
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced catalytic activity and stability of SOFC electrodes through plasma-driven surface modification†
Hyunduck
Shin‡
a,
Jongsu
Seo‡
b,
SungHyun
Jeon‡
 a,
Seung Jin
Jeong
a,
Jinwook
Kim
a,
Siwon
Lee
c,
Jeong Jin
Lee
a and
WooChul
Jung
*a
a,
Seung Jin
Jeong
a,
Jinwook
Kim
a,
Siwon
Lee
c,
Jeong Jin
Lee
a and
WooChul
Jung
*a
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea. E-mail: wcjung@kaist.ac.kr
bHydrogen Research Department, Korea Institute of Energy Research (KIER), Daejeon 34129, Republic of Korea
cDepartment of Materials Science and Engineering, Hanbat National University, Daejeon 34158, Republic of Korea
First published on 12th April 2024
Abstract
The commercialization of solid oxide fuel cells (SOFCs) relies heavily on the development of active and stable oxygen electrodes that can operate at intermediate temperatures. However, the presence of an oxidizing atmosphere during the annealing process commonly used to fabricate and treat conductive perovskite oxides, which serve as oxygen electrode materials, can lead to the accumulation of Sr and formation of Sr-rich clusters on their surfaces, thereby degrading electrode performance. To suppress this segregation, a new approach that utilizes Ar plasma to amorphize the surfaces of porous oxide electrodes is proposed. The effects of said plasma treatment on the morphology, chemical composition, and crystallinity of a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) electrode surface are investigated, and the resulting changes in its electrochemical properties are monitored. Remarkably, only 5 minutes of plasma exposure achieves a 43% reduction in initial LSCF electrode polarization resistance and significantly improves durability at 650 °C, compared to the bare LSCF. This study demonstrates the viability of plasma-driven surface modification for high-temperature processes, and represents the first application of an amorphization strategy to a porous electrode in the field of SOFCs.
1. Introduction
Solid oxide fuel cells (SOFCs) are emerging as promising next-generation energy conversion devices owing to their high conversion efficiencies and eco-friendliness.1,2 The key to the SOFC energy conversion process is the oxygen reduction reaction (ORR) at the cathode, and the rapid oxygen exchange at the electrode surface determines the overall reaction rate.3,4 Perovskite oxides (chemical formula: ABO3) such as (La,Sr)MnO3, (La,Sr)CoO3, and (La,Sr)(Co,Fe)O3 are widely used as SOFC cathodes on account of their high electrical conductivity and high catalytic activity for the ORR.5 However, annealing these materials in an oxidizing atmosphere often leads to surface structure and chemical instability caused by cation segregation, resulting in electrode performance degradation.6,7 For instance, Sr segregation encompasses the enrichment of Sr at the electrode surface and the formation of SrOx-like insulating 2nd phases that block active sites and create Sr-deficient regions near the surface, thereby inhibiting oxygen exchange kinetics.8,9 Therefore, several strategies have been proposed to produce durable SOFC cathodes by suppressing Sr segregation. These strategies include surface coating with less reducible cations, developing heterostructure electrodes, isovalent doping, and controlling oxide crystallinity.10–13Yildiz et al. fabricated La0.6Sr0.4CoO3−δ (LSC64) thin films via pulsed laser deposition (PLD) and observed that poorly crystalline LSC64 deposited at lower temperatures exhibited higher electrode activity and durability than fully crystalline LSC64 deposited at higher temperatures. The aforementioned authors attributed this phenomenon to the amorphous structure having more defects and open sites than the dense crystalline structure, enabling excess Sr accommodation and preventing surface Sr segregation.14 Similarly, Fleig et al. reported that the surface of amorphous LSC64 films differed from that of crystalline films and could be particularly active for oxygen incorporation. Accordingly, they suggested that electrode performance degradation was related to changes in surface structure and chemistry caused by the crystallization of LSC thin films at elevated temperatures.15 Skinner et al. also found that amorphous La0.8Sr0.2CoO3−δ (LSC82) has higher oxygen diffusivity (D*) and oxygen exchange rate constant (k*) values compared to crystalline LSC82.16 Nevertheless, all previous research on fabricating amorphous electrodes has been limited to thin-film model samples deposited at low temperatures, whereas conventional fabrication routes for SOFC electrodes always involve a high-temperature sintering process.13–17 In particular, oxides with perovskite structures typically require a high calcination temperature, which makes it impossible to maintain the surface of the electrode in an amorphous state. Consequently, there is a need to develop a cost-effective and mass-producible surface-modification technology that can be universally applied to porous electrode manufacturing processes.
Plasma treatment is a widely employed surface treatment technology in various industrial fields on account of its advantages concerning volume production, large area coverage, high reproducibility, and short processing time. This treatment not only etches the target surface but also influences oxygen vacancy and dopant concentration, hydrophilicity, and surface chemical composition.18–22 Plasma treatment can selectively modify surface regions of several to several tens of nanometers; as a result, it may be possible to impact Sr segregation without altering bulk properties. However, plasma treatment is predominantly used in low-temperature devices such as electrocatalysts for the oxygen evolution reaction (OER), catalytic synthesis, and polymer electrolyte membrane fuel cells (PEMFCs).23–25 To the best of our knowledge, there are no reports on the use of plasma treatment to suppress phase separation in high-temperature electrochemical devices.
