
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining NMR and impedance spectroscopy in situ to study the dynamics of solid ion conductors†
Sheyi Clement
Adediwura
 ,
Neeshma
Mathew
and
Jörn
Schmedt auf der Günne
*
,
Neeshma
Mathew
and
Jörn
Schmedt auf der Günne
*
Department of Chemistry and Biology, Faculty IV: School of Science and Technology, Inorganic Materials Chemistry and Center of Micro- and Nanochemistry and Engineering (Cμ), University of Siegen, Adolf-Reichwein-Straße 2, 57068 Siegen, Germany. E-mail: gunnej@chemie.uni-siegen.de
First published on 23rd May 2024
Abstract
Differences in activation energies of solid ion conductors as measured by different techniques can be either considered a consequence of experimental uncertainty or a valuable source of information about the local ion transport mechanism. Here, it is suggested that an in situ combination of solid-state nuclear magnetic resonance (NMR) and electrochemical impedance spectroscopy (EIS) helps to differentiate these two cases because the sample preparation and thermal history are then identical for the NMR and EIS measurement. To this end, an in situ NMR–EIS probe head is developed, calibrated and its performance is tested on the lithium-ion conductor Li8SnO6. For validation, the results were compared to carefully conducted ex situ measurements. The differences in activation energies as observed by NMR and EIS experiments could in this case be confirmed and rationalized by comparison to results from nudged elastic band calculation using density functional theory under periodic boundary conditions.
1 Introduction
The progress of solid-state batteries is strongly correlated to the discovery of solid ion conductors with high ionic conductivity of >10−2 S cm−1 at room temperature.1–3 Improving the ion transport efficiency and capacity retention in solid-state batteries requires a holistic understanding of the conduction mechanism, especially concerning ion transport across the electrolyte/electrode interface.2–4 Several techniques have been deployed to characterize the ion transport properties of solid materials at different length and time scales.5–7 The commonly used approaches to determine the ionic conductivity and diffusion properties of materials are: electrochemical impedance spectroscopy (EIS) which probes long-range charge transport in the bulk and at interfaces,8 NMR diffusometry which probes long-range diffusion,9,10 line-shape and spin-alignment echo solid-state nuclear magnetic resonance spectroscopy probe ionic motion on a short to medium range,11–13 and the conventional spin–lattice relaxometry solid-state nuclear magnetic resonance spectroscopy which probes short-range motion.11,14Together, these techniques provide a comprehensive understanding of the charge and mass transport associated with ionic motion. The respective activation energies for mass and charge transport, which are obtained from experiments conducted at different temperatures, provide an insight into whether the corresponding motional processes leading to mass and charge transport are identical. A literature review reveals in several cases differences in activation energies from impedance and nuclear magnetic resonance spectroscopy.13,15–17 While there is often a good agreement within error limits, differences remain a valuable source of information especially when techniques with different inherent time and length scales explore the complicated energy landscape featuring one or more barriers of disordered ionic conductors.18 While some of the differences can be explained by the different length and time scales of the techniques, even between different NMR techniques used to study macros- and microscopic transport,5,13 differences in activation energy can also just be a consequence of the experimental conditions (see below). According to a recent round-robin test19 conducted on six reference compounds in the form of a powder, the measured ionic conductivity for the same material varies by about one order of magnitude between eight participating laboratories around the world. It is not unreasonable to assume that also activation energies are subject to sizeable errors, especially because variable temperature and pressure conditions used in impedance spectroscopy can lead to processes modifying the grain boundaries.20–22 Furthermore, some of these changes are not detectable by certain routine structural characterisation techniques such as X-ray diffraction for after-effect analysis.20
Discrepancies between activation energies from different methods are in fact common place. As an example for the fast Li-ion conductor Li7La3Zr2O12, the reported values are 0.3–0.34 eV measured by impedance spectroscopy,23 0.53 eV by spin-alignment echo NMR,24 0.43 eV by line shape NMR and 0.32–0.48 eV by spin–lattice relaxometry NMR,25 while 0.33–0.36 eV and 0.26 eV were predicted by initio molecular dynamics simulation,26,27 and by nudged elastic band calculations,28 respectively. Another less prominent example is the Li-rich phase Li8SnO6,29,30 which will serve as a test case for this contribution, for which a consistent interpretation of the different reported activation energies (NMR: 0.31 eV,31 EIS: 0.91 eV,31 quantum chemistry: 0.43–1.40 eV,32 forcefield calculations: 0.20–1.06 eV (ref. 33)) is still missing. The structure of Li8SnO6 is surprisingly complicated in the sense that it features two different Li sites with an octahedral and a tetrahedral coordination polyhedron which are a potential source of dynamic heterogeneity of the Li motion.30,32,33
One way to overcome the experimental uncertainties related to the determination of activation energies is to change from ex situ to in situ NMR–EIS measurements so that differences cannot be blamed on sample related changes. To the best of our knowledge, such a setup has not been reported yet, which may be related to the problems associated with separating the required electromagnetic waves whose frequencies are only different by one order of magnitude. Technically much simpler is the separation of a direct current channel from an NMR frequency channel, which in the context with batteries is known as operando NMR. It provides insight into electrode materials while charging and discharging, so that metastable intermediates and structural changes could be identified:34–42 For the anode it could be shown that the structure of graphite intercalation compounds correlated with the charge state.35–39 Lithium dendrites, which need to be avoided can be traced in the early stages.42 Silicon as anode material or as an additive to carbon-based anodes could be studied40 to identify intermediates at different states of charging. In this context, the combination with ex situ EIS was important41 to get a better understanding of the cell resistance as a function of the charging state of the battery.
The target of this contribution is to develop an in situ NMR–EIS setup, evaluate its performance by comparing ex situ and in situ measurements and demonstrate its advantages on the example of the compound Li8SnO6.
2 Experimental part
2.1 3D printing
All models were designed using FreeCAD v0.20 software, then sliced with Ultimaker cura v5.1.0 and Intamsuite v3.7.0. The prototypes were developed using a Creality CR-10s 3D printer, (Cordol Technology Co., Limited) and the final version was printed with a high-temperature 3D printer, INTAMSYS FUNMAT HT (INTAMSYS). The printers were configured as shown in (Table S1†). All prototypes were printed with a hardened steel nozzle with 0.4 mm diameter at 0.1 mm layer height which is a compromise between printing quality and time. The high temperature version was printed with a nozzle with a 0.25 mm diameter using the same layer height as the prototypes. Parts with thermoplastic filaments were sintered in an oven at a temperature of 473 K for 2 hours to increase their thermal strength. The printed parts were afterwards incorporated into a home-built static probe head based on a McKay design.2.2 Sample preparation and characterization
to estimate the lithium ion jump rate 1/τ, where S0 is the final state correlation value and β is the stretching factor. The spin alignment echo measurements were conducted at sufficiently high temperatures to separate the contributions of quadrupolar spin–lattice relaxation T1Q from contributions from individual jumps of the spins between electrically non-equivalent crystallographic orbits. The NMR signals were deconvoluted and their second moments of the lineshape were determined using Deconv2DXY v0.7,50 while the lithium ion jump rates 1/τ from spin alignment echo decay curves were analysed using home written python script based on SciPy libraries.51
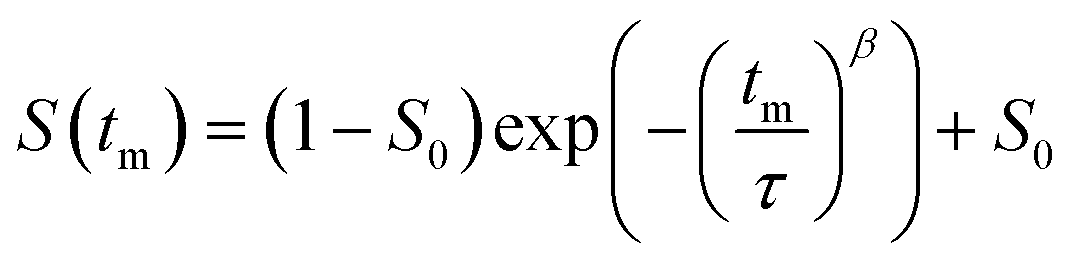
2.3 Quantum chemistry
The plane wave pseudopotential method based on density functional theory under periodic boundary conditions as implemented in Quantum Espresso (QE) 6.8 (ref. 52) was utilised to estimate activation energies of the lithium-ion diffusion pathways in Li8SnO6. The crystal structure of Li8SnO6 in the space group R3 (hexagonal axes, ICSD collection code 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 131)29 served as a structure model. The cif file of Li8SnO6 was converted into an input file using cif2cell 2.0.0 (ref. 53) for the QE calculations. Vacancy models were generated using a compensating background charge and all fractional coordinates were allowed to relax. The QE calculations employed pseudopotentials of the GBRV type54,55 and k-point selection followed the Monkhorst–Pack scheme.56 The presented simulations used a 2 × 2 × 3 k-mesh and a supercell containing 180 atoms (convergence error 0.001 eV). The supercells were optimized to the most stable configuration by setting up a kinetic energy cut-off of 480 eV and 60 eV for the charge density cut-off (convergence error ∼0.01 eV). Climbing Image-Nudged Elastic Band (CI-NEB) calculations using QE were performed to calculate the activation energy of a vacancy moving from one Li position to a neighbouring one.57 Eight intermediate images were used along with the first and last images to identify the diffusion pathway for the Li-ion transport. These calculations provide an estimate for the saddle-point of the movement of the vacancy between Li sites, i.e. the minimum energy path in the energy-hypersurface and thus an estimate for the activation energy.
