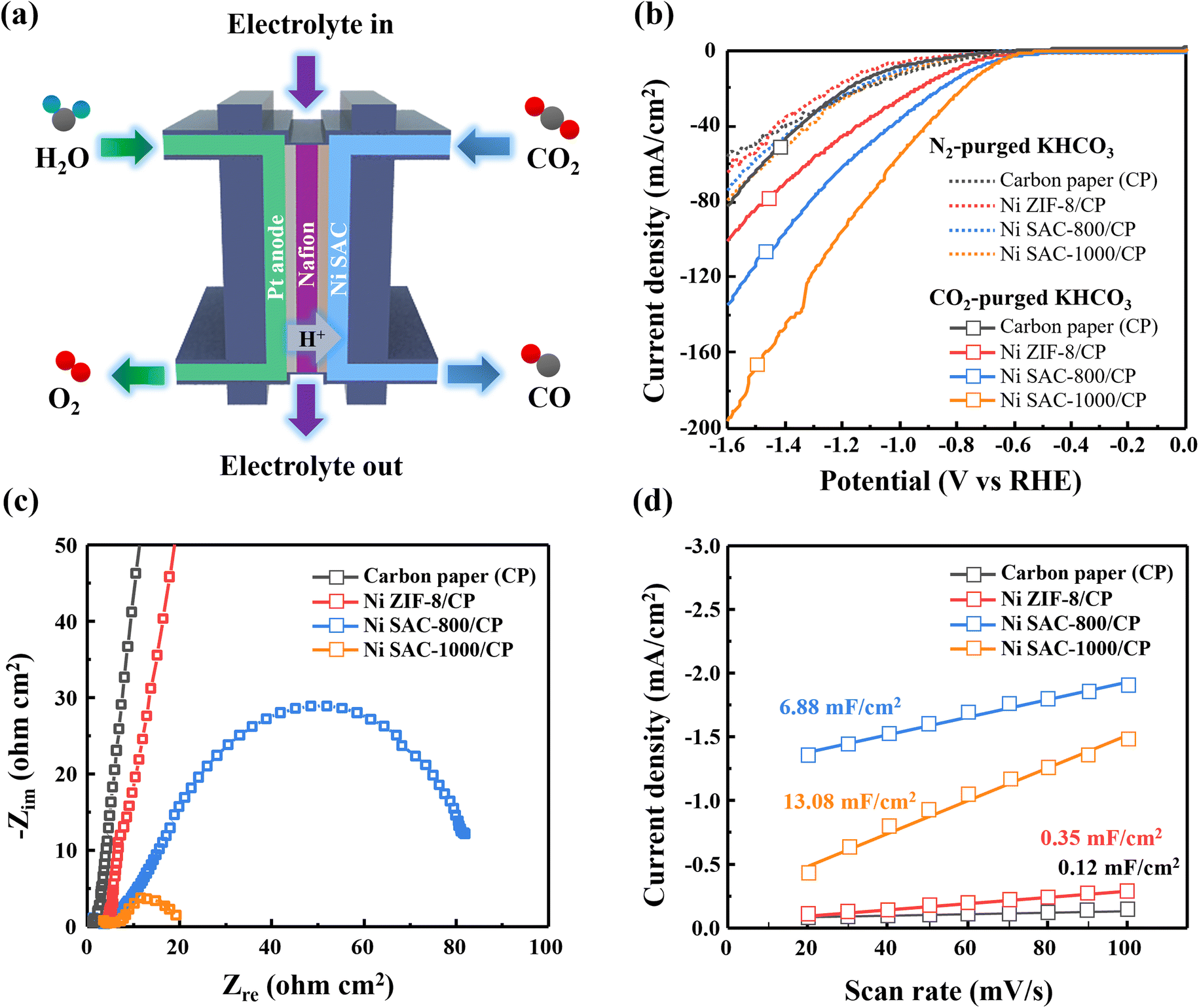 | ||
| Fig. 5 (a) High-rate CO2 electrolysis with a gas and electrolyte-feed flow cell system for Ni-based samples. The results of (b) LSV, (c) Nyquist impedance, and (d) ECSA measurements for Ni ZIF-8 (before pyrolysis) and the Ni SAC (after pyrolysis at 800 and 1000 °C). | ||
 | ||
| Fig. 6 (a) FECO and (b) jCO at various applied potentials for Ni-based samples in CO2-purged 0.5 M KHCO3 electrolytes. (c) Catalytic activities of Ni-based samples before and after pyrolysis at −0.8 VRHE. (d) Stability of Ni SAC-1000 at −0.8 VRHE in CO2-purged 0.5 M KHCO3. Current density (solid line) and FECO (symbol) at −0.8 VRHE were plotted as a function of reaction time. (e) Summary of the catalytic activities of the Ni-based SAC for the production of CO measured with a conventional H-cell and microfluidic flow cell system. | ||
Up to now, the study of transition metal-based SACs has been mainly conducted by using a conventional H-type cell system (Fig. 6e and Table 1). However, scaling up of the productivity of CO2 reduction products such as CO is limited in a traditional H-type cell because it requires larger overpotentials and the CO2 mass diffusion can be restricted in the electrolytes. In this work, we employed a 3-compartment microfluidic flow cell reactor, which contained a circulated anolyte, catholyte, and continuous gas-phase CO2 supply, to boost the current density while maintaining high CO selectivity. Ni SAC-1000 maintained FECO > 95% at potentials from −0.8 to −1.4 VRHE and showed significantly higher current density (∼200 mA cm−2) compared to that of the H-type cell system (∼20 mA cm−2) (Fig. S11†). Thus, the high performance of Ni SAC-1000 in the microfluidic flow cell system provides great opportunities for scaling up the productivity of CO2 reduction products toward the industrial level.
| Catalyst | FECO (%) | j CO (mA cm−2) | Potential (V) | Electrolyte | Cell system | Ref. |
|---|---|---|---|---|---|---|
| Ni–N–C | 94% | ∼10 mA cm−2 | −0.60 VRHE | 0.5 M KHCO3 | H-cell | Adv. Funct. Mater., 2022, 32, 2202351 |
