
High-rate and long-life flexible aqueous rechargeable zinc-ion battery enabled by hierarchical core–shell heterostructures†
Ziming
Xu
a,
Wenyuan
Zhang
ab,
Xianzhen
Wang
a,
Yuxin
Li
a,
Jinwen
Fu
a,
Yongbao
Feng
a,
Wenbin
Gong
b,
Jiabin
Guo
cd,
Pan
Xue
e and
Qiulong
Li
 *a
*a
aCollege of Materials Science and Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: qlli@njtech.edu.cn
bSchool of Physics and Energy, Xuzhou University of Technology, Xuzhou 221018, China
cKey Laboratory of Multifunctional Nanomaterials and Smart Systems, Advanced Materials Division, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China
dSchool of Electronic Science & Engineering, Southeast University, Nanjing 210096, China
eSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
First published on 13th December 2023
Abstract
Aqueous rechargeable zinc-ion batteries (ZIBs) are potential alternative candidates for current commercial lithium-ion batteries due to their cost-efficiency, safety and sustainable nature. As one of the prominent cathode materials, MnO2 exhibits high operating voltage and theoretical capacity. Yet, its poor electrochemical kinetics, low conductivity, and lifespan prevent its further application. Herein, an effective strategy for the construction of hierarchical TiN@MnO2 nanowire arrays (NWAs) core–shell heterostructures directly grown on carbon cloth (CC) is demonstrated to systematically solve the above issues. First-principles calculations reveal that decreased bandgap and Zn2+ diffusion barrier as well as more stable structure of the host material after Zn2+ insertion promote the electrochemical kinetics of TiN@MnO2. As a result, TiN@MnO2 NWAs/CC exhibits significantly increased capacity (385.1 vs. 310 and 194 mA h g−1 at 0.1 A g−1), rate performance (127.6 mA h g−1vs. 49.7 and 37.4 mA h g−1 at 4.0 A g−1) and cycling stability (101.6% capacity retention over 2300 cycles vs. 14.0% and 11.9%) compared with TiO2@MnO2 NWAs/CC and MnO2 NSs/CC, respectively. Finally, the as-assembled flexible ZIBs with TiN@MnO2 NWAs/CC cathode deliver an ultrahigh energy density of 327.7 W h kg−1 at 135.6 W kg−1. The proposition of the core–shell idea provides a novel strategy for development ZIBs.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) are widely used in the battery market because of their possessing high specific capacity and energy density.1,2 However, they suffer from severe challenges because of the safety concerns posed by flammable organic electrolytes and the scarcity of lithium feedstock.3,4 So, it is urgent to develop a new kind of rechargeable battery with high safety, low cost, environmental friendliness, and excellent electrochemical performances to replace LIBs. In recent years, aqueous rechargeable batteries possessing high safety and ionic conductivity have been receiving increasing attention as a promising solution to these problems.5,6 Aqueous rechargeable zinc-ion batteries (ZIBs) offer a research direction for energy-storage systems due to the use of mild aqueous electrolytes and natural resource-abundant zinc metal anodes.7–9 More importantly, as an important part of ZIBs, Zn metal anodes have many advantages such as high theoretical capacity (820 mA h g−1, 5855 mA h cm−3),10 low redox potential (−0.76 V vs. standard hydrogen electrode),11,12 low cost, and so on. Frustratingly, ZIBs are currently plagued by weak rate capability, poor cycling stability and low energy density.Therefore, the development of high-performance cathode materials is essential to improve the electrochemical performance of ZIBs. Within the last decade, various cathode materials have been reported, which mainly include manganese-based materials (ZnMn2O4,13 Mn3O4,14 Mn2O3,15 MnO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16,17etc.), vanadium-based materials (VS2,18 VOPO4,19 V2O520,21etc.) and Prussian blue analogs (CuHCF,22 ZnHCF23etc.). Although vanadium-based materials exhibit high specific capacity, they are limited by their toxicity, low operating voltage (≈0.7 V vs. Zn2+/Zn) and high raw material cost.24,25 Prussian blue analogs have high operating voltages (>1.4 V vs. Zn2+/Zn), but poor cycling stability and low specific capacity are non-negligible drawbacks.26 Manganese-based materials represent promising cathode materials because of their satisfactory operating voltages and specific capacity. Among them, manganese dioxide (MnO2) shows the advantages of high theoretical capacity (the theoretical capacity for single electron transfer is 308 mA h g−1 and for double electron transfer is 616 mA h g−1).27,28 high average discharge voltage (1.2–1.4 V vs. Zn2+/Zn),29 low cost, and environmental friendliness, but low capacity, poor cycle stability, and slow reaction kinetics have seriously restricted its practical applications in ZIBs.30,31
16,17etc.), vanadium-based materials (VS2,18 VOPO4,19 V2O520,21etc.) and Prussian blue analogs (CuHCF,22 ZnHCF23etc.). Although vanadium-based materials exhibit high specific capacity, they are limited by their toxicity, low operating voltage (≈0.7 V vs. Zn2+/Zn) and high raw material cost.24,25 Prussian blue analogs have high operating voltages (>1.4 V vs. Zn2+/Zn), but poor cycling stability and low specific capacity are non-negligible drawbacks.26 Manganese-based materials represent promising cathode materials because of their satisfactory operating voltages and specific capacity. Among them, manganese dioxide (MnO2) shows the advantages of high theoretical capacity (the theoretical capacity for single electron transfer is 308 mA h g−1 and for double electron transfer is 616 mA h g−1).27,28 high average discharge voltage (1.2–1.4 V vs. Zn2+/Zn),29 low cost, and environmental friendliness, but low capacity, poor cycle stability, and slow reaction kinetics have seriously restricted its practical applications in ZIBs.30,31
To address the above issues of MnO2, researchers have used various strategies to improve its electrochemical performances, including composite conductive materials,32,33 defect engineering,34 doping engineering,35 surface coating36 and construction of core–shell heterostructures.37 Among them, hierarchical core–shell heterostructures are known to be a promising strategy for improving the electrochemical performance of electrodes. For example, Wang et al. successfully prepared MnO2@PANI core–shell nanowire film by uniformly wrapping conductive PANI on the MnO2 core, which effectively provided a fast charge transport and improved reaction kinetics, improved reaction kinetics, and was able to reach a capacity of 342 mA h g−1 at a current density of 0.2 A g−1.38 Furthermore, Feng et al. successfully designed heteroatom cross-doped one-dimensional core–shell bimetallic oxides via the self-templated method and one-step hydrothermal reaction. The bimetallic oxides are composed of Mo-doped α-MnO2 nanoparticles as the shell and Mn-doped MoO3 nanobelts as the core, exhibiting the advantages of large specific surface area, resistance to mechanical deformation, and enlarged diffusion channels.39
Therefore, in order to improve the electrochemical performances for active materials with hierarchical core–shell heterostructures, selection of highly conductive core materials and high-capacity shell layers is a very effective way, which effectively addresses the issue of the low conductivity of MnO2 layers. Compared with oxides, transition metal nitrides have the advantages of high conductivity and stable physicochemical properties. Among them, titanium nitride (TiN) possesses high conductivity and excellent mechanical stability which has received wide attention and has a range of applications.40,41 Furthermore, in the electrode fabrication process, the active material, conductive agent and binder are mixed and directly coated on the substrate, which will cause “dead mass”, increase the interfacial resistance and lead to poor electrochemical performance.42 Thus, the construction of 3D self-standing electrodes is an effective strategy. Carbon cloth (CC), as a conductive current collector, has excellent flexibility and can be used as a self-standing flexible substrate to achieve improved electrochemical performances.43
Herein, a hierarchical TiN nanowire arrays (NWAs)@MnO2 nanosheets (NSs) core–shell heterostructure directly grown on CC (TiN@MnO2 NWAs/CC) was constructed as a self-standing aqueous ZIB cathode. Given the mechanical stability and high conductivity of the TiN core, combining with the MnO2 shell layer to form a hierarchical core–shell heterostructure can improve the mass loading of MnO2 and also enhance the capacity and rate performance. TiN as a core can reduce the bandgap and the diffusion barrier of Zn2+ in hierarchical core–shell heterostructures as determined using density functional theory (DFT). Meanwhile, the novel hierarchical core–shell heterostructure significantly optimizes the electrochemical kinetics of MnO2 and accommodates more zinc ions in this system. As a result, the prepared TiN@MnO2 NWAs/CC exhibits excellent electrochemical performance, which specifically shows ultrahigh capacity (385.1 mA h g−1vs. 310 and 194 mA h g−1 at 0.1 A g−1), high rate capability (33.1% vs. 16.0% and 20.3% at 4 A g−1), and long-life performance (101.6% capacity retention over 2300 cycles vs. 14.0% and 11.9%) compared with TiO2@MnO2 NWAs/CC and MnO2 NSs/CC, respectively. Importantly, the as-assembled quasi-solid-state flexible ZIBs with TiN@MnO2 NWAs/CC cathode achieve a high capacity of 241.7 mA h g−1 at 0.1 A g−1 and impressive cycling performance with 92.6% capacity retention after 100 cycles at 0.2 A g−1 and superhigh energy density of 327.7 W h kg−1 at 135.6 W kg−1. This work marks the successful preparation of a novel cathode material, which provides a new strategy for designing high-rate and long-life materials for next-generation flexible energy-storage devices.