131)29 served as a structure model. The cif file of Li8SnO6 was converted into an input file using cif2cell 2.0.0 (ref. 53) for the QE calculations. Vacancy models were generated using a compensating background charge and all fractional coordinates were allowed to relax. The QE calculations employed pseudopotentials of the GBRV type54,55 and k-point selection followed the Monkhorst–Pack scheme.56 The presented simulations used a 2 × 2 × 3 k-mesh and a supercell containing 180 atoms (convergence error 0.001 eV). The supercells were optimized to the most stable configuration by setting up a kinetic energy cut-off of 480 eV and 60 eV for the charge density cut-off (convergence error ∼0.01 eV). Climbing Image-Nudged Elastic Band (CI-NEB) calculations using QE were performed to calculate the activation energy of a vacancy moving from one Li position to a neighbouring one.57 Eight intermediate images were used along with the first and last images to identify the diffusion pathway for the Li-ion transport. These calculations provide an estimate for the saddle-point of the movement of the vacancy between Li sites, i.e. the minimum energy path in the energy-hypersurface and thus an estimate for the activation energy.
3 Results and discussion
An in situ NMR–EIS technique (Fig. 1) requires a setup in which nuclear magnetic resonance and impedance spectroscopy can be conducted at different temperatures on the same compound. To show such a setup is functional, in the first two parts of this chapter.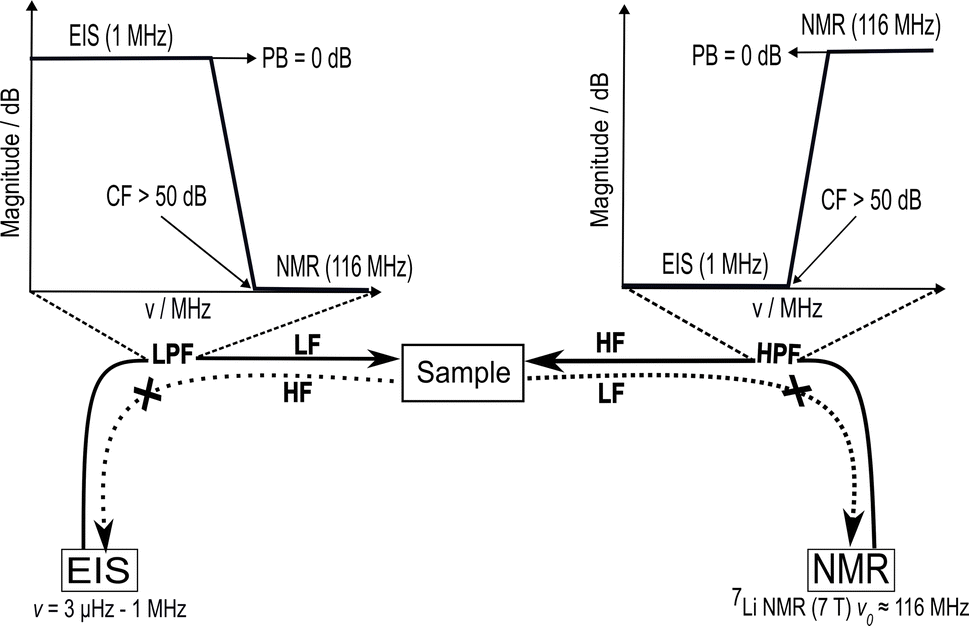 | ||
| Fig. 1 Sketch of the in situ NMR–EIS setup to perform electrochemical impedance spectroscopy (EIS) and solid-state nuclear magnetic resonance (NMR) spectroscopy on the same sample. This technique requires a sufficient electrical separation of the high-frequency (HF) channel, operating at the NMR (Larmor) frequency v0 of 116 MHz in the presented work, and the low-frequency (LF) channel for EIS operating in a frequency range between 3 μHz and 1 MHz. This separation requires low-pass and high-pass filters on the opposite channels to achieve an attenuation of at least 50 dB at the cut-off frequency while the pass-band remains unattenuated (<0.05 dB) up to frequencies required for the in situ measurements. | ||
1. Design, fabrication and calibration of the in situ NMR–EIS probe head and
2. Validation: ex situ versus in situ measurements on Li8SnO6 will be presented. Finally, the experimental results for the activation energies will be compared to
3. Activation energies for Li8SnO6 from quantum chemical calculations.
3.1 In situ NMR–EIS probe head
The design targets for the in situ NMR–EIS probe head can be described as follows. The probe head needs to separate a high (NMR) from a low (EIS) frequency channel, which is achieved by introducing electrical high and low pass filter elements into the circuits. The probe head needs to cope with an extended cable length of about 5 m on the EIS side which is a consequence of the required distance to the superconducting magnet. The sample cell must allow variable temperatures, provide a reproducible and stable contact for the EIS electrodes and have a sufficiently big frequency range to conduct impedance spectroscopy measurements. While the design targets may look similar to the ones of an in situ NMR battery cycler, the proximity of the frequency ranges of the two channels of the in situ EIS–NMR probe head requires other solutions, including a different design of the sample cell, calibration and filter specifications (see below).By design (Fig. 2), the sample sits in between two metallic electrodes connected to an impedance spectrometer inside a solenoid coil. Considering the skin depth associated with a radio frequency pulse and the magnetic susceptibility difference of the materials,58,59 this configuration is expected to have both an inhomogeneity in the pulse B1 and magnetic field B0.