| Ni–Zn–N–V | 99% | 17 mA cm−2 | −0.80 VRHE | 0.5 M KHCO3 | H-cell | Adv. Mater., 2021, 33, 2102212 |
| Ni-SAC@NC | 95% | 5.7 mA cm−2 | −0.60 VRHE | 0.5 M KHCO3 | H-cell | Appl. Catal. B: Environ., 2022, 304, 120997 |
| Ni–N–CNSs | 95% | 5.5 mA cm−2 | −0.75 VRHE | 0.5 M KHCO3 | H-cell | J. Colloid Interface Sci., 2020, 570, 31–40 |
| Ni(NC)-1 | 99% | ∼10 mA cm−2 | −0.75 VRHE | 0.5 M KHCO3 | H-cell | ACS Catal., 2020, 10, 1086–1093 |
| Ni@NCNTs | 99.1% | ∼13 mA cm−2 | −0.90 VRHE | 0.5 M KHCO3 | H-cell | Carbon, 2019, 150, 52–59 |
| Ni/NC | 96.5% | 12.6 mA cm−2 | −0.90 VRHE | 0.1 M KHCO3 | H-cell | ACS Sustainable Chemi. Eng., 2019, 7, 15030–15035 |
| Ni–NC_ATPA@C | 90% | ∼16 mA cm−2 | −0.90 VRHE | 0.5 M NaHCO3 | H-cell | Chem. Sci., 2018, 9, 8775–8780 |
| Ni–N–C | 80% | 11.2 mA cm−2 | −0.75 VRHE | 0.1 M KHCO3 | H-cell | Nat. Commun., 2017, 8, 944 |
| Ni–N–C | 97% | 22 mA cm−2 | −0.72 VRHE | 0.5 M KHCO3 | H-cell | Nat. Energy, 2018, 3, 140−147 |
| Ni–N–C | 81% | 15 mA cm−2 | −0.84 VRHE | 0.1 M KHCO3 | H-cell | Energy Environ. Sci., 2019, 12, 640–647 |
| Ni1–N–C | 96.8% | 27 mA cm−2 | −0.80 VRHE | 0.5 M KHCO3 | H-cell | Angew. Chem. Int. Ed., 2020, 59, 20589–20595 |
| NiN–graphene layers | 90% | 65 mA cm−2 | −0.79 VRHE | 0.5 M KHCO3 | Flow cell | Chem, 2017, 3, 950 |
| Ni3N/C | 92.5% | 23 mA cm−2 | −0.79 VRHE | 0.5 M NaCl | Flow cell | ACS Appl. Mater. Interfaces, 2018, 10, 38024–38031 |
| Ni–NG | 97% | 50 mA cm−2 | 2.80 Vcell | 0.5 M KHCO3 | Flow cell | Energy Environ. Sci., 2018, 11, 893–903 |
| ZnNi–DCN-1000 | 90% | ∼40 mA cm−2 | −0.50 VRHE | 1 M KOH | Flow cell | Adv. Funct. Mater., 2022, 32, 2203842 |
| Ni-SA/NC | 96.9% | ∼25 mA cm−2 | −0.66 VRHE | 1 M KOH | Flow cell | Nano Energy, 2021, 82, 105689 |
| Pyrrolic N-stabilized Ni SAC | 98.2% | 45.03 mA cm−2 | −0.80 VRHE | 0.5 M KHCO3 | Flow cell | This work |
| 95.1% | 148.25 mA cm−2 | −1.40 VRHE |
DFT calculations