2. Experimental section
2.1 Preparation of TiN NWAs/CC
In a typical preparation process, CC was immersed in 0.2 M TiCl4 aqueous solution. The solution was transferred to an autoclave and reacted at 100 °C for 1 h. After washing three times with deionized water, the treated CC was dried in a vacuum oven at 60 °C for 12 h. The treated CC was placed in a tubular furnace and annealed in air at 350 °C at a heating rate of 5 °C min−1 for 1 h, so that a seed layer is obtained on the CC. Then CC was immersed in a mixture of 0.5 mL TiCl4, 30 mL hydrochloric acid (36–38 wt%) and 30 mL deionized water, and the mixture was transferred to a 100 mL Teflon-lined stainless-steel autoclave and treated at 150 °C for 6 h to obtain TiO2 NWAs grown on the surface of CC. The obtained TiO2 NWAs/CC was washed three times with deionized water and dried in a vacuum oven overnight. The dried TiO2 NWAs/CC was placed in a tubular furnace and annealed with a mixed atmosphere of Ar and NH3 at 800 °C for 8 h to obtain TiN NWAs/CC.2.2 Preparation of TiN@MnO2 NWAs/CC cathode
TiN@MnO2 NWAs/CC was prepared via a simple electrochemical deposition method. Electrochemical deposition was conducted with a three-electrode system (CC as working electrode, Pt sheet as counter electrode and Ag/AgCl as reference electrode) in 50 mL of an aqueous solution containing 0.05 M MnSO4·H2O, 0.05 M CH3COONa and 10% volume fraction ethanol. The electrodeposition current density and time were 5 mA cm−2 and 300 s, respectively. TiO2@MnO2 NWAs/CC and MnO2 NSs/CC were also synthesized for comparison using an identical process. The average mass loading of MnO2 NSs on the CC surface is about 0.75 mg cm−2, and the average mass loading of MnO2 NSs on TiO2 NWAs/CC and TiN NWAs/CC is about 0.83 mg cm−2.2.3 DFT calculations
First-principles calculations were performed within the framework of DFT, implemented with the Vienna ab initio simulation package (VASP) code. The study describes the electron exchange–correlation potential via the Perdew–Burke–Ernzerhof (PBE) function. The Hubbard-type correction (Ueff = 2.1 eV) was applied to the 3d orbitals of Mn. The energy cutoff was 450 eV and the SCF tolerance was 1.0 × 10−6 eV per atom. In the calculations, a 15 Å vacuum region for MnO2 (001), TiN (111), TiO2 (110) and TiN@MnO2 was established based on experimental results. The k-points of the MnO2 (001), TiN (111), TiO2 (110) and TiN@MnO2 surfaces were set to 4 × 4 × 1, 5 × 5 × 1, 8 × 4 × 1 and 5 × 5 × 1. The optimized lattice constants are α = b = 5.74 Å, c = 21.40 Å for MnO2 (001); α = b = 5.95 Å, c = 23.51 Å for TiN (111); α = 2.95 Å, b = 6.50 Å, c = 27.30 Å for TiO2 (001) as well as TiN@MnO2 with the following lattice constants: α = b = 5.84 Å, c = 30.55 Å. The geometry structures are relaxed until the force on each atom is less than 0.03 eV Å−1 and the total energy convergence criterion is chosen as 10−5 eV. To investigate transition states and minimum-energy pathways of zinc ion transfer, the climbing-image nudged elastic band (CI-NEB) method was used. The NEB calculation convergence parameters for electronic and ionic relaxation were 10−5 eV and 10−2 eV Å−1, respectively. This formation energy was calculated using the expression Ef = (Eh + xEZn − Etot)/x, where Etot, Eh and EZn represent the total energy of the Zn ions adsorbed on the electrode, the energy of the electrode and the energy of the Zn ions, respectively, and x is the number of Zn ions.3. Results and discussion
In order to verify the rationality of the innovation and design, we have carried out scientifically theoretical calculations for the hierarchical TiN@MnO2 core–shell heterostructures through DFT. Additionally, TiN as a core material provides theoretical evidence for optimizing the zinc-ion storage performances of MnO2, further demonstrating the feasibility of constructing hierarchical core–shell heterostructures as cathodes for ZIBs. Fig. 1a–d display the density of states (DOS) of pure MnO2, TiO2, TiN and hierarchical TiN@MnO2 core–shell heterostructure, respectively. The top and side views of these crystal structures are displayed in Fig. S1–S4.† From Fig. 1a, it can be noticed that the valence band and conduction band of MnO2 at the Fermi level are obviously separated, with a large bandgap of ∼1.35 eV, exhibiting semiconductor characteristics. Therefore, to improve the electrochemical performance of MnO2, it is first necessary to optimize its conductivity. As is known, TiO2 and TiN are both chemically stable materials.41,44,45 From the calculation results, TiO2 exhibits a poor conductivity with a bandgap of ∼2.08 eV (Fig. 1b) while TiN exhibits metallic characteristics and preferable conductivity (Fig. 1c). This result can further reflect the advantage of using TiN as core material to construct hierarchical core–shell heterostructures, which may be more conducive to improve the Zn2+-storage performances of MnO2. Not surprisingly, the hierarchical TiN@MnO2 core–shell heterostructure also exhibits excellent conductivity (Fig. 1d), which inherits the metallic property of TiN and improves the conductivity of MnO2 as a shell material. These results are beneficial for charge transfer and promoting zinc ion reaction kinetics, resulting in an excellent rate performance. Considering the Zn2+-storage performances of the cathode materials, we optimized the structure of the diffusion path for Zn2+ in pure MnO2 and TiN@MnO2, as shown in Fig. 1e–h. The diffusion path for Zn2+ in pure MnO2 is presented in Fig. 1e with a diffusion barrier of 0.43 eV (Fig. 1f). The diffusion path for Zn2+ in TiN@MnO2 is shown in Fig. 1g with a diffusion barrier of only 0.21 eV (Fig. 1h), which is significantly lower than in pure MnO2, suggesting that it is easier for Zn2+ to migrate in TiN@MnO2.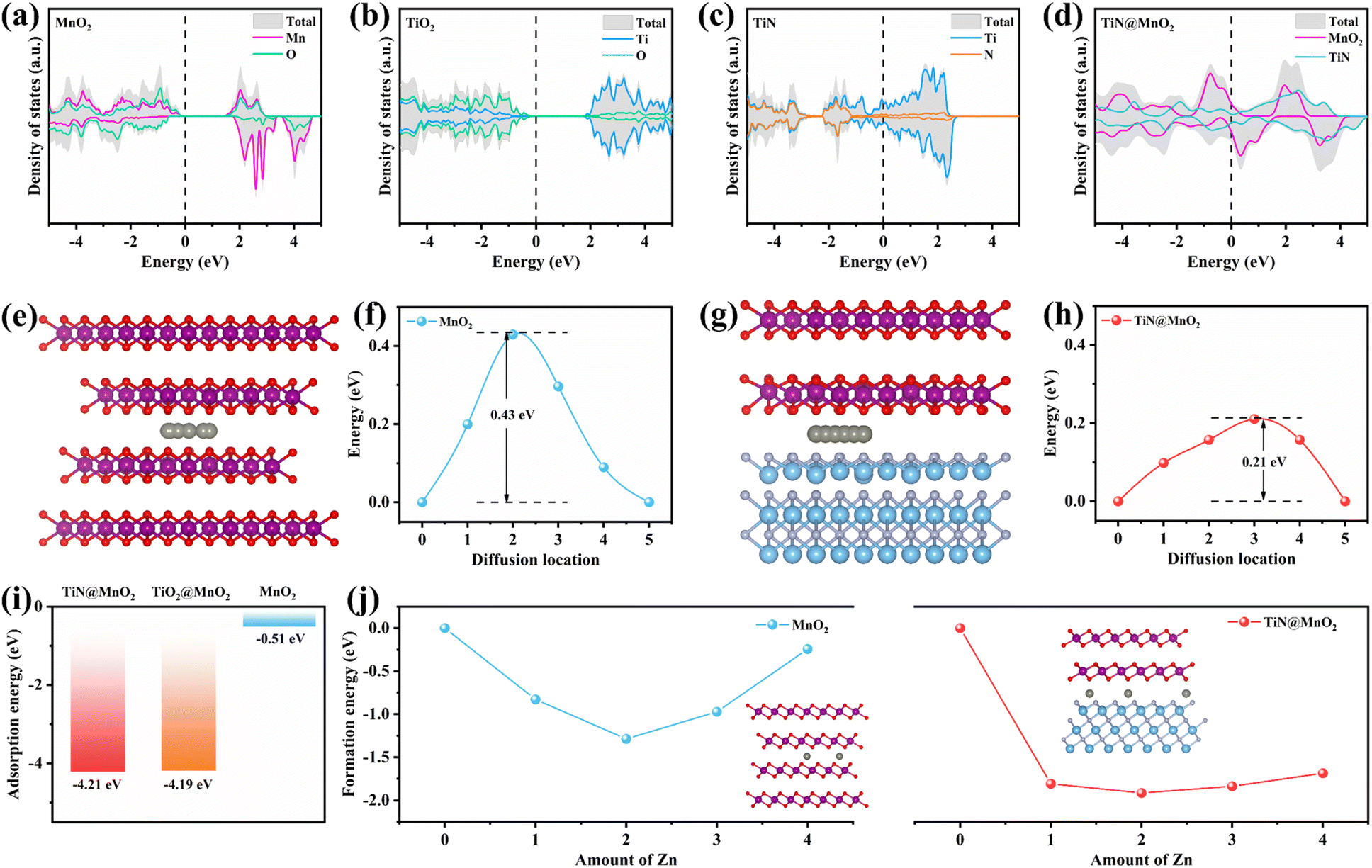 | ||
| Fig. 1 Calculated DOS of pure MnO2 (a), TiO2 (b), TiN (c) and TiN@MnO2 (d). Schematic diagram of pure MnO2 for diffusion path of Zn2+ (e) and diffusion barrier (f). Schematic diagram of TiN@MnO2 for diffusion path of Zn2+ (g) and diffusion barrier (h). (i) Calculated adsorption energy of Zn2+ in pure MnO2, TiO2@MnO2 and TiN@MnO2. (j) Calculated Zn2+ insertion formation energy in pure MnO2 and TiN@MnO2. Purple, red, blue, lavender and grey spheres represent Mn, O, Ti, N, and Zn atoms, respectively. | ||
Furthermore, the adsorption energy of Zn2+ in materials is also an important parameter to evaluate the capture capacity of Zn2+ during the charging and discharging process for the cathode materials. As exhibited in Fig. 1i, the adsorption energy of Zn2+ in pure MnO2 is only −0.51 eV, while the adsorption energies of TiN@MnO2 and TiO2@MnO2 are as high as −4.21 eV and −4.19 eV, respectively, which suggests that both TiN@MnO2 and TiO2@MnO2 possess a stronger ability to adsorb Zn2+ during the charge/discharge process. TiO2 can provide abundant nucleation sites for the deposition of Zn2+ and exhibits strong adsorption capability for Zn2+.46,47 However, due to the strong adsorption capability and poor conductivity of TiO2, the diffusion barrier of Zn2+ in TiO2@MnO2 is as high as 0.93 eV, higher than that of pure MnO2 (Fig. S5†). The high diffusion barrier of Zn2+ in TiO2@MnO2 is not favorable for the (de)intercalation of Zn2+, which further proves the weaker reaction kinetics of Zn2+ in the hierarchical core–shell heterostructure constructed with a TiO2 core in comparison with TiN@MnO2. To further assess the effect of Zn2+ storage for the structures, we calculated the energy of formation of Zn2+ inserted into pure MnO2 and TiN@MnO2, as presented in Fig. 1j. For pure MnO2, the formation energy achieves a minimum value of −1.29 eV when the intercalated Zn2+ reaches 2.0 per formula, and it starts to increase significantly with further Zn2+ insertion, suggesting that Zn ions insert into MnO2 with great difficulty when exceeding 2.0 per formula. The formation energy of Zn2+ insertion into TiN@MnO2 also reaches a minimum value of −1.91 eV at 2.0 per formula, which is far lower than that of pure MnO2. Furthermore, with further Zn2+ insertion into TiN@MnO2 reaching to 4.0 per formula, the formation energy is only −1.68 eV, still significantly lower than that for pure MnO2, which signally implies that TiN@MnO2 can accommodate more Zn ions. Additionally, lower formation energy means easier insertion of Zn ions into TiN@MnO2 during the discharging process, resulting in a higher capacity. Meanwhile, the greater absolute value of the formation energy indicates a more stable structure of the material after inserting Zn ions.48–50 In comparison, the absolute value of the formation energy of TiN@MnO2 is dramatically larger than that of pure MnO2, indicating that TiN as a core-layer material can significantly improve the structural stability for the MnO2 shell layer after inserting Zn ions, resulting in a superior cycling performance. The above theoretical simulations demonstrate that the construction of hierarchical core–shell heterostructures by introducing highly conductive TiN as the core layer can effectively improve the zinc ion storage performance of MnO2.
Fig. 2a schematically shows the preparation process for the hierarchical TiN@MnO2 NWAs core–shell heterostructure directly grown on CC surface as a self-standing cathode for flexible aqueous rechargeable ZIBs. Firstly, the TiO2 NWAs/CC precursors were synthesized and confirmed by X-ray diffraction (XRD) results, as displayed in Fig. S6.† The XRD pattern corresponds to the standard card of TiO2 (JCPDS Card No. 21-1276), indicating successful synthesis for TiO2. Then, after nitrogenization, the precursors were completely converted to TiN NWAs/CC, and finally TiN@MnO2 NWAs/CC was obtained by electrochemical deposition for constructing the hierarchical core–shell heterostructure. The XRD patterns of TiN NWAs/CC, MnO2 NSs/CC, and TiN@MnO2 NWAs/CC are presented in Fig. 2b. The XRD pattern of TiN NWAs/CC exhibits obvious characteristic diffraction peaks at 36.6°, 42.6°, 61.8°, 74.1° and 77.9° indexed to the (111), (200), (220), (311), and (222) lattice planes of TiN (JCPDS Card No. 38-1420), respectively.51 MnO2 NSs has relatively poor crystallinity, which may be due to the complex electric field environment and rapid nucleation-growth rate resulting in random directional growth during electrochemical deposition. Notably, the XRD pattern of TiN@MnO2 NWAs/CC corresponds to the standard card of TiN, while the characteristic diffraction peaks are not obvious for MnO2 NSs due to its low crystallinity. In addition, MnO2 NSs as a shell layer decreases the intensity of the diffraction peaks of TiN@MnO2 NWAs/CC, which are weaker than those of TiN NWAs/CC. Since TiO2@MnO2 NWAs/CC has a similar hierarchical core–shell heterostructure, its diffraction peak intensity is also weaker than that of TiO2 NWAs/CC (Fig. S6†). Fig. S7† displays scanning electron microscope (SEM) images of TiO2 NWAs/CC at different magnifications, and one is able to clearly see that the nanowire arrays are uniformly grown on the CC surface. After nitrogenization treatment, the nanowire arrays' structure was still maintained for TiN NWAs/CC, as shown in Fig. 2c and d. Fig. S8† depicts SEM images of MnO2 NSs/CC, which is uniformly wrapped around the CC surface. Fig. 2e and f display SEM images of TiN@MnO2 NWAs/CC. It can be seen that the surface of TiN NWAs is uniformly covered with MnO2 NSs, thus forming a representative three-dimensional hierarchical core–shell heterostructure. To further observe the core–shell heterostructure of a single TiN@MnO2 nanowire, transmission electron microscopy (TEM) was performed, the TiN nanowire being homogeneously covered by MnO2 NSs, as exhibited in Fig. 2g. Fig. 2h highlights the Ti and N uniformly distributed in the core and Mn and O homogeneously distributed in the shell layer. These results all reveal that TiN@MnO2 NWAs/CC was successfully prepared and exhibits a typical hierarchical core–shell heterostructure.
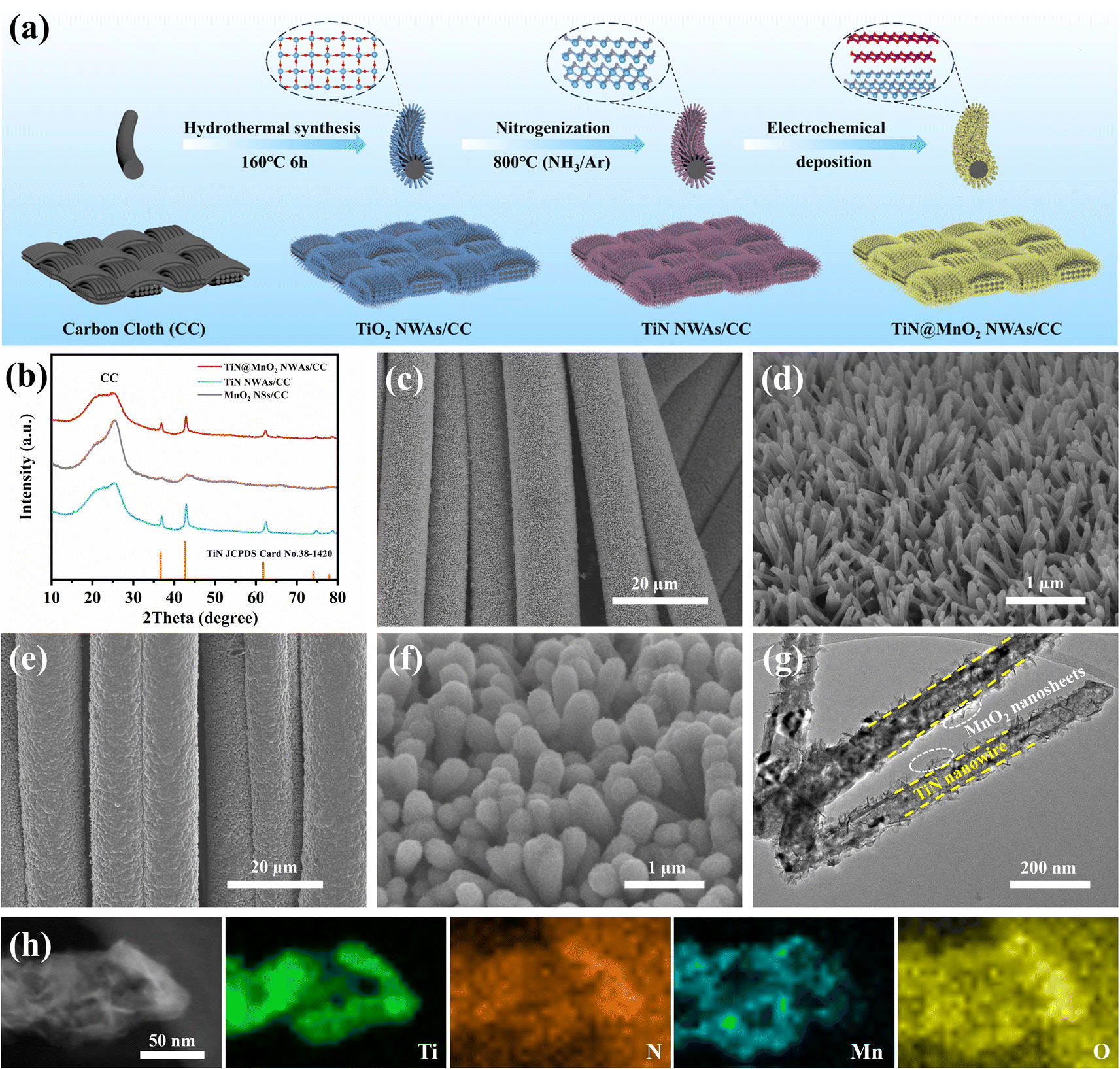 | ||
| Fig. 2 (a) Schematic diagram of preparation of TiN@MnO2 NWAs/CC. (b) XRD patterns of TiN@MnO2 NWAs/CC, TiN NWAs/CC and MnO2 NSs/CC. SEM images of TiN NWAs/CC (c, d) and TiN@MnO2 NWAs/CC (e, f) at different magnifications. TEM image (g) and EDS element mapping (h) of single TiN@MnO2 nanowires. | ||
X-ray photoelectron spectroscopy (XPS) is commonly used to analyze the elemental composition and the chemical valence states of materials. Fig. 3a shows the full XPS survey spectrum of TiN@MnO2 NWAs/CC, which shows the presence of Ti, N, Mn, O and C elements, where C is mainly derived from the CC. The high-resolution Ti 2p XPS spectrum (Fig. 3b) exhibits multiple peaks, which are attributed to Ti–O, Ti–N–O and Ti–N bonds. As shown in Fig. 3c, the peaks of N 1 s are at 396.3 eV, 397.3 eV and 399.8 eV, attributed to Ti–N, Ti–N–O and adsorbed N, respectively.51 For Mn and O included in the shell material, the high-resolution Mn 2p XPS spectrum is displayed in Fig. 3d. The Mn 2p spectrum exhibits two characteristic peaks of Mn 2p3/2 and Mn 2p1/2. The Mn 2p3/2 and Mn 2p1/2 spin–orbit peaks are resolved corresponding to Mn4+ (2p3/2, 642.8 eV; and 2p1/2, 654.7 eV) and Mn3+ (2p3/2, 642.5 eV; and 2p1/2, 653.3 eV);32,35 therefore the Mn state in the hierarchical core–shell heterostructures exists in the form of Mn4+ and Mn3+. The O 1s spectrum in Fig. 3e consists of a peak corresponding to the Mn–O–Mn bond at 530.1 eV, which can be attributed to the MnO2 directly grown on the TiN NWAs surface.32,52 Fig. S9† presents the high-resolution C 1s XPS spectrum.13,53 Moreover, the Raman spectrum (Fig. 3f) shows that the peak located at 641 cm−1 is assigned to the vibration of the Mn–O bond for MnO2.54
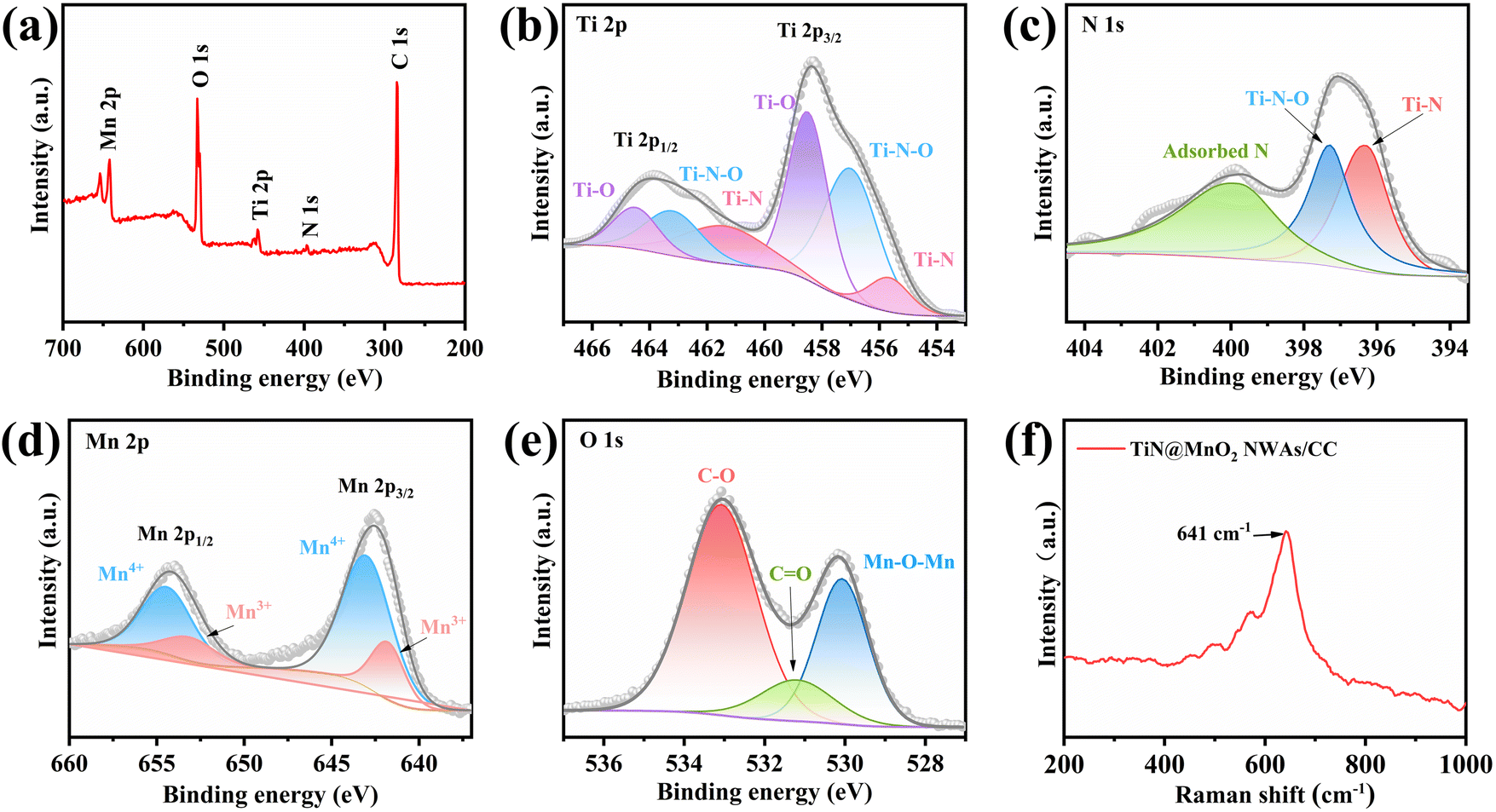 | ||
| Fig. 3 (a) Full XPS survey spectrum of TiN@MnO2 NWAs/CC and high-resolution Ti 2p (b), N 1s (c), Mn 2p (d), and O 1s (e) XPS spectra. (f) Raman spectrum of TiN@MnO2 NWAs/CC. | ||
To verify the advantages in terms of electrochemical performance of choosing the TiN core, the electrochemical performances of the three electrodes (TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC) were investigated (Fig. 4). The cyclic voltammetry (CV) curves of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC at a scan rate of 0.2 mV s−1 are displayed in Fig. 4a. Notably, the TiN@MnO2 NWAs/CC electrode possesses a larger response current and more distinct redox peaks, indicating a stronger Zn2+ storage ability. Additionally, for the TiN@MnO2 NWAs/CC electrode, the oxidation peaks located at 1.57 and 1.61 V are slightly shifted to lower potentials, and the redox peaks form smaller potential differences, indicating better redox kinetic behaviors of the electrode. The galvanostatic charge–discharge (GCD) curves for the three electrodes at a current density of 0.1 A g−1 are shown in Fig. 4b. The potential gap for TiN@MnO2 NWAs/CC is ∼0.19 V, while the TiO2@MnO2 NWAs/CC and MnO2 NSs/CC electrodes exhibit potential gaps of ∼0.24 V and ∼0.22 V, further proving the excellent redox kinetic behaviors. Moreover, the TiN@MnO2 NWAs/CC electrode exhibits higher capacity capable of reaching 385.1 mA h g−1, far exceeding the 310.6 mA h g−1 of TiO2@MnO2 NWAs/CC and 194.4 mA h g−1 of MnO2 NSs/CC electrodes. It is noteworthy that, according to DFT, TiN@MnO2 exhibits a stronger adsorption energy and lower formation energy for Zn ions, as well as more easily accommodating Zn2+, in comparison with pure MnO2, thus resulting in a higher capacity for the TiN@MnO2 NWAs/CC electrode (Fig. 1i and j). The GCD curves of the TiN@MnO2 NWAs/CC electrode and the rate performance of the three electrodes at different current densities are displayed in Fig. 4c and d. The discharging and charging platforms of the TiN@MnO2 NWAs/CC electrode correspond well to the positions of the redox peaks in its CV curves (Fig. 4a). The GCD curves of MnO2 NSs/CC and TiO2@MnO2 NWAs/CC electrodes are presented in Fig. S10 and S11,† respectively. From Fig. 4d, the capacity of the TiN@MnO2 NWAs/CC electrode increases continuously at 0.1 A g−1 and reaches 385.1 mA h g−1 at the fifth cycle, which may be due to the fact that the active materials were activated at low current density and gradual permeation of the electrolyte resulting in higher capacity. The reversible capacities of 322.1, 265.0, 226.4, 202.0, 185.5, 159.0 and 127.6 mA h g−1 were achieved at 0.2, 0.4, 0.6, 0.8, 1.0, 2.0 and 4.0 A g−1, respectively. When the current density is restored to 0.1 A g−1, the capacity of the TiN@MnO2 NWAs/CC electrode can immediately return to the initial state, which reveals that TiN@MnO2 NWAs/CC exhibits outstanding high rate tolerance and better rate performance. This is attributed to the introduction of the highly conductive TiN core material, thereby lowering the band gap of the MnO2 shell layer, which is beneficial for charge transfer and promotes zinc ion reaction kinetics (Fig. 1d), further demonstrating the superiority of the TiN core. Furthermore, TiN NWAs provide a larger specific surface area for MnO2 NSs growth, which enables MnO2 to contact the electrolyte more efficiently and improve utilization of active substances. Besides, the highly conductive core material redistributes the electrode material charge, which solves the problem of the low conductivity of MnO2. Although TiO2 NWAs as a core layer can also offer considerable specific surface area for the growth of MnO2 NSs and form hierarchical core–shell heterostructures, the low conductivity of TiO2 and high diffusion barrier cause slow ion diffusion kinetics, not conducive for the improvement of rate performance for TiO2@MnO2 NWAs/CC. The above considerations are the important reasons for TiN as a core material to improve the electrochemical performance of novel hierarchical core–shell heterostructures. Fig. 4e exhibits the CV curves of the TiN@MnO2 NWAs/CC electrode for the first five cycles at a scan rate of 0.1 mV s−1. The oxidation peak in the first cycle is at 1.6 V, while in the second cycle there is a significant change in the CV curve with two oxidation peaks appearing at 1.58 and 1.62 V, respectively. Notably, the CV curves from the third to the fifth cycles almost overlap, implying that the electrodes possess great stability and reversibility. Besides, the cycle stability of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC was evaluated at different current densities, as presented in Fig. 4f and g. After 100 cycles at 0.2 A g−1, the capacity of TiN@MnO2 NWAs/CC changes from 345 mA h g−1 to 250.6 mA h g−1 with a capacity retention of 72.6%, while the capacity retention of TiO2@MnO2 NWAs/CC and MnO2 NSs/CC was only 58.7% and 50.6%, respectively (Fig. 4f). At a current density of 0.2 A g−1, it can be clearly seen that the coulombic efficiency (CE) of TiO2@MnO2 NWAs/CC is lower than that of TiN@MnO2 NWAs/CC, suggesting that the Zn2+ reaction kinetics in TiO2@MnO2 NWAs/CC is slow, and the existence of irreversible reactions results in a low CE, which is unfavorable for the cycling of the electrode.5,55–58 For long-term cycle life, TiN@MnO2 NWAs/CC maintains outstanding cycling performance at an ultrahigh current density of 2.0 A g−1, where the initial capacity is 186.3 mA h g−1 and then the capacity increases continuously during cycling, reaching 236.1 mA h g−1 at the 2000th cycle and returning to 189.4 mA h g−1 at the 2300th cycle, where the capacity retention is 101.6%. In contrast, the capacity of TiO2@MnO2 NWAs/CC decayed continuously after the 1000th cycle, and the capacity retention rates of TiO2@MnO2 NWAs/CC and MnO2 NSs/CC were only 14.0% and 11.9% after 2300 cycles, respectively (Fig. 4g). This is due to the greater absolute value of the formation energy during Zn2+ insertion into TiN@MnO2, implying a more stable structure and thus improved cycling stability of the electrode material (Fig. 1j). The above electrochemical measurements in combination with theoretical calculation results demonstrate that the novel hierarchical core–shell heterostructure constructed with the TiN core possesses the advantages of high rate and long life for zinc ion storage features.
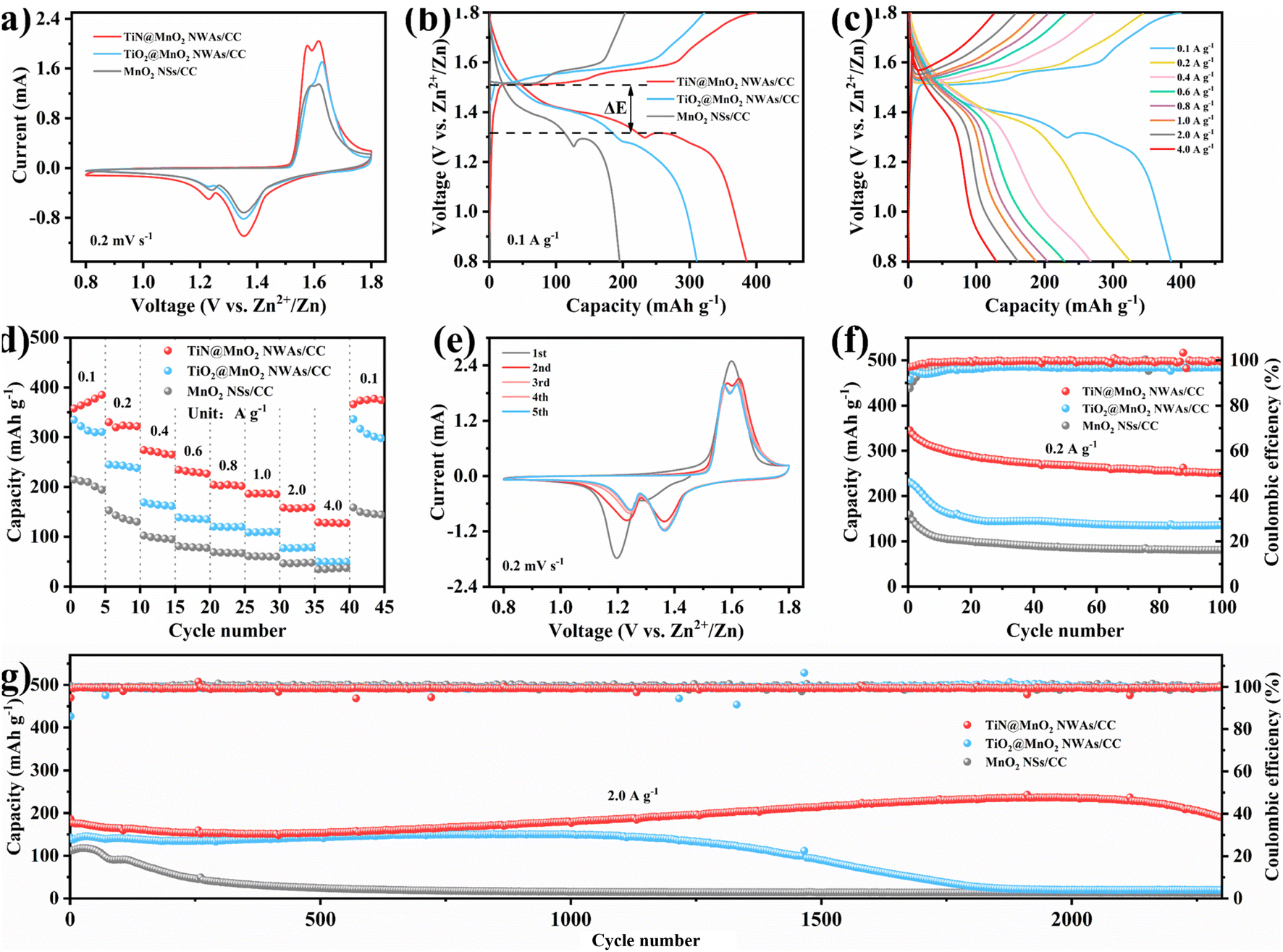 | ||
| Fig. 4 Electrochemical performance of ZIBs based on the cathode materials of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC. CV curves at a scan rate of 0.2 mV s−1 (a) and GCD profiles at 0.1 A g−1 (b) of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC electrodes. (c) GCD curves of the TiN@MnO2 NWAs/CC electrode at different current densities. (d) Rate performance of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC electrodes. (e) CV curves for 5 initial cycles of the TiN@MnO2 NWAs/CC electrode at a scan rate of 0.2 mV s−1. Cycle performance of TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC at 0.2 A g−1 (f) and 2.0 A g−1 (g). | ||
To evaluate the effect on the reaction kinetics of TiN@MnO2 NWAs/CC by TiN core materials, its Zn2+ storage behavior was profiled by CV curves at different scan rates (Fig. 5a). It is evident that the CV curves at different scan rates maintain a similar shape, with the redox peaks becoming wider with increasing scan rates. Analysis of the capacitive contribution in the TiN@MnO2 NWAs/CC electrode can be reflected according to the following equation:
| i = avb |
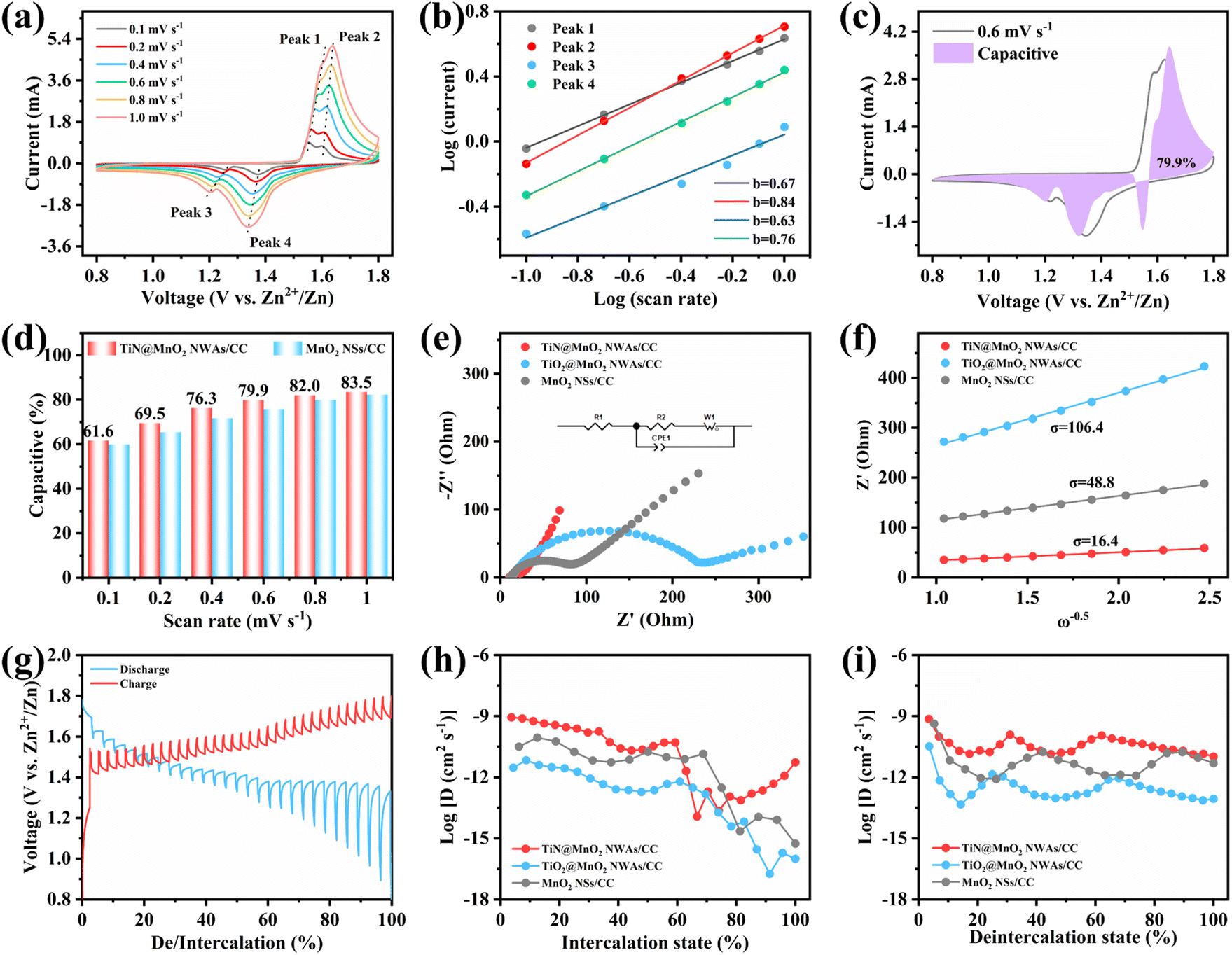 | ||
| Fig. 5 The electrochemical kinetics of TiN@MnO2 NWAs/CC during the charge/discharge process. (a) CV curves at different scan rates. (b) The relationship between log(i) and log(v) for scan rates from 0.1 to 1 mV s−1. (c) CV curves with a capacitive contribution at 0.6 mV s−1. (d) Capacitive contribution at different scan rates. (e) EIS results for the TiN@MnO2 NWAs/CC, TiO2@MnO2 NWAs/CC and MnO2 NSs/CC electrodes. (f) Relationship between Z′ and ω−1/2 for the electrodes in the low-frequency region. (g) The charge/discharge GITT curves of the TiN@MnO2 NWAs/CC electrode. (h, i) The ion diffusion coefficient during the charge–discharge process. | ||
The above equation can be rearranged as follows:
| log(i) = blog(v) + log(a) |
| i = k1v + k2v1/2 |
The formula can be transformed into:
| i/v1/2 = k1v1/2 + k2 |
To further investigate the effect of the hierarchical core–shell heterostructures on the electrochemical performance of MnO2, electrochemical impedance spectroscopy (EIS) was carried out to gain insight into the reaction kinetics of the electrode, as displayed in Fig. 5e. The diameter of the semicircle in the high-frequency region indicates the charge transfer resistance (Rct), where the TiN@MnO2 NWAs/CC electrode exhibits an Rct value of 21.64 Ω, this being significantly lower than that of TiO2@MnO2 NWAs/CC (230.8 Ω) and MnO2 NSs/CC (65.03 Ω), demonstrating that the hierarchical core–shell heterostructure constructed with TiN can significantly accelerate ion diffusion and thus improve the charge transfer. Additionally, the ion diffusion coefficient (D) could be calculated based on the relationship between low frequency and impedance, and the Warburg factor is obtained by fitting for Z′–ω−1/2, where a smaller slope of the linear tendency means a larger ion diffusion coefficient. The diffusion coefficient D of TiN@MnO2 NWAs/CC can be calculated according to the equation as 1.61 × 10−12 cm2 s−1, while those of TiO2@MnO2 NWAs/CC and MnO2 NSs/CC are only 3.8 × 10−14 cm2 s−1 and 1.82 × 10−13 cm2 s−1, respectively (Fig. 5f). This is due to the lower diffusion barrier of Zn2+ in TiN@MnO2, which means that ions more easily migrate and diffuse (Fig. 1e–h). Furthermore, the galvanostatic intermittent titration technique (GITT) is an important method to measure the diffusion coefficient of electrodes (Fig. 5g). The calculation equation is presented below:14,59,60
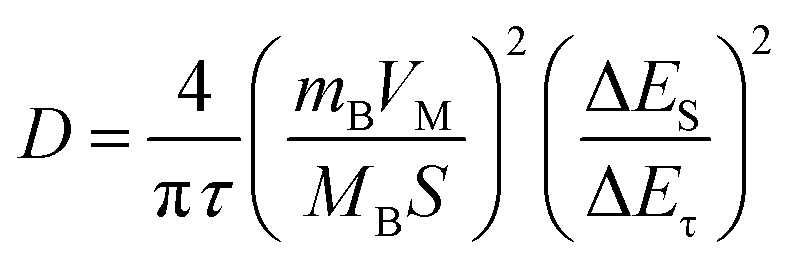
Quasi-solid-state flexible ZIBs were successfully assembled, when coupling with TiN@MnO2 NWAs/CC as the cathode and Zn NSs/CC as the anode, to verify the remarkable electrochemical performances and potential practical applications for the as-prepared cathode. For such, the Zn NSs/CC anode was obtained by a simple electrochemical deposition method. All the characteristic diffraction peaks in the XRD pattern were able to be matched with pure Zn (JCPDS Card No. 04-0831), indicating that Zn was successfully directly deposited on the CC surface (Fig. S17†). A schematic diagram of the as-assembled flexible ZIBs is illustrated in Fig. 6a. The CV curves of our as-assembled flexible ZIBs at different scan rates always maintain a similar shape with increasing scan rates, revealing an excellent electrochemical stability, as displayed in Fig. 6b. Fig. 6c exhibits the GCD curves for the device at different current densities and two obvious charge/discharge voltage plateaus can still be observed at low current density. There is a slight decrease of the capacity in comparison with solution electrolyte because of the slower ion diffusion in the gel electrolyte. Fig. 6d presents the rate performance of the as-assembled flexible ZIBs. Reversible capacities of 241.7, 203.8, 171.4, 152.2, 139.7, 129.6, 102.1 and 73.9 mA h g−1 were achieved at 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 2.0 and 4.0 A g−1, respectively, exhibiting a favorable rate capability. Notably, the capacity instantly returns to the initial state when the current density is restored to 0.1 A g−1, also demonstrating a robust tolerance to the rapid (de)intercalation of Zn2+ even though in a gel electrolyte. Notably, the assembled flexible ZIBs can provides a maximum energy density of 327.7 W h kg−1 with a power density of 135.6 W kg−1, while at a maximum power density of 5.2 kW kg−1, the as-assembled device still exhibits an impressive energy density of 100.2 W h kg−1, as shown in the Ragone plots in Fig. 6e. More importantly, the electrochemical performances of our as-assembled flexible ZIBs greatly exceed those of many previously reported aqueous ZIBs using different cathode materials, such as Mn2O3,15 V2O5,61 ZnMn2O4,62 N-ZMO,63 MoS2,64 VOPO4,19 MnO2@rGO,65 NixCay-VO2@C66 and NiCo–MnO2.67 Cycling stability, as one of the essential indexes for assessing the performance, plays a critical role during practical applications. Thus we evaluated the cycling stability of the assembled flexible ZIBs, as presented in Fig. 6f and g. The initial capacity was 208.5 mA h g−1 with the capacity retention reaching 92.6% after 100 cycles at a low current density of 0.2 A g−1 (Fig. 6f). At a high current density of 2.0 A g−1, the assembled ZIBs show a brief and obvious capacity decay for the initial 100 cycles, and then tend to be stable, achieving a capacity retention of 68.7% through 3000 cycles and the CE is maintained at around 100%, further exhibiting outstanding cycling stability (Fig. 6g). Finally, two assembled flexible ZIBs connected in series can light LEDs, as displayed in Fig. S18.† Therefore, TiN@MnO2 NWAs/CC has great potential as a ZIB cathode for wearable electronic devices. Such flexible energy-storage devices exhibit high rate and long life, demonstrating that novel hierarchical core–shell heterostructures have prominent potential in practical applications.
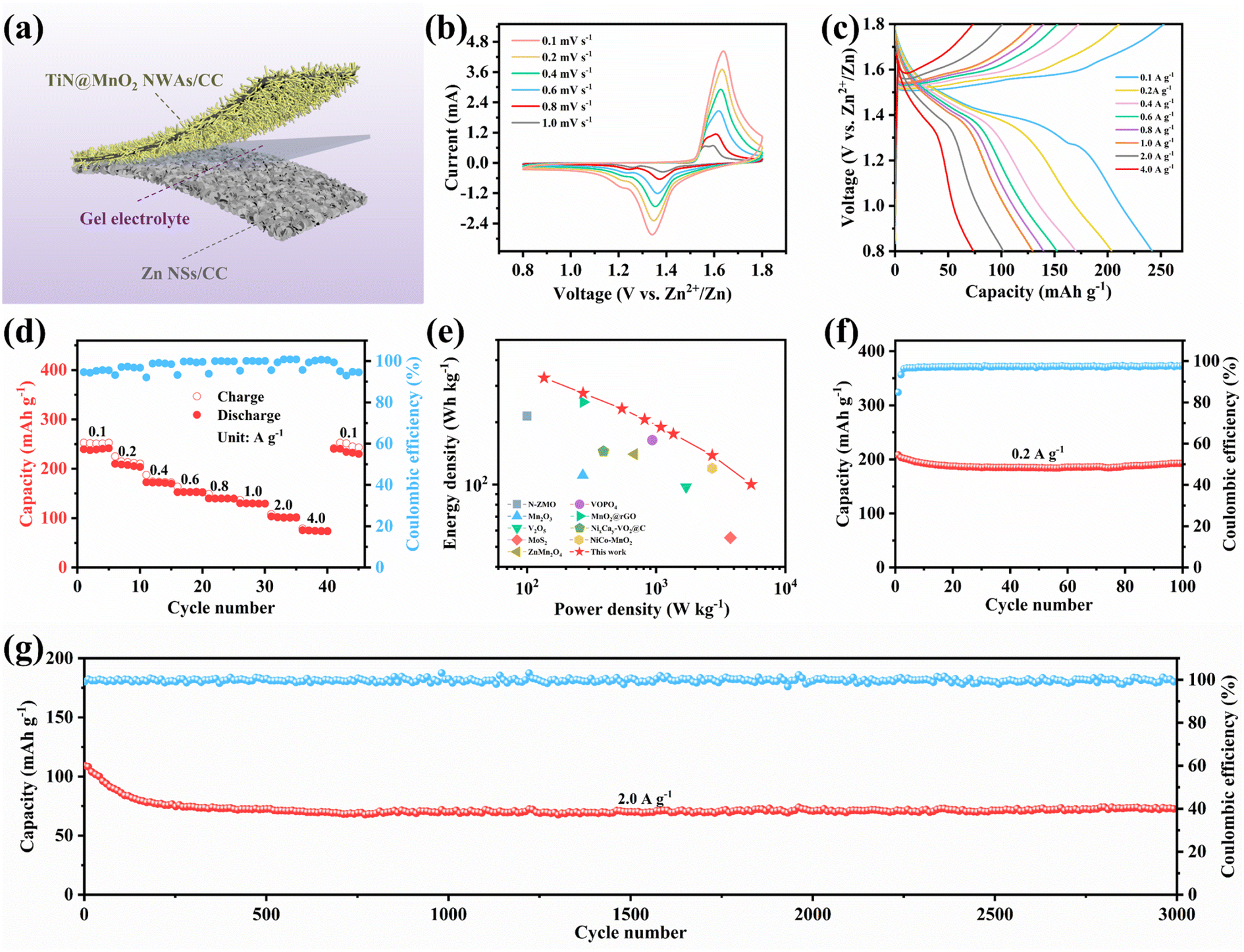 | ||
| Fig. 6 Electrochemical performances of the as-assembled flexible ZIBs. (a) Schematic illustration. (b) CV curves at different scan rates. (c) GCD curves at various current densities. (d) Rate performance at various current densities from 0.1 to 4.0 A g−1. (e) Ragone plot of TiN@MnO2 NWAs/CC//Zn NSs/CC battery compared with previously reported flexible ZIBs. Cycling performance at 0.2 A g−1 (f) and 2.0 A g−1 (g). | ||
4. Conclusion
In summary, we successfully constructed a novel and advanced cathode by anchoring MnO2 NSs on TiN NWAs directly grown on CC via a simple and economical hydrothermal method as well as carrying out electrochemical deposition to produce flexible ZIBs. The as-prepared TiN@MnO2 NWAs/CC exhibits a typical hierarchical core–shell heterostructure, which is conducive to accelerate reaction kinetics, possessing a more stable structure during charge/discharge, and improving rate capability and cycling performance in ZIBs. The DFT results also demonstrate these points. As a result, the TiN@MnO2 NWAs/CC core–shell electrode exhibits an ultrahigh capacity of 385.1 mA h g−1, while MnO2 NSs/CC can only deliver a low capacity of 194.4 mA h g−1 at a current density of 0.1 A g−1. Furthermore, to highlight the rate capability of the core–shell heterostructure using TiN as the core, the TiN@MnO2 NWAs/CC core–shell heterostructure provided a higher capacity of 127.6 mA h g−1 (33.1%), while the TiO2@MnO2 NWAs/CC core–shell and MnO2 NSs/CC electrodes only sustain a low capacity of 49.7 mA h g−1 (16.0%) and 39.6 mA h g−1 (20.3%) when the current density was increased to 4.0 A g−1, respectively. More importantly, the TiN@MnO2 NWAs/CC electrode achieves a capacity retention of 101.6% under a current density of 2.0 A g−1 after 2300 cycles, far surpassing that of the TiO2@MnO2 NWAs/CC (14.0%) and MnO2 NSs/CC electrodes (11.9%), further evidence of the superiority of TiN core materials. Benefiting from the remarkable electrochemical performance of the flexible TiN@MnO2 NWAs/CC electrode, the as-assembled quasi-solid-state flexible ZIBs achieve a high capacity of 241.7 mA h g−1 at 0.1 A g−1 and prominent energy density of 327.7 W h kg−1 at a power density of 135.6 W kg−1. Furthermore, our flexible ZIBs also exhibit an excellent cycling performance, with a capacity retention of 92.6% after 100 cycles at 0.2 A g−1, and a long cycle life of 68.7% capacity retention after 3000 cycles at 2.0 A g−1. The above results highlight that a novel cathode material has been successfully prepared, providing ideas for the design of high-capacity and high-energy-density cathode materials for next-generation flexible energy-storage devices.Author contributions
Ziming Xu: conceptualization, investigation, methodology, writing – original draft, writing – review & editing. Wenyuan Zhang: data curation, writing – review & editing. Xianzhen Wang: data curation, methodology. Yuxin Li: validation, data curation, investigation. Jinwen Fu: validation, data curation. Yongbao Feng: resources, funding acquisition, project administration, writing – review & editing. Wenbin Gong: software, resources. Jiabin Guo: resources. Pan Xue: investigation, resources. Qiulong Li: supervision, resources, funding acquisition, project administration, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 51703241 and 12004324), the Scientific Research Start Foundation of Nanjing Tech University (grant number 39803163), the Science and Technology Project of Jiangxi Province (20202ACBL211005), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (22KJA140003) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- P. Xue, C. Guo, L. Li, H. Li, D. Luo, L. Tan and Z. Chen, Adv. Mater., 2022, 34, 2110047 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu, Z. Liu, X. Wu, Y. Wen, H. Chen, X. Ni, G. Liu, J. Huang and S. Peng, J. Energy Chem., 2022, 64, 23–32 CrossRef CAS.
- S. Wang, G. Zeng, Q. Sun, Y. Feng, X. Wang, X. Ma, J. Li, H. Zhang, J. Wen, J. Feng, L. Ci, A. Cabot and Y. Tian, ACS Nano, 2023, 17, 13256–13268 CrossRef CAS PubMed.
- Y. Zeng, Y. Wang, Q. Jin, Z. Pei, D. Luan, X. Zhang and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 25793–25798 CrossRef CAS PubMed.
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- X. Wang, G. Ding, Z. Ma, Z. Xu, Y. Feng, W. Gong, C. Liu, K. Tian, Z. Yong and Q. Li, Chem. Eng. J., 2023, 472, 144996 CrossRef CAS.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y. W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 2001769 Search PubMed.
- Y. Liu, K. Wang, X. Yang, J. Liu, X.-X. Liu and X. Sun, ACS Nano, 2023, 17, 14792–14799 CrossRef CAS PubMed.
- Z. Zhang, B. Xi, X. Wang, X. Ma, W. Chen, J. Feng and S. Xiong, Adv. Funct. Mater., 2021, 31, 2103070 CrossRef CAS.
- S. Deng, Z. Tie, F. Yue, H. Cao, M. Yao and Z. Niu, Angew. Chem., Int. Ed., 2022, 61, e202115877 CrossRef CAS PubMed.
- Q. Tan, X. Li, B. Zhang, X. Chen, Y. Tian, H. Wan, L. Zhang, L. Miao, C. Wang, Y. Gan, J. Jiang, Y. Wang and H. Wang, Adv. Energy Mater., 2020, 10, 2001050 CrossRef CAS.
- D. Zhang, J. Cao, X. Zhang, N. Insin, S. Wang, J. Han, Y. Zhao, J. Qin and Y. Huang, Adv. Funct. Mater., 2021, 31, 2009412 CrossRef CAS.
- H. Moon, K. H. Ha, Y. Park, J. Lee, M. S. Kwon, J. Lim, M. H. Lee, D. H. Kim, J. H. Choi, J. H. Choi and K. T. Lee, Adv. Sci., 2021, 8, 2003714 CrossRef CAS PubMed.
- X. Xiao, Z. Zhang, Y. Wu, J. Xu, X. Gao, R. Xu, W. Huang, Y. Ye, S. T. Oyakhire, P. Zhang, B. Chen, E. Cevik, S. M. Asiri, A. Bozkurt, K. Amine and Y. Cui, Adv. Mater., 2023, 35, 2211555 CrossRef CAS PubMed.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- Z. Wu, C. Lu, F. Ye, L. Zhang, L. Jiang, Q. Liu, H. Dong, Z. Sun and L. Hu, Adv. Funct. Mater., 2021, 31, 2106816 CrossRef.
- F. Wan, Z. Hao, S. Wang, Y. Ni, J. Zhu, Z. Tie, S. Bi, Z. Niu and J. Chen, Adv. Mater., 2021, 33, 2102701 CrossRef CAS PubMed.
- M. Bao, Z. Zhang, X. An, J. Liu, J. Feng, B. Xi and S. Xiong, Nano Res., 2023, 16, 2445–2453 CrossRef CAS.
- R. Trócoli and F. L. Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- Z. Feng, Y. Zhang, J. Sun, Y. Liu, H. Jiang, M. Cui, T. Hu and C. Meng, Chem. Eng. J., 2022, 433, 133795 CrossRef CAS.
- S. Kong, Y. Li, X. Zhang, Z. Xu, X. Wang, Y. Feng, W. Gong, C. Liu, K. Tian and Q. Li, Small, 2023, 2304462 CrossRef CAS PubMed.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- M. Mateos, N. Makivic, Y. S. Kim, B. Limoges and V. Balland, Adv. Energy Mater., 2020, 10, 2000332 CrossRef CAS.
- B. Sambandam, V. Mathew, S. Kim, S. Lee, S. Kim, J. Y. Hwang, H. J. Fan and J. Kim, Chem, 2022, 8, 924–946 CAS.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- Y. Zhang, S. Deng, Y. Li, B. Liu, G. Pan, Q. Liu, X. Wang, X. Xia and J. Tu, Energy Storage Mater., 2020, 29, 52–59 CrossRef.
- J. Li, N. Luo, L. Kang, F. Zhao, Y. Jiao, T. J. Macdonald, M. Wang, I. P. Parkin, P. R. Shearing, D. J. L. Brett, G. Chai and G. He, Adv. Energy Mater., 2022, 12, 2201840 CrossRef CAS.
- G. Wang, Y. Wang, B. Guan, J. Liu, Y. Zhang, X. Shi, C. Tang, G. Li, Y. Li, X. Wang and L. Li, Small, 2021, 17, 2104557 CrossRef CAS PubMed.
- J. Yang, G. Yao, Z. Li, Y. Zhang, L. Wei, H. Niu, Q. Chen and F. Zheng, Small, 2022, 19, 2205544 CrossRef PubMed.
- Y. Wang, Y.-Z. Zhang, Y.-Q. Gao, G. Sheng and J. E. ten Elshof, Nano Energy, 2020, 68, 104306 CrossRef CAS.
- Y. Ma, M. Xu, R. Liu, H. Xiao, Y. Liu, X. Wang, Y. Huang and G. Yuan, Energy Storage Mater., 2022, 48, 212–222 CrossRef.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- Q. Li, M. Liu, F. Huang, X. Zuo, X. Wei, S. Li and H. Zhang, Chem. Eng. J., 2022, 437, 135494 CrossRef CAS.
- N. Li, Z. Hou, S. Liang, Y. Cao, H. Liu, W. Hua, C. Wei, F. Kang and J.-G. Wang, Chem. Eng. J., 2023, 452, 139408 CrossRef CAS.
- H. Wang, R. Guo, Y. Ma and F. Zhou, Adv. Funct. Mater., 2023, 2301351 CrossRef CAS.
- L. Zhang, J. Bi, T. Liu, X. Chu, H. Lv, D. Mu, B. Wu and F. Wu, Energy Storage Mater., 2023, 54, 410–420 CrossRef.
- X. Lu, G. Wang, T. Zhai, M. Yu, S. Xie, Y. Ling, C. Liang, Y. Tong and Y. Li, Nano Lett., 2012, 12, 5376–5381 CrossRef CAS PubMed.
- A. Dennyson Savariraj, C. Justin Raj, A. M. Kale and B. C. Kim, Small, 2023, 19, 2207713 CrossRef CAS PubMed.
- H. Wang, J. Deng, C. Xu, Y. Chen, F. Xu, J. Wang and Y. Wang, Energy Storage Mater., 2017, 7, 216–221 CrossRef CAS.
- M.-S. Balogun, C. Li, Y. Zeng, M. Yu, Q. Wu, M. Wu, X. Lu and Y. Tong, J. Power Sources, 2014, 272, 946–953 CrossRef CAS.
- L. Han, J. Luo, R. Zhang, W. Gong, L. Chen, F. Liu, Y. Ling, Y. Dong, Z. Yong, Y. Zhang, L. Wei, X. Zhang, Q. Zhang and Q. Li, ACS Nano, 2022, 16, 14951–14962 CrossRef CAS PubMed.
- B. Li, J. Xue, X. Lv, R. Zhang, K. Ma, X. Wu, L. Dai, L. Wang and Z. He, Surf. Coat. Technol., 2021, 421, 127367 CrossRef CAS.
- P. Xue, C. Guo, N. Wang, K. Zhu, S. Jing, S. Kong, X. Zhang, L. Li, H. Li, Y. Feng, W. Gong and Q. Li, Adv. Funct. Mater., 2021, 31, 2106417 CrossRef CAS.
- Y. Yang, J. Xiao, J. Cai, G. Wang, W. Du, Y. Zhang, X. Lu and C. Li, Adv. Funct. Mater., 2021, 31, 2005092 CrossRef CAS.
- J. Yang, X. Xiao, W. Gong, L. Zhao, G. Li, K. Jiang, R. Ma, M. H. Rummeli, F. Li, T. Sasaki and F. Geng, Angew. Chem., Int. Ed., 2019, 58, 8740–8745 CrossRef CAS PubMed.
- S. Wang, Z. Yuan, X. Zhang, S. Bi, Z. Zhou, J. Tian, Q. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2021, 60, 7056–7060 CrossRef CAS PubMed.
- Q. Li, Q. Zhang, C. Liu, Z. Zhou, C. Li, B. He, P. Man, X. Wang and Y. Yao, J. Mater. Chem. A, 2019, 7, 12997–13006 RSC.
- L. Ding, J. Gao, T. Yan, C. Cheng, L.-Y. Chang, N. Zhang, X. Feng and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 17570–17577 CrossRef CAS PubMed.
- T. Li, R. Guo, L. Meng, X. Sun, Y. Li, F. Li, X. Zhao, L. An, J. Peng and W. Wang, Chem. Eng. J., 2022, 446, 137290 CrossRef CAS.
- G. Cui, Y. Zeng, J. Wu, Y. Guo, X. Gu and X. W. Lou, Adv. Sci., 2022, 9, 2106067 CrossRef CAS PubMed.
- H. Jiang, X. Han, X. Du, Z. Chen, C. Lu, X. Li, H. Zhang, J. Zhao, P. Han and G. Cui, Adv. Mater., 2022, 34, 2108665 CrossRef CAS PubMed.
- P. Li, Y. Wang, Q. Xiong, Y. Hou, S. Yang, H. Cui, J. Zhu, X. Li, Y. Wang, R. Zhang, S. Zhang, X. Wang, X. Jin, S. Bai and C. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202303292 CrossRef CAS PubMed.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
- Z. Li, Y. Ren, L. Mo, C. Liu, K. Hsu, Y. Ding, X. Zhang, X. Li, L. Hu, D. Ji and G. Cao, ACS Nano, 2020, 14, 5581–5589 CrossRef CAS PubMed.
- X. Wang, Z. Zhang, M. Huang, J. Feng, S. Xiong and B. Xi, Nano Lett., 2022, 21, 119–127 CrossRef PubMed.
- Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Energy Storage Mater., 2022, 47, 424–433 CrossRef.
- J. Zhao, H. Ren, Q. Liang, D. Yuan, S. Xi, C. Wu, W. Manalastas, J. Ma, W. Fang, Y. Zheng, C.-F. Du, M. Srinivasan and Q. Yan, Nano Energy, 2019, 62, 94–102 CrossRef CAS.
- F. Gao, B. Mei, X. Xu, J. Ren, D. Zhao, Z. Zhang, Z. Wang, Y. Wu, X. Liu and Y. Zhang, Chem. Eng. J., 2022, 448, 137742 CrossRef CAS.
- W. Qiu, H. Xiao, H. Feng, Z. Lin, H. Gao, W. He and X. Lu, Chem. Eng. J., 2021, 422, 129890 CrossRef CAS.
- S. Li, Y. Liu, X. Zhao, K. Cui, Q. Shen, P. Li, X. Qu and L. Jiao, Angew. Chem., Int. Ed., 2021, 60, 20286–20293 CrossRef CAS PubMed.
- Z.-F. Liu, C.-Y. Zhu, Y.-W. Ye, Y.-H. Zhang, F. Cheng and H.-R. Li, ACS Appl. Mater. Interfaces, 2022, 14, 25962–25971 CrossRef CAS PubMed.
- X. Zhao, L. Mao, Q. Cheng, F. Liao, G. Yang and L. Chen, Carbon, 2022, 186, 160–170 CrossRef CAS.
- J. Yin, R. Zhu, L. Xia, H. Liu, Y. Gao, Z. Gan, X. Feng, M. Wang, G. Meng, Y. Su, Y. Cheng and X. Xu, J. Mater. Chem. A, 2023, 11, 11436–11444 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06183c |
| This journal is © The Royal Society of Chemistry 2024 |