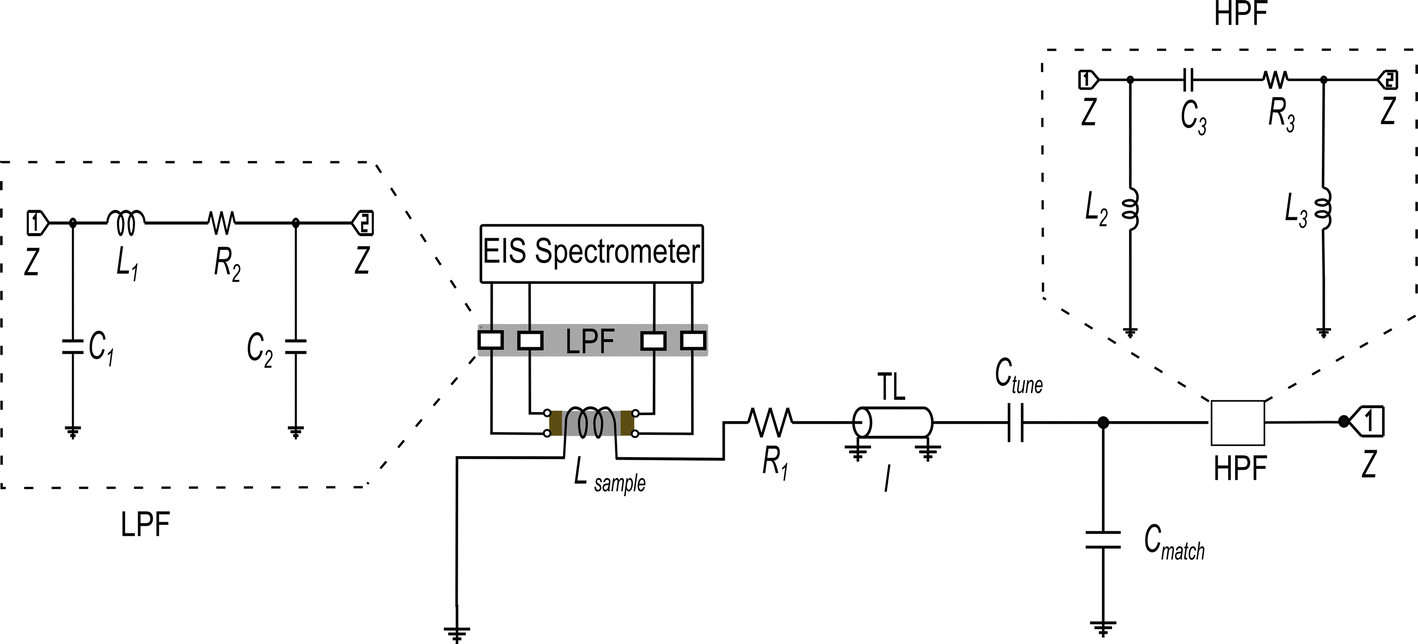 | ||
| Fig. 2 Schematic circuit diagram of 7Li single resonance in situ NMR–EIS probe with a solenoid coil for the NMR wide-bore magnet. The NMR circuit was tuned and matched with the help of the variable capacitors Ctune and Cmatch to the resonance frequency of 7Li of 116.64 MHz. Inductor Lsample represents the sample coil, the resistor R1 represents losses in the coil and remaining circuit, and l is the length of a transmission line (TL). The low-pass and high-pass filter circuits (LPF and HPF) are shown with dotted outline, connected to the EIS and NMR channels, respectively. The circuits' components consist of capacitors C1–C3, resistors R2–R3, and inductors L1–L3. Details about simulation and fabrication of the filters are in the ESI (Section S2, and Fig. S1(a–f)).† The impedance Z of the in situ NMR–EIS probe circuit was matched to 50 Ω. | ||
Additionally, the sample cell design is problematic because a sensitive NMR resonator with a high filling factor would typically require a solenoid coil while impedance spectroscopy would imply the sample is inserted between two electrodes. An in situ NMR–EIS sample cell was developed with good mechanical properties for a temperature range from 160 K to 473 K (Fig. 3), which provides a good electrical contact for impedance measurements and a high filling factor for NMR. To ensure the in situ sample cell is air-tight and suitable for moisture-sensitive samples, high-temperature o-rings were incorporated into the sample cell. The development profited from rapid prototyping which was possible because the cell was produced using 3D printing of thermoplastic filaments (i.e. polyether ether ketone and polyphenylene sulfone). The latter was especially relevant to optimize gas and temperature flow within the probe head to achieve low temperature gradients over the sample and a quick response of the sample temperature on a temperature change of the flowing N2 gas. To this end, the wall thickness of the sample cell was reduced to only 0.7 mm. The 3D-printed top parts for the variable-temperature experiment allow temperatures up to 473 K.
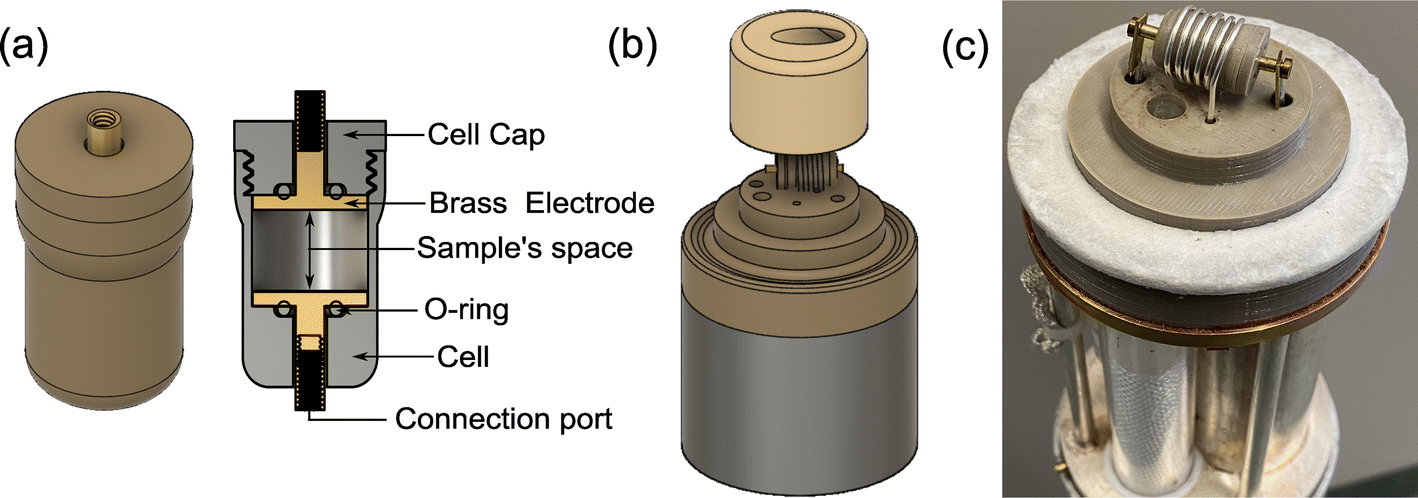 | ||
| Fig. 3 Photos and diagrams for the in situ NMR–EIS probe head. (a) 3D model of the in situ sample cell, printed with polyether ether ketone filament. (b) 3D model of the in situ probe head top part, showing the location of the sample cell in the solenoid coil for the detection of the NMR signal and the top insulation cap for variable-temperature experiments, printed with polyphenylene sulfone filament. (c) Photograph of the mounted sample cell already connected to the EIS channel. | ||
The presented in situ NMR–EIS probe head employs McKay design: a remote tunable single coil transmission line NMR probe head, similar to other probe heads used in the group including a symmetrizing capacitor (Fig. 3c).60 The in situ NMR–EIS probe head was used only in single channel mode. Because in the in situ probe head, both channels are connected to the same sample, a good separation of the high-frequency (NMR: 116.6 MHz) and of the low-frequency (EIS: 1 MHz–30 mHz) channel is compulsory: Both channels will mutually add noise and artefacts to the other channel which is not wanted. A simple solution is to add passive LCR filters which block the unwanted channel with at least 50 dB attenuation and have a flat passband (<0.05 dB). In fact losses in the EIS channel are less critical than ripple in the passband. The filters were realized as passive 3-element filters and are described in detail in the ESI (low pass-filter Fig. S1(a), high-pass filter Fig. S1(d)).† Each of the four cables connecting the impedance spectrometer and the measurement cell was equipped with a shielded separate filter box connected via BNC connectors. These cables needed to be longer (length 5 m) than in the ex situ setup to position the EIS spectrometer in at sufficient distance from the magnet.