To identify the reason for the increased catalytic activity of Ni SAC-1000 compared to that of Ni SAC-800 in the CO2RR, we have to consider the effect of the coordination environment of Ni species (Ni–N) and the ratio of pyrrolic N/pyridinic N in the carbon matrix. Thus, DFT calculations were employed to understand the enhanced selectivity of CO on Ni SAC-1000 (Fig. 7). For the study, we considered possible reaction mechanisms for the CO2RR in the 2e− reduction pathway to produce CO.42 First, adsorbed gaseous CO2 on the catalytic surface is reduced to the *COOH intermediate by a proton-coupled electron transfer reaction. The *COOH can be further reduced to the *CO intermediate by an additional proton/electron from the electrolyte and lastly, the *CO intermediate detached from the catalytic surface, resulting in the formation of gaseous CO products.43 Previous studies suggested that the rate-determining step (RDS) could be the reaction (1), (2), or (4) depending on the binding energy of the reaction intermediate.44| CO2 (g) + * → *CO2 | (1) |
| *CO2 + H+ (aq) + e− → *COOH | (2) |
| *COOH + H+ (aq) + e− → *CO + H2O (l) | (3) |
| *CO → CO (g) + * | (4) |
 | ||
| Fig. 7 (a) Free-energy profiles for the CO2RR to CO on Ni SACs with different Ni–Nx coordination environments. (b) Optimized calculation models for pyrrolic N stabilized Ni SACs with Ni–N2V2 sites. (c) Charge density difference plot on CO adsorbed Ni–N2V2 with pyrrolic N configuration, where yellow-colored and cyan-colored isosurfaces represent the boundary of the charge depletion region and charge accumulation region, respectively. (d) Sliced 2-dimensional charge density difference plot of a CO adsorbed single atom Ni site. | ||
Especially, the step for desorption of the *CO intermediate is critical because the binding energy of *CO on the catalytic surface can tune the selectivity of the CO2RR products. The relatively weak binding of *CO (low desorption energy of *CO) can produce gaseous CO; meanwhile strongly bonded *CO (high desorption energy of *CO) could suppress CO production. Therefore, it is important to reduce the energy barrier of the *CO desorption step to achieve high selectivity of CO production.45
In our DFT calculations, we compared the Gibbs free energy change (ΔG) of reaction intermediates of *COOH and *CO on the Ni SAC on various Ni–N configurations (Fig. 7a).