In this study, we aimed to modify the surface of La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF), a state-of-the-art cathode material,26 using Ar plasma treatment to create an oxygen electrode with high catalytic activity and durability. The effects of the Ar plasma treatment on the surface phase separation phenomenon and electrochemical properties were investigated by analyzing the corresponding changes in surface structure and chemical composition. We observed that exposure to plasma creates an amorphous layer on the surface of the LSCF (at approximately 2.5 nm deep), which successfully accommodates Sr cations and suppresses surface phase separation, resulting in a stable electrode performance without the formation of insulating secondary particles and/or Sr-deficient regions on the surface. Furthermore, the Ar plasma created excess oxygen vacancies and enriched the B-site transition metal cations at the surface. We then evaluated the electrochemical properties of a porous LSCF electrode fabricated using a screen-printing method. A reduction in the area-specific resistance (ASR) of the porous LSCF (0.13 Ω cm2) was observed after a 5 minutes plasma treatment at 650 °C, compared to the bare LSCF (0.23 Ω cm2). Additionally, the plasma-treated LSCF exhibited a stable performance after 100 hours of operation. This case study presents the successful implementation of a novel strategy for manufacturing an SOFC electrode by applying a first-of-its-kind approach to a porous electrode. The proposed strategy opens a promising pathway towards achieving unique amorphous characteristics that were previously confined to laboratory model studies.
2. Results and discussion
2.1 Plasma-driven surface amorphization
To reduce the electrode surface's crystallinity and alter composition, Ar plasma treatment was conducted at room temperature employing an inductively coupled plasma (ICP) Etcher. The LSCF sample was loaded onto a wafer stage and introduced into a vacuum chamber using a load lock. By introducing Ar gas and applying radio-frequency power, electrons gain kinetic energy and collide with neutral Ar gas atoms. Collisions resulting in electron energies surpassing the ionization voltage cause said neutral atoms to become positive ions (Ar+). Electrons expelled from the outermost shell collide with other neutral atoms under an electric field, resulting in a rapid increase in the number of electrons and ions. Furthermore, electrons, on account of their smaller mass and higher mobility, reach the LSCF sample surface before ions do, charging said surface. This forms a dark space in which electrons are depleted between the plasma region and the sample surface, accelerating ions towards the LSCF surface while retarding electrons.27,28 The ion energy gain depends on the applied bias power; we observed a high bias power caused electrode surface etching whereas low bias power did not induce significant changes in electrode surface properties or crystal structure. Moreover, source power affects ion density. To achieve surface modification while minimizing surface etching, we carefully selected the source and bias power for this study.When LSCF perovskite undergoes Ar plasma treatment, energetic ion bombardment can lead to local heating and the sputtering of surface atoms, resulting in changes in the crystal structure and surface chemistry of the material. The transmission electron microscopy (TEM) image in Fig. 1b shows the alterations in the surface crystallinity of the LSCF porous electrode after plasma exposure. While the bulk region remained crystalline, physical ion bombardment resulted in the formation of a 2.5 nm thick amorphous layer on the surface. This structure enables the preservation of excellent electron conductivity by maintaining crystallinity in the bulk region while introducing amorphousness solely on the surface. The amorphous layer exhibits an increased abundance of defect sites, including oxygen vacancies, compared to the crystalline layer. Moreover, the amorphous structure has a higher concentration of unoccupied spaces within the lattice. These distinct characteristics have a discernible impact on phase separation and the associated electrochemical properties.
 | ||
| Fig. 1 (a) Schematic illustrations of plasma-induced La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) surface modification and (b) transmission electron microscopy (TEM) image of porous LSCF before and after plasma exposure. | ||
Fig. 2a displays the TEM image of the plasma-treated LSCF porous electrode following heat treatment at 650 °C for 30 hours; the persistence of an amorphous layer on the electrode surface despite the high-temperature exposure can be observed. This is consistent with the findings of Fleig et al., who reported a low degree of crystallinity in LSC films deposited at 470 °C, and observed no increase in X-ray diffraction (XRD) peak intensity following heat treatment at 600 °C for 72 hours.15 Hence, the temperature range of 600 to 650 °C appears insufficient for crystallizing LSC or LSCF materials with low-crystallinity.