The modifications required for the in situ setup make calibration of the impedance spectrometer compulsory. This was achieved using a short/load calibration61 against standards fabricated for the developed measurement cell (Fig. S3†). The validation results of the calibration procedures (Section S3†) confirm that the residual fixtures of the extended measurement cables and low pass filters were effectively isolated from the sample's impedance measurement and that the in situ NMR–EIS probe head was successfully calibrated. We did not succeed in calibrating the cell with 2-element passive filters but only with the aforementioned 3-element filters (Fig. S1†). The calibration was further tested using the compound Li1.5Al0.5Ge1.5(PO4)3, for which impedance spectra were obtained with an ex and in situ cell which only differed by 0.3% (estimate from five separate measurements, Fig. S4, and Table S2†).
Finally, the performance of the new in situ NMR–EIS probe head (Fig. S5†) was experimentally tested on standard parameters including impedance measurement stability, nutation frequency, in situ NMR–EIS measurement stability, interference suppression, operating temperature range (160–473 K) and extended time thermal stability with good results. A nutation frequency of 19 kHz was estimated from a 7Li NMR nutation curve measurement for LiCl (Fig. S6†). The nutation curve shows a rather fast decay which is due to a pulse field inhomogeneity as expected from the use of a metallic electrode necessary for the EIS measurement. Thermal equilibrium could be achieved within 5 min for variable temperature measurements.
3.2 Ex situ versus in situ EIS–NMR experiments on Li8SnO6
The test compound Li8SnO6 was synthesised via a solid state synthesis route to demonstrate the in situ EIS–NMR technique and to validate the setup. The crystalline phase was identified by X-ray diffraction (Fig. S7†). The compound purity is determined to be higher than 99% with a side phase of Li2O, according to a Rietveld refinement of X-ray diffraction data. Ex situ7Li single pulse NMR spectra of Li8SnO6 were recorded in a static mode at different temperatures (Fig. 4), to monitor the effect of lithium ion jumps on the 7Li spectral lineshape. At low temperatures below 300 K, the central transition peak is broadened by dipolar interactions. The central transition of the spectrum in the first approximation is independent of the quadrupolar interaction and determined by the dipolar interaction.11 As the temperature increases, the 7Li NMR line shape of the central transition starts to narrow as a consequence of translational ionic motion. This motional narrowing effect in the central transition occurs when the jump rate of the mobile species is of the order of the square root of the dipolar second moment.11 Along with the broad central transition, satellite transitions from the two different Li sites are also visible which in principle could allow study of lithium ion motion on octa- and tetrahedral Li sites independently. Because of the limited resolution and only small changes, we have not further investigated temperature induced changes in the satellite transitions.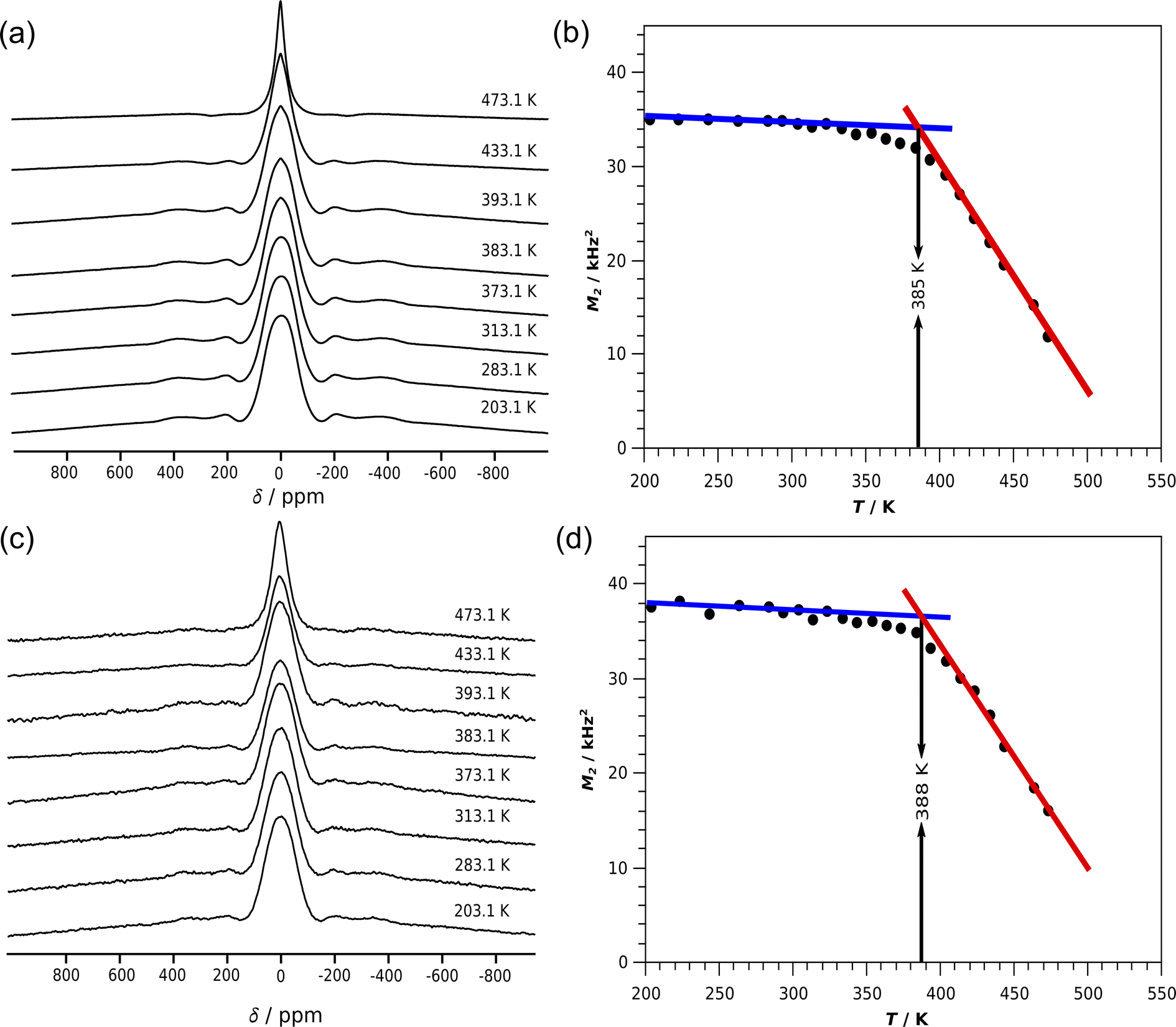 | ||
| Fig. 4 7Li NMR spectra of Li8SnO6 of static powder samples measured at a frequency of 116.643 MHz (a) using ex situ NMR probe head (c) using the developed in situ NMR–EIS probe head at different (selected) temperatures. The second moment M2 calculated from the central transition of a baseline corrected spectrum for temperatures ranging from 203.1 to 473.1 K (b) using an standard NMR probe head for ex situ measurements and (d) using the developed in situ NMR–EIS probe head. The onset temperatures were estimated from the crossover of the red and blue lines. | ||
The line-width of the central transitions of the 7Li atoms was estimated from the second moment of the central transition peak as a function of temperature (Fig. 4b). For Li8SnO6 a flat low-temperature plateau is observed, which justifies the simplified treatment according to Waugh and Fedin,62,63 who found that the onset temperature Tonset of line-narrowing is linearly related to the activation energy EA = 1.617 meV K−1·Tonset with an estimated error of 10%. The in situ NMR line shape analysis is similar to the ex situ measurements (Fig. 4). The obtained activation energies from ex situ and in situ lineshape analysis of Li8SnO6 agree within error limits (Table 1).
| NMR line shape analysis technique | T onset/K | E A/eV |
|---|---|---|
| Ex situ | 385 | 0.62(6) |
| In situ | 388 | 0.63(6) |
An independent source of information to study Li-motion are spin-alignment experiments.48,49,64 The 7Li spin alignment echo NMR amplitudes S(T,tm) of Li8SnO6 were recorded for both ex situ and in situ at different mixing times tm and temperatures T. The decay curves are not affected by the quadrupolar spin–lattice relaxation T1Q, but rather solely by lithium ion jumps between different crystallographic orbits in the Li8SnO6.49,65 The quadrupolar spin–lattice relaxation induces two-step decay curves at low temperatures; thus, spin alignment echo measurements were performed at higher temperatures (Fig. 5a and c). The lithium ion jump rates 1/τ determined from ex situ and in situ measurements follow an Arrhenius law τ−1 = τ0−1exp(−EA/(kBT)).49,65 Here, EA is the estimated activation energy, τ0 is the pre-exponential factor and kB is the Boltzmann constant.
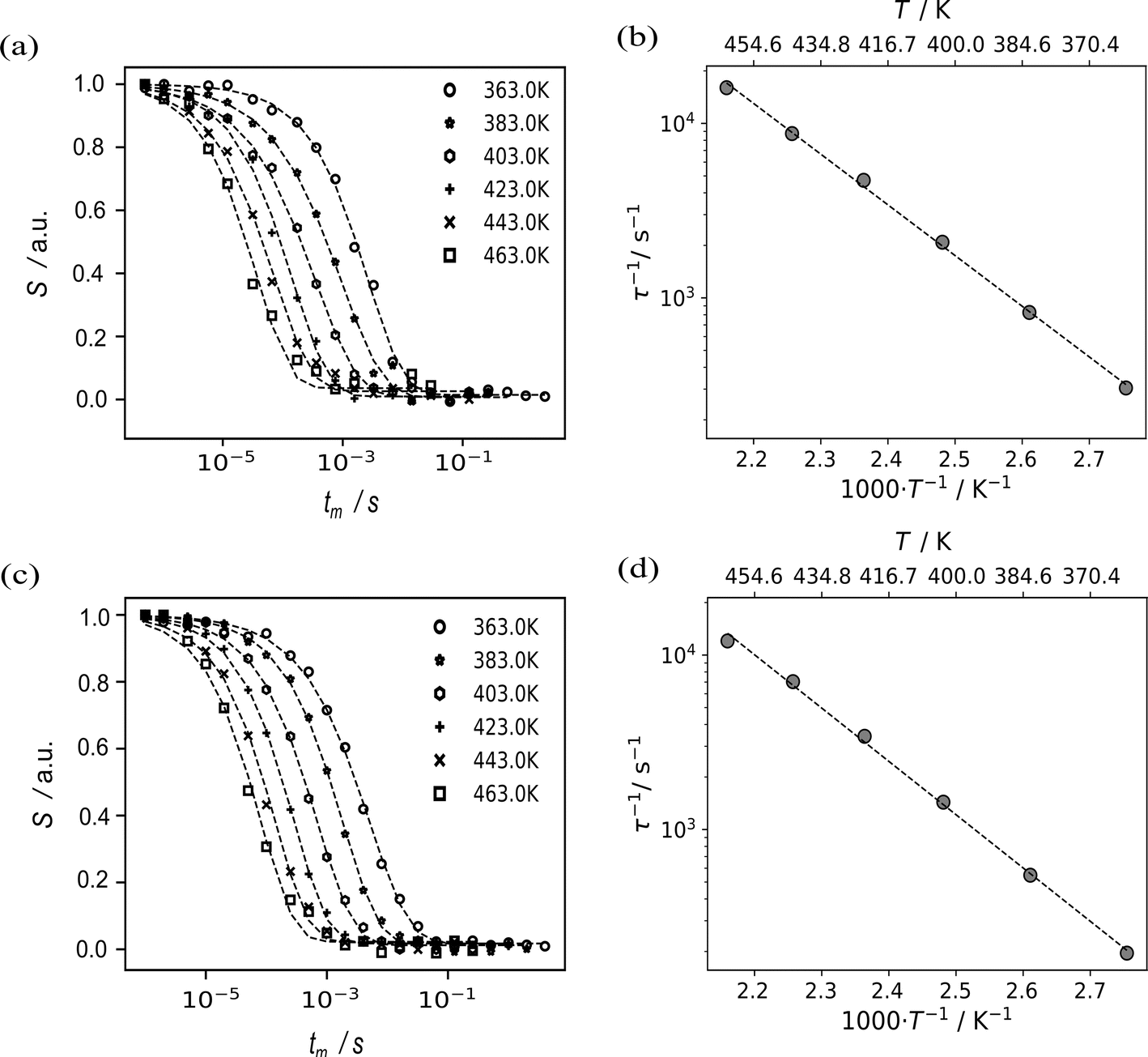 | ||
| Fig. 5 Results from 7Li spin alignment echo NMR of Li8SnO6 at a Larmor frequency of 116.643 MHz. The diagrams on the left show 7Li spin-alignment echo amplitude S(T,tm) as a function of the mixing time tm at different temperatures T in the range from 363 to 463 K for the ex situ (bottom, c) and the in situ setup (top, a). From those measurements, jump rates 1/τ were estimated by fitting a stretched exponential (dashed lines, left column) into the data at individual temperatures. Plots of the resulting jump rate as a function of the inverse temperature are shown on the right (b and d). The dashed fitted lines on the right correspond to an Arrhenius function (see main text).The pre-exponential factor τ0−1 for both ex situ and in situ is 5.26(4)·1010 s−1 and 3.20(3)·1010 s−1, respectively. | ||
The ex situ activation energy is 0.61(1) eV while the in situ activation energy is also 0.58(1) eV. The calculated jump rates τ−1 vary from 196 s−1 to 16667 s−1 at temperatures from 363 to 463 K, respectively. The pre-exponential factor is consistent with low diffusion coefficients and the results from impedance spectroscopy (vide infra). An excellent agreement between the results from the in situ and the ex situ setup is observed (Fig. 5).
Macroscopic lithium ion conductivities were investigated by impedance spectroscopy using two ion blocking gold electrodes. The complex impedance spectra (example Fig. 6) exhibit typical characteristics (with up to two half-circles at sufficiently low temperature) of an electrically inhomogeneous solid ion conductor, where the grain boundary resistance dominates the overall impedance.8 The impedance spectra were modelled with an equivalent circuit (Fig. 6 inset) consisting of a series connection of two parallel arrangements of a resistor R and a constant phase element Q, depicting the conductive and capacitive characteristics of the bulk and grain boundary regions of the solid ion conductor, respectively.8 The electrode polarisation is modelled using additional constant phase element Q.8 The effective capacitance Ceff of the constant-phase element is calculated using Brug's formula Ceff = (Q·R(α−1))(1/α).66 From the equivalent circuit analysis of the impedance data, the high frequency arc (Fig. 6) corresponds to a capacitance of 7.61 pF, thus depicting the bulk region of the sample while the mid-frequency arc with a capacitance of 0.02 nF corresponds to the grain boundary region of the material. This analysis is consistent with the established bricklayer model in terms of the capacitance value for bulk and grain boundary regions.8
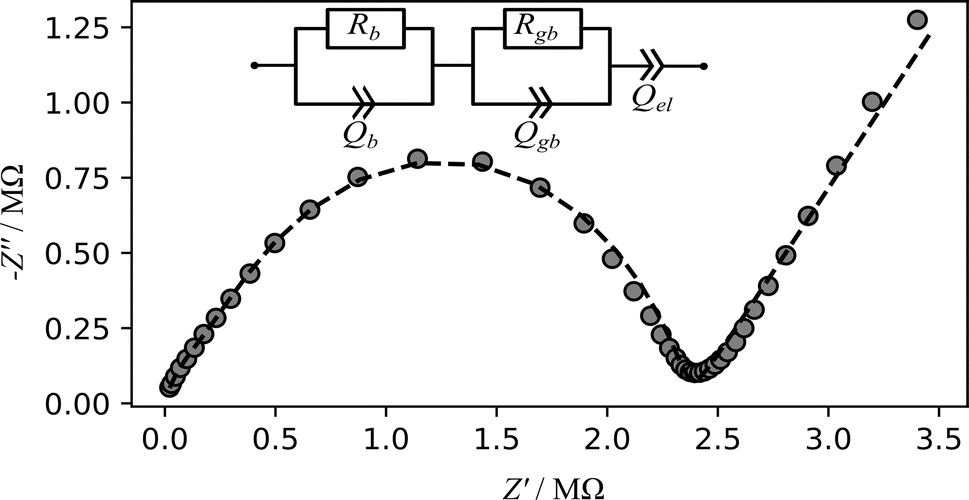 | ||
| Fig. 6 Exemplary Nyquist (imaginary part of the impedance against real part) plot of impedance measurements for the in situ setup conducted on a Li8SnO6 pellet (diameter = 8 mm, thickness = 2.1 mm) at a temperature of 393 K. The grey cycles represent the experimental data while dashed line represents the result of fit using the shown model. Rb and Rgb are the bulk and grain boundary resistance while Qb, Qgb and Qel represent the constant phase elements for the bulk, grain boundary and the electrode polarisation, respectively. | ||
The conductivities of Li8SnO6 were determined for both ex situ and in situ impedance measurements (Fig. 7). The ex situ and in situ impedance results demonstrate consistency within error limits (Table S3†), and the respective extrapolated overall conductivities at room temperature (≈10−9 S cm−1) are two orders of magnitude higher than previously reported conductivity of 3.7·10−11 S cm−1,31 which can be ascribed to differences in the sample preparation.19
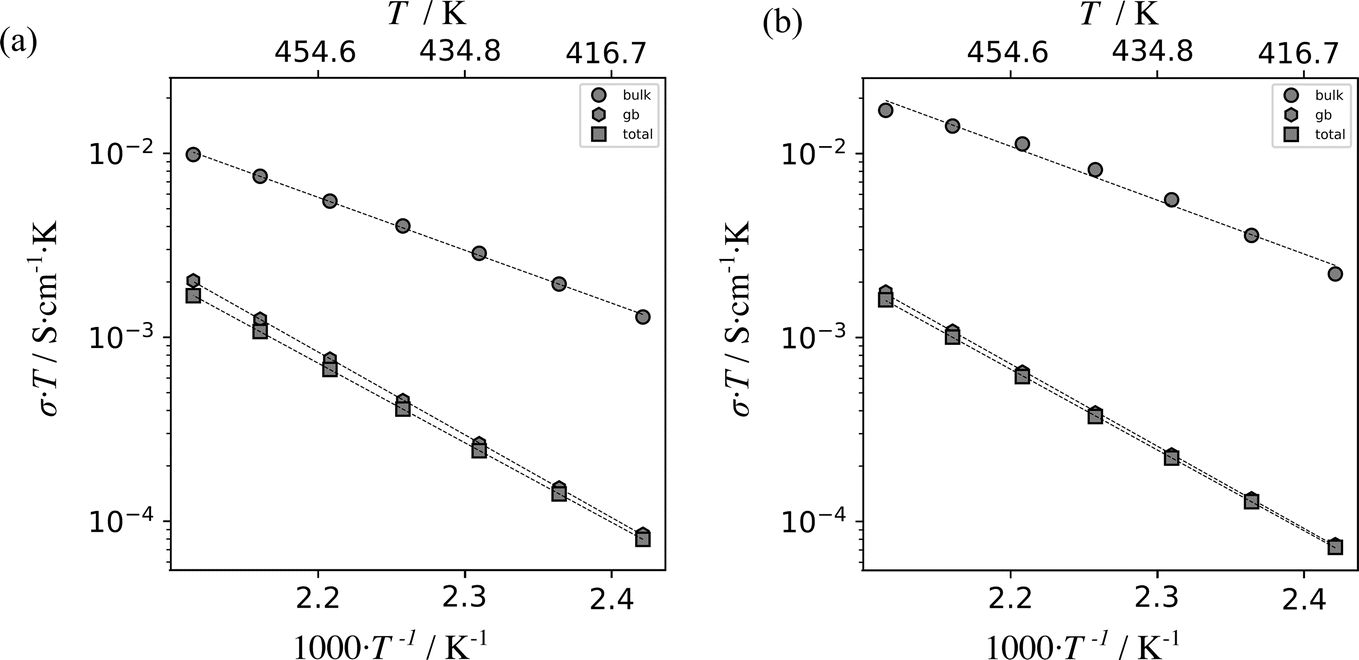 | ||
| Fig. 7 Plot of the product of electrical conductivity σ and temperature T versus inverse temperature for Li8SnO6 as measured using a standard, ex situ impedance spectroscopy setup (a) and the described in situ NMR–EIS setup (b). The results were obtained from impedance spectra in temperature range from 413 K to 473 K. Linear fits are indicated by dashed lines. The data points are indicated by grey circles, hexagons and squares which correspond to bulk, grain boundary and total ionic conductivity values, respectively. The calculated activation energies are shown in Table S3.† | ||
An excellent agreement of results from the ex situ and in situ setup was observed for impedance spectroscopy, 7Li NMR spin-alignment measurements and 7Li NMR line-shape analysis (Tables 2, and S4†), which means that the target of the study to establish a reliable in situ EIS–NMR setup was achieved.
| Technique | E in situ A/eV | E ex situ A/eV |
|---|---|---|
| 7Li NMR line shape analysis of Tonset | 0.63(6) | 0.62(6) |
| 7Li NMR spin-alignment echo | 0.58(1) | 0.61(1) |
| Impedance spectroscopy of bulk conductivity | 0.58(3) | 0.569(8) |
What remains to be done is to give meaning to the different observations. Qualitatively, spin-alignment echo NMR and impedance spectroscopy show low jump rates and a rather disappointingly bulk low ion conductivity. An excellent agreement between the activation energies of about 0.60 eV of these two techniques is observed, which indicates that the long-range ion-transport as probed by EIS is limited by the local Li hopping motion probed by NMR.
3.3 Activation energies for Li8SnO6 from quantum chemical calculations
Nudged elastic band (NEB) calculations as implemented in quantum espresso (QE) were employed to identify and analyse potential pathways of lithium vacancy migration and to estimate the activation energies of Li8SnO6.31 Lithium cations are distributed over repeating units of one octahedral crystallographic orbit Liocta and one tetrahedral orbit Litetra in the crystal structure of Li8SnO6 (Fig. 8). Macroscopic conduction requires pathways to cover the complete unit cell for example in c-direction (Fig. 8) that is crossing the tetrahedral Li-layer like arrangement and passing the octahedral Li sites, or in a/b-direction that is either within the tetrahedral layer (Fig. S13(b)†) or within the octahedral layer (Fig. S14†).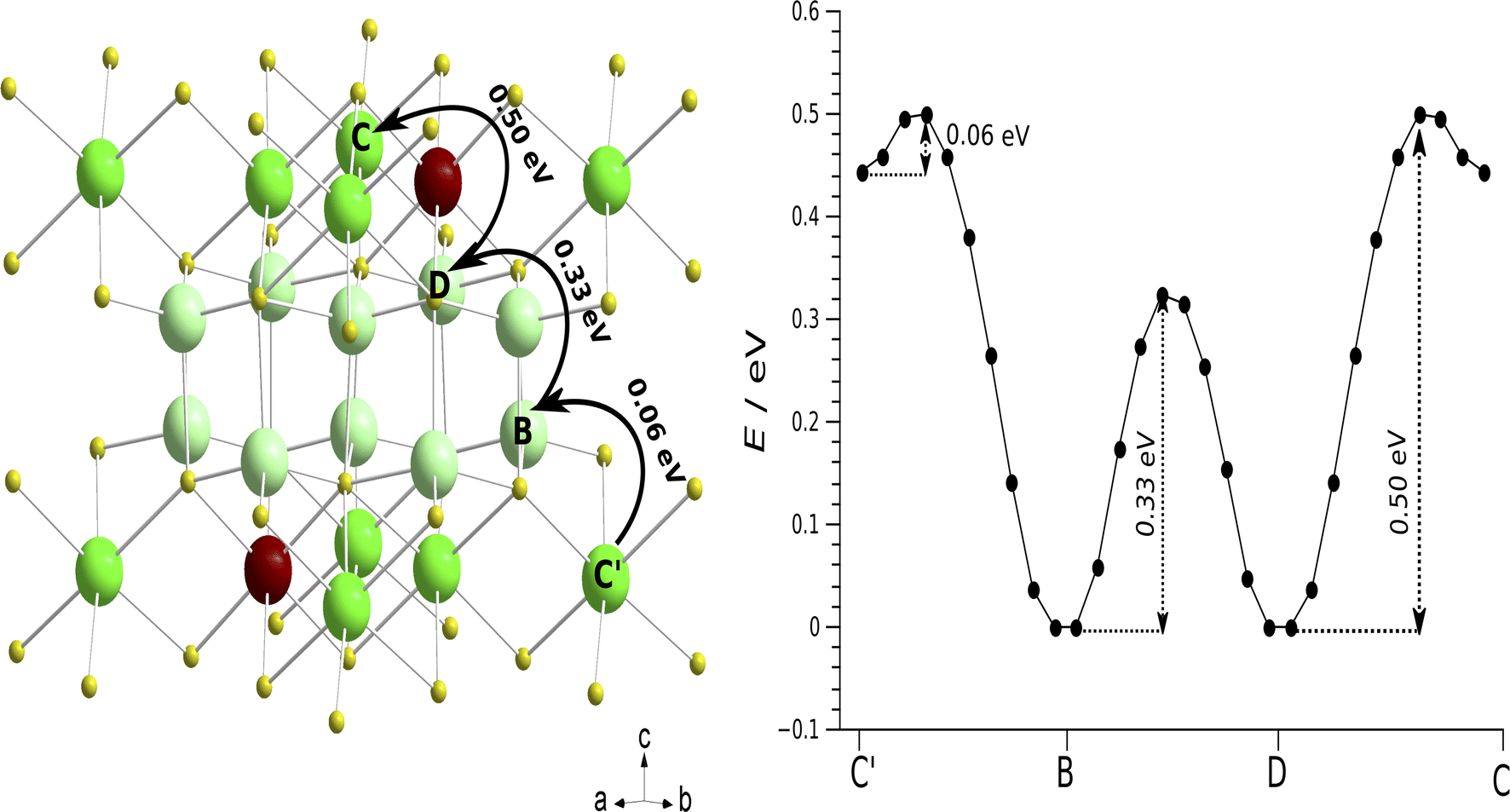 | ||
| Fig. 8 Potential pathways of vacancy-based Li migration and the associated activation energies determined by Nudged Elastic Band (NEB) calculations. Left: Li-diffusion transport routes from A → B → C → D through a complete unit cell of Li8SnO6. The solid arrows represent the most favorable long-range Li ion diffusion pathway in the sense of featuring the lowest maximum activation energy. Right: The graph shows the energy barriers corresponding to the path depicted on the left and the points in the graph correspond to different NEB images along the path. | ||
While the results from the nudged elastic band (NEB) calculation as implemented in quantum espresso (QE) used in this work, are consistent with previously obtained activation energies as implemented in Vienna ab initio simulation package (VASP)32 and classical force field simulations as established in the general utility lattice program (GULP)33 code (Table 3), we were not able to reconstruct the pathways over a complete unit cell from the information presented in these articles.
| Techniques | Direction | E A/eV | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Litetra1 → Litetra1 C → Gb | Litetra2 → Litetra2 B → Eb | Litetra1 → Litetra2 B → C | Liocta → Litetra1 A → B | Litetra2 → Liocta C → D | Liocta → Liocta F → Fb | |||
| a The dashes indicate the data in the corresponding literature are missing. b The potential pathways of vacancy-based Li vacancy migration and the associated activation energies determined by Nudged Elastic Band calculations are shown in Fig. S13–S15. | ||||||||
| NEB-QE | Forward | 0.72 | 0.72 | 0.33 | 0.06 | 0.5 | 0.98 | This work |
| Reverse | 0.72 | 0.72 | 0.33 | 0.5 | 0.06 | 0.98 | ||
| NEB-VASPa | Forward | — | — | 0.4 | — | 0.51 | 0.88 | 32 |
| Reverse | — | — | 0.43 | — | 0.04 | 0.88 | ||
| GULP | Forward | 0.6 | 0.6 | 0.2 | 0.59 | 0.59 | 1.06 | 33 |
| Reverse | 0.6 | 0.6 | 0.2 | 0.59 | 0.59 | 1.06 | ||
From a comparison of three possible pathways covering a complete unit cell (Fig. 8, S13, and S14†), it can be concluded that the preferred lithium vacancy diffusion pathway is along the c-direction through Liocta → Litetra1 → Litetra2 → Liocta (A → B → C → D) with a maximum activation energy of 0.50 eV (Fig. 8), while the next lower activation energy of 0.72 eV corresponds to diffusion within the layer-like arrangement of tetrahedrally coordinated Li atoms and the most unlikely motion is motion only within the layer-like arrangement of octahedrally coordinated Li atoms (0.98 eV). This conclusion is only valid if it is assumed that the bottleneck is determined by the activation energies and not by the attempt frequencies. Further, it should be noted that the quantum-chemical calculations are performed at a temperature of 0 K which is far from the experimental conditions and may explain the observed differences. Qualitatively, the experimental activation energy of about 0.60 eV for a macroscopic Li motion is in good agreement with the activation energy of 0.5 eV found by the NEB calculations.
4 Conclusions
In this work, a new setup for in situ7Li NMR–EIS spectroscopy is presented and validated against ex situ experiments. It is shown in results on Li8SnO6 and Li1.5Al0.5Ge1.5(PO4)3 that in situ NMR–EIS experiments can be conducted to exclude the sample preparation and thermal history as possible explanations for differences between different measurement techniques including NMR line-shape analysis, spin-alignment echo NMR, impedance spectroscopy and in principle also NMR relaxometry. NMR–EIS spectroscopy was applied to the material Li8SnO6 and consistent results for the activation energy were obtained by three independent experimental techniques. With the help of nudged elastic band calculations for the first time, a consistent interpretation of obtained activation energies of Li8SnO6 is provided. It is anticipated that NMR–EIS spectroscopy will help to resolve discrepancies based on sample related changes such as thermally induced irreversible changes or ageing effects in solid ion conductors. Furthermore, it can be applied to study metastable intermediaries in solid ion conductors.Author contributions
Conceptualization: JSADG; investigation: SCA and NM; writing original draft: SCA and NM; writing – review and editing: JSADG; visualization: SCA and NM; funding acquisition: JSADG, SCA; supervision: JSADG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Andreas Neuberger and Xu Ke for their help in getting started with the electrical parts and 3D printing, respectively. The brass parts were produced by the mechanical workshop of the University of Siegen. Financial support by the House of Young Talents, University of Siegen is acknowledged.References
- J. B. Goodenough, J. Solid State Electrochem., 2012, 16, 2019–2029 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- P. Heitjans and S. Indris, J. Phys.: Condens. Matter, 2003, 15, R1257 CrossRef CAS.
- Y. Gao, A. M. Nolan, P. Du, Y. Wu, C. Yang, Q. Chen, Y. Mo and S.-H. Bo, Chem. Rev., 2020, 120, 5954–6008 CrossRef CAS PubMed.
- Z.-H. Fu, X. Chen and Q. Zhang, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13, e1621 CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 2004, 42, 288–292 CrossRef.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- R. Böhmer, K. R. Jeffrey and M. Vogel, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 87–174 CrossRef.
- Y.-X. Xiang, G. Zheng, G. Zhong, D. Wang, R. Fu and Y. Yang, Solid State Ionics, 2018, 318, 19–26 CrossRef CAS.
- M. Wilkening, R. Amade, W. Iwaniak and P. Heitjans, Phys. Chem. Chem. Phys., 2007, 9, 1239–1246 RSC.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- M. Wilkening, C. Mühle, M. Jansen and P. Heitjans, J. Phys. Chem. B, 2007, 111, 8691–8694 CrossRef CAS.
- M. Wilkening, A. Kuhn and P. Heitjans, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 054303 CrossRef.
- M. Tatsumisago, C. A. Angell and S. W. Martin, J. Chem. Phys., 1992, 97, 6968–6974 CrossRef CAS.
- K. L. Ngai and C. León, J. Non-Cryst. Solids, 2003, 315, 124–133 CrossRef CAS.
- S. Ohno, T. Bernges, J. Buchheim, M. Duchardt, A.-K. Hatz, M. A. Kraft, H. Kwak, A. L. Santhosha, Z. Liu, N. Minafra, F. Tsuji, A. Sakuda, R. Schlem, S. Xiong, Z. Zhang, P. Adelhelm, H. Chen, A. Hayashi, Y. S. Jung, B. V. Lotsch, B. Roling, N. M. Vargas-Barbosa and W. G. Zeier, ACS Energy Lett., 2020, 5, 910–915 CrossRef CAS.
- P. G. Bruce and A. R. West, J. Solid State Chem., 1982, 44, 354–365 CrossRef CAS.
- H. Y.-P. Hong, Mater. Res. Bull., 1978, 13, 117–124 CrossRef CAS.
- J. G. Kamphorst and E. E. Hellstrom, Solid State Ionics, 1980, 1, 187–197 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS.
- A. Kuhn, V. Epp, G. Schmidt, S. Narayanan, V. Thangadurai and M. Wilkening, J. Phys.: Condens. Matter, 2011, 24, 035901 CrossRef.
- A. Kuhn, S. Narayanan, L. Spencer, G. Goward, V. Thangadurai and M. Wilkening, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094302 CrossRef.
- K. Meier, T. Laino and A. Curioni, J. Phys. Chem. C, 2014, 118, 6668–6679 CrossRef CAS.
- R. Jalem, Y. Yamamoto, H. Shiiba, M. Nakayama, H. Munakata, T. Kasuga and K. Kanamura, Chem. Mater., 2013, 25, 425–430 CrossRef CAS.
- M. Xu, M. S. Park, J. M. Lee, T. Y. Kim, Y. S. Park and E. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 052301 CrossRef.
- M. Troemel and J. Hauck, Z. Anorg. Allg. Chem., 1969, 368, 248–253 CrossRef CAS.
- R. Hoppe and R. M. Braun, Z. Anorg. Allg. Chem., 1977, 433, 181–188 CrossRef CAS.
- J. Senegas, A. M. Villepastour and C. Delmas, J. Solid State Chem., 1980, 31, 103–112 CrossRef CAS.
- N. Luo, Z. Hou, C. Zheng, Y. Zhang, A. Stein, S. Huang and D. G. Truhlar, Chem. Mater., 2021, 33, 834–844 CrossRef CAS.
- N. Kuganathan, A. L. Solovjov, R. V. Vovk and A. Chroneos, Heliyon, 2021, 7, e07460 CrossRef CAS PubMed.
- O. Pecher, J. Carretero-González, K. J. Griffith and C. P. Grey, Chem. Mater., 2017, 29, 213–242 CrossRef CAS.
- F. Chevallier, M. Letellier, M. Morcrette, J.-M. Tarascon, E. Frackowiak, J.-N. Rouzaud and F. Béguin, Electrochem. Solid-State Lett., 2003, 6, A225 CrossRef CAS.
- K. Märker, C. Xu and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 17447–17456 CrossRef PubMed.
- F. Chevallier, F. Poli, B. Montigny and M. Letellier, Carbon, 2013, 61, 140–153 CrossRef CAS.
- M. Letellier, F. Chevallier, F. Béguin, E. Frackowiak and J.-N. Rouzaud, J. Phys. Chem. Solids, 2004, 65, 245–251 CrossRef CAS.
- M. Letellier, F. Chevallier and M. Morcrette, Carbon, 2007, 45, 1025–1034 CrossRef CAS.
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed.
- J. Arai, K. Gotoh, R. Sayama and K. Takeda, J. Electrochem. Soc., 2017, 164, A6334–A6340 CrossRef CAS.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- J. Nasir, N. Steinbrück, K. Xu, B. Engelen and J. Schmedt auf der Günne, J. Appl. Crystallogr., 2022, 55, 1097–1103 CrossRef CAS PubMed.
- A. A. Coelho, J. Evans, I. Evans, A. Kern and S. Parsons, Powder Diffr., 2011, 26, S22–S25 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Concepts Magn. Reson., 2002, 14, 326–346 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- J. Jeener and P. Broekaert, Phys. Rev., 1967, 157, 232–240 CrossRef CAS.
- F. Qi, G. Diezemann, H. Böhm, J. Lambert and R. Böhmer, J. Magn. Reson., 2004, 169, 225–239 CrossRef CAS PubMed.
- S. Faske, B. Koch, S. Murawski, R. Küchler, R. Böhmer, J. Melchior and M. Vogel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 024202 CrossRef.
- D. Jardón-Álvarez and J. Schmedt auf der Günne, Solid State Nucl. Magn. Reson., 2018, 94, 26–30 CrossRef PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa and P. van Mulbregt, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- T. Björkman, Comput. Phys. Commun., 2011, 182, 1183–1186 CrossRef.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
- J. W. Bennett, B. G. Hudson, I. K. Metz, D. Liang, S. Spurgeon, Q. Cui and S. E. Mason, Comput. Mater. Sci., 2019, 170, 109137 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- A. J. Ilott, S. Chandrashekar, A. Klöckner, H. J. Chang, N. M. Trease, C. P. Grey, L. Greengard and A. Jerschow, J. Magn. Reson., 2014, 245, 143–149 CrossRef CAS PubMed.
- B. J. Walder, M. S. Conradi, J. J. Borchardt, L. C. Merrill, E. G. Sorte, E. J. Deichmann, T. M. Anderson, T. M. Alam and K. L. Harrison, Sci. Adv., 2021, 7, eabg8298 CrossRef CAS PubMed.
- J. Schaefer, and R. A. McKay, Multi-Tuned Single Coil Transmission Line Probe for Nuclear Magnetic Resonance Spectrometer, US Pat., US5861748A, 1999 Search PubMed.
- N. Meddings, M. Heinrich, F. Overney, J.-S. Lee, V. Ruiz, E. Napolitano, S. Seitz, G. Hinds, R. Raccichini, M. Gaberšček and J. Park, J. Power Sources, 2020, 480, 228742 CrossRef CAS.
- J. S. Waugh and E. I. Fedin, Fiz. Tverd. Tela, 1962, 4, 2233–2237 CAS.
- J. S. Waugh and E. I. Fedin, Phys. Solid State, 1963, 4, 1633–1636 Search PubMed.
- F. Qi, T. Jörg and R. Böhmer, Solid State Nucl. Magn. Reson., 2002, 22, 484–500 CrossRef CAS PubMed.
- S. Faske, H. Eckert and M. Vogel, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 104301 CrossRef.
- G. J. Brug, A. L. G. van den Eeden, M. Sluyters-Rehbach and J. H. Sluyters, J. Electroanal. Chem. Interfacial Electrochem., 1984, 176, 275–295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Information about 3D printing, synthesis, powder X-ray diffraction, technical details about alternating current filters, electronic details of the in situ cell, probe head, detailed fitting results of impedance spectra and details about nudged elastic band calculations. See DOI: https://doi.org/10.1039/d3ta06237f |
| This journal is © The Royal Society of Chemistry 2024 |