In terms of the *CO desorption step, it showed that the reaction barrier of desorption of the *CO intermediate is relatively smaller in pyrrolic N sites compared to those of pyridinic N sites regardless of containing vacancies. Notably, Ni–N2V2 with pyrrolic N sites revealed the favorable formation of CO products due to its lowest desorption energy of the *CO intermediate (Fig. 7b). In our study, when the pyrolysis temperature increased from 800 °C (Ni SAC-800) to 1000 °C (Ni SAC-1000), the content ratio of pyrrolic N/pyridinic N increased from 0.37 to 1.01 as well as the coordination number of Ni in Ni–Nx sites decreased from 3.14 to 2.63. We additionally conducted charge density generation calculation on *CO adsorbed Ni–N2V2-pyrrolic (adsorbate + slab), CO (adsorbate), and Ni–N2V2-pyrrolic (slab). Then we calculated the charge density difference induced by the CO adsorption by volumetric subtraction of the spatial charge density of Ni–N2V2-pyrrolic (slab) and CO (adsorbate) from that of *CO adsorbed Ni–N2V2-pyrrolic (adsorbate + slab), which was then plotted as a charge density difference plot in Fig. 7. The yellow-colored isosurface of Fig. 7c represents the boundary of the charge depletion region, which matches with the positive oxidation state of the Ni single atom center. The cyan-colored isosurface represents the boundary of the charge accumulation region and the strong bond bridged with accumulated electron density between Ni and C of the CO adsorbate was confirmed in the 2-dimensional sliced charge density plot in Fig. 7d. As a result, we conclude that the synergistic effect between the high content ratio of pyrrolic N and low-coordinated Ni can decrease the energy barrier for the desorption of the *CO intermediate during the CO2RR, leading to high production of CO in Ni SAC-1000 (FECO = 98.24% at −0.8 VRHE and jCO = 148.25 mA cm−2 at −1.4 VRHE) compared to that of Ni SAC-800 (FECO = 40.76% at −0.8 VRHE and jCO = 4.41 mA cm−2 at −1.4 VRHE).
Spin-polarized partial density of states (PDOS) calculations on 5 configurations without adsorbed intermediates, namely pristine (a) Ni–N2V2-pyrrolic, (b) Ni–N3V1-pyrrolic, (c) Ni–N4-pyrrolic, (d) Ni–N3V1-pyridinic, and (e) Ni–N4-pyridinic, are plotted in Fig. S12.† We calculated the Ni d-band center position and it turned out that if the Ni d-band center position was closer to the Fermi-level, the reaction energy barrier of the CO2 to CO pathway was reduced. The Ni d-band center of Ni–N2V2-pyrrolic configuration was calculated to be −0.71 eV, which was the lowest among the 5 pristine configurations without adsorbed intermediates and showed the lowest reaction energy barrier of 0.149 eV toward the CO pathway. In contrast, Ni–N4-pyridinic configuration showed the largest reaction energy barrier of 1.112 eV toward the CO pathway and the lowest Ni d-band center position of −2.09 eV. The closer Ni d-band center to the Fermi level induces the optimal *CO binding strength via orbital coupling with the C and O orbitals of the adsorbate near the Fermi level, which eventually leads to the facile adsorption and desorption of *CO at the same time. We also conducted partial density of states calculations on 5 different configurations with adsorbed CO on the Ni site and calculated the Ni d-band center in Fig. S13.†
The HCOOH production pathway and H2 evolution reaction pathway are the two representative competing reactions toward the electroreduction of CO2 to CO pathway, and we revisited our structural models and placed adsorbates for those competing reactions (Fig. S14†). It turned out that the reaction energy barrier for both HCOOH production and H2 evolution reaction pathways is higher than that of the CO2 to CO pathway, which ensured outstanding selectivity and faradaic efficiencies toward CO higher than 95%.
Conclusions
In summary, we have demonstrated a pyrrolic N-stabilized Ni SAC with low-coordinated Ni–Nx sites by using a thermal activation of Ni ZIF-8. We found that Ni SAC-800 and -1000 showed atomically dispersed Ni species with low-coordinated N sites in the carbon matrix and inhibited the formation of Ni nanoparticles as confirmed by HAADF-STEM and XAS. As the pyrolysis temperature increased from 800 to 1000 °C, the coordination number of Ni–Nx sites decreased from 3.14 to 2.63 and increased to a relative content ratio of pyrrolic N/pyridinic N from 0.37 to 1.01. The synergistic effect between the low-coordinated Ni–Nx sites and the high content ratio of pyrrolic N/pyridinic N decreases the energy barrier for the desorption of the *CO intermediate during the CO2RR, which enhanced the catalytic performances in Ni SAC-1000 (FECO = 98.24% at −0.8 VRHE) compared to that of Ni SAC-800 (FECO = 40.76% at −0.8 VRHE). Moreover, we employed a microfluidic flow cell system to boost catalytic productivity at the industrial level. Ni SAC-1000 showed relatively outstanding current density (∼200 mA cm−2) in the flow cell system compared to that of conventional H-type cell configuration (∼20 mA cm−2). The FECO keeps well above 95% at potentials from −0.8 to −1.4 VRHE and it showed excellent stability to produce CO during long-term operation for 24 h. This work experimentally demonstrated the relationship between local N configuration in Ni–Nx sites and catalytic activities for the CO2RR in a flow cell system, providing guidelines for designing highly productive CO selective Ni SACs.Author contributions
J. W. Lim, D. H. Choo, and J. H. Cho conceptualized the idea and experimental design. J. W. Lim and J. H. Cho, with support from S. Lee, K. Kim, J. Son, and S. Y. Choi, prepared the catalysts and assessed the electrochemical CO2 reduction performances. W. S. Cho and O. F. N. Okello contributed to the material characterization of the catalysts. J. Kim conducted the DFT calculations under the supervision of H. W. Jang and S. Y. Kim. The overall work was supervised by J. K. Kim and J.-L. Lee, with all authors actively participating in the discussion of the results.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (20212010100040), and in part by the Brain Korea 21 PLUS project for Education and Research center for future materials (F21YY7105002), and in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00209139).References
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Energy Technol., 2017, 5, 834–849 CrossRef.
- W. S. Cho, D. M. Hong, W. J. Dong, T. H. Lee, C. J. Yoo, D. Lee and J.-L. Lee, Energy Environ. Mater., 2024, 7, e12490 CrossRef CAS.
- F. M. Baena-Moreno, M. Rodríguez-Galán, F. Vega, B. Alonso-Fariñas, L. F. V. Arenas and B. Navarrete, Energy Sources, Part A, 2019, 41, 1403–1433 CrossRef CAS.
- J. W. Lim, W. J. Dong, J. Y. Park, D. M. Hong and J.-L. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 22891–22900 CrossRef CAS PubMed.
- J. W. Lim, W. J. Dong, W. S. Cho, C. J. Yoo and J.-L. Lee, ACS Catal., 2022, 12, 13174–13185 CrossRef CAS.
- M. E. Dry, J. Chem. Technol. Biotechnol., 2002, 77, 43–50 CrossRef CAS.
- J. Y. Park, W. J. Dong and J.-L. Lee, ACS Appl. Energy Mater., 2022, 5, 1627–1634 CrossRef CAS.
- J. Y. Park, W. J. Dong, S.-M. Jung, Y.-T. Kim and J.-L. Lee, Appl. Catal., B, 2021, 298, 120586 CrossRef CAS.
- W. J. Dong, J. W. Lim, J. Y. Park, C. J. Yoo, S. Baek, W. S. Cho, W. Kim and J.-L. Lee, Appl. Surf. Sci., 2021, 565, 150460 CrossRef CAS.
