
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Delving into Fe-content effects on surface reconstruction of Ba0.50Sr0.50Co1−xFexO3−δ for the oxygen evolution reaction†
Dino
Aegerter
 *a,
Emiliana
Fabbri
*a,
Mario
Borlaf‡
b,
Nur Sena
Yüzbasi
b,
Nataša
Diklić
a,
Adam H.
Clark
c,
Vladyslav
Romankov
c,
Cinthia
Piamonteze
c,
Jan
Dreiser
c,
Thomas
Huthwelker
c,
Thomas
Graule
b and
Thomas J.
Schmidt
ad
*a,
Emiliana
Fabbri
*a,
Mario
Borlaf‡
b,
Nur Sena
Yüzbasi
b,
Nataša
Diklić
a,
Adam H.
Clark
c,
Vladyslav
Romankov
c,
Cinthia
Piamonteze
c,
Jan
Dreiser
c,
Thomas
Huthwelker
c,
Thomas
Graule
b and
Thomas J.
Schmidt
ad
aElectrochemistry Laboratory, Paul Scherrer Institute, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland. E-mail: dino.aegerter@psi.ch; emiliana.fabbri@psi.ch
bLaboratory for High Performance Ceramics, Empa – Swiss Federal Laboratories for Materials Science and Technology, CH-8600 Dübendorf, Switzerland
cPhoton Science Division, Paul Scherrer Institute, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland
dInstitute of Molecular Physical Science, ETH Zurich, CH-8093 Zürich, Switzerland
First published on 31st January 2024
Abstract
Surface reconstruction of cobalt-based oxides is recognized as a key to efficiently electrocatalyze the oxygen evolution reaction (OER) in alkaline environment. Identifying material features that promote surface reconstruction is crucial to rationally improve OER electrocatalysts. Here, the Fe-content effects on the surface reconstruction of flame-spray synthesized Ba0.50Sr0.50Co1−xFexO3−δ (BSCo1−xFex) is systematically investigated by gradually substituting Co with Fe (0 < x < 1). The electrochemical characterization reveals a volcano-shaped trend of the OER activity and stability as a function of the Fe-content, and identifies BSCo0.80Fe0.20 as the best performing electrocatalyst. This Fe-content dependent performance trend directly correlates with the extent of surface reconstruction, as unveiled by combining ex situ surface and operando bulk X-ray absorption spectroscopy. More specifically, the increasing electrocatalytic performance from x = 0.01 to 0.20 is explained by the ability of Fe to stabilize surface Co2+-atoms in the pristine material. This enhances the electrochemically triggered irreversible surface Co oxidation, leading to a more extensive formation of a Co- and Fe-based (oxyhydr)oxide layer that reaches deep into the electrochemically metastable bulk. The decreasing performance trend for x > 0.20 is related to the increasing oxygen content in the pristine material, leading to a stabilization of the bulk structure and preventing the (oxyhydr)oxide from growing into the bulk. Moreover, a high Fe-content (x > 0.40) stabilizes the surface Co2+-atoms in such an extent that the irreversible surface Co oxidation is increasingly suppressed, limiting the reconstruction process even on the surface. Overall, this study provides a fundamental understanding of the Fe-content effects on surface reconstruction in BSCo1−xFex and deciphers the highest electrocatalytic performance of BSCo0.80Fe0.20 as a combination of optimally, neither too weakly nor too strongly, stabilized surface Co2+-atoms and bulk structure, leading to the most extensive surface reconstruction.
Introduction
The industrial production of sustainable hydrogen with alkaline water electrolyzers (AWE's) requires highly active and stable electrocatalysts, which are synthesized from low-cost precursors and with a scalable method.1 However, the sluggish kinetics of the anodic oxygen evolution reaction (OER) in AWE's indicates that the OER electrocatalyst still needs to be improved to lower the overpotential, and thus the hydrogen production costs.1Ba0.50Sr0.50Co0.80Fe0.20O3−δ (BSCo0.80Fe0.20) is a member of the perovskite-type oxide family ABO3−δ (δ = oxygen vacancy concentration)2 and a possible candidate as an OER electrocatalyst in AWE's.3 BSCo0.80Fe0.20 became particularly interesting after scaling down the particle size into the nanoscale-range by using the scalable flame-spray synthesis technique,4–8 which enables the production of high-surface area nanoparticles on the industrial scale.9,10 The high OER activity and stability of BSCo0.80Fe0.20 is associated with extensive irreversible structural changes (surface reconstruction), as observed initially by ex situ11 and later by operando X-rays absorption spectroscopy (XAS) measurements.5 Moreover, post-mortem high-resolution transmission electron microscopy (HRTEM) experiments revealed an irreversible surface amorphization process by leaching the A-site metal cations (Ba2+ and Sr2+) of BSCo0.80Fe0.20, which is beneficial for the OER charge transfer kinetics.12–14
One drawback of this type of material is the relatively expensive cobalt, which is also associated with additional negative aspects such as future supply shortages due to the massive increasing demand of lithium-ion batteries15 or problematic mine locations.16 One possible strategy to further decrease the cobalt content in BSCo0.80Fe0.20 is to increase the substitution with iron above the existing 20 mol% (B-site) while preserving the high electrocatalytic performance. Exploring the feasibility of this strategy requires a fundamental understanding of the Fe-content effect on the electrocatalytic performance in such perovskite-type oxides. Previous studies have shown that BSCo0.80Fe0.20 and La0.20Sr0.80Co0.80Fe0.20O3−δ with 20 mol% of Fe on the B-site have a higher OER activity and stability in comparison to materials with lower Fe-content but similar surface area.5,6 This improved OER activity in BSCo0.80Fe0.20 is explained by the ability of a higher Fe-content to enhance the stabilization of Co in an initially lower oxidation state, leading to a more extensive electrochemical formation of a self-assembled (oxyhydr)oxide layer as observed from oxidation state and local geometry changes of cobalt by operando XAS during electrochemical oxidation.6 Moreover, the increase of the Fe-content up to x = 0.20 in BSCo1−xFex transforms an electrochemically unstable material into a metastable BSCo0.80Fe0.20 with an improved OER stability.6 These findings indeed foster the prospect of increasing the Fe-content in BSCo0.80Fe0.20 while preserving the high electrocatalytic performance and indicate a significant influence of the Fe-content on the surface reconstruction process. Besides of these OER beneficial Fe-content effects, non-beneficial effects were revealed in Co-Fe-based (oxyhydr)oxides with a high Fe-content, where the unfavorable electrically insulating properties of Fe-based (oxyhydr)oxides limited the electrocatalytic performance.17 These overall antagonistic Fe-content effects lead typically to volcano-shaped activity and stability trends as a function of the Fe-content in Co-Fe-based OER electrocatalysts.17–19
To explore the possibility of further replacing the relatively expensive Co with cost-effective Fe in BSCo0.80Fe0.20 and to better understand the Fe-content effects on the surface reconstruction of Co(-Fe)-based oxide OER electrocatalysts, this study systematically investigates a flame-spray synthesized BSCo1−xFex series by gradually substituting Co with Fe (0 < x < 1). The materials were characterized electrochemically by cyclic voltammetry using the rotating-disk electrode (RDE) methodology in alkaline liquid electrolyte, and their electrocatalytic performances were tested by performing activity and stability protocols. To rationalize the observed electrochemical behavior and electrocatalytic performance trends as a function of the Fe-content, (ex situ) surface-sensitive and (operando) bulk XAS measurements were combined to analyze the as-synthesized materials as well as their electrochemically triggered changes, which are crucial to determine the extent of the surface reconstruction process. The performed experiments enable to reveal a correlation between the extent of the surface reconstruction and the volcano-shaped OER activity and stability trend as a function of the Fe-content in the BSCo1−xFex series. Conclusively, the best performing material of BSCo0.80Fe0.20 exhibits the most extensive electrochemically triggered surface reconstruction. Finally, the ideal Fe-content of x = 0.20 in BSCo1−xFex is revealed as the best compromise between optimally stabilized surface Co2+-atoms and bulk structure, enabling the formation of the most extensive Co- and Fe-based (oxyhydr)oxide during irreversible surface Co oxidation.
Results and discussion
As-synthesized properties
An unsupported perovskite-type oxide series of Ba0.50Sr0.50Co1−xFexO3−δ (BSCo1−xFex) was synthesized with the scalable flame-spray technique by using cost-efficient inorganic metal precursors (refer to the Methods section for more details).4,5,7 The material series has a constant A-site metal (Ba and Sr) composition of 0.50 ± 0.01 and eight different B-site metal (Co and Fe) compositions with an Fe-content of x = 0.01, 0.10, 0.20, 0.30, 0.40, 0.50, 0.79, and 0.99 as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) experiments (Tables S1 and S2†). To better distinguish the lowest (x = 0.01) and highest (x = 0.99) Fe-content materials from the remaining of the series, the minor metal component on the B-site of these two materials are written in brackets in the abbreviated chemical formula such as BSCo0.99(Fe0.01) and BS(Co0.01)Fe0.99. The morphology of the as-synthesized BSCo0.80Fe0.20 and BSCo0.60Fe0.40 powders was investigated with (scanning) transmission electron microscopy ((S)TEM) as representatives of the series and revealed agglomerated spherical nanoparticles with ∼5–40 nm diameter at the lower end of the size range, whereas the corresponding energy-dispersive X-ray (EDX) spectroscopy maps confirmed a uniform distribution of all present elements on a particle size resolution level (Fig. S1–S9†). The similarity of the morphology within the BSCo1−xFex series is supported by the comparable specific surface area (SSA), and thus surface area-to-bulk volume ratio, for all the materials (∼8–13 m2 g−1) as determined with the Brunauer–Emmett–Teller (BET) methodology (Fig. S7†).The as-synthesized bulk properties of the BSCo1−xFex series were investigated by powder X-ray diffraction (XRD) and transition metal (TM) K-edge XAS (Fig. 1a–c, S10–S12, Tables S3 and S4†). All XRD patterns of the BSCo1−xFex series show the crystalline long-range bulk structure of a cubic perovskite-type oxide with the space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (Fig. 1a). The secondary phases are mainly assigned either to carbonates/nitrates for the A-site metals or to oxides for the B-site metals, and are typically present in flame-spray synthesized perovskite-type oxides (Fig. S10a and S11a, and Note S1†).4–7 A closer look at the patterns reveals a gradual shift of the primary phase diffraction peaks towards lower 2θ, indicating a unit cell expansion and thus changes in the metal oxidation states, when the Fe-content increases in the BSCo1−xFex series (Fig. 1b and Note S2†).
m (Fig. 1a). The secondary phases are mainly assigned either to carbonates/nitrates for the A-site metals or to oxides for the B-site metals, and are typically present in flame-spray synthesized perovskite-type oxides (Fig. S10a and S11a, and Note S1†).4–7 A closer look at the patterns reveals a gradual shift of the primary phase diffraction peaks towards lower 2θ, indicating a unit cell expansion and thus changes in the metal oxidation states, when the Fe-content increases in the BSCo1−xFex series (Fig. 1b and Note S2†).
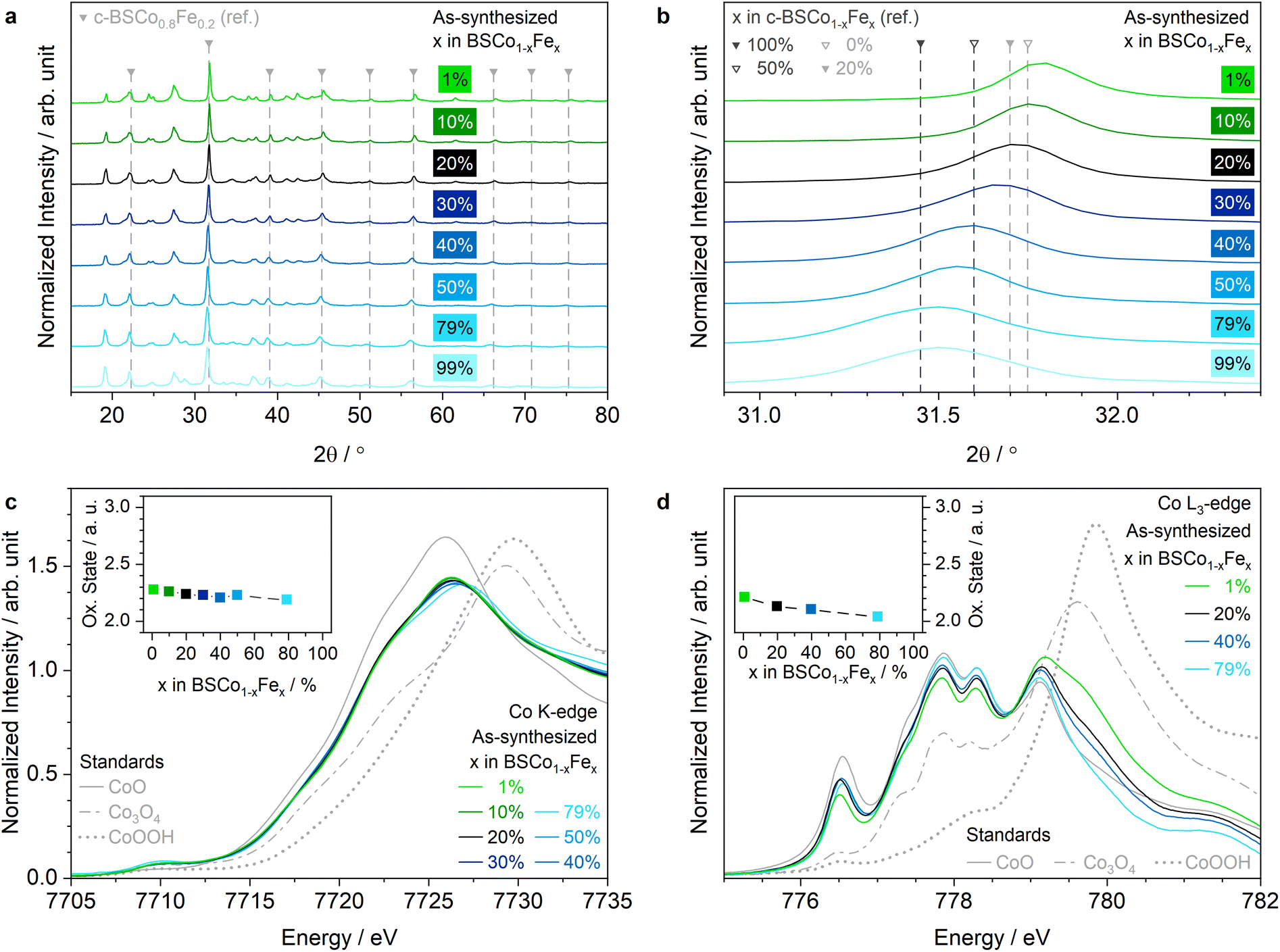 | ||
| Fig. 1 As-synthesized properties of BSCo1−xFex series. (a) XRD patterns measured in Bragg–Brentano mode. The peaks of the shown reference pattern (ref.) correspond to the following crystal planes (from low to high 2θ): (100), (110), (111), (200), (210), (211), (220), (221)/(300), and (310). (b) XRD pattern peak position as a function of the Fe-content exemplarily shown for the (110) crystal plane. The ICSD collection codes of all shown ref. patterns are listed in ‘Detailed Methods’ of ESI.† (c) Co K-edge XANES spectra. Inset (c) Co oxidation state in the bulk as a function of the Fe-content. (d) Co L3-edge TEY-XAS spectra. Inset (d) surface Co oxidation state as a function of the Fe-content. The Ba M5-edge contribution in the Co L3-edge XAS spectra were subtracted for all materials in the BSCo1−xFex series. | ||
The bulk oxidation state of the B-site metals (Co and Fe) was experimentally assigned with the corresponding K-edge X-ray absorption near-edge structure (XANES) spectra according to their absorption edge positions at the half-normalized intensity: The bulk oxidation state of Co and Fe averaged over all materials is ∼+2.25 and ∼+3, respectively, and decreases slightly for both elements towards the higher Fe-content materials (Fig. 1c and S11b†). The lower average oxidation state of Co relative to Fe is explained by the lower reduction temperature of Co (>∼1200 K for +2.67 → +2)20 in comparison to Fe (>∼1400 K for +3 → +2.67).21 The bulk oxidation state of the A-site metals (Ba and Sr) was assumed to be +2 as typically for oxidic alkaline earth metal cations. The assigned bulk metal oxidation states in combination with the individual stoichiometry of the metals reveal an increasing oxygen content towards the higher Fe-content materials in the BSCo1−xFex series, which is explained by the substitution of Co2.25+-cations (on average) by more positively charged Fe3+-cations (Fig. S10b and Note S3†).
To better understand the consequence of the increasing oxygen content on the bulk short-range order around the TMs, the corresponding K-edge Fourier-transformed extended X-ray absorption fine structures (FT-EXAFS) spectra were analyzed (Fig. S10c–e, S11c–e, and S12, and Tables S3 and S4†). The first peak at a phase uncorrected interatomic bond distance of ∼1.5 Å corresponds to the TM–O coordination shell for both edges and is present in any TM-based oxide. Noteworthy is the second peak at ∼2.6 Å (phase uncorrected), which is typically attributed to the TM–TM coordination shell of edge-sharing polyhedra (esp) in such materials.5,6,22,23 However, an esp-peak does not exist in BSCo1−xFex as a cubic perovskite-type oxide with corner-sharing polyhedra (csp), which has the TM–TM and TM–Ba/Sr coordination shell typically at longer phase uncorrected distances (>3 Å).5,11 Despite this anomaly, an esp-peak has been already observed in Co–Mn-based perovskite-type oxides with high oxygen vacancy concentrations22,23 and is a key characteristic of such materials when synthesized with the flame-spray technique at high temperatures (refer to Note S4† for a possible explanation of the esp-peak).5,6 A closer look at the maximal esp-peak intensity (∼2.6 Å) unveils a decreasing trend at both TM K-edges towards the higher Fe-content materials in the BSCo1−xFex series (Fig. S10c–e and S11c–e†). The decreasing trend of the esp-peak correlates not only with the increasing oxygen content, as estimated from the bulk metal oxidation states, but also with the unit cell expansion, as observed in the XRD patterns, when the Fe-content increases. These overlapping trends indicate, on one side, a coherent structure from the short- to long-range order for each single material and, on the other side, a gradual change in the bulk properties as a function of the Fe-content in the BSCo1−xFex series.
After focusing on the bulk properties of the as-synthesized BSCo1−xFex series, the characterization is completed by investigating their surface with TM L3-edge XAS using the total-electron-yield (TEY) detection mode (Fig. 1d, S13 and S14†). In comparison to the bulk, the surface Co oxidation state decreases more significantly from ∼+2.2 (x = 0.01) to ∼+2 (x = 0.79) towards the higher Fe-content materials, indicating the ability of a higher Fe-content to better stabilize surface Co2+-atoms (Fig. 1d and S13a†). The general tendency of having a slightly lower Co oxidation state on the surface than in the bulk for each material can possibly originate from the reductive environment at low pressure, which is required for the TEY mode.24 Unlike Co, the surface Fe oxidation state is close to ∼+3 in all materials and comparable with the bulk (Fig. S14a and b†). Consequently, the estimated mole fraction weighted average oxidation state trend of the surface B-site metals is also comparable with the bulk and increases towards the higher Fe-content materials in the BSCo1−xFex series (Fig. S14e†). Moreover, the surface-sensitive investigation reveals that the Fe signal in BSCo0.99(Fe0.01) and Co signal in BS(Co0.01)Fe0.99 are barely visible in the corresponding L3-edge TEY-XAS spectra of the as-synthesized materials (Fig. S13b and S14c, d†). Beside of oxidation state and stoichiometry of the elements, the Co L3-edge XAS spectra can also reveal electronic and local structural information. More specifically, the first peak at 776.5 eV in the Co L3-edge XAS spectra is characteristic for a high-spin (hs) Co2+, which is octahedrally coordinated by oxygen atoms (Oh-sites).25 This surface faction of hs Co2+ in Oh-sites significantly increases from x = 0.01 to 0.20, remains constant until x = 0.40, and slightly decreases then towards x = 0.79 of the as-synthesized BSCo1−xFex series (Fig. 1d, S13c, and Note S5†).
In summary, the bulk properties and compositions of the as-synthesized BSCo1−xFex series are comparable to those on the surface, and an overall increase in the oxygen content and a stabilization/accumulation of Co2+-atoms is observed towards the higher Fe-content materials.
OER performance
The OER activity and stability of the BSCo1−xFex series was investigated in 0.1 M KOH electrolyte solution with a rotating-disk electrode (RDE) setup. The results of chronoamperometric (CA) measurements are summarized in the Tafel plot (Fig. 2a) and reveal a volcano-shaped relationship between OER activity and Fe-content in the BSCo1−xFex series as typically observed in Co-Fe-based electrocatalysts.17–19,26–30 The OER current, no matter if normalized by the electrocatalyst- (Fig. 2a) or Co-mass (Fig. S15a†), strongly increases from x = 0.01 to 0.20 and then decreases again steadily until x = 0.99. Consequently, the trend is inverted (U-shaped) when plotting the OER potential as a function of the Fe-content. A similar U-shaped relationship is also observed between the Tafel slope and the Fe-content, whereas the lowest slope was determined for x = 0.20 (Fig. S15a†). Noteworthy, BSCo0.99(Fe0.01) is significantly more OER active than BS(Co0.01)Fe0.99, which confirms the challenge to design Co-free and pure Fe-based electrocatalysts with high OER activity (Fig. 2a). Moreover, the most OER active BSCo1−xFex has a relative low Fe-content (x = 0.20), which indicates the difficulty to extensively replace Co with Fe in this material series. | ||
| Fig. 2 Electrochemical characterization of BSCo1−xFex series. (a) Tafel plot of electrocatalyst-mass normalized OER currents from CA measurements, including linear fits (dotted lines) and error bars from four independent measurements (refer to inset (a) for complete color code). Inset (a) OER activity from the Tafel plot as a function of the Fe-content. (b) OER stability measurements of BSCo0.80Fe0.20 with absolute OER current density changes (ΔJ) at 1.6 VRHE as a function of the cycling number between 1.0 and 1.6 VRHE from CA measurements, including error bars from three independent measurements. Inset (b) OER stability as a function of the Fe-content for the maximum (Max) and final ΔJ at 1.6 VRHE. (c and d) Last (25th) CV cycle measured with 10 mV s−1 before performing the CA experiments for the Tafel plot. Inset (c and d) magnification of the last (25th) CV cycle to highlight the pre-OER potential range. The cycling direction is indicated with arrows: first, the positive (orange) and afterwards the negative going potential scan (red). All electrochemical measurements were performed with an RDE setup in 0.1 M KOH at room temperature. | ||
The OER stability behavior of the BSCo1−xFex series was investigated by cycling 505 times between the CA potentials of 1 and 1.6 VRHE (Fig. 2b and S16†). The absolute OER current density change at 1.6 VRHE in comparison to the initial value (ΔJ) is exemplary shown for BSCo0.80Fe0.20 as a function of the cycling number (Fig. 2b). The OER stability behavior within the BSCo1−xFex series is compared by plotting the maximum value (Max ΔJ) and the value of the final cycle (Final ΔJ) for each material as a function of the Fe-content (inset Fig. 2b). Both curves show a volcano-shaped relationship as already observed for the OER activity: Max ΔJ and Final ΔJ increases from x = 0.01 to 0.20 and decreases again until x = 0.99 (Note S6†). Moreover, the cycle number, where Max ΔJ is recorded for each material, has also a volcano-shaped trend as a function of the Fe-content and also there has BSCo0.80Fe0.20 the highest value (Fig. S15b†). Conclusively, BSCo0.80Fe0.20 shows the best OER stability behavior among all materials in the BSCo1−xFex series, as BSCo0.80Fe0.20 not only experiences the most extended surface activation process (Max ΔJ > 0), lasting the most cycles and leading to the largest Max ΔJ, but also exhibits the smallest performance drop in the remaining cycles after Max ΔJ is reached (Max ΔJ − Final ΔJ is minimal). Surprising is that one single material from the BSCo1−xFex series such as BSCo0.80Fe0.20 is able to unify the highest OER activity and stability, particularly when using the RDE methodology, where the accumulation of bubbles from the produced oxygen on the electrocatalyst surface is more likely in higher active materials.31–33 Here, bubble accumulation is understood as a continuous process during the entire stability protocol, which decreases the OER current density by masking the surface of the electrocatalyst. Contrarily, a surface activation process is an opposing phenomenon but with a decreasing contribution during the stability protocol. Conclusively, the most stable material (x = 0.20) experiences the largest surface activation process, thus the largest increase in OER charge transfer kinetics, and enables BSCo0.80Fe0.20 to overcompensate the negative effects of bubble accumulation for the longest duration, resulting in the largest Max ΔJ and Final ΔJ values.
To better understand the OER activity and stability trend as a function of the Fe-content, all materials were also analyzed with cycling voltammetry (CV), which were measured directly before recording the CA's for the Tafel plot in the same protocol (Fig. 2c, d and S15c, d†). BSCo0.99(Fe0.01) shows two redox couples, typically assigned to Co2+ ⇄ Co3+ (A1|C1) and Co3+ ⇄ Co4+ (A2|C2),34–36 in the pre-OER potential range of the last (25th) CV (Fig. 2c). These Co redox couples significantly change when the Fe-content increases, which is a typically observed phenomenon in Co-Fe-based materials.17,18,27 Here, the Co2+ oxidation (A1) and reduction peak (C1) increases in the BSCo1−xFex series from x = 0.01 to 0.20 but in a significantly larger extent for A1 than for C1, which indicates an enhancing irreversible surface Co2+ oxidation towards the higher Fe-content material of BSCo0.80Fe0.20 (Fig. 2c). This trend changes when the Fe-content further increases in the BSCo1−xFex series so that the Co redox couples and thus the irreversible surface Co2+ oxidation decreases again from x = 0.20 to 0.99 (Fig. 2c and d). Differently, the A2|C2 Co redox couple decrease straight from the beginning when the Fe-content increases from x = 0.01 to 0.99. The trend of the two redox couples (A1|C1 and A2|C2) leads to a complete vanishing of these processes when reaching the highest Fe-content material of BS(Co0.01)Fe0.99, indicating the limited ability of surface Fe-atoms to change their oxidation states at least before OER, and justifies the assignment of the observed redox processes in the remaining materials (0.10 ≤ x ≤ 0.79) exclusively to surface Co-atoms (Fig. 2c and d). The change of the Co redox couples in the BSCo1−xFex series (excluding BS(Co0.01)Fe0.99 with minor Co-content) can be even better visualized by comparing the Co-mass normalized net current density (Jnet), representing the difference between the positive (ΔE·t−1 > 0) and negative (ΔE·t−1 < 0) going CV scan in the pre-OER potential range (Fig. S17b and c†). In addition, the irreversible surface Co oxidation vs. Fe-content trend can be quantified with the Co-mass normalized cumulated net charge (Qnet), which is proportional to the integral of Jnet (Fig. S17a†). The positive value of Qnet confirms the accumulation of irreversibly oxidized surface Co-atoms in all analyzed materials and shows a volcano-shaped trend as a function of the Fe-content with BSCo0.80Fe0.20 at the top, resembling the OER activity vs. Fe-content trend. The correlation of those two trends suggests that the accumulation of oxidized surface Co-atoms is beneficial for high OER activity. Moreover, the observed trends of the Co redox couples in the BSCo1−xFex series clearly indicate that a higher Fe-content has not only the ability to stabilize surface Co2+-atoms in the as-synthesized material (Fig. 1d), but has also the ability to stabilize them electrochemically (Fig. 2c and d). This electrochemical stabilization can then lead to a shift of the Co redox process towards higher potentials into OER, resulting in a decrease of the Co redox couples in the pre-OER potential range towards the Fe-content materials as typically observed in Co-Fe-based electrocatalyst series.17,18,37
The OER potential range of all CV ‘s (excluding BS(Co0.01)Fe0.99 with minor OER activity) is analyzed with special attention to which of the two CV scanning directions, the positive or the negative going afterwards, presents the higher OER current density: BSCo0.99(Fe0.01) presents the higher OER activity during the positive going potential scan, whereas all the other materials (0.10 ≤ x ≤ 0.79) have the higher OER activity during the negative going potential scan afterwards (Fig. 2c and d). The latter observation indicates an extensive surface activation process during the positive going potential scan, leading then to a higher OER activity in the negative going afterwards. This extensive surface activation process during OER has already been observed in another flame-spray synthesized Co-Fe-based oxide series, but also there only in the higher Fe-content materials with initially stabilized surface Co2+-atoms and the ability to oxidize irreversibly, which then led to an improved OER activity and stability.37
After considering only the last (25th) CV cycle, a closer look at the changes of the CV as a function of the cycling number reveals a visible growth of the Co redox couples in each cycle for all materials excluding BS(Co0.01)Fe0.99 (Fig. S15c and d†). This observation typically indicates a surface reconstruction process with an increasing exposure of Co-atoms to the electrolyte, which is accompanied with a surface composition and/or number of active sites (surface area) changes, leading to higher OER activity. However, the extent of the Co redox couple growth with cycling, and thus the surface reconstruction process, gradually decreases towards the higher Fe-content materials in the BSCo1−xFex series, leading to a smaller increase of the OER activity. This indicates a structural stability increase towards the higher Fe-content materials, which is not beneficial for the OER activity, and correlates with the decreasing esp-peak in the FT-EXAFS spectra of the as-synthesized materials as well as with the increasing oxygen content (Fig. S10b–e, S11c–e, and S14e†). Moreover, the observed enhanced structural stability towards the higher Fe-content materials in the BSCo1−xFex series is supported by density-functional theory (DFT) calculated Pourbaix diagrams, which predict a lower A-site metal dissolution of BSCo1−xFex when the Fe-content increases.6
In summary, the irreversible oxidation of initial stabilized surface Co2+-atoms and the bulk stability are identified as two crucial aspects of the Fe-content dependent surface reconstruction process, responsible for the volcano-shaped OER activity and stability trend with BSCo0.80Fe0.20 as the best performing material in the BSCo1−xFex series. To support the interpretation of the corresponding CV's and the relevance of these two aspects for OER, spectroscopy measurements were performed with a special focus on the surface and bulk changes.
Surface changes vs. OER performance
The surface changes were investigated by ex situ TM L3-edge TEY-XAS, which is able as a surface- and oxidation state-sensitive technique to detect irreversible surface oxidations in a rather reductive environment, particularly for Co(-Fe)-based oxides,24 at low pressures (Fig. 3 and S18–S22†). All four representatives of the BSCo1−xFex series (x = 0.01, 0.20, 0.40, and 0.79) have a significantly higher surface Co oxidation state ex situ after OER in comparison to the as-synthesized powder or as-prepared electrode, indicated by the decreasing peaks at ∼778 eV and the increasing peak at ∼780 eV (Fig. 3a–e). An electrochemically triggered irreversible surface Fe oxidation is also revealed for the three materials with x = 0.20, 0.40, and 0.79 when comparing the corresponding ex situ after OER Fe L3-edge TEY-XAS spectra with the spectra of the as-synthesized powders or as-prepared electrodes, but these oxidations are less extensive than for Co (Fig. S21†). In contrast to the higher Fe-content materials, BSCo0.99(Fe0.01) with the lowest Fe-content and a barely visible surface Fe signal in the as-synthesized powder shows a significant increase of the Fe signal after the electrode preparation and even stronger after OER (Fig. S22†). Moreover, the mole fraction weighted average oxidation state of the surface B-site metals is after OER above +2.67 in all materials, which rules out the possibility that a spinel phase is the OER active surface layer of BSCo1−xFex and supports previous findings in the literature (Fig. 3e, S18b and S21f†).5,6,11,13 | ||
| Fig. 3 Ex situ surface changes of Co. ‘As-synthesized’ (same as in Fig. 1d), ‘as-prepared’ and ex situ ‘after OER’ Co L3-edge TEY-XAS spectra of BSCo1−xFex with x = 0.01 (a), 0.20 (b), 0.40 (c), and 0.79 (d). The Ba M5-edge contribution in the Co L3-edge XAS spectra were subtracted for all materials in the BSCo1−xFex series. (e) ‘As-synthesized’, ‘as-prepared’ and ex situ ‘after OER’ surface Co oxidation state as a function of the Fe-content, including the definition of the irreversible surface Co oxidation (Δ in CoΔ+) as the difference between the after OER and as-synthesized surface Co oxidation state. (f) Δ in CoΔ+ (black: left axis and stars) and Co-mass normalized OER activity (red: right axis and circles, same as in inset Fig. 2a) as a function of the Fe-content. | ||
The increase of the surface Co oxidation state from the as-synthesized powder to the ex situ after OER sample is defined here as Δ in CoΔ+ and represents the extent of the irreversible surface Co oxidation. The analysis of Δ in CoΔ+ as a function of the Fe-content in the BSCo1−xFex series reveals a volcano-shaped relationship: Δ in CoΔ+ increases from x = 0.01 to 0.20, which has a similar value as x = 0.40, and decreases again to x = 0.79 (Fig. 3f). The initially increasing trend of Δ in CoΔ+ from x = 0.01 to 0.20 correlates with the previous electrochemical findings such as Jnet and Qnet from the in-depth CV analysis and supports the previous interpretation of an enhanced irreversible Co oxidation when the Fe-content increases in the BSCo1−xFex series (Fig. 3f and S17†). The similar Δ in CoΔ+ of BSCo0.80Fe0.20 and BSCo0.60Fe0.40 does not correlate with the trend of the Co redox couples in the pre-OER potential range of the CV, which decreases towards the latter material (Fig. 2c, d and 3f). This observation indicates that the stabilization of the surface Co2+-atoms towards the higher Fe-content materials leads indeed to a shift of the corresponding Co redox process towards higher potentials into OER. Therefore, the surface Co2+-atoms of BSCo0.60Fe0.40 oxidize irreversibly not only before but also during OER, resulting in a similar Δ in CoΔ+ as for BSCo0.80Fe0.20. Consequently, the decrease of Δ in CoΔ+ from BSCo0.60Fe0.40 to BSCo0.21Fe0.79 is explained by the ability of a high Fe-content in the BSCo1−xFex series to stabilize surface Co2+-atoms in such an extent that the corresponding Co redox couples are even gradually shifted out of the upper OER potential scanning range. These suppressed Co redox couples lead then to a decrease of the irreversible surface Co oxidation and thus to a lower Δ in CoΔ+ as observed for x = 0.79. Beside of Co oxidation state changes, the analysis of the ex situ ‘after OER’ Co L3-edge TEY-XAS spectra reveals also that mainly hs Co2+ in Oh-sites are oxidized irreversibly (Fig. S18–S20†). The decrease of this specific Co fraction after OER directly correlates with the overall trend of Δ in CoΔ+ as a function of the Fe-content and indicates the importance of having initially hs Co2+ in Oh-sites for an extensive irreversible surface Co oxidation (Fig. S18a†).
The correlation between ex situ measured changes in the Co L3-edge TEY-XAS spectra and the electrochemical behavior of the BSCo1−xFex series allows to directly compare Δ in CoΔ+ (‘after OER’ vs. ‘as-synthesized’) with the Co-mass normalized OER activity as a function of the Fe-content (Fig. 3f). The similarity of the volcano-shaped trends clearly supports the importance of the irreversible oxidation of initially stabilized surface Co2+-atoms for a high OER activity: the increase of the Fe-content shifts the Co redox couples towards higher potentials and enhances irreversible surface Co oxidation, which promotes the formation of a higher active surface layer as long as the redox couples remain in the upper OER potential scanning range. Despite the similarity of the volcano-shaped trends, the lower Fe-content materials, specifically BSCo0.80Fe0.20, have a significantly higher OER activity than expected from the surface-sensitive spectroscopic measurement (Δ in CoΔ+). This demonstrates the partial limitation of considering only irreversible ex situ surface changes, particularly when significant changes deep into the bulk are expected in the low Fe-content materials. To better understand the extent of the reconstruction process, bulk changes were measured by operando K-edge XAS and ex situ XRD.
Bulk changes vs. OER performance
The operando bulk changes of BSCo1−xFex with x = 0.20 and 0.40 were investigated with TM K-edge XAS between 1 VRHE and 1.55 VRHE (Fig. 4, S23–S31, and Tables S5–S10†). Both materials show an increasing Co oxidation state towards the higher potentials in the operando Co K-edge XANES spectra but in a significantly larger extent for BSCo0.80Fe0.20 than for BSCo0.60Fe0.40 (Fig. 4a, b and S23a–d†). The comparison of the Co oxidation state at 1 VRHE measured before and after OER reveals that the large operando changes in BSCo0.80Fe0.20 are mostly irreversible (Fig. S23a and c†). A similar trend is observed in the operando Fe K-edge XANES spectra but in a significantly smaller extent than at the Co K-edge (Fig. S28a, b and S29a, b†): BSCo0.80Fe0.20 shows a relatively small increase of the Fe oxidation state at higher potentials, whereas BSCo0.60Fe0.40 exhibits even smaller changes. In contrast to Co, the operando Fe K-edge XANES spectra changes seem to be more reversible in BSCo0.80Fe0.20 based on the small differences at 1 VRHE measured before and after OER, but has also to be considered with caution due to overall small changes (Fig. S28a and b†). In addition to operando changes, the influence of the electrode preparation and alkaline electrolyte (0.1 M KOH) on the Co and Fe oxidation state relative to the as-synthesized powder has been investigated revealing only marginal changes in both materials (Fig. 4a, b and S23e, f, S28c, and S29c†). Overall, these results clearly indicate that the electrochemistry triggers mainly oxidation state changes of surface Co-atoms rather than Fe, and supports to assign the peaks in the pre-OER potential range of the CV's as Co redox couples (Fig. 2c and d). Moreover, the decreasing operando bulk changes from x = 0.20 to 0.40 correlates not only with the smaller growth of the Co redox couples during CV cycling and increasing structural stability towards the higher Fe-content materials in the BSCo1−xFex series (Fig. S15c and d†), but also with the decreasing OER activity and stability trend from x = 0.20 to 0.99 (Fig. 2a and b). This correlation confirms the OER non-beneficial effect of having a large Fe-content in the BSCo1−xFex series, which prevents the active surface layer from growing deeper into the bulk. | ||
| Fig. 4 Operando bulk changes of Co. ‘As-synthesized’ (same as in Fig. 1c), at 1.00 VRHE, and at 1.55 VRHE Co K-edge XANES spectra of BSCo1−xFex with x = 0.20 (a) and 0.40 (b). Inset (a) and (b) Co oxidation state in the bulk as a function of the applied potential in the positive going scan. Magnitude of k2-weighted FT-EXAFS of ‘as-synthesized’, at 1.00 VRHE, and at 1.55 VRHE Co K-edge XAS spectra for BSCo1−xFex with x = 0.20 (c) and 0.40 (d). The intensities of the standards in (c) and (d) were halved (×0.5). | ||
To better understand the electrochemically triggered short-range order changes around the B-site metals, the corresponding operando FT-EXAFS spectra at the TM K-edge were analyzed. Both materials show an increase of the maximum peak intensity at a lower phase uncorrected interatomic bond distances for the Co–O (∼1.4 Å) and Co–TM (∼2.4 Å) coordination shells, but again in a significantly larger extent for BSCo0.80Fe0.20 than for BSCo0.60Fe0.40 (Fig. 4c, d, S24 and S25†). Additionally, BSCo0.80Fe0.20 shows similar local structure changes at the Fe K-edge but also here in a smaller extent than at the Co K-edge, whereas the Fe-atoms in BSCo0.60Fe0.40 experience only minor short-range order changes (Fig. S30 and S31†). The large electrochemically triggered changes in the FT-EXAFS spectra of BSCo0.80Fe0.20 are mostly irreversible at both edges as revealed by comparing the spectrum at 1 VRHE measured before and after OER (Fig. S24d and S30d†). The electrode preparation and the alkaline electrolyte trigger also changes in the short-range order around the Co- and Fe-atoms in both materials, but are smaller than the electrochemically induced changes (Fig. 4c, d, S24e, S25d, S30e and S31d†). Overall, the results clearly indicate an electrochemically triggered growth of a similar surface layer in both materials but in a larger extent for BSCo0.80Fe0.20 than in BSCo0.60Fe0.40. The comparison of FT-EXAFS standard spectra with the electrochemically triggered increasing peaks in both materials indicates Co1−χFeχOy(OH)z as the growing OER active surface layer and is supported by the best fits of the FT-EXAFS spectra (Fig. 4c, d, S26 and S27, and Tables S5–S10†). This observation is in agreement with the literature,5,6,11,38 but shows in contrast to them FT-EXAFS changes also at the Fe–K-edge, clearly indicating the presence of Fe within the (oxyhydr)oxide phase, particularly for BSCo0.80Fe0.20. The identification of Co1−χFeχOy(OH)z is also in agreement with the mole fraction weighted average oxidation state of the surface B-site metals as estimated from the ex situ after OER TM L3-edge TEY-XAS spectra, which are all close to +3 (Fig. S18b†). These local structural insights and the previous finding in the TM oxidation state changes allow to further refine the OER non-beneficial effect of a higher Fe-content in the BSCo1−xFex series: An Fe-content above the optimal value of x = 0.20 stabilizes the bulk structure in such an extent that the formed surface Co1−χFeχOy(OH)z layer is prevented from growing deeper into the bulk and limits the electrocatalytic performance (Fig. 5).
 | ||
| Fig. 5 Surface vs. bulk: Fe-content dependent extent of surface reconstruction in the BSCo1−xFex series. (a) BSCo0.80Fe0.20 has the highest OER activity and stability by forming Co1−χFeχOy(OH)z in the largest extent resulting in similar surface and bulk changes. (b) Co oxidation state (changes) on the surface (ex situ after OER vs. as-synthesized with L3-edge TEY-XAS) and in the bulk (operando at 1.55 VRHE during OER vs. as-synthesized with K-edge XAS) for BSCo0.80Fe0.20 and BSCo0.60Fe0.40. (c) BSCo0.60Fe0.40 has similar surface but smaller bulk changes than BSCo0.80Fe0.20, which limits the OER activity and stability of BSCo0.60Fe0.40. | ||
The decreasing electrochemically triggered changes in the bulk TM oxidation state and short-range order from BSCo0.80Fe0.20 to BSCo0.60Fe0.40 is also supported by ex situ XRD measurements after OER. The crystalline cubic perovskite-type oxide peaks decrease in the XRD pattern after OER relative to the ‘as-synthesized’ or ‘as-prepared’ in both materials but again in a larger extent for BSCo0.80Fe0.20 (Fig. S28d and S29d, and Note S7†). This indicates a more extensive transformation of the initial BSCo0.80Fe0.20 phase in comparison to BSCo0.60Fe0.40 and correlates with the higher OER activity and stability of BSCo0.80Fe0.20. The Co1−χFeχOy(OH)z phase itself was not detected in the ex situ XRD pattern of the sample after OER and is most likely explained by being amorphous.
Conclusions
This study reveals a correlation between the OER electrocatalytic performance and extent of surface reconstruction as a function of the Fe-content in the flame-spray synthesized and unsupported BSCo1−xFex series (0 < x < 1). Both trends exhibit a volcano-shape with BSCo0.80Fe0.20 as the best performing material and thus with the most extensive surface reconstruction. These correlating trends clearly demonstrate the importance of surface reconstruction for high OER activity and stability of Co(-Fe)-based OER electrocatalysts, and emphasizes the crucial influence of the Fe-content on both processes. Furthermore, the study elucidates that the Fe-content influences these processes by controlling the stabilization of the surface Co2+-atoms and bulk structure. More specifically, an increasing Fe-content leads to an accumulation of surface Co2+-atoms in the as-synthesized materials, which is beneficial for the electrochemically triggered irreversible surface Co oxidation and thus for the surface reconstruction, leading to higher OER charge transfer kinetics. However, the increase in oxygen-content, and thus the decrease in oxygen vacancy concentration (δ) towards the higher Fe-content materials enhances the stability of the bulk structure, preventing the surface reconstruction process from growing deeper into the bulk. Consequently, the ideal Fe-content in BSCo0.80Fe0.20 represents the best compromise of neither too weakly nor too strongly stabilized surface Co2+-atoms and bulk structure, leading to the formation of the most extensive amorphous Co- and Fe-based (oxyhydr)oxide layer in the BSCo1−xFex series. This result clearly highlights the challenge of extensively replacing Co with Fe in BSCo1−xFex while preserving the high electrocatalytic performance. Overall, this study provides a fundamental understanding of the Fe-content effects on the surface reconstruction of the BSCo1−xFex series and emphasizes the importance of combining spectroscopic surface-sensitive and bulk techniques with an in-depth electrochemical analysis to properly rationalize the electrocatalytic performance of unsupported Co(-Fe)-based oxides.Methods
Material syntheses
The Ba0.50Sr0.50Co1−xFexO3−δ (BSCo1−xFex) series was flame-spray synthesized with a nominally A-site metal composition of Ba0.50Sr0.50 (0.50 ± 0.01 from ICP-OES) and x = 0.00 (0.01 from ICP-OES), 0.10, 0.20, 0.30, 0.40, 0.50, 0.80 (0.79 from ICP-OES), and 1.00 (0.99 from ICP OES).4,7 Briefly, stoichiometric amounts of barium carbonate (≥99.0%, Sigma Aldrich), strontium nitrate (≥98%, Sigma Aldrich), cobalt nitrate hexahydrate (99.9%, Sigma Aldrich), and iron nitrate nonahydrate (≥98%, Sigma Aldrich) precursors were dissolved in an ultrapure water (MicroPure UV, Thermo Scientific) and acetic acid (≥99.0%, Roth) solution mixture (3-to-1 vol. ratio) to have finally a total metal concentration of 0.1 M. This precursor solution mixture was then injected with 50 mL min−1 by a three-piston pump (C-610, Büchi) into the acetylene (13 L min−1, 99.6%, PanGas) and oxygen (17 L min−1, 99.5%, PanGas) combusted flame. The precursor solution was dispersed into small droplets by using oxygen also as dispersion gas (25 L min−1). Finally, the formed particles were collected on ashless filter papers (Whatman®, GE Healthcare) by using vacuum pumps (VACFOX VC 50, Rietschle Thomas) and were then removed from the papers with the help of a spatula resulting in the so-called as-synthesized materials.The standard material of β-cobalt oxyhydroxide (CoOOH) was synthesized by chemically oxidizing cobalt hydroxide (Co(OH)2) with hydrogen peroxide (35%, Sigma-Aldrich) in a hot and highly alkaline aqueous solution (NaOH, 99.99% semiconductor grade, Sigma-Aldrich) following the same procedure as in the literature.39,40 The required Co(OH)2 was freshly prepared by precipitation reaction, using cobalt nitrate hexahydrate (≥98% ACS reagent, Sigma-Aldrich) and a hot alkaline aqueous solution, following the same procedure as in the literature cited above.39 The successful synthesis of CoOOH was verified by XRD (Fig. S32a†).
Physicochemical characterization of as-synthesized materials
The metal content of the as-synthesized BSCo1−xFex series was determined by ICP-OES (5110, Agilent) in axial viewing mode. Each as-synthesized powder (∼50 mgcat) was digested in a hot (<100 °C) nitric acid (65% AnalaR NORMAPUR, VWR Chemicals) and hydrochloric acid (37% fuming EMSURE®, Supelco Merck) solution mixture (1-to-3 vol. ratio) at atmospheric pressure, and diluted in 0.2 M nitric acid solution (ultrapure water from PURELAB Chorus 1, Elga Veolia). Commercial standard solutions for Ba, Sr, Co, and Fe (CertiPrep, Spex) were diluted with ultrapure water to assign the metal concentration in the digested solution. The concentrations were assigned by a quadratic fit of the standard signals and using the blank (0.2 M nitric acid) intensity as 0 ppm. The electrocatalyst mass, which was not assigned to Ba, Sr, Co, and Fe, is called ‘Rest’ and includes beside of the mass of oxygen also the mass of anions from secondary phases (carbonates and nitrates), of adsorbed water as well as the measurement error if cation were lost during the digestion. The relative standard deviation including only ICP-OES measurement errors is estimated to be less than 2% for the relative mole fractions.The XRD patterns of the as-synthesized BSCo1−xFex series were measured as powder in Bragg–Brentano mode (SmartLab, Rigaku) using a Si-zero-background holder and a Kβ-filter (but not Kα2). The ICSD collection code or PDF number of the reference patterns are listed in ‘Detailed Methods’ of ESI.†
The EDX maps, TEM and ADF-STEM images were recorded with JEM-ARM200F NEOARM (JEOL) using a cold field emission gun (FEG) at 200 kV for BSCo0.80Fe0.20 and BSCo0.60Fe0.40. The as-synthesized powders were directly put on the continuous ultrathin carbon film coated lacey carbon side (400 mesh) of the copper grid and were stabilized by sputtering a thin film of carbon on the top (MED010, BalTec).
The SSABET (Autosorb-1, Quantachrome Instruments) of the as-synthesized BSCo1−xFex series was normalized by the weighed-in mass before the outgassing step (85 °C for 1.5 h) and the reported values are the average of three independent measurements per material.
The Co and Fe K-edge XAS spectra of the as-synthesized BSCo1−xFex series and the standard materials were measured as pellets at the SuperXAS beamline of the Swiss Light Source (SLS, PSI, Switzerland). The hard X-rays were produced by a 2.9 T superbend magnet source. This polychromatic photon beam got then collimated with a Si-coated mirror at 2.9 mrad and were monochromatized with a liquid N2 cooled channel-cut Si(111) crystal oscillating with a 1 Hz frequency. Finally, the beam is focused on a spot size of 1 × 0.2 mm2 with a Rh-coated toroidal mirror. All as-synthesized Co and Fe K-edge XAS spectra were detected in transmission using three ionization chambers filled with 1 bar N2 by recording both edges in a continuous scan for one minute with the quick extended X-ray absorption fine structure (QEXAFS) mode (120 spectra per min).41 The 120 XAS spectra per edge were energy calibrated (Fig. S32b†) using commercial reference foils of Co or Fe metal (99.9%, GoodFellow), normalized, and averaged with the ProQEXAFS software.42 The bulk Co oxidation state was determined by linearly interpolating the energy of the half-normalized edge step between the CoO, Co3O4 and CoOOH standard XAS spectra. All commercial standards are listed in ‘Detailed Methods’ of ESI.† The magnitude of the FT-EXAFS spectra at the Co K-edge of BSCo0.80Fe0.20 and BSCo0.60Fe0.40 were fitted over a k-range of 3 to 11.5 Å−1 with a k2-weighting using Artemis from the Demeter software package.43 The amplitude reduction factor for each sample fit was received by fitting the 1st coordination shell of the corresponding reference foil. The ICSD collection code of the CIF-files used to calculate the models for the fits are listed in ‘Detailed Methods’ of ESI.† The magnitude of the FT-EXAFS spectra was not phase corrected as indicated with R + ΔR on the x-axis.
The Co and Fe L3-edge XAS spectra of the as-synthesized BSCo1−xFex series and the standard materials were measured as powder on carbon tape at the X-Treme44 and PHOENIX beamlines of SLS. All as-synthesized Co and Fe L3-edge XAS spectra were detected in TEY mode, at room temperature, and at low pressure (10−11 mbar (UHV) at X-Treme and 10−5 mbar (HV) at PHOENIX) and were recorded three times. The as-purchased CoO and FeO standards were sputtered twice (1st: 1 h, 1 keV, and 2.2 μA and 2nd: 10 min, 1 keV, and 12 μA) with Ar-ions to remove the (oxidized) passivation layer. The XAS spectra were then divided by I0 (taken as TEY signal from an Au grid), averaged, energy calibrated, subtracted by the baseline, and normalized (‘Detailed Methods’ in ESI†). The Ba M-edge was removed in the Co L3-edge of the as-synthesized BSCo1−xFex series by subtracting the XAS spectrum of BS(Co0.01)Fe0.99, which was Co-free at the surface (Fig. S32c†). The surface Co oxidation state was estimated by linearly interpolating the normalized maximum δ peak intensity between the CoO, Co3O4, and CoOOH standard XAS spectra. The surface high-spin Co2+ in Oh-sites fraction (sum of all cobalt-sites fraction is 100%) was determined by linearly interpolating the normalized maximum α peak intensity between the CoO (100%) and CoOOH (0%) standard XAS spectra. The mole fraction weighted average oxidation state of the surface B-site metals and the remaining surface Co fractions are estimated as described in ‘Detailed Methods’ of ESI.† A systematic error of ∼10% is assumed in all these calculations.
Electrochemical characterization
The thin-film RDE methodology was applied to characterize the OER activity and stability of the BSCo1−xFex series (VMP-300 Potentiostat, BioLogic Science Instruments) and were all performed at room temperature, with a RDE speed rate of 900 rpm (MSR, Pine Research Instrumentation), and in daily freshly prepared 0.1 M KOH electrolyte solution (KOH, 99.99%, Sigma Aldrich and ultrapure water, PURELAB Chorus 1, Elga Veolia) saturated with synthetic air (5.6, PanGas).45 The fluorinated ethylene propylene (FEP) cell was covered with a Teflon cap and cleaned before use in piranha solution (H2SO4 and H2O2 in 3-to-1 vol. ratio). The working electrodes (WEs) were prepared by drop-casting an electrocatalyst containing ink suspension on freshly polished (0.3 μm and 0.05 μm MicroPolish alumina solution and MicroCloths, Buehler) glassy carbons (GC, SIGRADUR G, 5.0 mm OD, HTW GmbH) with a total electrocatalysts loading of 0.1 mgcat cmgeom−2. The ultrasonicated (30 min, Ultrasonic Cleaner, VWR) ink suspension was a 2-propanol (99.9% (HPLC Plus), Sigma Aldrich), ultrapure water and Na+-exchanged Nafion solution mixture (NaOH, 99.99% and Nafion® perfluorinated resin solution, 5 wt% in lower aliphatic alcohols and water, contains 15–20% water, Sigma Aldrich).46 The counter electrode (CE) was a freshly flame-annealed gold mesh (Advent Research Materials Ltd). The reference electrode (RE) was a commercial Hg/HgO electrode (RE-61AP, ALS), which was filled with 0.1 M KOH electrolyte solution, and was experimentally calibrated against a polycrystalline platinum disk insert (Ptpc, 5.0 mm OD (0.196 cm2), Pine Research Instrumentation) in hydrogen (5.0, PanGas) saturated 0.1 M KOH electrolyte solution to convert the potentials to the reversible hydrogen electrode (VRHE) scale (Fig. S32e†).47–49The OER activity protocol included impedance spectroscopy measurements (Fig. S32f†) at 1.2 VRHE (at beginning and end), and 1.4 VRHE (only at end) with a 10 mV amplitude and a frequency range of 1 MHz to 1 Hz to assign the uncompensated ohmic resistance (Ru) for correcting the ohmic drop (EIR-free = Eapplied − I·Ru), 25 CV's between 1.0 and 1.7 VRHE measured with a scan rate of 10 mV s−1, and 15 CA measurements between 1.3 and 1.7 VRHE (30 s per step). The current density (J) from the CV and CA experiments were calculated by normalizing the measured current (I) either by the electrocatalyst- (gcat) or Co-mass (gCo from ICP-OES). The Tafel plot shows the averaged steady-state current density of four independent measurements in the linear potential range.
The OER stability protocol included mainly 505 CA potential cycles between 1.0 and 1.6 VRHE holding each for 10 s. Refer to ‘Detailed Methods’ in ESI† for the complete protocol. The OER stability measurement was performed three times and is plotted as electrocatalyst-mass normalized average steady-state current at 1.6 VRHEvs. cycle number. Three different representations are shown: (1) current density in A gcat−1 (J@1.6 VRHE), (2) absolute current density change in A gcat−1 (ΔJ@1.6 VRHE), where all values were subtracted by the value of the first cycle, and (3) relative current density change in % (ΔJ@1.6 VRHE), where all values were divided by the value of the first cycle. The calculations of Jnet and Qnet are described in ‘Detailed Methods’ of ESI.†
Operando/ex situ XAS and ex situ XRD
BSCo0.80Fe0.20 and BSCo0.60Fe0.40 containing ultrasonicated (30 min, Ultrasonic Cleaner, VWR) ink suspensions (ultrapure water and Na+-exchanged Nafion) were spray-coated on Au sputtered (∼100 nm) and carbon coated Kapton® foils (DuPont) to produce as-prepared electrodes for operando Co and Fe K-edge XAS and ex situ XRD experiments with final loadings of 4.0 mgcat cmgeom−2 (XPE206DR, Mettler Toledo).50 The operando XAS experiments were performed at the SuperXAS beamline by simultaneously detecting the signal in transmission and fluorescence mode,41,51 using the same beamline setup as already described for the as-synthesized measurements. The shown operando Co K-edge spectra were measured in transmission, whereas the operando Fe K-edge spectra were measured in fluorescence (500 μm thick passivated implanted planar silicon (PIPS) detector, Mirion Technologies).51 The slight calibration offset of the operando Fe K-edge fluorescence spectra were aligned to the corresponding transmission spectra (Fig. S33e and f†). An in-house developed PEEK flow cell was used to execute the operando XAS measurements,50 which included CA measurements between 1.00 and 1.55 VRHE and then CV cycles between 1.00 and 1.60 VRHE (Fig. S33a and b†). Refer to ‘Detailed Methods’ in ESI† for the complete protocol. The operando XAS spectra were recorded for 1 min (∼120 spectra) per CA potential step after stabilizing the current for 1 min. The CE was a high-surface area carbon black (Black Pearls 2000, Cabot Corporation) electrocatalyst, which was spray-coated on Au sputtered and carbon coated Kapton® foil (DuPont).50 The RE was an Ag/AgCl electrode (Harvard Apparatus), which was experimentally calibrated before and after each operando XAS experiment against the previously used Hg/HgO RE. A 0.1 M KOH solution was used as electrolyte. The same fitting approach was applied for the operando FT-EXAFS spectra as for the as-synthesized powders. No edge jump loss was detected between 1.00 and 1.55 VRHE, which confirms the reliability of the measurement (Fig. S33c and d†).52An as-prepared electrode and one after being used for the operando XAS measurement (ex situ ‘after OER’) were analyzed with XRD in transmission mode with the same equipment as used to characterize the as-synthesized powders. A bare Au-sputtered and carbon coated Kapton® foil was characterized as well to assign the additional Au peaks (ICSD collection code 52700) from the used electrodes. The background was subtracted for all XRD patterns measured in transmission.
GC electrodes covered either with BSCo0.99(Fe0.01), BSCo0.80Fe0.20, BSCo0.60Fe0.40, or BSCo0.21Fe0.79 were used to measure the ‘as-prepared’ and ex situ ‘after OER’ Co and Fe L3-edge TEY-XAS spectra. The method used to obtain the ‘as-prepared’ GC electrodes was identical as already described in the ‘Electrochemical Characterization’ section. The ex situ ‘after OER’ samples represent the GC electrodes after performing the OER activity protocol in the RDE setup. The only difference in this protocol, compared to the one described in the ‘Electrochemical characterization’ section, was that the CA part was repeated at the end, so that the last applied potential was 1.7 VRHE. These ‘after OER’ electrodes were then cleaned with ultrapure water and stored together with the as-prepared GC electrodes at 0.1 mbar for few days before being analyzed either at X-Treme44 or PHOENIX beamline of SLS. The measurement conditions and analysis methods for the as-prepared and ‘after OER’ Co and Fe L3-edge XAS spectra were identical as for the as-synthesized powders. The only difference was that the F K-edge background, originating from the Na+-exchanged Nafion, had to be removed from the Fe L3-edge XAS spectra. The background was removed by subtracting the Fe L3-edge XAS spectra of a GC electrode, where an electrocatalyst-free ink (only ultrapure water and Na+-exchanged Nafion) was drop-casted (Fig. S32d†). The amount of irreversible surface Co oxidation (Δ in CoΔ+) was calculated by subtracting the appropriate quantities of the ‘as synthesized’ or ‘as prepared’ XAS spectra from the quantities of the ‘after OER’ XAS spectra. Also in this analysis, a systematic error of ∼10% is assumed.
Author contributions
D. A.: conceptualization (equal), formal analysis, investigation (lead), and writing of original draft; E. F.: conceptualization (equal), draft editing (lead), and project supervision (supporting); M. B.: draft editing (supporting) and synthesis (lead); N. S. Y.: draft editing (supporting) and synthesis (supporting); N. D.: draft editing (supporting) and XAS investigation (supporting); A. H. C.: draft editing (supporting) and XAS investigation (supporting); V. R.: draft editing (supporting) and XAS investigation (supporting); C. P.: draft editing (supporting), XAS investigation (equal), and XAS supervision (equal); J. D.: draft editing (supporting) and XAS investigation (equal); T. H.: draft editing (supporting) and XAS supervision (equal); T. G.: draft editing (supporting) and synthesis supervision; T. J. S.: conceptualization (supporting), draft editing (supporting), funding acquisition, and project supervision (lead). All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge SCCER Heat & Electricity Storage and Innosuisse for the funding. Moreover, the authors thank SLS at PSI for the beamtimes at the PHOENIX, SuperXAS and X-Treme beamlines as well as EMF at PSI for using the TEM. Moreover, the authors are grateful to Dr Elisabeth Müller and Dr Emiliya Poghosyan for their help during the TEM sessions and Dr Camelia N. Borca for her help during the PHOENIX beamtimes.Notes and references
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- Z. Shao, W. Yang, Y. Cong, H. Dong, J. Tong and G. Xiong, J. Membr. Sci., 2000, 172, 177–188 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- A. Heel, P. Holtappels, P. Hug and T. Graule, Fuel Cells, 2010, 10, 419–432 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–932 CrossRef CAS PubMed.
- B. J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS PubMed.
- D. Aegerter, M. Borlaf, E. Fabbri, A. H. Clark, M. Nachtegaal, T. Graule and T. J. Schmidt, Catalysts, 2020, 10, 984 CrossRef CAS.
- B. J. Kim, X. Cheng, D. F. Abbott, E. Fabbri, F. Bozza, T. Graule, I. E. Castelli, L. Wiles, N. Danilovic, K. E. Ayers, N. Marzari and T. J. Schmidt, Adv. Funct. Mater., 2018, 28, 1804355 CrossRef.
- S. E. Pratsinis, AIChE J., 2010, 56, 3028–3035 CrossRef CAS.
- A. J. Grohn, S. E. Pratsinis, A. Sanchez-Ferrer, R. Mezzenga and K. Wegner, Ind. Eng. Chem. Res., 2014, 53, 10734–10742 CrossRef CAS.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CrossRef CAS.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- T.-H. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, J. Am. Chem. Soc., 2020, 142, 15876–15883 CrossRef CAS PubMed.
- Y. Inoue, Y. Miyahara, K. Miyazaki, Y. Kondo, Y. Yokoyama and T. Abe, J. Electrochem. Soc., 2022, 169, 010518 CrossRef CAS.
- A. Zeng, W. Chen, K. D. Rasmussen, X. Zhu, M. Lundhaug, D. B. Müller, J. Tan, J. K. Keiding, L. Liu, T. Dai, A. Wang and G. Liu, Nat. Commun., 2022, 13, 1341 CrossRef CAS PubMed.
- R. Deberdt and P. L. Billon, Extr. Ind. Soc., 2023, 14, 101232 Search PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- X. Cheng, B. J. Kim, E. Fabbri and T. J. Schmidt, ACS Appl. Mater. Interfaces, 2019, 11, 34787–34795 CrossRef CAS PubMed.
- S. Lee, A. Moysiadou, Y.-C. Chu, H. M. Chen and X. Hu, Energy Environ. Sci., 2022, 15, 206–214 RSC.
- V. Bartunek, S. Huber, D. Sedmidubsky, Z. Sofer, P. Simek and O. Jankovsky, Ceram. Int., 2014, 40, 12591–12595 CrossRef CAS.
- G. Ketteler, W. Weiss, W. Ranke and R. Schlogl, Phys. Chem. Chem. Phys., 2001, 3, 1114–1122 RSC.
- P. D. Battle, T. C. Gibb and R. Strange, J. Solid State Chem., 1989, 81, 217–229 CrossRef CAS.
- T. C. Gibb, J. Mater. Chem., 1992, 2, 387–393 RSC.
- J. Simböck, M. Ghiasi, S. Schönebaum, U. Simon, F. M. F. de Groot and R. Palkovits, Nat. Commun., 2020, 11, 652 CrossRef PubMed.
- C. F. Chang, Z. Hu, H. Wu, T. Burnus, N. Hollmann, M. Benomar, T. Lorenz, A. Tanaka, H. J. Lin, H. H. Hsieh, C. T. Chen and L. H. Tjeng, Phys. Rev. Lett., 2009, 102, 116401 CrossRef CAS PubMed.
- T. Grewe, X. H. Deng and H. Tuysuz, Chem. Mater., 2014, 26, 3162–3168 CrossRef CAS.
- L. Calvillo, F. Carraro, O. Vozniuk, V. Celorrio, L. Nodari, A. E. Russell, D. Debellis, D. Fermin, F. Cavani, S. Agnoli and G. Granozzi, J. Mater. Chem. A, 2018, 6, 7034–7041 RSC.
- X. T. Wang, T. Ouyang, L. Wang, J. H. Zhong, T. Y. Ma and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 13291–13296 CrossRef CAS PubMed.
- S. Saddeler, G. Bendt, S. Salamon, F. T. Haase, J. Landers, J. Timoshenko, C. Rettenmaier, H. S. Jeon, A. Bergmann, H. Wende, B. R. Cuenya and S. Schulz, J. Mater. Chem. A, 2021, 9, 25381–25390 RSC.
- W. Xiang, N. Yang, X. Li, J. Linnemann, U. Hagemann, O. Ruediger, M. Heidelmann, T. Falk, M. Aramini, S. DeBeer, M. Muhler, K. Tschulik and T. Li, Nat. Commun., 2022, 13, 179 CrossRef CAS PubMed.
- A. C. Garcia and M. T. M. Koper, ACS Catal., 2018, 8, 9359–9363 CrossRef CAS PubMed.
- M. Bernt, A. Hartig-Weiss, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schroter, C. Gebauer and H. A. Gasteiger, Chem. Ing. Tech., 2020, 92, 31–39 CrossRef CAS.
- M. F. Tovini, A. Hartig-Weiss, H. A. Gasteiger and H. A. El-Sayed, J. Electrochem. Soc., 2021, 168, 014512 CrossRef CAS.
- A. Bergmann, T. E. Jones, E. M. Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711–719 CrossRef CAS.
- J. Chivot, L. Mendoza, C. Mansour, T. Pauporte and M. Cassir, Corros. Sci., 2008, 50, 62–69 CrossRef CAS.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC.
- D. Aegerter, E. Fabbri, N. S. Yüzbasi, N. Diklić, A. H. Clark, M. Nachtegaal, C. Piamonteze, J. Dreiser, T. Huthwelker, T. Graule and T. J. Schmidt, ACS Catal., 2023, 13, 15899–15909 CrossRef CAS.
- A. Boucly, L. Artiglia, E. Fabbri, D. Palagin, D. Aegerter, D. Pergolesi, Z. Novotny, N. Comini, J. T. Diulus, T. Huthwelker, M. Ammann and T. J. Schmidt, J. Mater. Chem. A, 2022, 10, 2434–2444 RSC.
- J. Yang, H. W. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- B. Seo, Y. J. Sa, J. Woo, K. Kwon, J. Park, T. J. Shin, H. Y. Jeong and S. H. Joo, ACS Catal., 2016, 6, 4347–4355 CrossRef CAS.
- O. Muller, M. Nachtegaal, J. Just, D. Lutzenkirchen-Hecht and R. Frahm, J. Synchrotron Rad., 2016, 23, 260–266 CrossRef PubMed.
- A. H. Clark, J. Imbao, R. Frahm and M. Nachtegaal, J. Synchrotron Radiat., 2020, 27, 551–557 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. Piamonteze, U. Flechsig, S. Rusponi, J. Dreiser, J. Heidler, M. Schmidt, R. Wetter, M. Calvi, T. Schmidt, H. Pruchova, J. Krempasky, C. Quitmann, H. Brune and F. Nolting, J. Synchrotron Radiat., 2012, 19, 661–674 CrossRef CAS PubMed.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stab, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS.
- W. C. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- P. J. Rheinlander, S. Henning, J. Herranz and H. A. Gasteiger, ECS Trans., 2013, 50, 2163–2174 CrossRef.
- P. J. Rheinlander, J. Herranz, J. Durst and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, F1448–F1457 CrossRef.
- T. Binninger, E. Fabbri, A. Patru, M. Garganourakis, J. Han, D. F. Abbott, O. Sereda, R. Kotz, A. Menzel, M. Nachtegaal and T. J. Schmidt, J. Electrochem. Soc., 2016, 163, H906–H912 CrossRef CAS.
- A. H. Clark, P. Steiger, B. Bornmann, S. Hitz, R. Frahm, D. Ferri and M. Nachtegaal, J. Synchrotron Radiat., 2020, 27, 681–688 CrossRef CAS PubMed.
- N. Diklic, A. H. Clark, J. Herranz, J. S. Diercks, D. Aegerter, M. Nachtegaal, A. Beard and T. J. Schmidt, ACS Energy Lett., 2022, 7, 1735–1740 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed methods, ESI Fig. S1–S33 including (S)TEM, EDX, BET, K-edge & L-edge XAS, XRD, and additional electrochemistry data, ESI Notes S1–S7, and ESI Tables S1–S10 including ICP-OES data and EXAFS best fits values. See DOI: https://doi.org/10.1039/d3ta06156f |
| ‡ Present Address: Universidad Autónoma de Madrid (UAM). Department of Inorganic Chemistry. C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain. |
| This journal is © The Royal Society of Chemistry 2024 |