 | ||
| Fig. 2 (a) Transmission electron microscopy (TEM) image, (b) scanning transmission electron microscopy (STEM), and energy-dispersive X-ray spectroscopy (EDS) line-scanning analysis of plasma-treated La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) after heat treatment at 650 °C for 30 hours. | ||
2.2 Evolution of surface chemical composition
Scanning transmission electron microscopy (STEM) combined with energy-dispersive X-ray spectroscopy (EDS) line-scanning analysis was used to investigate the compositional evolution from the bulk of the electrode to its outermost surface. Fig. 2b illustrates the EDS line scan profiles of the plasma-treated LSCF porous electrode after heat treatment at 650 °C for 30 hours. A noticeable change in the spatial distribution of the A- and B-site cations on the LSCF surface was observed at a depth of approximately 3 nm from the surface. Specifically, an enhancement in the B-site/A-site cation ratio was observed on the surface of the LSCF electrode. This tendency can be attributed to factors such as the preferential sputtering of A-site cations or migration of B-site cations to the surface.29–31 Indeed, B-site transition metals have been widely reported as key sites for the active ORR. Therefore, the increase in the number of these transition metals is expected to improve the catalytic activity of the electrode and facilitate efficient charge transfer during fuel cell operation.32–34The ratio of Sr species in the non-lattice region (Srnon-lattice) to Sr in the bulk lattice (Srlattice) on the electrode surface has been considered an indicator of the degree of Sr separation;35,36 thus, one frequent approach to examine the effect of plasma exposure on Sr segregation is to determine said Srnon-lattice/Srlattice value. As shown in Fig. 3, we utilized X-ray photoelectron spectroscopy (XPS) to investigate the chemical composition of the surface of the LSCF porous electrodes. Fig. 3a shows the Sr 3d spectra of the bare and plasma-treated LSCF porous electrodes after heat treatment at 650 °C for 30 hours. These spectra were deconvoluted into two sets of spin–orbit split doublets. As per the data in Table S1,† the XPS spectra for Sr 3d was deconvoluted into 3d5/2 and 3d3/2, respectively, with a peak ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The main Sr 3d doublets located at a lower binding energies (i.e., 131.7 eV for 3d5/2 and 133.5 eV for 3d3/2) correspond to Sr in the bulk lattice (Srlattice), whereas those located at higher binding energies (i.e., 133.3 eV and 135.1 eV) originate from contributions of other surface Sr species (Srnon-lattice), such as strontium oxides, carbonates, and/or hydroxides.37 Based on the deconvolution results, Fig. 3c shows the Srnon-lattice/Srlattice ratio on the bare and plasma-treated LSCF, respectively; results showed a lower value after plasma exposure, indicating a smaller amount of non-lattice Sr species. This observation indicates that the Ar plasma treatment effectively suppressed Sr segregation.
:2. The main Sr 3d doublets located at a lower binding energies (i.e., 131.7 eV for 3d5/2 and 133.5 eV for 3d3/2) correspond to Sr in the bulk lattice (Srlattice), whereas those located at higher binding energies (i.e., 133.3 eV and 135.1 eV) originate from contributions of other surface Sr species (Srnon-lattice), such as strontium oxides, carbonates, and/or hydroxides.37 Based on the deconvolution results, Fig. 3c shows the Srnon-lattice/Srlattice ratio on the bare and plasma-treated LSCF, respectively; results showed a lower value after plasma exposure, indicating a smaller amount of non-lattice Sr species. This observation indicates that the Ar plasma treatment effectively suppressed Sr segregation.
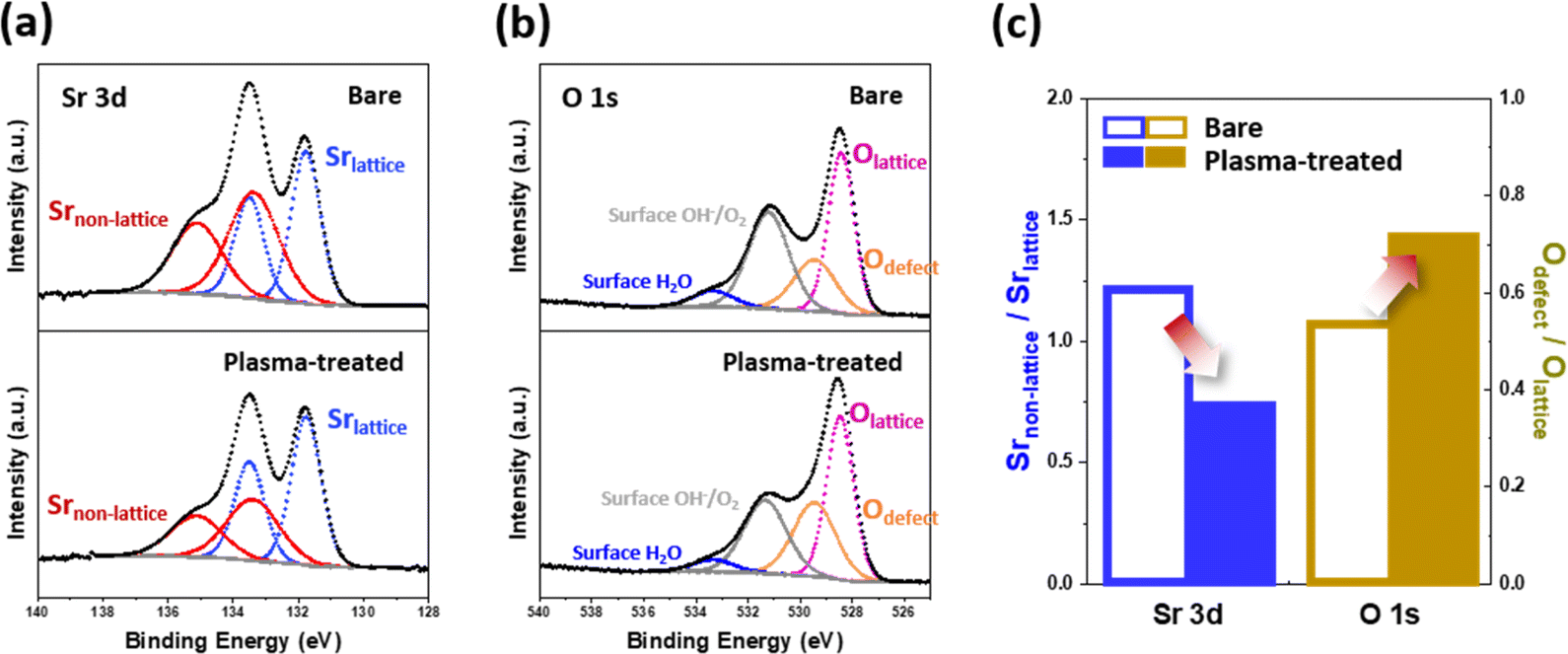 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) results of (a) Sr 3d and (b) O 1s, and (c) summarized graph of bare and plasma-treated La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) surfaces after heat treatment at 650 °C for 30 hours. | ||