- C. J. Yoo, W. J. Dong, J. Y. Park, J. W. Lim, S. Kim, K. S. Choi, F. O. O. Ngome, S.-Y. Choi and J.-L. Lee, ACS Appl. Energy Mater., 2020, 3, 4466–4473 CrossRef CAS.
- T. S. Safaei, A. Mepham, X. Zheng, Y. Pang, C.-T. Ding, M. Liu, D. Sinton, S. O. Kelley and E. H. Sargent, Nano Lett., 2016, 16, 7224–7228 CrossRef PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- W. J. Dong, C. J. Yoo, J. W. Lim, J. Y. Park, K. Kim, S. Kim, D. Lee and J.-L. Lee, Nano Energy, 2020, 78, 105168 CrossRef CAS.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chem., Int. Ed., 2016, 55, 9297–9300 CrossRef CAS PubMed.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- W. J. Dong, J. W. Lim, D. M. Hong, J. Kim, J. Y. Park, W. S. Cho, S. Baek and J.-L. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 18905–18913 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301–1306 CrossRef CAS PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- R. Boppella, M. A. P, Y. Kim, E. Kim, I. Song, Y. Eom, P. Kumar, M. Balamurugan, E. Sim, D. H. Kim and T. K. Kim, Adv. Funct. Mater., 2022, 32, 2202351 CrossRef CAS.
- B. Wulan, X. Cao, D. Tan, X. Shu and J. Zhang, Adv. Funct. Mater., 2022, 32, 2203842 CrossRef CAS.
- Q. Wang, T. Ina, W.-T. Chen, L. Shang, F. Sun, S. Wei, D. S. Waterhouse, S. G. Telfer, T. Zhang and G. I. N. Waterhouse, Sci. Bull., 2020, 65, 1743–1751 CrossRef CAS PubMed.
- Y. Guo, S. Yao, Y. Xue, X. Hu, H. Cui and Z. Zhou, Appl. Catal., B, 2022, 304, 120997 CrossRef CAS.
- S. Li, M. Ceccato, X. Lu, S. Frank, N. Lock, A. Roldan, X.-M. Hu, T. Skrydstrup and K. Daasbjerg, J. Mater. Chem. A, 2021, 9, 1583–1592 RSC.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, ACS Catal., 2020, 10, 10920–10931 CrossRef CAS.
- S. A. Abbas, J. T. Song, Y. C. Tan, K. M. Nam, J. Oh and K.-D. Jung, ACS Appl. Energy Mater., 2020, 3, 8739–8745 CrossRef CAS.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., Int. Ed., 2015, 127, 13905–13909 CrossRef.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem, 2018, 1, 0105 CrossRef.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitaski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifaitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Q. Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- B. Shen, B. Wang, L. Zhu and L. Jiang, Nanomaterials, 2020, 10, 1636 CrossRef CAS PubMed.
- X. Wu, J. Chen, M. Wang, X. Li, L. Yang, G. Li, L. Shan, X. Li, Y. Lin and J. Jiang, Chem. Commun., 2022, 58, 2914–2917 RSC.
- D. Muñoz-Gil and F. M. L. Figueiredo, Nanomaterials, 2019, 9, 1369 CrossRef PubMed.
- B. Hirschorn, M. E. Orazem, B. Tribollet, V. Vivier, I. Frateur and M. Musiani, Electrochim. Acta, 2010, 55, 6218–6227 CrossRef CAS.
- A. Sacco, J. CO2 Util., 2018, 27, 22–31 CrossRef CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- X. Wang, Z. Mao, G. Wei, Y. Zhang, B. Li, L. Zhu, S.-M. Xu, J. Jiang and S. Tang, Appl. Surf. Sci., 2023, 639, 158243 CrossRef CAS.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv.Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- Y. Zhang, T. Liu, X. Wang, Q. Dang, M. Zhang, S. Zhang, X. Li, S. Tang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 9073–9083 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06399b |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |