
Synergistic effect of diatomic materials on efficient formaldehyde sensing and degradation†
Zhouhao
Zhu
ab,
Hengcong
Tao
ad,
Renkun
Zhang
*a,
Liyong
Gan
 *b and
Yingtang
Zhou
*ac
*b and
Yingtang
Zhou
*ac
aSchool of Petrochemical Engineering & Environment, Zhejiang Ocean University, Zhoushan 316022, China. E-mail: zhouyingtang@zjou.edu.cn
bCollege of Physics and Center of Quantum Materials and Devices, Chongqing University, Chongqing 401331, China
cZhejiang Key Laboratory of Petrochemical Environmental Pollution Control, National Engineering Research Center for Marine Aquaculture, Marine Science and Technology College, Zhejiang Ocean University, Zhoushan 316004, China
dNational & Local Joint Engineering Research Center of Harbor Oil & Gas Storage and Transportation Technology, Zhoushan 316022, China
First published on 20th November 2023
Abstract
A high-sensitivity sensor for formaldehyde (HCHO) is crucial in environmental detection and human health studies. However, the development of sensing materials with remarkable adsorption capacity remains a challenge. In this study, we employed density functional theory to screen twenty-seven transition metals as potential single atom (SA) and diatomic (DA) adsorption materials for detecting HCHO. Among them, Hf2-C2N exhibited excellent HCHO adsorption capability among fifty-four candidate materials due to its more negative adsorption energy (−7.32 eV). Through extensive electronic structure and orbital analyses, we elucidated that the enhanced activity of Hf DAs, acting as the active site for HCHO adsorption, was attributed to the introduction of an additional Hf atom in Hf2-C2N. Furthermore, we discussed in detail the degradation process of HCHO on Hf2-C2N using transition state analysis which revealed a low potential barrier (2.1 eV), indicating its potential as a high-performance catalyst. This work not only screens a bifunctional catalyst for efficient adsorption and degradation of HCHO but also provides valuable insights for further experimental exploration.
1. Introduction
Formaldehyde (HCHO) is a hazardous volatile organic compound (VOC) commonly found indoors, characterized by its colourless and odourless nature, which poses an undetectable threat to human health and acts as a serious carcinogen.1,2 Moreover, prolonged exposure to HCHO can lead to genetic mutations.3 The World Health Organization (WHO) has set a stringent exposure threshold of 0.1 mg m−3 for HCHO,4 underscoring the urgency in detecting this compound. Gas sensors assume a pivotal role in the detection of HCHO. However, conventional sensors often suffer from limitations such as poor performance in high humidity environments known as water poisoning.5,6 Another challenge lies in the selectivity of traditional sensors towards detecting HCHO gas under complex environmental conditions.7 Therefore, it is imperative to explore efficient materials for sensing HCHO and catalysts for its degradation.In recent decades, a range of metal oxide semiconductors (MOSs) have proven to be effective in the detection of formaldehyde (HCHO), such as TiO2,8 In2O3,9 and SnO2.10 However, the low sensitivity of these materials in parts per million (ppm) limits their application in HCHO sensing.11 Two-dimensional materials, owing to their distinct chemical and physical characteristics (layered structure, large surface area, and changeable chemical properties), are considered to be an alternative to conventional sensing materials.12 Due to their superior surface sensitivity, 2D materials have the most potential for chemical sensing applications, especially in highly sensitive environments.13 To further improve the detection sensitivity of HCHO for 2D materials, a lot of interest has been drawn to SAs owing to their enormous specific surface area, high adsorption capacity and potentially low cost.14,15 Wang et al. prepared V-SAs on ultrathin carbon nitride, which exhibited high activity HCHO oxidation performance (over a span of 180 minutes, the concentration of HCHO decreased from an initial level of 300 ppm to 57 ppm). The experiments and theoretical calculations were combined to explain that the V-SA as the active site promoted the oxidation of HCHO.16 Shin et al. trapped a Pt SA in a carbon nitrogen/SnO2 heterostructure, and this 1D nano-heterostructure supported Pt SA system exhibited high sensitivity (response = 33.9 at 5 ppm) and selectivity for the detection of HCHO.17 In our previous work, Zr-C3N6 and Hf-C3N6 were identified to exhibit efficient adsorption of HCHO.18 However, SAs as the HCHO detection material remain short of industrial requirements and the single active site of SAs limit their adsorption for HCHO.
Recently, diatomic (DA) materials have received significant attention, presenting a promising avenue in catalysis beyond SA catalysts. DA materials exhibit increased metal loading and greater adaptability in the configuration of active sites, resulting in enhanced adsorption and catalytic capacity compared to SA catalysts due to synergistic effects between metals.19,20 The interaction of two adjacent atoms in DA structures effectively stabilizes the intermediates, leading to improved activity and selectivity.21,22 At present, although the adsorption detection and detection of HCHO by SAs has been demonstrated in experiments23,24 and simulations,25 DA materials that possibly offer higher performance are still lacking in reports. As a crucial component of DA systems, the substrate plays a vital role. C2N, a graphene-like material with distributed pores, large specific surface area, and suitable band gap values, is widely employed as a substrate for investigating adsorption and catalytic performances of DA materials.26–28 However, further research progress has been impeded by the high cost and time requirements associated with standard experimental methods. Therefore, there is an urgent need to employ theoretical simulation methods for screening transition metals anchored on C2N substrates in both SA and DA configurations for formaldehyde (HCHO) adsorption. More importantly, although there are numerous newly discovered novel materials with highly efficient properties, for practical applications, it is more feasible to prioritize materials that can be easily synthesized rather than those existing solely in theoretical realms.
In this study, we conducted a comprehensive simulation analysis on twenty-seven transition metals anchored on C2N to investigate their thermodynamic stability and adsorption capacity for HCHO in two different configurations. Among the candidates, Hf2-C2N exhibited strong adsorption of HCHO. To understand the underlying mechanism, we analyzed various properties including projected density of states (PDOS), d-band center, electron localization function (ELF), charge density difference (CDD), Bader charge transfer, and crystal orbital Hamiltonian population (COHP). Additionally, the potential degradation ability of Hf2-C2N towards HCHO was discussed using transition state theory. This theoretical investigation not only presents a promising material for efficient sensing and degradation of diatomic molecules like HCHO but also provides valuable insights into its adsorption mechanism.
2. Calculation method
All spin-polarized DFT simulations were performed using with the generalized gradient approximation (GGA) method with the Perdew–Burke–Ernzerhof (PBE)29 functional as implemented in the Vienna ab initio simulation package (VASP)30 with van der Waals (vdW) correction (DFT+D3)31 based on the MedeA platform. The projector-augmented plane-wave (PAW)32 method was employed to describe the ion–electron interactions and the cutoff energy was set as 450 eV. For the calculation of structural relaxation and electronic information, the K-points were set to 3 × 3 × 1 and 6 × 6 × 1 in the Monkhorst–Pack meshes, respectively. A vacuum layer with a length of 15 Å in the z direction was used to avoid interactions between periodic structures. Up until the convergence thresholds of energy and the Hellmann–Feynman force, which are 10−5 eV and 0.03 eV Å−1, respectively, structures relaxed. Post-processing of electronic structure information was carried out with the VASPKIT core.33 The climbing image nudged elastic band (CI-NEB) method was employed to search for the conformation and energy of the transition state in the degradation process of HCHO.34The binding energy (Eb) of transition metals anchored on pores of C2N is defined as:
| Eb = E(TMn-C2N) − E(C2N) − nE(TM) | (1) |
| ECoh = E(TM-bulk)/N − E(TM) | (2) |
| Eads = E(TMn-C2N@HCHO) − E(TMn-C2N) − E(HCHO) | (3) |
3. Results and discussion
3.1 Structures of SA-C2N and DA-C2N
The C2N structure was initially relaxed, as shown in Fig. 1a. The homogeneous pore of C2N is formed by 12C and 6 N atoms into four units. The inter-hole distance for C2N is 8.300 Å, which agrees with earlier reports.35 Monomolecular C2N has two different forms of N atoms (pyridine-type and pyrazine-type).36 According to the report by Du et al., the bond lengths of C–C(1) (1.465 Å), C–C(2) (1.428 Å), and C–N (1.335 Å) of C2N are all in agreement.37 In addition, it is found that monolayer C2N exhibits electronic properties indicative of a semiconductor, characterized by a direct band gap of 1.67 eV, consistent with prior theoretical investigations.26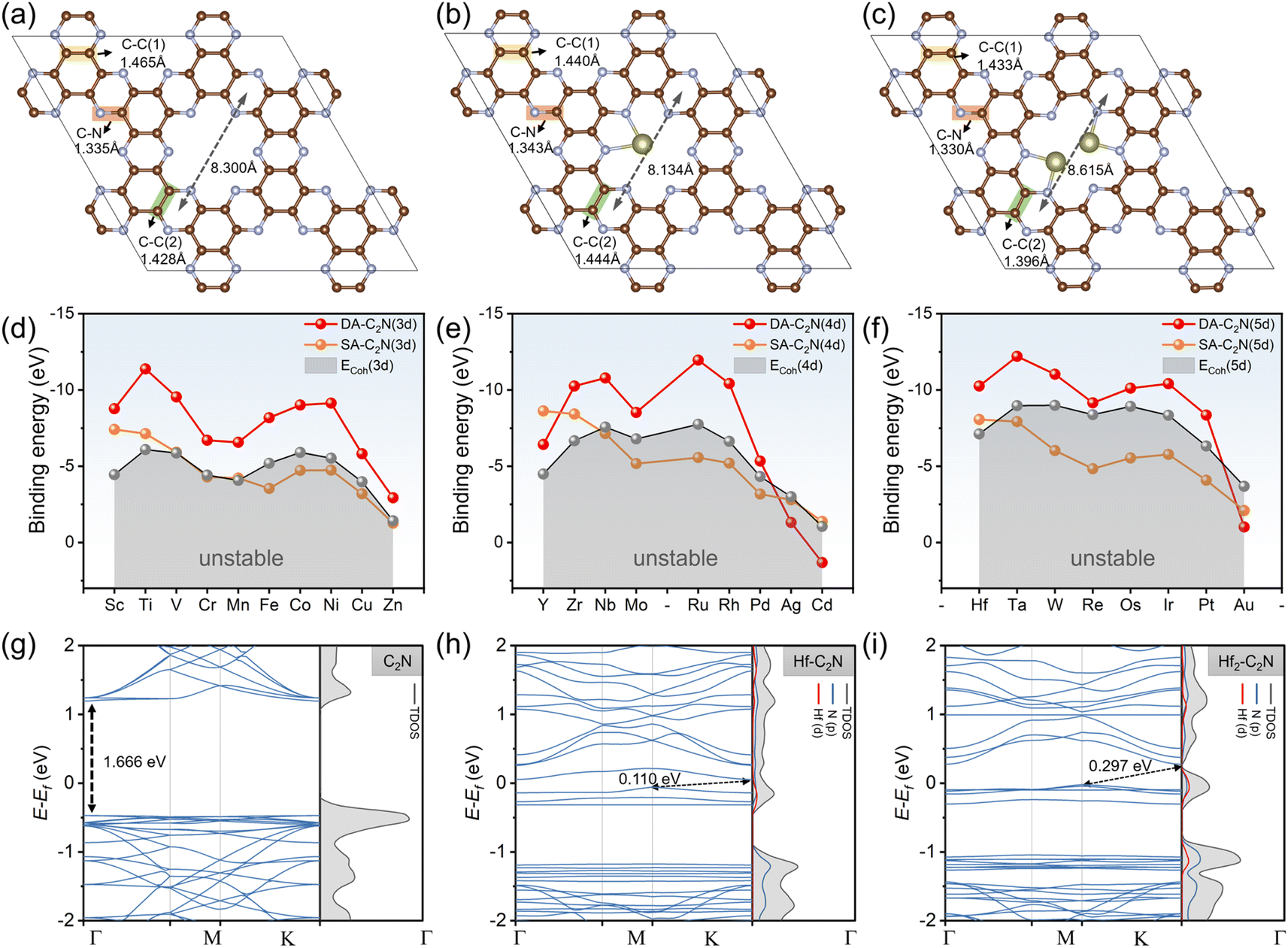 | ||
| Fig. 1 (a–d) The optimised structures of pure C2N, SA-C2N and DA-C2N. (d–f) The binding energies and cohesive energies of third, fourth and fifth period transition metals. (g–i) Band structure and projected density of states of C2N, Hf-C2N and Hf2-C2N. | ||
Following this, we investigated twenty-seven transition metals anchored on the pores of monolayer C2N in two forms (SA-C2N and DA-C2N), displaying Hf-C2N and Hf2-C2N as representative structures as shown in Fig. 1b and c The transition metal SA prefers to anchor the two nitrogen atoms around the C2N pore, and the two transition metal atoms of DA incline to anchor the two pairs of nitrogen atoms facing each other in the C2N pore. The transition metals SA and DA are anchored on the same plane as C2N. In addition, the pore length of C2N shortens after single-atom anchoring (from 8.300 Å to 8.134 Å); however, it lengthens when double-atom anchoring occurs (from 8.300 Å to 8.615 Å).
To study the stability of the substrate materials for HCHO adsorption, we investigated the Eb of SAs and DAs anchored on C2N and the ECoh of transition metals, and the detailed energies of twenty-seven SAs and DAs are shown in the Tables S2 and S3,† respectively. As shown in Fig. 1d–f, for the twenty-seven transition metals considered, a more negative Eb to ECoh value indicates more stability for the structure.38 Except for Ag-C2N, Au-C2N and Cd-C2N, Eb values of DA-C2N are all more negative than their corresponding ECoh values, demonstrating the thermodynamic stability of DA-C2N. The Eb values of DA-C2N are generally more negative than that of SA-C2N, suggesting that anchoring DAs on C2N pores is more stable than anchoring SAs (except for Y-C2N, Ag-C2N, Cd-C2N and Au-C2N). SA-C2N, particularly the fifth-period transition metals, has more positive Eb values than the corresponding ECoh values, suggesting that the stability of some SA-C2N materials is a non-negligible synthetic hindrance. In summary, except for Y-C2N, Ag-C2N, Au-C2N and Cd-C2N, DA-C2N materials are stable and universally more stable than SA-C2N. In addition, the band structure of C2N shows that the direct band gap of C2N was 1.666 eV (Fig. 1g), and with the introduction of metal atoms, the band gap decreases sharply, switching from a direct to an indirect band gap (using Hf-C2N and Hf2-C2N as examples, as shown in Fig. 1h and i), which is consistent with previous theoretical and experimental reports.26,28 The identical band gaps indicate that the structures are efficiently a universal structure for the ensuing adsorption.
3.2 Adsorption characteristics
The adsorption of HCHO on both SA-C2N and DA-C2N is of great importance in evaluating their potential as highly sensitive HCHO sensing materials. Subsequently, the adsorption of HCHO on SA-C2N and DA-C2N was explored in two distinct adsorption configurations, namely, side-on and end-on orientations. Schematic diagrams of the adsorption configurations are shown in Fig. 2a, where the side-on configuration is HCHO adsorbed on the transition metal site in a parallel way on C2N and the end-on configuration for vertical direction. According to previous reports, HCHO in the end-on adsorption configuration favoured bound metal atoms with an O atom.39 The results of the adsorption energy of SA-C2N and DA-C2N for HCHO are shown in Fig. 2b–d and e–g, respectively. A more negative adsorption energy implies stronger material adsorption for HCHO.40,41 It was found that the Eads values of materials are negative except for Mn-C2N and Mn2-C2N, indicating that for these materials the adsorption of HCHO is a thermodynamically spontaneous reaction. Note that for transition metals of the same period, the metals of IIIB and IVB groups anchored on C2N are more strongly adsorbed to HCHO. In addition, HCHO adsorption on SA-C2N and DA-C2N generally favours the side-on configuration owing to the more negative Eads. For the same transition metal, DA-C2N generally showed a stronger adsorption of HCHO than SA-C2N. In these materials above, Hf-C2N (−3.14 eV) and Hf2-C2N (−7.32 eV) showed the most negative Eads values for HCHO on SA-C2N and DA-C2N, respectively. The strong adsorption capacity is suitable for high-sensitivity HCHO sensing materials, and a more negative adsorption energy than −0.5 eV indicates that they adsorb HCHO by chemisorption.42,43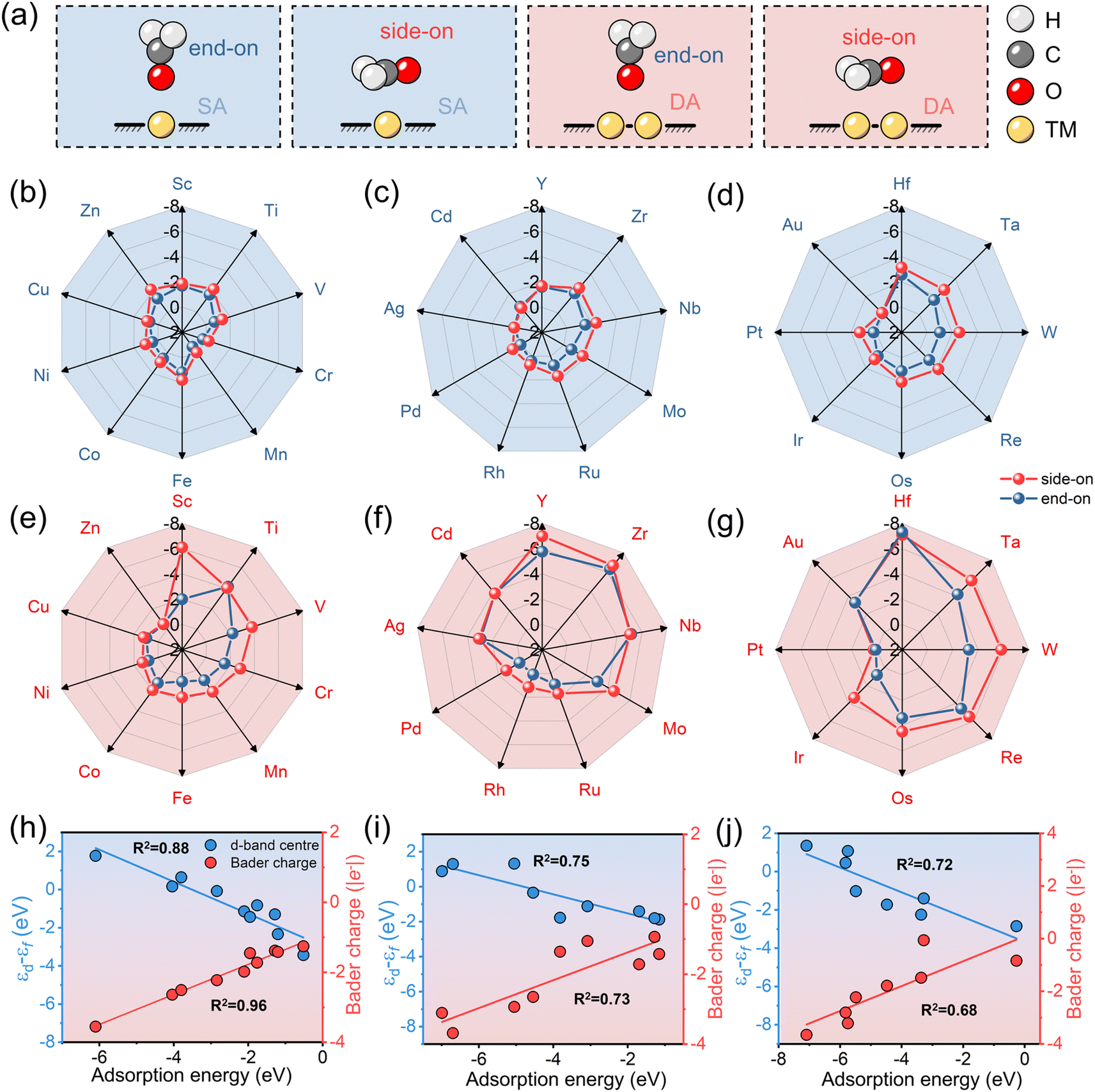 | ||
| Fig. 2 (a) The schematic diagram of HCHO absorbed on SA-C2N and DA-C2N with different configurations, respectively. (b–d) The adsorption energy of HCHO absorbed on SA-C2N with side-on and end-on configurations. (e–g) The adsorption energy of HCHO absorbed on DA-C2N with side-on and end-on configurations. (h–j) The linear relationship of the d-band centre and Bader charge with adsorption energy for different periods of metal anchored DA-C2N, h, j and k represent 3d, 4d and 5d transition metal anchored C2N structures, respectively. | ||
Then we investigated the metal d-band centre and Bader charge of HCHO adsorption by DA with different periodic metal anchoring, and the results revealed a linear correlation between the d-band centre and Bader charge concerning adsorption energy, as shown in Fig. 2h–j. The d-band centre and Bader charge of 3d metal-anchored C2N have a strong linear relationship with the corresponding adsorption energy (R2 is 0.88 and 0.96, respectively). However, the correlation of the d-band centre and Bader charge with adsorption energy gradually decreases with increasing metal period. The correlation coefficient (R2) values of the d-band centre and adsorption energy for 3d, 4d, and 5d transition metal anchored C2N structures are 0.88, 0.75, and 0.72, respectively. The R2 values of the Bader charge and adsorption energy for 3d, 4d, and 5d transition metal anchored C2N structures are 0.96, 0.73, and 0.68, respectively. Thus, the d-band centre and Bader charge are potential descriptors of HCHO adsorption for 3d metals, but are not as satisfactory for 4d and 5d metals.
The electron localization function (ELF) was employed to further reveal the interactions between Hf-C2N, Hf2-C2N and HCHO. (Fig. 3a and b) ELF values close to 0 indicate ionic bonds characterised by completely delocalised electrons, 0.5 indicates metallic bonds with electron gas-like behaviour and 1.0 indicates covalent bonds characterised by complete electron localisation. The ELF of Hf-C2N and Hf2-C2N revealed that Hf has a metal bond with C with electron gas-like behaviour. This further illustrates Hf as an adsorption active site for Hf-C2N and Hf2-C2N. Notably, the distance between the C and O atoms of HCHO was longer on Hf2-C2N than on Hf-C2N (1.455 Å vs. 1.428 Å), suggesting that HCHO adsorption on Hf2-C2N was more activated and favoured subsequent degradation reactions.
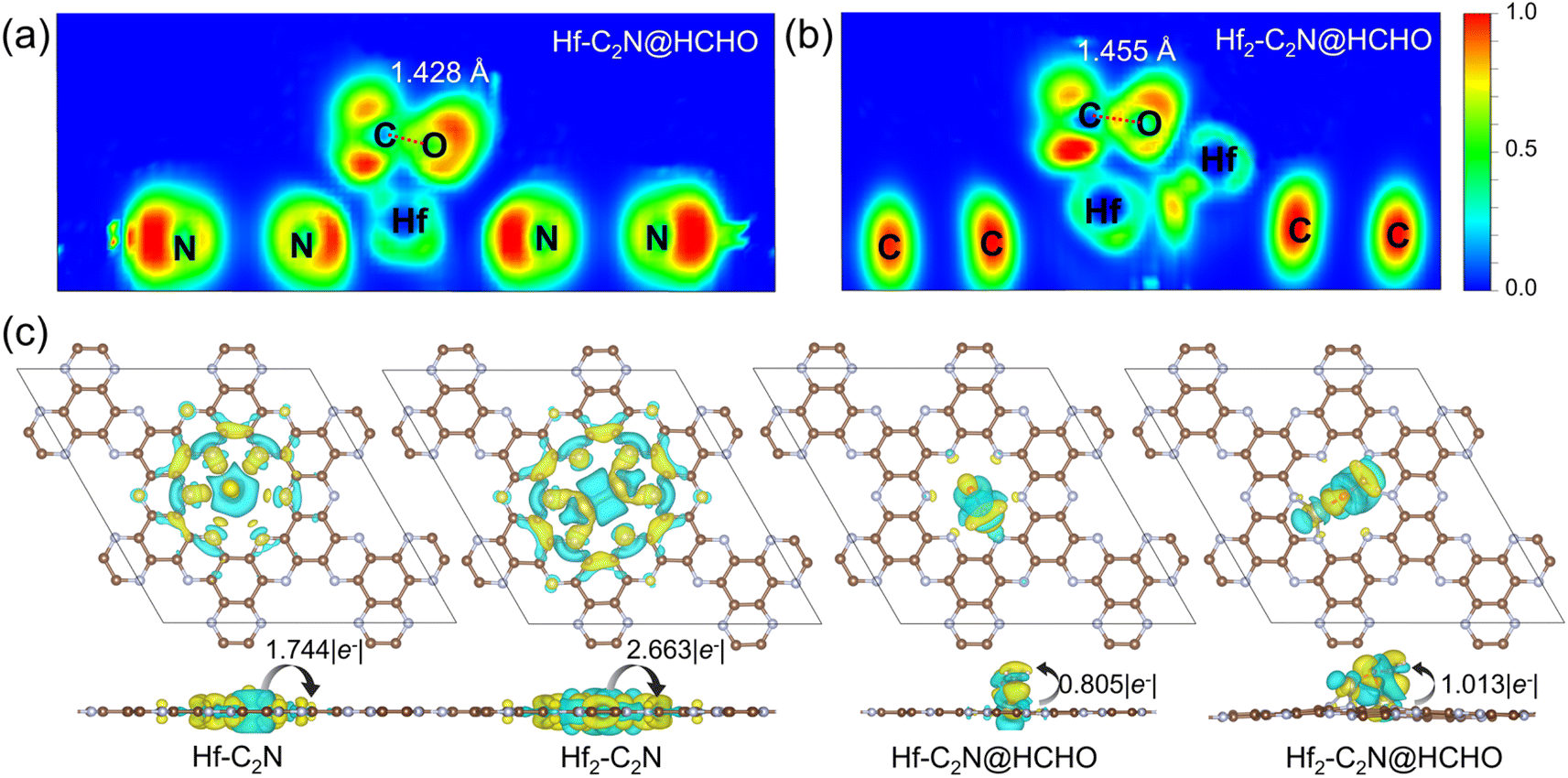 | ||
| Fig. 3 (a and b) The electron localization function of Hf-C2N@HCHO and Hf2-C2N@HCHO. (c) The charge density difference of Hf-C2N, Hf2-C2N, Hf-C2N@HCHO and Hf2-C2N@HCHO, respectively. The isosurface value is 0.003 e Å−3. | ||
To further explore the charge density transfer, we calculated the Bader charge and CDD (Fig. 3c) for Hf-C2N and Hf2-C2N, with cyan representing charge dissipation and yellow for charge aggregation. In both Hf-C2N and Hf2-C2N, significant charge transfer occurs from the introduced Hf atom to the C2N substrate. The Hf atom of Hf-C2N exhibits electron transfer and interaction mainly with the three N atoms on the surrounding C2N pore. Each Hf atom of Hf2-C2N exhibts electron transfer with the two N atoms closer to the C2N pore. A higher amount of electron transfer occurred in Hf2-C2N (2.663 |e−|) compared with Hf-C2N (1.744 |e−|), indicating the stronger binding ability of Hf2 and C2N. When Hf-C2N and Hf2-C2N adsorbed HCHO, electrons were mainly transferred from the Hf active site to HCHO, with a greater amount of electron transfer by Hf2-C2N (1.013 |e−|) relative to Hf-C2N (0.805 |e−|) showing a stronger adsorption capacity for HCHO.
Projected density of states (PDOS) analysis was applied to gain deeper insights into the enhanced adsorption mechanism of HCHO by Hf-C2N and Hf2-C2N, with a focus on orbital interactions. As shown in Fig. 4a–c, for Hf-C2N and Hf2-C2N, Hf d is clearly hybridized with N p for the substrate C2N above the Fermi level, suggesting that Hf has robust orbital coupling with C2N, which favours the stability of Hf-C2N and Hf2-C2N. The PDOS of HCHO adsorbed by Hf2-C2N is shown in Fig. 4c, Hf2-C2N@HCHO possesses abundant states around the Fermi level suggesting a superior adsorption activity of Hf2-C2N for HCHO.44 The coupling of the Hf d to O p and C p orbitals of HCHO illustrated Hf as the active site for Hf2-C2N. The d-band centre developed by Nørskov was employed to unravel the mechanism behind the superior adsorption capacity of Hf2-C2N.45 As a measure of the possible transition metal reactions, generally the increase of the d-band centre results in strong adsorption interaction.46 As shown in Fig. 4b and c, for Hf2-C2N, after adsorption of HCHO, its d-band centre increased from 1.338 to 1.354 eV, indicating that Hf2-C2N favors a strong adsorption interaction. The cause of the raised Hf d of Hf2-C2N is presented in Fig. 4d; after adsorption of HCHO, the bandwidth of Hf d is shrunken, inducing an increasing level of the d-band centre, therefore resulting in a strong adsorption of HCHO by Hf2-C2N.
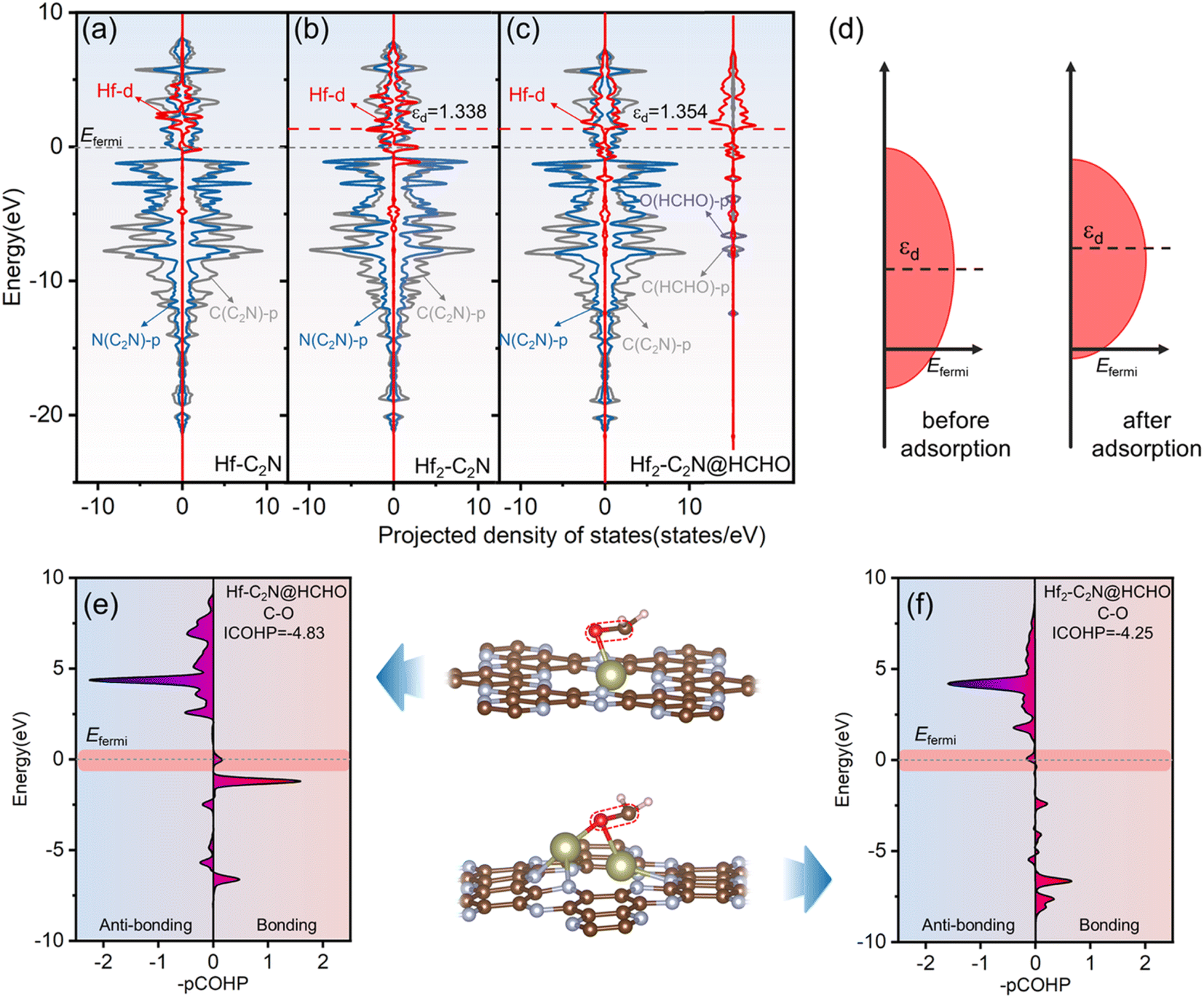 | ||
| Fig. 4 (a and b) (a–c) The projected density of states of Hf-C2N, Hf2-C2N and Hf2-C2N@HCHO, respectively. (d) Schematic diagram of the d-band centre bandwidth of Hf2-C2N before and after adsorption of HCHO. (e and f) The crystal orbital Hamiltonian population of C–O of HCHO for Hf-C2N@HCHO and Hf2-C2N@HCHO, respectively. | ||
The crystal orbital Hamiltonian population (COHP) was used to further expose the bonding and anti-bonding states between C and O atoms of HCHO. As shown in Fig. 4e and f, negative values of –pCOHP represent anti-bonding and positive values represent bonding. Near the Fermi energy level, the –pCOHP values of C and O atoms of HCHO adsorbed on Hf-C2N are positive, indicating that HCHO is not sufficiently activated due to the filling of the bonding state near the Fermi energy level. However, the negative value of –pCOHP for HCHO adsorption on Hf2-C2N indicates that the antibonding state is populated around the Fermi energy level and that HCHO is sufficiently activated to favour the ensuing degradation. By integrating the energy up to the Fermi level, we obtain the ICOHP, which serves as a quantitative indicator. Generally, the more negative the ICOHP, the greater the combining strength.47 The ICOHP of the C and O atoms of HCHO adsorbed to Hf2-C2N (−4.25) is more positive than that of Hf-C2N (−4.83), indicating that the C and O atoms of HCHO interact more weakly, which facilitates the subsequent HCHO degradation reaction. The results are consistent with the previous results for PDOS, d-band centre and ELF.
In summary, following the screening of twenty-seven transition metals for stability and adsorption capacity in SA-C2N and DA-C2N, we have identified Hf2-C2N as a highly promising candidate with significant potential for HCHO adsorption. The electronic structure and orbital analysis revealed that the superior adsorption of HCHO on Hf2-C2N can be attributed to the additional introduction of Hf atoms, which enhances their activity. Additionally, we demonstrated that Hf2-C2N is more favorable for the subsequent degradation reaction of HCHO.
3.3 Possible catalytic degradation of HCHO
As described above, we carried out a systematic screening of SA-C2N and DA-C2N for the adsorption of HCHO and revealed that Hf2-C2N showed strong adsorption of HCHO and could be potentially used as a HCHO sensing material. Hydroxyl (˙OH) and superoxide (O2˙−) radicals are essential in the degradation of HCHO.48 As sources of ˙OH and O2˙−, H2O and O2, it is worthwhile to study their adsorption on Hf2-C2N. Thus, we simulated the adsorption of H2O and O2 on Hf2-C2N in different configurations, respectively. As shown in Fig. 5a, the original configurations are presented on the left side of the figures, while the optimized configurations are displayed on the right side. It was noticeable that the two adsorption configurations of H2O were induced by the strong adsorption of Hf2-C2N to form an –OH group and H atom, and the –OH group was bonded to two Hf atoms. Similarly, the strong adsorption of Hf2-C2N caused the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of both adsorbed configurations of O2 to break, forming two O atoms chemically bonded to Hf atoms, and one of the O atoms is in between the two Hf atoms. Thus, the strong adsorption capacity of Hf2-C2N facilitates the adsorption of H2O and the generation of hydroxyl radicals. On the other hand, Hf2-C2N also shows a high potential to activate O2 molecules into superoxide radicals, thus facilitating the ensuing degradation reactions.
O bond of both adsorbed configurations of O2 to break, forming two O atoms chemically bonded to Hf atoms, and one of the O atoms is in between the two Hf atoms. Thus, the strong adsorption capacity of Hf2-C2N facilitates the adsorption of H2O and the generation of hydroxyl radicals. On the other hand, Hf2-C2N also shows a high potential to activate O2 molecules into superoxide radicals, thus facilitating the ensuing degradation reactions.
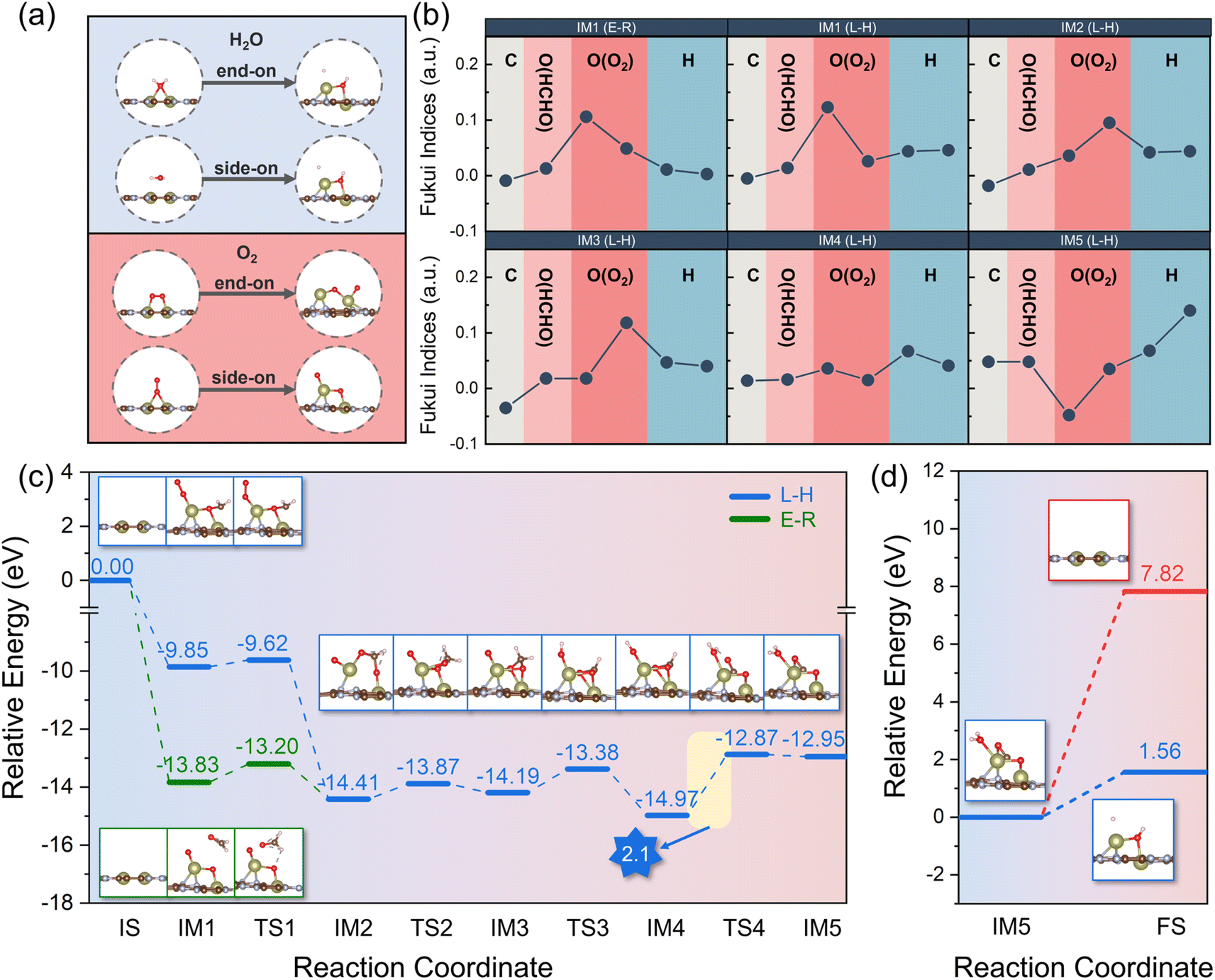 | ||
| Fig. 5 (a) Before and after optimisation of configurations of H2O and O2 adsorption on Hf2-C2N and (b) Fukui index for intermediates of L–H and E–R mechanisms. (c) Potential energy profile of HCHO degradation in L–H and E–R mechanisms. (d) Potential energy profile of H2O and CO2 desorption. | ||
The primary pathways for the catalytic oxidation of HCHO involve the Langmuir–Hinshelwood (L–H) and Eley–Rideal (E–R) mechanisms. The L–H mechanism involves HCHO adsorbing on the catalyst surface and reacting with a nearby reactive oxygen molecule to decompose into H2O and CO2, while the E–R mechanism involves HCHO reacting with O2 on the catalyst surface to decompose into H2O and CO2.49 To further investigate the reaction mechanism, we calculated the Fukui function f0 of HCHO and O2 for intermediates (IMs) in the process of the initial state (IS) and final state (FS) in both L–H and E–R mechanisms. In general, the higher the value of f0, the more inclined to the radical reaction.50–52 As shown in Fig. 5b, IM2, IM3, IM4, and IM5 were the share intermediates for the L–H and E–R mechanisms. For IM1 to IM4 intermediates, the O atom from O2 had higher f0 values, indicating that O atoms were more inclined to radical reactions. For IM5, the f0 value of the H atom was higher, indicating that H2O was more favourable to the formation of –OH and a H atom. Furthermore, we investigated the transition state (TS) of the intermediates for both L–H and E–R mechanisms; as shown in Fig. 5c, the maximum potential barrier for both the L–H and E–R mechanisms was the step IM4 → TS4 with a potential barrier of 2.1 eV, which was lower than that in previously reported theoretical studies for a potential Pt/TiO2 catalyst (230.45 kJ mol−1).53 In the step IM5 → FS (Fig. 5d), CO2 tended to desorb from the catalyst; however H2O preferred to decompose into –OH and a H atom due to the lower potential barrier (1.56 eV vs. 7.82 eV). The production of –OH radicals was more favourable for the subsequent degradation of HCHO. In summary, Hf2-C2N is a potential catalyst for the degradation of HCHO due to its strong adsorption capacity which is more favourable to the formation of –OH and superoxide and the low reaction potential (2.1 eV) in the catalytic HCHO degradation steps.
The stability of the structure is an essential indicator of the synthesis and performance of the catalyst. To further investigate the potential ability of Hf2-C2N as the catalyst for HCHO detection adsorption and degradation, ab initio molecular dynamics (AIMD) simulations were subsequently performed for Hf2-C2N at 300 K, 500 K and 1000 K, respectively. As shown in Fig. 6, it was found that Hf2-C2N maintained its structure throughout and was not significantly damaged. The C2N substrate structure of Hf2-C2N remained stable with some fluctuations during 6 ps. The detailed configuration structures are shown in Fig. S1.† Two Hf atoms were shifted in the pores of C2N during 6 ps, and finally two Hf atoms protruded from the surface of C2N, and the protruding Hf was favorable for the adsorption of HCHO.
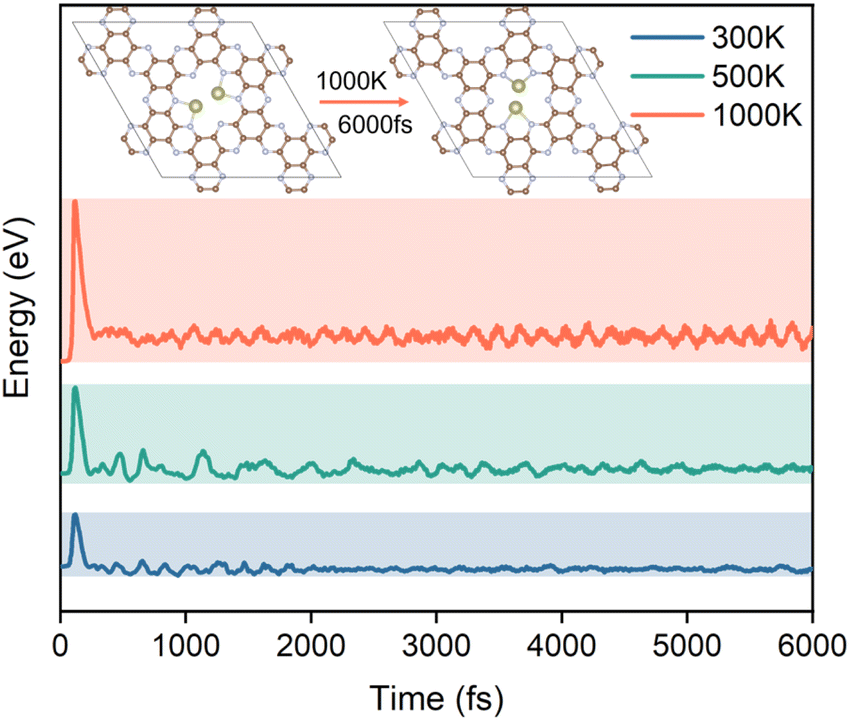 | ||
| Fig. 6 The potential energy of Hf2-C2N at 300 K, 500 K and 1000 K for 6000 fs. | ||
4. Conclusion
In summary, we conducted a systematic density functional theory screening of twenty-seven transition metal single atom (SA) and double atom (DA) candidates for highly efficient formaldehyde adsorption. Among them, Hf2-C2N exhibited superior HCHO adsorption capacity with a more negative adsorption energy (−7.32 eV). Through various electronic structure and orbital analyses including partial density of states, d-band centre, CDD, Bader charge analysis, and ELF, we discovered that the introduction of an additional Hf atom into C2N enhanced the activity of Hf atoms, resulting in strong HCHO adsorption activity. Furthermore, we investigated the degradation mechanism of HCHO on Hf2-C2N and found that it can potentially be degraded through strong adsorption interactions with water molecules and oxygen molecules to generate hydroxyl radicals (–OH) and superoxide radicals. The low reaction potential (2.1 eV) further supports the capability of Hf2-C2N for efficient degradation of HCHO. This study screens a promising diatomic material for highly efficient sensing and degradation while providing valuable insights for future experimental exploration.Author contributions
Zhouhao Zhu: conceptualization, simulation, data analysis, writing – original draft. Hengcong Tao: supervision, funding acquisition. Renkun Zhang: supervision, writing – review & editing. Liyong Gan: supervision, writing – review & editing. Yingtang Zhou: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2023C01191) and the National Natural Science Foundation of China (no. 22005269 and 22262024).References
- J. van den Broek, D. Klein Cerrejon, S. E. Pratsinis and A. T. Güntner, J. Hazard. Mater., 2020, 399, 123052 CrossRef CAS PubMed.
- C. Lou, G. Lei, X. Liu, J. Xie, Z. Li, W. Zheng, N. Goel, M. Kumar and J. Zhang, Coord. Chem. Rev., 2022, 452, 214280 CrossRef CAS.
- J. Bellat, I. Bezverkhyy, G. Weber, S. Royer, R. Averlant, J.-M. Giraudon and J.-F. Lamonier, J. Hazard. Mater., 2015, 300, 711–717 CrossRef CAS PubMed.
- P.-R. Chung, C.-T. Tzeng, M.-T. Ke and C.-Y. Lee, Sensors, 2013, 13, 4468–4484 CrossRef CAS PubMed.
- Y. Xu, L. Zheng, C. Yang, W. Zheng, X. Liu and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 20704–20713 CrossRef CAS PubMed.
- Y. Xu, C. Lou, L. Zheng, W. Zheng, X. Liu, M. Kumar and J. Zhang, Sens. Actuators, B, 2020, 307, 127616 CrossRef CAS.
- D. Wang, Z. Li, J. Zhou, H. Fang, X. He, P. Jena, J.-B. Zeng and W.-N. Wang, Nano-Micro Lett., 2017, 10, 4 CrossRef PubMed.
- X. Li, X. Li, J. Wang and S. Lin, Sens. Actuators, B, 2015, 219, 158–163 CrossRef CAS.
- D. Zhang, Y. Cao, Z. Yang and J. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 11979–11989 CrossRef CAS PubMed.
- G. Zhang, S. Zhang, L. Yang, Z. Zou, D. Zeng and C. Xie, Sens. Actuators, B, 2013, 188, 137–146 CrossRef CAS.
- A. Dey, Mater. Sci. Eng. B, 2018, 229, 206–217 CrossRef CAS.
- Y. Ravi Kumar, K. Deshmukh, T. Kovářík and S. K. Khadheer Pasha, Coord. Chem. Rev., 2022, 461, 214502 CrossRef CAS.
- Y. Zhou, H. Yin and S. Ai, Coord. Chem. Rev., 2021, 447, 214156 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- R. Riaz, M. Ali, I. A. Sahito, A. A. Arbab, T. Maiyalagan, A. S. Anjum, M. J. Ko and S. H. Jeong, Appl. Surf. Sci., 2019, 480, 1035–1046 CrossRef CAS.
- K. Wang, L. Jiang, T. Xin, Y. Li, X. Wu and G. Zhang, Chem. Eng. J., 2022, 429, 132229 CrossRef CAS.
- H. Shin, W.-G. Jung, D.-H. Kim, J.-S. Jang, Y. H. Kim, W.-T. Koo, J. Bae, C. Park, S.-H. Cho, B. J. Kim and I.-D. Kim, ACS Nano, 2020, 14, 11394–11405 CrossRef CAS PubMed.
- M. Chen, H. Wang, J. Wang, M. Sun, Y. Hu, X. Zhao and Y. Zhou, Phys. Chem. Chem. Phys., 2023, 25, 25353–25360 RSC.
- X. Zhao, J. Chen, Z. Bi, S. Chen, L. Feng, X. Zhou, H. Zhang, Y. Zhou, T. Wågberg and G. Hu, Adv. Sci., 2023, 10, 2205889 CrossRef CAS PubMed.
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. G. Humphrey, Q. Fu and J. Huang, ACS Catal., 2021, 11, 1952–1961 CrossRef CAS.
- B. Kim, D. Kwon, J.-O. Baeg, M. Austeria P, G. H. Gu, J.-H. Lee, J. Jeong, W. Kim and W. Choi, Adv. Funct. Mater., 2023, 33, 2212453 CrossRef CAS.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- H. Zhang, G. Guo, Z. Wang, Q. He, X. He and H. Ji, Appl. Catal., B, 2023, 333, 122774 CrossRef CAS.
- Z. Liu, J. Wei, G. Zhang, D. Zhang, J. Zhang, W. Yang, C. Wu and I. D. Gates, J. Mater. Chem. A, 2023, 11, 1978–1990 RSC.
- H. Tang, J. Zhang, M. Huang, J. Zhang, Y. Zhou, G. Wang, R. Wang and J. Chen, J. Colloid Interface Sci., 2022, 624, 527–536 CrossRef CAS PubMed.
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I.-Y. Jeon, S.-M. Jung, H.-J. Choi, J.-M. Seo, S.-Y. Bae, S.-D. Sohn, N. Park, J. H. Oh, H.-J. Shin and J.-B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed.
- L. Zhu, Q. Xue, X. Li, T. Wu, Y. Jin and W. Xing, J. Mater. Chem. A, 2015, 3, 21351–21356 RSC.
- W. Zhong, Y. Qiu, H. Shen, X. Wang, J. Yuan, C. Jia, S. Bi and J. Jiang, J. Am. Chem. Soc., 2021, 143, 4405–4413 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- X. Zhao, M. Chen, Z. Bi, H. Zhang, G. Hu and Y. Zhou, Small, 2023, 8, 2304854 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- Y. Yang, M. Guo, G. Zhang and W. Li, Carbon, 2017, 117, 120–125 CrossRef CAS.
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I.-Y. Jeon, S.-M. Jung, H.-J. Choi, J.-M. Seo, S.-Y. Bae, S.-D. Sohn, N. Park, J. H. Oh, H.-J. Shin and J.-B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed.
- J. Du, C. Xia, W. Xiong, X. Zhao, T. Wang and Y. Jia, Phys. Chem. Chem. Phys., 2016, 18, 22678–22686 RSC.
- P. Janthon, S. M. Kozlov, F. Viñes, J. Limtrakul and F. Illas, J. Chem. Theory Comput., 2013, 9, 1631–1640 CrossRef CAS PubMed.
- H. Liu, X. Wang, C. Pan and K. M. Liew, J. Phys. Chem. C, 2012, 116, 8044–8053 CrossRef CAS.
- Z. Zhu, H. Tao, J. Fu, Y. Zhou, J. Guo and C. Zhai, Chin. Chem. Lett., 2023, 34, 107476 CrossRef CAS.
- T. Jia, L. Wang, Z. Zhu, B. Zhu, Y. Zhou, G. Zhu, M. Zhu and H. Tao, Chin. Chem. Lett., 2023, 108692, DOI:10.1016/j.cclet.2023.108692.
- D. Mei, N. Aaron Deskins and M. Dupuis, Surf. Sci., 2007, 601, 4993–5001 CrossRef CAS.
- Z. Zhu, M. Chen, M. Sun, J. Wang, Y. Zhou, X. Li and H. Tao, Phys. Chem. Chem. Phys., 2022, 24, 26776–26784 RSC.
- J. Wang, Y.-P. Liu, H. Zhang, D.-J. Huang and K. Chu, Catal. Sci. Technol., 2019, 9, 4248–4254 RSC.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- X. Liao, R. Lu, L. Xia, Q. Liu, H. Wang, K. Zhao, Z. Wang and Y. Zhao, Energy Environ. Mater., 2022, 5, 157–185 CrossRef CAS.
- X. Guo, J. Gu, X. Hu, S. Zhang, Z. Chen and S. Huang, Catal. Today, 2020, 350, 91–99 CrossRef CAS.
- Y. Su, W. Li, G. Li, Z. Ao and T. An, Chin. J. Catal., 2019, 40, 664–672 CrossRef CAS.
- C. Mang, G. Li, M. Rao, X. Zhang, J. Luo and T. Jiang, Appl. Surf. Sci., 2023, 608, 154964 CrossRef CAS.
- Y. Ma, J. Liang, D. Zhao, Y.-L. Chen, J. Shen and B. Xiong, RSC Adv., 2014, 4, 17262–17264 RSC.
- Z. Nie, M. Chen, L. Zhang, Q. Feng, J. Hu, X. Huang, C. Zhou, Y. Zhou, T. Wågberg and G. Hu, Chem. Eng. J., 2023, 463, 142411 CrossRef CAS.
- J. Wang, M. Sun, C. Liu, Y. Ye, M. Chen, Z. Zhao, Y. Zhang, X. Wu, K. Wang and Y. Zhou, Adv. Mater., 2023, 35, 2306103 CrossRef CAS PubMed.
- J. Ding, Y. Yang, J. Liu and Z. Wang, Chemosphere, 2020, 248, 125980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06132a |
| This journal is © The Royal Society of Chemistry 2024 |