To gain further insight into the surface characteristics of the porous electrode, we investigated the changes in the O 1s spectra induced by the plasma treatment (Fig. 3b and Table S2†). The O 1s spectra were deconvoluted and fitted to four components: lattice oxygen (Olattice, ∼528.4 eV), defective oxygen (Odefect, ∼529.4 eV), hydroxyl groups or surface-adsorbed oxygen (Osurface, ∼531.1 eV), and adsorbed molecular water (H2Oad, ∼533.3 eV).18,38 Proceeding in a similar way to the previous Sr separation assessment, we utilized the Odefect/Olattice ratio to estimate the extent to which plasma treatment alters the electrode surface. Fig. 3c shows that the plasma-treated LSCF had a higher Odefect/Olattice (0.71) compared to that of the bare LSCF (0.51), which in turn suggests that plasma treatment generates more defective oxygen species on the electrode surface. Additionally, as illustrated in Fig. S1,† the plasma-treated LSCF demonstrated an increased Odefect/Olattice ratio (1.12) in comparison to the bare LSCF (0.77). This disparity indicates an elevated presence of defective oxygen species on the electrode surface following plasma treatment, prior to any subsequent heat treatment. Although defective oxygen is not limited solely to oxygen vacancies (Ovacancy), they are commonly regarded as the predominant defects in oxygen-depleted oxide materials, including perovskite oxides.39 Therefore, as will be discussed later, these surface oxygen vacancies could serve as active sites for electrochemical reactions, facilitating the adsorption of oxygen species and enhancing the transport of oxygen ions during the ORR.
2.3 Suppressing surface phase separation
To investigate the effect of plasma exposure on the suppression of surface phase separation, thin films were employed to facilitate direct observation of the Sr precipitates, as they offer better visibility than porous structures. Single-crystal Al2O3 (0001) substrates were used to fabricate LSCF thin films via PLD as the model mechanism. The Sr-rich surface layer formed during the PLD process was eliminated by chemical etching with a 0.1 M HCl aqueous solution for 15 s at room temperature, resulting in bare LSCF. The XRD patterns of the LSCF films (Fig. S1†) indicated a successful fabrication was accomplished without the development of a secondary phase. While oxygen vacancies typically cause lattice expansion and a lower two-theta value in the lattice constant, no discernible changes in lattice constant were observed before and after plasma exposure.40 This can be attributed to the fact that, while the Ar plasma treatment predominantly modifies the surface of the thin film, the XRD technique primarily probes the bulk region. Nevertheless, the XRD results substantiate that the Ar plasma treatment had no significant impact on the bulk region of the thin film, which is consistent with the TEM image shown in Fig. 1. Furthermore, atomic force microscopy (AFM) analysis corroborated a decrease in root mean square (RMS) surface roughness following plasma treatment (Fig. S3†). Hence, the forthcoming discussion in the electrochemical performance section illustrates that the observed results concerning the electrode's electrochemical performance are not attributable solely to the augmented specific surface area prompted by Ar plasma treatment.Scanning electron microscopy (SEM) analysis was conducted on bare and plasma-treated LSCF films after heat treatment at 650 °C for 30 hours to examine the phase separation phenomenon, as depicted in Fig. 4. The top-view SEM images revealed distinct differences in the surface morphologies of the two film types. Although phase separation was observed on the bare LSCF surface, the plasma-treated LSCF exhibited a smooth, cluster-free surface. Analyzing the EDS line scan profile in Fig. S4b† reveals a notable increase in Sr in the area where precipitated particles exist, unlike other elements. Additionally, examining the EDS point analysis in Fig. S4c,† which compares the precipitated particles with the non-precipitated clean surface area, indicates a higher Sr ratio in the precipitated particles. Furthermore, cross-sectional STEM and EDS elemental maps indicated that the precipitated clusters on the bare LSCF surface were Sr-rich phases (Fig. 4c). This observation suggests that plasma exposure is effective in suppressing Sr segregation, which is in good agreement with the XPS results shown in Fig. 3a.
 | ||
| Fig. 4 Scanning electron microscopy (SEM) top-view images of (a) bare and (b) plasma-treated La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) films after heat treatment at 650 °C for 30 hours. (c) Cross-sectional scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDS) elemental maps of bare LSCF film. | ||
The absence of phase separation on the plasma-treated LSCF surface after high-temperature heat treatment can be attributed to the amorphous layer created by the Ar plasma, as observed in the TEM image in Fig. 1. Upon annealing in an oxidizing atmosphere, the perovskite oxide surface was enriched with Sr cations. When Sr cations accumulate beyond the solubility limit, they cannot be accommodated within the perovskite oxide lattice, leading to phase separation on the surface.14,41 Crystalline perovskite oxides with well-defined and organized structures restrict the accommodation of Sr ions due to steric hindrance or electrostatic repulsion. As a result, dopants, such as Sr2+, tend to migrate to the surface. In contrast, amorphous materials offer more open spaces, including defect sites, vacancies, nanopores, and strain fields, than crystalline materials with close-packed structures.14 Furthermore, the lack of long-range order in amorphous materials implies there are more available interstitial sites for atoms or molecules to occupy, which can help prevent phase separation by reducing the concentration of atoms or molecules at any given location. Consequently, the structural flexibility of amorphous materials increases the solubility limit, effectively suppressing phase separation by accommodating more Sr cations within the lattice.
2.4 Electrochemical performance evaluation of porous LSCF electrodes
The electrochemical properties of the porous electrode were evaluated using electrochemical impedance spectroscopy (EIS) analysis of the oxygen reduction reaction (ORR) in relation to the aforementioned findings. To examine the changes in the porous electrode, EIS measurements were performed on a symmetric cell configuration featuring LSCF electrodes of identical sizes on both sides of a single-crystal Y2O3-stabilized ZrO2 (YSZ) electrolyte fabricated through a typical screen-printing process. To mitigate chemical side reactions at the interface between the LSCF and YSZ, a 100 nm Gd-doped CeO2 (GDC) layer was deposited on both sides of the electrode via PLD. Fig. 5a presents typical Nyquist plots at 650 °C for bare and plasma-treated LSCF. The Nyquist plot is composed of an offset resistance at higher frequencies and a continuous semicircle at lower frequencies, representing the electrode response. This indicates that plasma exposure resulted in a significant reduction (by approximately half) in the initial LSCF electrode resistance (0.23 → 0.13 Ω cm2). Notably, this effectiveness extends beyond laboratory-made electrode materials, as demonstrated in commercial powders (Fig. S5†). Moreover, the Arrhenius plot shown in Fig. 5b indicates a slight decrease in the activation energy (1.36 → 1.29 eV) and the absolute magnitudes of ASRs are consistently lower in the intermediate temperature range (450–650 °C). These results indicate that Ar plasma treatment can enhance the ORR catalytic activity of LSCF. | ||
| Fig. 5 Electrochemical impedance spectroscopy (EIS) results of bare and plasma-treated La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) (a) Nyquist plot measured at 650 °C, (b) Arrhenius plot, (c) distribution of relaxation time (DRT) analysis, and (d) short-term stability test at 650 °C. Pristine LSCF was added to stability graph for the comparison. | ||
To gain a more comprehensive understanding of these outcomes, a distribution of relaxation time (DRT) analysis was performed. This analysis enables the isolation of the different steps involved in the complex ORR process into high-frequency (HF), medium-frequency (MF), and low-frequency (LF) states, thereby assisting in understanding the improved electrode performance. When comparing the resistance results for each frequency with temperature and oxygen partial pressure (pO2) dependencies, as illustrated and elucidated in Fig. S6,† to existing literature reports on LSCF electrodes, it can be inferred that the resistances at HF, MF, and LF correlate with charge transfer, surface oxygen exchange, and gas diffusion phenomena, respectively.42,43 An examination of the DRT results presented in Fig. 5c and S6c† confirms that the Ar plasma treatment decreased the resistance in both the HF and MF regions, primarily HF. Considering the O 1s XPS results (Fig. 3b) and the TEM results (Fig. 2a), we deduce that the reduction in MF resistance can be attributed to the enhancement of the surface oxygen exchange reaction resulting from the presence of a defect-rich amorphous layer on the surface. Additionally, the decrease in HF resistance can be attributed to the enrichment of transition metal elements located at the B-sites on the surface of the electrode, as depicted in Fig. 2b, which leads to an enhanced charge-transfer reaction.
In addition to the initial performance, the effect of the amorphous layer on the short-term stability of the LSCF was evaluated. Fig. 5d displays the change in electrode resistance over a 100 hour period at 650 °C, under open circuit voltage (OCV) conditions, for pristine, bare, and plasma-treated LSCF samples. Pristine LSCF, without chemical etching, displays a higher initial electrode resistance compared to bare LSCF, attributed to the surface-formed Sr segregation during electrode fabrication. After 100 hours of operation, the electrode resistance increases by approximately 30%. Bare LSCF subjected to chemical etching exhibits an initially lower resistance due to the elimination of the Sr-rich layer. However, with extended operation, the resistance experiences a rapid surge, escalating by 50% after 100 hours; a phenomenon commonly associated with the performance decline attributed to Sr segregation. Notably, it's reported that electrode resistance increases more rapidly post-chemical etching.14 In contrast, the plasma-treated LSCF exhibited a sustained maintenance of the improved initial electrode resistance throughout the 100 hour operation. This enhanced stabilization can be attributed to the introduced amorphous layer, which contains an excess free volume compared with the crystalline layer, facilitating an efficient accommodation of Sr ions and preventing their exposure to the surface. Moreover, Sr, characterized by a larger diameter, typically coordinates with 12 oxygen atoms in the perovskite lattice, thereby experiencing significant compressive stress. Jung et al. attributed this lattice strain to the chemical instability observed in Sr-containing perovskites, considering it a primary contributing factor.8 Amorphous structures demonstrate thermodynamic stability against Sr segregation due to their reduced compressive strain when compared to their crystalline counterparts.7,8,44–46 Consequently, LSCF-plasma treatment addresses two factors commonly associated with electrode performance deterioration due to Sr segregation: (1) the presence of a Sr-rich phase on the surface and (2) a Sr-deficient layer near the surface. By mitigating these factors, the potential of LSCF plasma-treatment to prevent electrode degradation during short-term operation is validated.
Based on the discussion thus far, we report for the first time that Ar plasma treatment activates the surface sites of transition metals and oxygen defects while suppressing the surface separation of Sr-rich phases, thereby enhancing the reactivity and durability of the LSCF electrode. Additionally, we verified that the process of amorphization through plasma treatment can be extended to LSCF sintered at 1100 °C, which is an industrial sintering temperature (Fig. S7†). The results of this study clearly demonstrate that attempts to amorphize the electrode surface, which were previously limited to thin-film-based control experiments, can be applied in porous SOFC electrode manufacturing. The application of plasma treatment for electrode development in high-temperature operating environments is a relatively unexplored area in the SOFC field. Its potential extends to diverse high-temperature systems, including protonic ceramic fuel cells (PCFCs) and solid oxide electrolytic cells (SOECs). Recently, Mariotti et al. and Tsampas et al. successfully implemented high-density exsolution of nanoparticles from perovskite oxide through plasma exposure.30,31 This pioneering development holds great promise, as it is anticipated to be applicable not only to electrochemical catalysts but also to various thermochemical catalysts. In the future, further research should be dedicated to exploring the application of plasma treatment to restore the performance of electrodes deteriorated over prolonged operation or due to Cr poisoning, utilizing various gases.
Plasma treatment exhibits a remarkable versatility as a platform technology for surface modification, offering broad applicability across various materials. Furthermore, the unique attributes of plasma treatment, such as low-temperature processing and ion linearity, allow for its targeted application to the cathode after cell fabrication, ensuring minimal impact on other components. This approach holds significant potential for porous SOFC applications, marking a crucial milestone towards successful commercialization. Consequently, we firmly believe that plasma treatment not only holds significant industrial value, owing to its capacity for volume production and large-scale implementation, but also serves as an exceptional method with immense potential to tackle diverse challenges in the field of SOFC.
3. Conclusions
This study investigated the effects of Ar plasma treatment on the morphology, chemical composition, crystallinity, and electrochemical properties of the La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) electrode. Plasma exposure leads to surface modifications including amorphization, increased oxygen vacancy concentration, and B-site cation enrichment through physical ion bombardment. Plasma-induced surface amorphization increased the structural flexibility of the LSCF surface, which inhibited surface phase separation by accommodating Sr cations within the lattice during high-temperature heat treatment. The enrichment of B-site transition metals and increasing concentration of oxygen vacancies are believed to enhance the ORR activity by promoting the adsorption and activation of oxygen molecules. Furthermore, the plasma-treated porous LSCF electrode showed high initial electrode activity and stable short-term performance at 650 °C compared to bare LSCF, even after 5 minutes of processing time. These findings suggest that surface amorphization strategies, previously limited to model systems, are also effective for porous SOFC electrodes and that plasma treatment is a promising approach for high-temperature processes.4. Experimental section
4.1 Sample preparation
La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) films were deposited on single-crystal Al2O3 (0001) substrates (10 mm × 10 mm × 0.65 mm) by the pulsed laser deposition (PLD) technique with a KrF excimer laser of 248 nm wavelength. The PLD system was operated with a laser beam energy of 300 mJ per pulse and a repetition rate of 10 Hz at a heater setting temperature of 700 °C with an oxygen pressure of 10 mTorr. The resulting films had thicknesses of approximately 200 nm.To evaluate the electrochemical properties, symmetrical structures with identically sized LSCF electrodes on both sides of a single-crystal Y2O3-stabilized ZrO2 (YSZ) electrolyte ((100), 10 × 10 × 0.5 mm) were fabricated using the screen-printing method. To prevent chemical side reactions at the LSCF/YSZ interface, a 100 nm Gd-doped CeO2 (GDC) layer was deposited on both sides of the electrode using PLD. After screen printing, the samples underwent a 1 hour sintering process at 950 °C to form a porous structure and improve adhesion between the electrode and the electrolyte.
The Sr-rich layer on the surface formed by the PLD or sintering process was removed by wet etching with a 0.1 M HCl aqueous solution at room temperature for 15 s, resulting in bare LSCF.
4.2 Plasma treatment
To reduce surface crystallinity, the LSCF samples were plasma-treated using an inductively coupled plasma (ICP) Etcher. A 13.56 MHz radio-frequency (RF) power supply was connected to the induction coil and wafer stage. A RF power supply (source power) connected to an induction coil affects ion density, and the ion energy was controlled through the RF power supply (bias power) connected to the wafer stage and the ion energy is controlled independently of the plasma discharge. For surface modification, Ar plasma exposure was performed for 5 minutes at room temperature with a source power of 50 W and a bias power of 10 W at a pressure of 15 mTorr in a vacuum chamber.4.3 Physical characterization and surface morphological changes
X-ray diffraction (XRD, Ultima IV, RIGAKU) measurements were performed to investigate the changes in the crystal structure and lattice parameters caused by the Ar plasma treatment. The surface morphology and phase-separated clusters on the LSCF thin films were examined using atomic force microscopy (AFM, XE-100, Park Systems) and scanning electron microscopy (SEM, JSM-IT800, JEOL).4.4 Surface chemistry characterization
X-ray photoelectron spectroscopy (XPS, Nexsa G2, Thermo Scientific) measurements were performed in an ultrahigh vacuum to confirm the surface composition of the polycrystalline LSCF thin films. CasaXPS software was used for peak-fitting and chemical quantification. The binding energy values were calibrated against the C 1s peak at 284.8 eV. Transmission electron microscopy (TEM; Talos F200X, FEI) was performed at 200 kV to observe the disordered layer on the surface of the plasma-treated LSCF. Scanning transmission electron microscopy (STEM, Talos F200X, FEI) coupled with energy-dispersive X-ray spectroscopy (EDS) was conducted to confirm the surface composition.4.5 Electrochemical measurement
Electrochemical impedance spectroscopy (EIS, VSP-300, Biologic) measurements, covering the frequency range from 0.01 Hz–1 MHz, with an amplitude of 40 mV, were conducted using a mesh-type alumina reactor at 650 °C for 100 hours under a synthetic air atmosphere. Synthetic air (21% O2 and 79% Ar), with a total flow rate of 100 sccm, was controlled using a mass flow controller.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by SK Hynix Inc. and National R&D Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science, ICT (NRF-2020M3H4A3105824).References
- Z. Shao and S. M. Halle, Nature, 2004, 431, 170–173 CrossRef CAS PubMed.
- M. Choi, S. Hwang, D. Byun and W. Lee, J. Mater. Chem. A, 2016, 4, 4420–4424 RSC.
- N. P. Brandon, S. Skinner and B. C. H. Steele, Annu. Rev. Mater. Res., 2003, 33, 183–213 CrossRef CAS.
- W. C. Chueh and S. M. Haile, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 313–341 CrossRef CAS PubMed.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979–7988 RSC.
- W. Jung and H. L. Tuller, Energy Environ. Sci., 2012, 5, 5370–5378 RSC.
- D. Kim, J. W. Park, B.-H. Yun, J. H. Park and K. T. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 31786–31792 CrossRef CAS PubMed.
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Nat. Mater., 2016, 15, 1010–1016 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, R. Ran, Y. Chen, Z. Shao and M. Liu, Nano Lett., 2016, 16, 512–518 CrossRef CAS PubMed.
- B. Koo, J. Seo, J. K. Kim and W. Jung, J. Mater. Chem. A, 2020, 8, 13763–13769 RSC.
- M. Kubicek, A. Limbeck, T. Frömling, H. Hutter and J. Fleig, J. Electrochem. Soc., 2011, 158, B727–B734 CrossRef CAS.
- Z. Cai, M. Kubicek, J. Fleig and B. Yildiz, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS.
- J. Januschewsky, M. Ahrens, A. Opitz, F. Kubel and J. Fleig, Adv. Funct. Mater., 2009, 19, 3151–3156 CrossRef CAS.
- A. Cavallaro, S. S. Pramana, E. Ruiz-Trejo, P. C. Sherrell, E. Ware, J. A. Kilner and S. J. Skinner, Sustainable Energy Fuels, 2018, 2, 862–875 RSC.
- K. Develos-Bagarinao, O. Celikbilek, R. A. Budiman, G. Kerherve, S. Fearn, S. J. Skinner and H. Kishimoto, J. Mater. Chem. A, 2022, 10, 2445–2459 RSC.
- Y. Zhu, X. Zhong, S. Jin, H. Chen, Z. He, Q. Liu and Y. Chen, J. Mater. Chem. A, 2020, 8, 10957–10965 RSC.
- H. J. Shim, J. S. Kim, D. W. Ahn, J. H. Choe, E. Jung, D. Oh, K. S. Kim, S. C. Lee and S. G. Pyo, Electron. Mater. Lett., 2022, 18, 321–329 CrossRef CAS.
- G. Lee, D. B. Go and C. P. O'Brien, ACS Appl. Mater. Interfaces, 2021, 13, 56242–56253 CrossRef CAS PubMed.
- A. Nipane, D. Karmakar, N. Kaushik, S. Karande and S. Lodha, ACS Nano, 2016, 10, 2128–2137 CrossRef CAS PubMed.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, ACS Catal., 2019, 10780–10793 CrossRef CAS PubMed.
- J. Choi, D. Kim, J. E. Chae, S. Lee, S. M. Kim, S. J. Yoo, H. J. Kim, M. Choi and S. Jang, ACS Appl. Mater. Interfaces, 2022, 14, 50956–50965 CrossRef CAS PubMed.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- H. Bae, Y. Shin, L. Mathur, D. Lee and S.-J. Song, J. Korean Ceram. Soc., 2022, 59, 876–888 CrossRef CAS.
- M. A. Lieberman and A. J. Lichtenberg, MRS Bull., 1994, 30, 899–901 Search PubMed.
- K. Nojiri, Dry Etching Technology for Semiconductors, Springer, 2015 Search PubMed.
- K. J. Kanarik, S. Tan and R. A. Gottscho, J. Phys. Chem. Lett., 2018, 9, 4814–4821 CrossRef CAS PubMed.
- V. Kyriakou, R. K. Sharma, D. Neagu, F. Peeters, O. De Luca, P. Rudolf, A. Pandiyan, W. Yu, S. W. Cha, S. Welzel, M. C. M. van de Sanden and M. N. Tsampas, Small Methods, 2021, 5, 2100868 CrossRef CAS PubMed.
- H. Khalid, A. ul Haq, B. Alessi, J. Wu, C. D. Savaniu, K. Kousi, I. S. Metcalfe, S. C. Parker, J. T. S. Irvine, P. Maguire, E. I. Papaioannou and D. Mariotti, Adv. Energy Mater., 2022, 12, 2201131 CrossRef CAS.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- D. N. Mueller, M. L. MacHala, H. Bluhm and W. C. Chueh, Nat. Commun., 2015, 6, 6097 CrossRef CAS PubMed.
- B. Koo, H. Kwon, Y. Kim, H. G. Seo, J. W. Han and W. Jung, Energy Environ. Sci., 2018, 11, 71–77 RSC.
- H. Chen, Z. Guo, L. A. Zhang, Y. Li, F. Li, Y. Zhang, Y. Chen, X. Wang, B. Yu, J. M. Shi, J. Liu, C. Yang, S. Cheng, Y. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 39785–39793 CrossRef CAS PubMed.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y. L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 6081–6088 RSC.
- B. Hua, M. Li, Y. Q. Zhang, Y. F. Sun and J. L. Luo, Adv. Energy Mater., 2017, 7, 1700666 CrossRef.
- M. Choi, I. A. M. Ibrahim, K. Kim, J. Y. Koo, S. J. Kim, J. W. Son, J. W. Han and W. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 21494–21504 CrossRef CAS.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. C. Jung, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- Y. Chen, Y. M. Choi, S. Yoo, Y. Ding, R. Yan, K. Pei, C. Qu, L. Zhang, I. Chang, B. Zhao, Y. Zhang, H. Chen, Y. Chen, C. Yang, B. deGlee, R. Murphy, J. Liu and M. Liu, Joule, 2018, 2, 938–949 CrossRef CAS.
- S. H. Jeon, J. Seo, J. W. Shin, S. Lee, H. G. Seo, S. Lee, N. Tsvetkov, J. Kim, J. An and W. C. Jung, Chem. Eng. J., 2023, 455, 140611 CrossRef CAS.
- B. Koo, H. Kwon, Y. Kim, H. G. Seo, J. W. Han and W. Jung, Energy Environ. Sci., 2018, 11, 71–77 RSC.
- Z. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 17696–17704 CrossRef CAS PubMed.
- H. Jalili, J. W. Han, Y. Kuru, Z. Cai and B. Yildiz, J. Phys. Chem. Lett., 2011, 2, 801–807 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06111f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |