
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ALD-made noble metal high entropy alloy nanofilm with sub-surface amorphization for enhanced hydrogen evolution†
Yiming
Zou
,
Lin
Jing
,
Jianghong
Zhang
,
Songzhu
Luo
 ,
Leyan
Wang
,
Yun
Li
,
Ronn
Goei
,
Kwan W.
Tan
* and
Alfred Iing
Yoong Tok
*
,
Leyan
Wang
,
Yun
Li
,
Ronn
Goei
,
Kwan W.
Tan
* and
Alfred Iing
Yoong Tok
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: kwtan@ntu.edu.sg; miytok@ntu.edu.sg
First published on 22nd February 2024
Abstract
Noble metal-based high entropy alloys (NM-HEAs) have been shown to have optimized catalytic properties through compositional adjustments. Recently, an amorphous HEA, known as high-entropy metallic glass (HEMG), has gained attention for its potential in surface modification and atomic rearrangement. In this work, RhRuPtPdIr HEA thin films (Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru:Pt:Pd:Ir = 26.1:28.7:8.6:16.3:20.3) were synthesized on glassy carbon (GC) electrodes using precisely controlled sequential atomic layer deposition (ALD) process of each noble metal layer, followed by electrical Joule heating (EJH) alloying at 1000 °C for 5 seconds. Cross-sectional HR-TEM imaging revealed a thickness of 20 nm and the surface microstructure composed of nanocrystallites and amorphous structures, suggesting explosive crystallization during the EJH process. The HEA thin film achieved outstanding HER performance, exhibiting overpotentials of 13, 77, and 65 mV at a current density of 10 mA cm−2 and Tafel slopes of 14, 45, and 78 mV dec−1 in 0.5 M H2SO4, 1.0 M PBS, and 1.0 M KOH electrolytes, respectively. Remarkably, HEA/GC in an acidic environment reached strikingly top-level kinetics, which was mainly contributed by intrinsic activity and surface amorphization. The corresponding DFT study revealed a modified electronic structure of the HEA surface that facilitates surface–hydrogen interaction. The study demonstrates the potential of NM-HEA nanofilm as catalysts for highly efficient HER in harsh environments. This study also demonstrates that ALD-EJH is a novel and reliable method for synthesizing, manipulating, and tuning complex high-entropy nanomaterials.
:Ru:Pt:Pd:Ir = 26.1:28.7:8.6:16.3:20.3) were synthesized on glassy carbon (GC) electrodes using precisely controlled sequential atomic layer deposition (ALD) process of each noble metal layer, followed by electrical Joule heating (EJH) alloying at 1000 °C for 5 seconds. Cross-sectional HR-TEM imaging revealed a thickness of 20 nm and the surface microstructure composed of nanocrystallites and amorphous structures, suggesting explosive crystallization during the EJH process. The HEA thin film achieved outstanding HER performance, exhibiting overpotentials of 13, 77, and 65 mV at a current density of 10 mA cm−2 and Tafel slopes of 14, 45, and 78 mV dec−1 in 0.5 M H2SO4, 1.0 M PBS, and 1.0 M KOH electrolytes, respectively. Remarkably, HEA/GC in an acidic environment reached strikingly top-level kinetics, which was mainly contributed by intrinsic activity and surface amorphization. The corresponding DFT study revealed a modified electronic structure of the HEA surface that facilitates surface–hydrogen interaction. The study demonstrates the potential of NM-HEA nanofilm as catalysts for highly efficient HER in harsh environments. This study also demonstrates that ALD-EJH is a novel and reliable method for synthesizing, manipulating, and tuning complex high-entropy nanomaterials.
1. Introduction
Hydrogen produced through hydrogen evolution reaction (HER), a clean and abundant energy source from water, has been extensively studied over the years. To reduce the energy barrier of HER and achieve low overpotential (η) and Tafel slope, various electrocatalysts have been developed.1 Among monometallic catalysts, platinum (Pt) stands out for its exceptional activity owing to the optimal adsorption/desorption energy of hydrogen on the Pt surface, placing it at the peak of the “volcano” plot.2–4 However, the application of Pt catalyst is restricted by its poor durability and high cost.5 To address this issue, alloying Pt with other transition metals is a widely adopted strategy to develop alternative catalysts with modified electronic structures and enhanced stability.6–9High entropy alloys (HEAs) have gained significant attention and have been applied to the electrocatalytic field in recent years due to their remarkable stability and synergistic effect.10–13 The unique structural stability of HEA arises from a combination of the high entropy effect and sluggish diffusion effect, driven by the variations in the atomic radii of the constituent elements and their highly disordered arrangement in the systems.14–16 Notably, noble metal-based HEAs (NM-HEAs) consisting of Pt, Pd, Rh, Ru, and Ir exhibit superior activity owing to their modified electronic structure, which has been demonstrated through both experimental17–20 and simulation21–23 studies. For example, Wu et al. demonstrated the broad and continuous valence band of the IrPdPtRhRu HEA, indicating the diverse localized density of states.18 The catalytic kinetics of NM-HEA can be further enhanced by amorphization. Amorphous NM-HEA, termed noble metal-based high-entropy metallic glass (NM-HEMG) has emerged as an area of great scientific interest. Unlike crystalline NM-HEA with well-defined atomic structure, NM-HEMG with high homogeneity, short-term and long-term disordered atomic structure24 was found to possess a reorganized surface, contributing to more electrochemical active surface area and enhanced intrinsic activity.25–27 For example, Jia et al. showed that Pt25Pd25Ni25P25 HEMG exhibited the overpotential of 19.8 mV at a current density of 10 mA cm−2 in KOH solution,28 surpassing that of other Pt- and Pd-based catalysts. It was demonstrated that the outstanding HER performance was due to the combination of synergistic effect and unsaturated atomic configuration, which optimized the adsorption and desorption of hydrogen. These findings were also corroborated by other studies on HEMG in alkaline electrolytes,24,25,29 while the discussion in acidic electrolytes remains limited.
Technically, a conformal and uniform NM-HEA coating, either crystalline or amorphous, can be achieved by atomic layer deposition (ALD) on a complex surface. ALD has been extensively applied to synthesize 2D/3D coatings with large active surface areas, serving as either substrates30–33 or catalysts34 for electrocatalytic HER. Owing to the intrinsic self-limiting growth, ALD can deposit thin films with precise thickness and high uniformity.35 Studies on surface evolution reveal that the thin film growth during the ALD process relies on the surface diffusion of atoms and smaller nanoparticles, followed by their growth.36–38 As such, the morphology of the deposited thin film usually exhibits nanoparticle-like features, which has been observed in numerous studies on ALD-made thin films.19,39–47 Therefore, the NM-HEA thin film deposited by ALD could expose an enlarged surface area. Nonetheless, the complexity of materials deposited by ALD is constrained by the limited deposition temperature window permissible for the deposition of various metals.48 Specifically, to deposit multi-element NM-HEA through a single straightforward ALD process, a fixed deposition temperature for all the constituent elements is required. However, to the best of our knowledge, this temperature has not been found among the reported noble metal ALD processes.49
In this study, a two-step method combining atomic layer deposition and electrical Joule heating (ALD-EJH) was developed to synthesize a RhRuPtPdIr HEA nanofilm on a glassy carbon substrate, as depicted in Fig. 1. A 20 nm five-layer Ir/Pd/Pt/Ru/Rh thin film was deposited on a glassy carbon electrode by sequential ALD processes of the selected noble metals, followed by 5 second EJH treatment at 1000 °C to achieve alloying. The feasibility of this method was verified in our previous works.50,51 The sub-surface area of HEA exhibited partial amorphization and nanocrystallites, which was caused by the explosive crystallization that occurred during the cooling phase of the EJH process.52,53 The ultrafast ramping/cooling rate also enabled the alloying of multiple layers without encountering phase separation and precipitation.54,55 Using the combined strategy, the limitation of ALD, as illustrated above, was overcome. The as-prepared HEA thin film demonstrated high activity and stability in all-pH environments, particularly in acidic electrolytes. This work highlights the potential of amorphized NM-HEA as a robust and efficient electrocatalyst for HER. Moreover, through this work, we expand the ALD synthesis to a broader array of materials containing more complex compounds and structures.
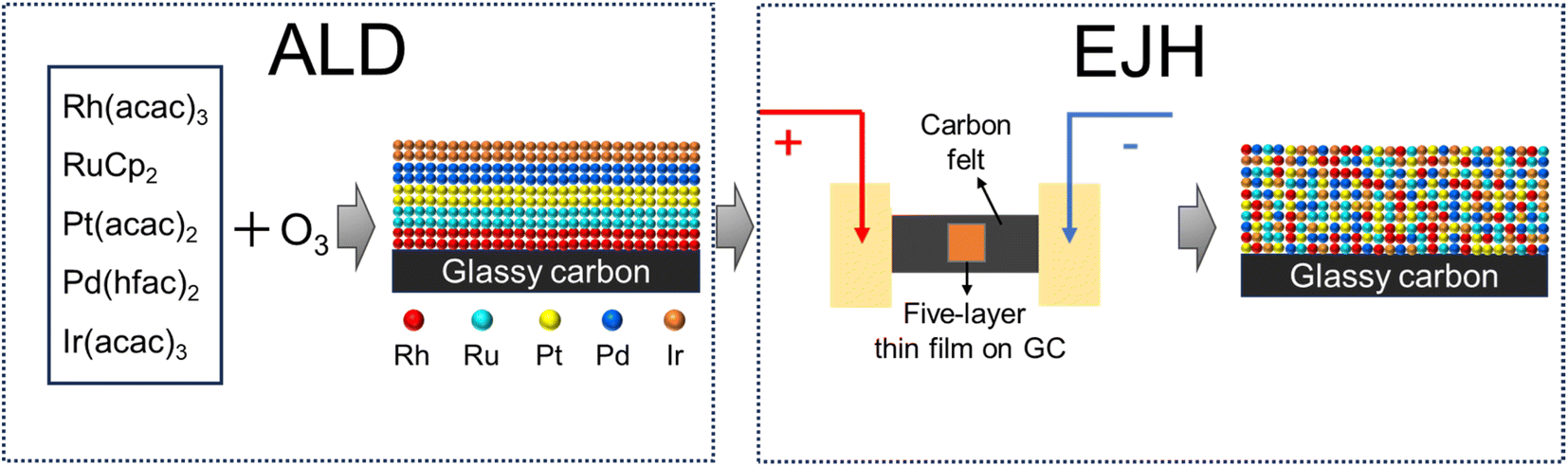 | ||
| Fig. 1 The schematic of synthesizing RhRuPtPdIr HEA/GC by the ALD/EJH method. | ||
2. Materials and methods
2.1. ALD of 5-layer Rh/Ru/Pt/Ir/Pd nanofilm
Sequential ALD processes of metallic Rh, Ru, Pt, Ir, and Pd were employed to deposit a five-layer Rh/Ru/Pt/Ir/Pd thin film on 1 × 1 cm2 glassy carbon substrates using a custom-built ALD system. Rh(acac)3 (97%, Sigma-Aldrich), RuCp2 (97%, Sigma-Aldrich), Pt(acac)2 (97%, Sigma-Aldrich), Ir(acac)3 (97%, Sigma-Aldrich), and Pd(hfac)2 (95%, Strem Chemicals) were used as precursors. Each precursor was stored separately in a dedicated bubbler. Prior to each ALD process, the bubbler, gas tube line, and reaction chamber were held at the temperatures, as shown in Table 1, to facilitate the precursor vaporization and reaction initiation. Ozone generated at 1.0 mbar partial pressure using an ozone generator (Nanofrontier, XLK-G20) was applied as the co-reactant for all the ALD processes. A continuous flow of 150 sccm high-purity nitrogen was applied as the purging gas to maintain a base pressure of 1.0 mbar within the reaction chamber and to eliminate the gaseous reactants and by-products after each pulse. Nitrogen was also used as the carrier gas and pulsed into the bubbler before each precursor pulse to assist in the delivery of the precursor from the bubbler to the reaction chamber.| Rh | Ru | Pt | Pd | Ir | |
|---|---|---|---|---|---|
| Bubbler temperature (°C) | 170 | 65 | 140 | 65 | 185 |
| Tube line temperature (°C) | 175 | 100 | 150 | 100 | 190 |
| Deposition temperature (°C) | 190 | 240 | 150 | 200 | 180 |
| Pulse length of precursor (s) | 2.0 | 0.5 | 2.0 | 0.5 | 2.0 |
| Pulse length of ozone (s) | 3 | 1 | 3 | 1 | 3 |
Before deposition, the glassy carbon substrates were sequentially washed with deionized water, ethanol, acetone, and isopropyl alcohol for 5 minutes in an ultrasonic bath. After drying using the nitrogen flow, the substrates were treated with 1 minute of oxygen plasma from a plasma gun (Piezobrush® PZ3, RELYON Plasma), to generate more anchoring sites on the surface for precursor adsorption.35,37 During the ALD process, vaporized precursor and ozone were alternately and repeatedly pulsed until the assigned cycle number was reached. The length of each step is provided in Table 1.
2.2. EJH of RhRuPtIrPd HEA thin film
The as-deposited 5-layer Rh/Ru/Pt/Ir/Pd thin film was fixed on the carbon felts, with their two ends connected to the electrodes, as shown in Fig. 1. By adjusting the electrical output power, the peak temperature could be controlled. The detailed setup and operational mechanism are illustrated elsewhere.50,54,55 The EJH process was conducted under low vacuum conditions (below 1.0 mbar). During the EJH process, the temperature was ramped up to 1000 °C and maintained for 5 seconds before the sample naturally cooled to ambient temperature. The peak temperature of the carbon felt were measured using a Raytek Raynger thermal infrared sensor.2.3. Characterization
A transmission electron microscope (TEM, JEM-2100F) operating at an accelerating voltage of 200 kV was used to obtain high-resolution TEM images and selected area electron diffraction (SAED) patterns of the cross sections of the HEA nanofilm. The elemental distributions were studied using high-angle annular dark-field scanning TEM (HAADF-STEM) and corresponding windowless energy dispersive X-ray (EDX) spectroscopy (Oxford EDX detector) on the same TEM. The cross-sectional lamellae for TEM imaging were prepared using a focused ion beam (FIB, Zeiss Crossbeam 540). The top-view morphology and structure were characterized using a field emission scanning electron microscope (FESEM, JEOL 7600F), and the atomic concentration of each noble metal element was measured by EDX spectroscopy (Oxford EDX detector). The elemental composition and chemical states were analyzed using an X-ray photoelectron spectrometer (XPS, Shimadzu Kratos Axis Supra) operating at 15 kV and 15 mA with Al Kα radiation. All spectra were calibrated with C 1s = 284.8 eV and fitted by the mixed 50% Gaussian–50% Lorentzian function. The crystal structure was analyzed by grazing incidence X-ray diffractometer XRD (GI-XRD, Bruker D8 Discover) with Cu Kα radiation source (λ = 1.54 Å).2.4. Electrochemical measurements
All electrochemical measurements were conducted with a three-electrode system on a CHI 750E electrochemical workstation at room temperature (24.0 °C). 0.5 M H2SO4, 1.0 M PBS, and 1.0 M KOH solutions were prepared as electrolytes for HER. A pristine glassy carbon (GC) substrate, five-layer/GC, and HEA/GC were clipped with a Pt electrode as the working electrode (1 × 0.5 cm2). A Pt plate was used as the counter electrode. An Ag/AgCl electrode was used as the reference electrode and was calibrated before use according to the ref. 56 All potentials in this work were referenced to a reversible hydrogen electrode (RHE). Linear sweep voltammetry (LSV) was conducted at a scan rate of 5 mV s−1. iR compensation was applied to the LSV test,57 where R is the solution resistance (Rs) obtained from electrochemical impedance spectroscopy (EIS) ranging from 0.1 to 105 Hz with an amplitude of 0.01 V at open circuit potential. Cyclic voltammetry (CV) curves were obtained from 0 to 0.1 V (vs. RHE) with sweep rates of 20, 40, 60, 80, and 100 mV s−1 to measure the electrochemical capacitance. To study the stability, chronoamperometry tests were conducted at an overpotential of 100 mV for a duration of 12 h.2.5. DFT calculations
Density functional theory (DFT) calculations were conducted using the Vienna ab initio simulation package (VASP).58,59 The exchange and correlation were described using the generalized gradient approximation (GGA) based on the Perdew, Burke, and Ernzerhof (PBE) functional.60 Specific parameters were adopted for precise calculations, including a plane wave cut-off energy of 400 eV, total energy convergence of 10−5 eV, and residual force convergence of 0.03 eV Å−1. The average adsorption free energy was obtained from the sampling tests at different adsorption sites. The adsorption free energy G was calculated from the equation G = Gtot − G* − Gas, where Gtot, G*, and Gas are the total free energy of the substrate with the adsorbed intermediate, free energy of the clean substrate, and free energy of the adsorbed species, respectively.For the crystalline Ir (5L) and HEA (RhRuPdPtIr), a 2 × 2 supercell of the (111) surface was built as the reaction substrate. The atomic number for each element was determined according to the EDX results (Fig. S3†). Atomic models were established using ab initio molecular dynamic (AIMD) simulations. For the generation of an amorphous structure, crystalline models were heated from 300 K to 3500 K with a 2 ps interval. The temperature was maintained at 3500 K for 5 ps using the Nosé–Hoover thermostat, with a time step of 2 fs. Subsequently, the sample was cooled back to 300 K within 0.5 ps. To minimize energy and stabilize the structure, the post-cooling model was equilibrated at 300 K for 1.0 ps. AIMD simulations were conducted at the Γ point.
3. Results and discussion
3.1. Materials and characterization
To investigate the elemental composition and morphology of the as-deposited five-layer Rh/Ru/Pt/Pd/Ir on glassy carbon (5L/GC) and HEA thin film after EJH treatment (HEA/GC), top-view field-emission scanning electron microscopy (FESEM) and energy dispersive X-ray (EDX) spectroscopy were performed. FESEM images shown in Fig. 2a–f elucidate the morphological evolution during the EJH process. The morphology of 5L/GC exhibits the typical island-coalesced pattern resulting from the diffusion and growth of clusters during the ALD processes. After the EJH process, the uniform and continuous ridge-and-valley morphology with a protrusion size of 6.5 ± 2.2 nm was observed. The formation of this morphology can be attributed to the rapid recrystallization and nucleation that occurred during the cooling phase of the EJH process. These protrusions also provide the additional active surface area for HER. According to EDX mappings (Fig. S1†), the presence of selected noble metals was verified, and the atomic ratio was determined to be Rh:Ru:Pt:Pd:Ir = 26.1:28.7:8.6:16.3:20.3.
 | ||
| Fig. 2 Top-view FESEM images of (a and b) pristine GC, (c and d) 5L/GC, and (e and f) HEA/GC. The morphology evolution from featureless surface to uniform coating, and to finalized ridges and valleys caused by ALD and EJH processes can be observed. | ||
The microstructure of the as-prepared HEA thin film was characterized using grazing-incidence X-ray diffraction (GI-XRD) patterns, as shown in Fig. 3a. A comparison of the XRD patterns of pristine GC, 5L/GC, and HEA/GC revealed no characteristic peaks associated with either crystalline RhRuPtPdIr HEA or its individual components. However, only the substrate peaks originating from GC were observed. The comparison implies the formation of amorphous phases or nanocrystallites within the HEA thin film. XRD patterns of HEA/GC at varying incidence angles (0.1, 0.3, and 0.5°) showed no discernible differences (Fig. S2†), indicating the uniform crystallinity throughout the film's depth and the diffusion of carbon into the thin film.
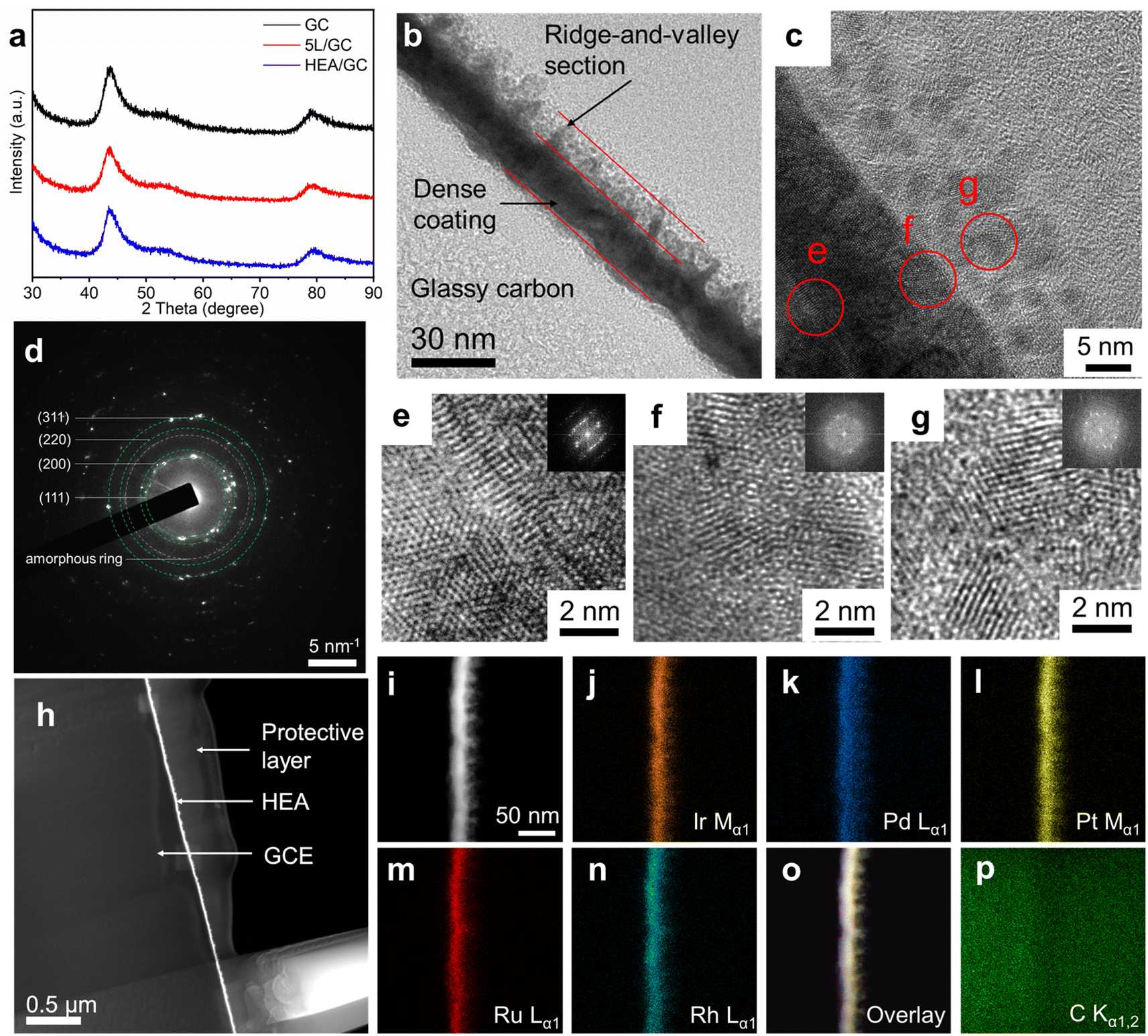 | ||
| Fig. 3 (a) GI-XRD patterns of GC, 5L/GC, and HEA/GC; (b) cross-sectional HR-TEM image of HEA/GC. The GC substrate, dense coating, and the ridge-and-valley section can be distinguished according to the brightness difference; (c) HR-TEM image of the HEA/GC sub-surface area, (d) corresponding SAED pattern, and (e–g) magnified images of sections e, f, and g. Inserts are FFT patterns; (h) STEM-HAADF images of HEA/GC. Except for GC substrate and HEA coating, a carbon layer was deposited by FIB to prevent damage from the etching and polishing steps during the TEM lamellae preparation; (i–p) cross-sectional STEM-HAADF-EDX mappings of Rh, Ru, Pt, Pd, Ir and their overlays, and C of HEA/GC. The distribution of noble metals is homogeneous throughout the thin film. | ||
High-resolution transmission electron microscopy (HR-TEM) was performed to characterize the microstructure of the cross-section of HEA/GC. As shown in Fig. 3b, the thickness of the HEA thin film was directly measured at 20 ± 5 nm, comprising two sections: the dense coating and the ridge-and-valley topography. This observation aligns with the findings from FESEM images, as shown in Fig. 2. Fig. 3c shows a magnified cross-sectional image of the sub-surface area of the as-prepared HEA/GC after fine polishing. This revealed that the close-arranged crystalline globules, measuring approximately 5 nm in size, constitute the ridge-and-valley section. The corresponding selected area electron diffraction (SAED) pattern shown in Fig. 3d displays diffraction rings corresponding to (111), (200), (220), and (311) planes, suggesting the crystalline face-centered cubic (fcc) structure with a lattice constant of 0.406 nm. To analyze the atomic arrangement within each section, specific HR-TEM images of the dense coating, interface between coating and globules, and globules are retrieved from the highlighted area (e, f, and g) in Fig. 3d, presented in Fig. 3e–g, respectively. The crystalline structure and ordered atomic arrangement are identified in the deep section of the dense coating (Fig. 3e), whereas, the highly disordered atomic arrangement was found at the coating-globule interface (Fig. 3f). Above the interface, as shown in Fig. 3g, the 5 nm globules are composed of smaller crystallites with the average grain size of 2 nm, surrounded by amorphous structures. The inserted FFT patterns also support the various amorphization at different depths of the thin film. These findings explain the absence of characteristic peaks in the XRD patterns, which is attributed to low crystallinity and small grain size. Such nano-crystallization and amorphization are likely a result of liquid phase nucleation during explosive crystallization when the heat is rapidly extracted during the cooling phase of the EJH process.52,53
To further confirm the formation of HEA and the distribution of elements within the thin film, high-angle annular dark-field scanning TEM (HAADF-STEM) and EDX were performed. As shown in Fig. 3i–p, the color-highlighted regions representing selected noble metal elements (i.e., Rh, Ru, Pt, Pd, and Ir) are highly overlapped, providing strong evidence for the homogeneous distribution of these elements throughout the thin film. The atomic ratio, as determined from the EDX spectrum (Fig. S3†) was Rh:Ru:Pt:Pd:Ir = 26.5:30.1:7.8:12.1:23.5, which is basically consistent with top-view EDX analysis.
To investigate the chemical states of the noble metal elements in the HEA thin film, X-ray photoelectron spectroscopy (XPS) was conducted with calibration using adventitious C–C at 284.8 eV. According to the wide scan covering a binding energy range from 0 to 1200 eV (Fig. 4a), all noble metals were confirmed from their characteristic peaks. To resolve the highly overlapping peaks of C, O, Rh, Ru, Pt, Pd, and Ir, deconvolution was applied to the fine-scan spectra shown in Fig. 4b–f. In the Rh 3d spectrum, apart from the asymmetric components assigned to metallic Rh at 307.2/312.0 eV and Rh–O at 308.1/312.9 eV, wide peaks of Pt 4d5/2 and Ir 4d3/2 were identified at 315.0 and 313.2 eV, respectively. In the spectra of Ru 3d, Pt 4f, Pd 3d, and Ir 4f, as shown in Fig. 4c–f, similar doublets representing the separate spin–orbit components were deconvoluted as the combination of metallic states and metal–oxygen bindings. To elaborate, the binding energies of metallic Ru 3d, Pt 4f, Pd 3d, and Ir 4f electrons were measured at 280.0/284.3, 71.2/74.5, 335.4/340.7, and 60.7/63.7 eV, while the corresponding peaks representing metal–oxygen binding were identified at 280.5/284.7, 72.1/75.4, 336.3/341.8, and 61.4/64.5 eV, respectively. The presence of oxygen on the surface of the HEA/GC could result from either the limited vacuum conditions during the EJH process or the exposure to ambient air during storage (adventitious adsorbed oxygen). In addition to adventitious carbon and oxygen, adventitious hydroxyl was identified at 531.9 eV in the O 1s spectrum.61,62 In brief, the HEA/GC surface is primarily composed of metallic states of all deposited noble metals, with a small fraction of the adsorbed contaminations.
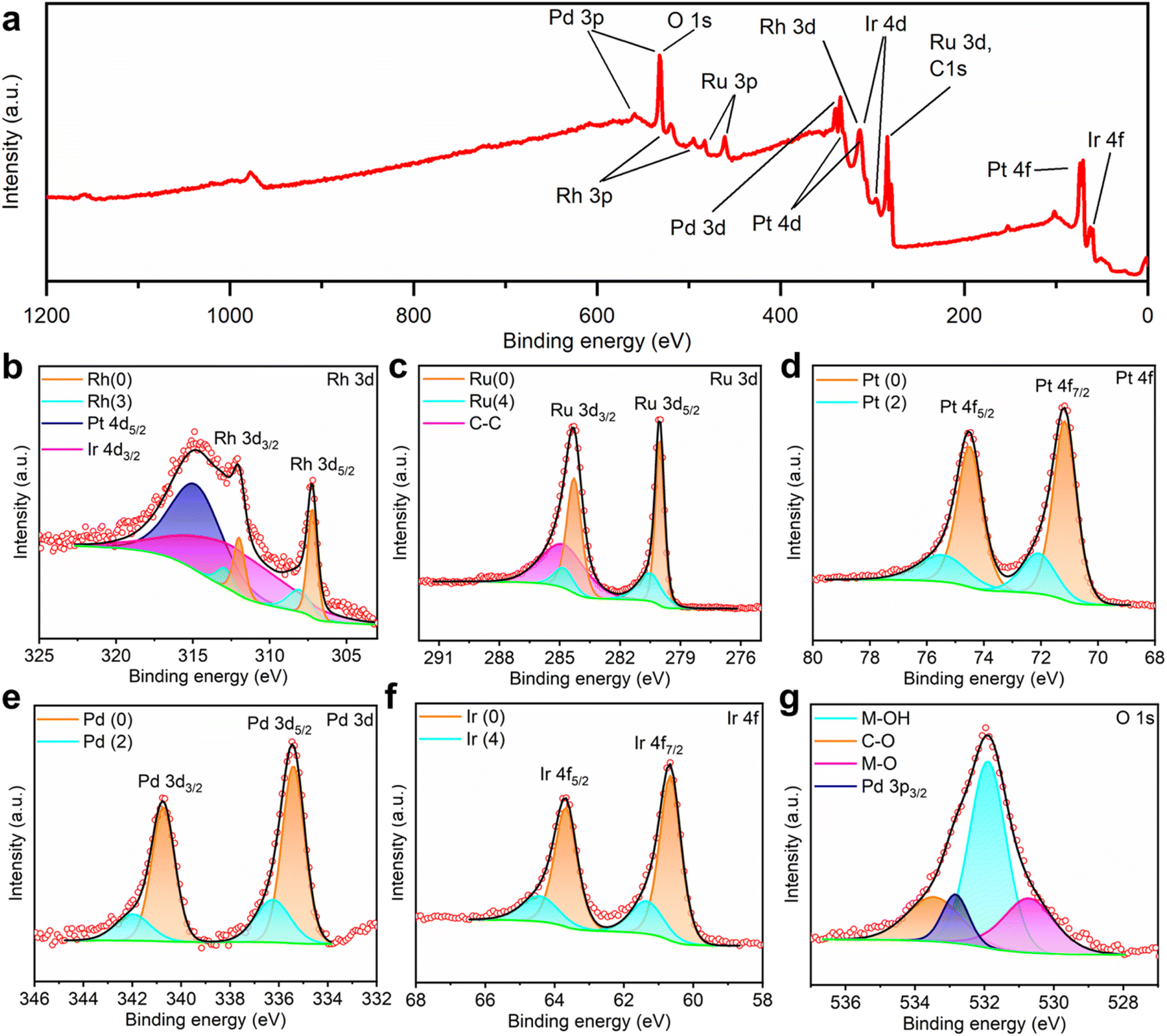 | ||
| Fig. 4 (a) Wide XPS spectrum in a binding energy range from 0 to 1200 eV. (b–g) XPS spectra and corresponding deconvoluted profiles of Rh 3d, Ru 3d, Pt 4f, Pd 3d, Ir 4f, and O 1s spectra. | ||
3.2. Electrochemical performance
To assess the catalytic activity of HEA/GC towards HER, linear sweep voltammetry (LSV) tests were conducted in acidic (0.5 M H2SO4 solution, pH = 0.28), neutral (1.0 M PBS solution, pH = 7.40), and alkaline (1.0 KOH solution, pH = 13.92) medium using a three-electrode system at a scan rate of 5 mV s−1. iR correction was applied based on electrochemical impedance spectroscopy (EIS) results (Fig. S5†). As a comparison, GC and 5L/GC were also tested. In 0.5 M H2SO4 solution, 5L/GC and HEA/GC exhibited overpotentials of 30 and 13 mV at a current density of 10 mA cm−2, respectively (Fig. 5a). To further study the HER kinetics, Tafel plots were extracted from the corresponding LSV curves and fitted using the Tafel equation (η = blogj + a). As shown in Fig. 5b, the Tafel slopes of GC, 5L/GC, and HEA/GC are 153, 31, and 14 mV dec−1, respectively. The remarkably low Tafel slope of HEA/GC indicates the high HER kinetics and efficient hydrogen production in an acidic environment.63 Compared with the other noble metal-based catalysts listed in Fig. 6 and Table S2,† RhRuPtPdIr HEA/GC showed top-level HER performance, particularly in acidic environment.8,9,24,25,64–70 To investigate the impact of electrochemically active surface area (ECSA) on HER activity, electrochemical double-layer capacitances (Cdl) were extracted from cyclic voltammetry (CV) curves (Fig. S4†). The capacitance of GC was 0.40 mF cm−2, whereas the capacitances of 5L/GC and HEA/GC were 0.91 and 0.91 mF cm−2, respectively, which increased by 127.5% (Fig. 5c). The boosting capacitances observed for both 5L/GC and HEA/GC provide evidence of enhanced ECSA resulting from the surface modification through the ALD and EJH processes. Notably, the capacitances of 5L/GC and HEA/GC were identical, whereas, the overpotential of HEA/GC was less than half of that of 5L/GC. This result suggests that the superior HER performance of HEA/GC is contributed not only by the enlargement of ECSA but also by its intrinsic activity.
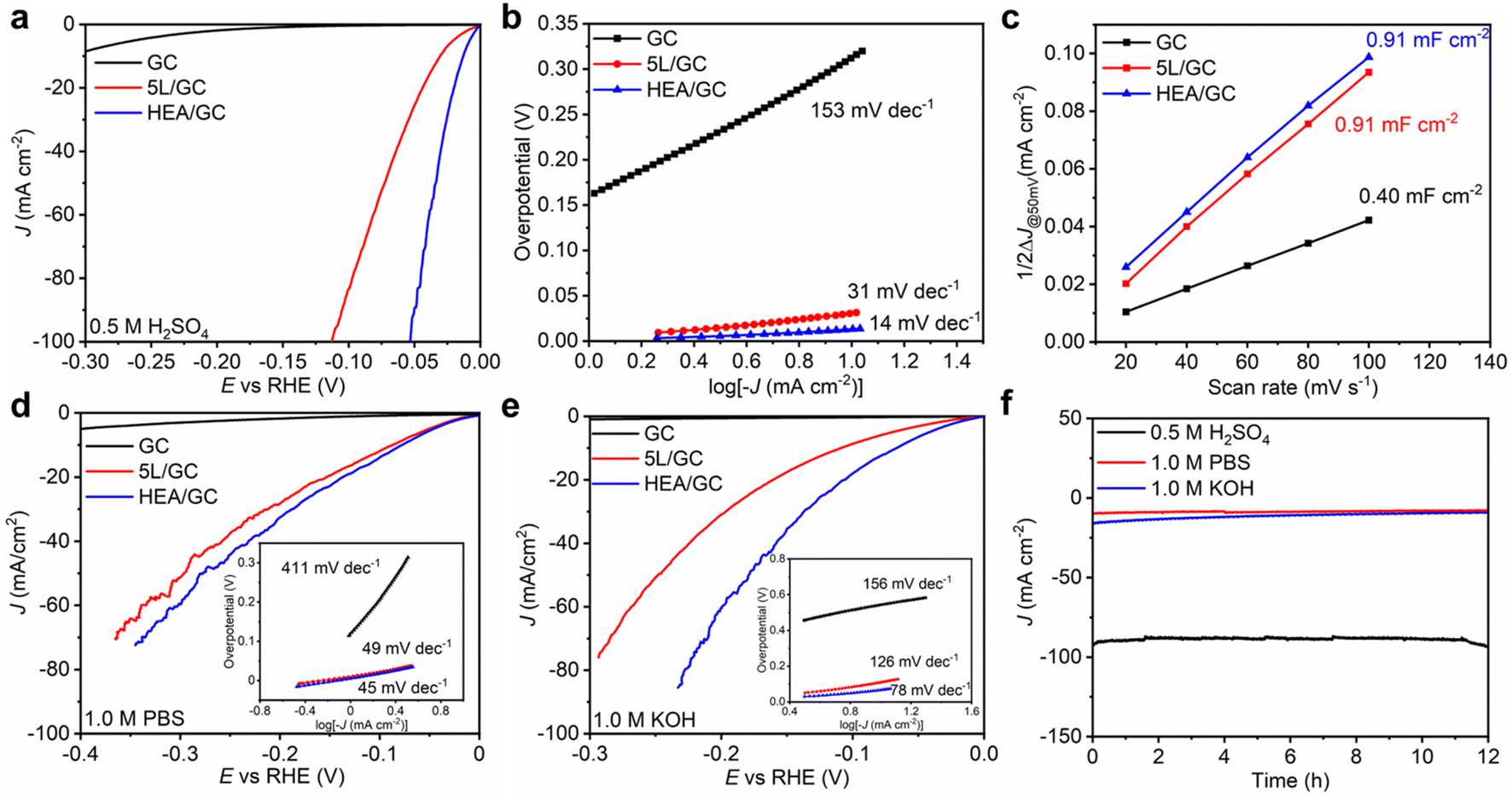 | ||
| Fig. 5 Electrochemical performances of GC, 5L/GC, and HEA/GC measured in acidic, neutral, and alkaline solutions. (a) LSV curves in 0.5 M H2SO4, (b) Tafel slopes in 0.5 M H2SO4, and (c) the capacitive current density ΔJ at 0.05 V as a function of the scan rate in the range of 0 to 0.1 V vs. RHE in 0.5 M H2SO4. (d) LSV curves in 1.0 M PBS (inserts: Tafel slopes) and (e) 1.0 M KOH solutions (insets: Tafel slopes). (f) i–t curves of HEA/GC at an overpotential of −100 mV vs. RHE for 12 hours in 0.5 M H2SO4, 1.0 PBS, and 1.0 M KOH electrolytes. | ||
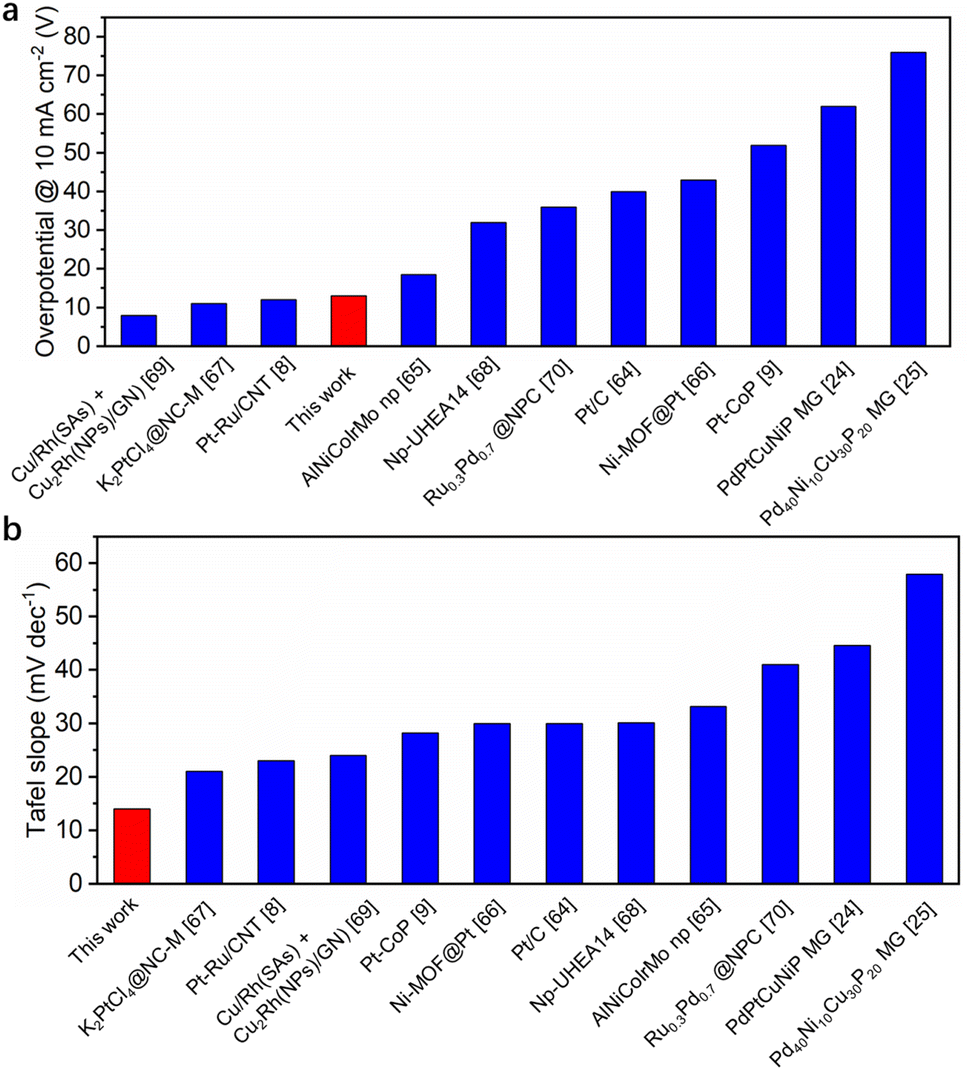 | ||
| Fig. 6 Comparison of HER overpotential at 10 mA cm−2 and Tafel slope in 0.5 M H2SO4 for best-performing noble-metal-based catalysts. | ||
In 1.0 M PBS and 1.0 M KOH electrolytes, the HEA/GC catalyst exhibited overpotentials of 77 mV and 65 mV, and Tafel slopes of 45 mV dec−1 and 78 mV dec−1, respectively. In contrast, the 5L/GC catalyst exhibited overpotentials of 85 mV and 107 mV, and 49 mV dec−1 and 126 mV dec−1 in the corresponding environments (Fig. 5d and e). When analyzing the LSV curves and Tafel plots of HEA/GC and 5L/GC, the HEA catalyst displayed improved HER kinetics in alkaline electrolytes, while no significant enhancement was observed in neutral solutions. The EIS measurements showed reduced solution resistance (Rs) and charge-transfer resistance (Rct) for HEA/GC, suggesting higher charge-transfer properties and HER kinetics (Fig. S5†). In alkaline electrolytes, the activity and stability were enhanced by HEA, but barely comparable to other works (Table S3†).
To examine the synergistic effect of RhRuPtPdIr HEA, the HER performances of quaternary RhRuPdIr and RhRuPtPd alloy thin films were investigated and compared. By simply removing either the Pt or Ir ALD process from sequential deposition, quaternary HEAs were obtained, without alternating the atomic ratios of the other four elements. The LSV curves and Tafel slopes of the three materials are presented in Fig. S6a and b.† RhRuPdIr and RhRuPtPd exhibit overpotentials of 20 and 16 mV at 10 mA cm−2, along with Tafel slopes of 25 and 19 mV dec−1, respectively. The HER performance of the quaternary alloys does not surpass RhRuPtPdIr HEA but outperforms that of 5L/GC. Such results demonstrate that the synergistic function of Pt and Ir atoms plays an essential role in enhancing HER activity.
Following electrochemical experiments, both 5L/GC and HEA/GC were characterized by FESEM and EDX to examine the possible changes in catalysts. As depicted in Fig. 7, the morphology of both 5L/GC and HEA/GC showed no obvious changes, compared with the FESEM images of the as-prepared samples. However, the EDX results revealed a substantial change in the chemical composition of 5L/GC. Although the atomic ratio between Rh and Pt remained unchanged, other noble metals, including Pd, Ru, and Ir were eliminated. In contrast, HEA/GC retained all the selected noble metals and their atomic ratios, while the sample was contaminated with elements from the electrolytes, such as K and S. The characterization results demonstrate the exceptional stability of HEA/GC across a wide range of pH environments.
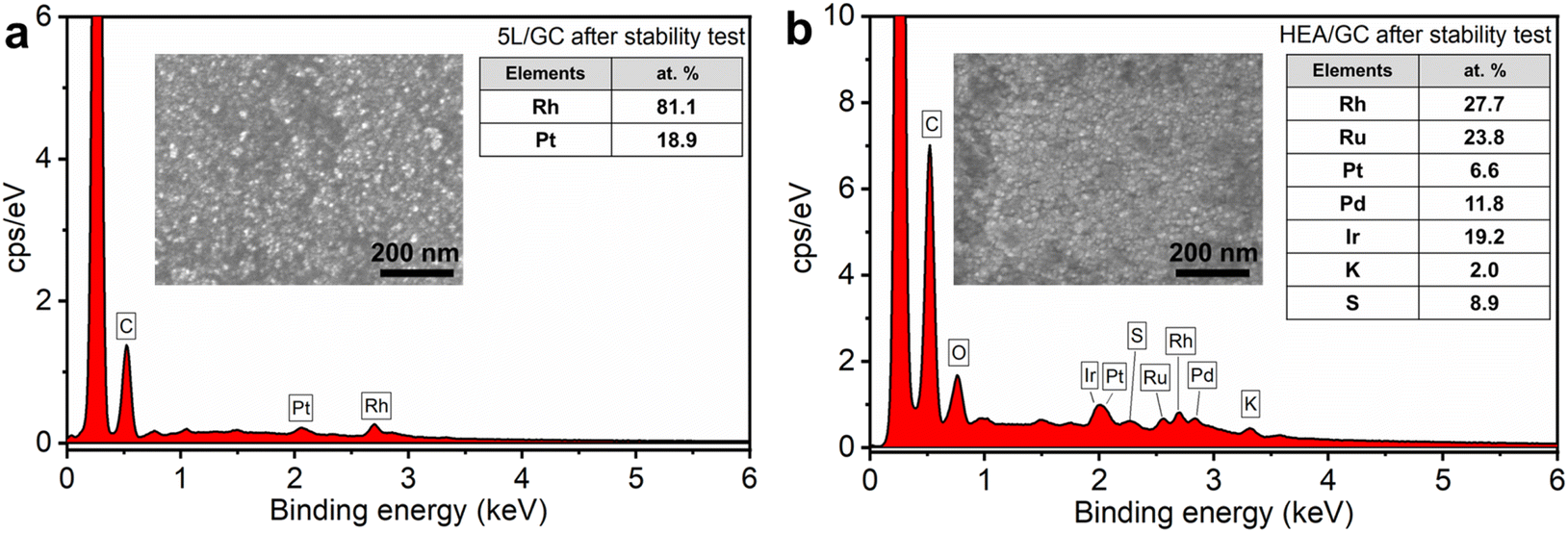 | ||
| Fig. 7 EDX spectra of (a) 5L/GC and (b) HEA/GC after the stability test inserted with corresponding FESEM images. | ||
3.3. DFT calculation results
Density functional theory (DFT) calculations were employed to study the mechanisms accounting for the enhanced HER performance of HEA (Rh:Ru:Pt:Pd:Ir = 12:18:18:10:6). Fig. 8a presents the models of amorphous, crystalline HEA, and Ir layers, corresponding to amorphous and crystalline sites on the HEA surface after the EJH process, and the 5L/GC surface before the EJH process. Their derived structures were also subjected to thermodynamic stability tests. The energy fluctuation within 0.008 eV validates the structures stability at 300 K for 2 ps (Fig. S7†).
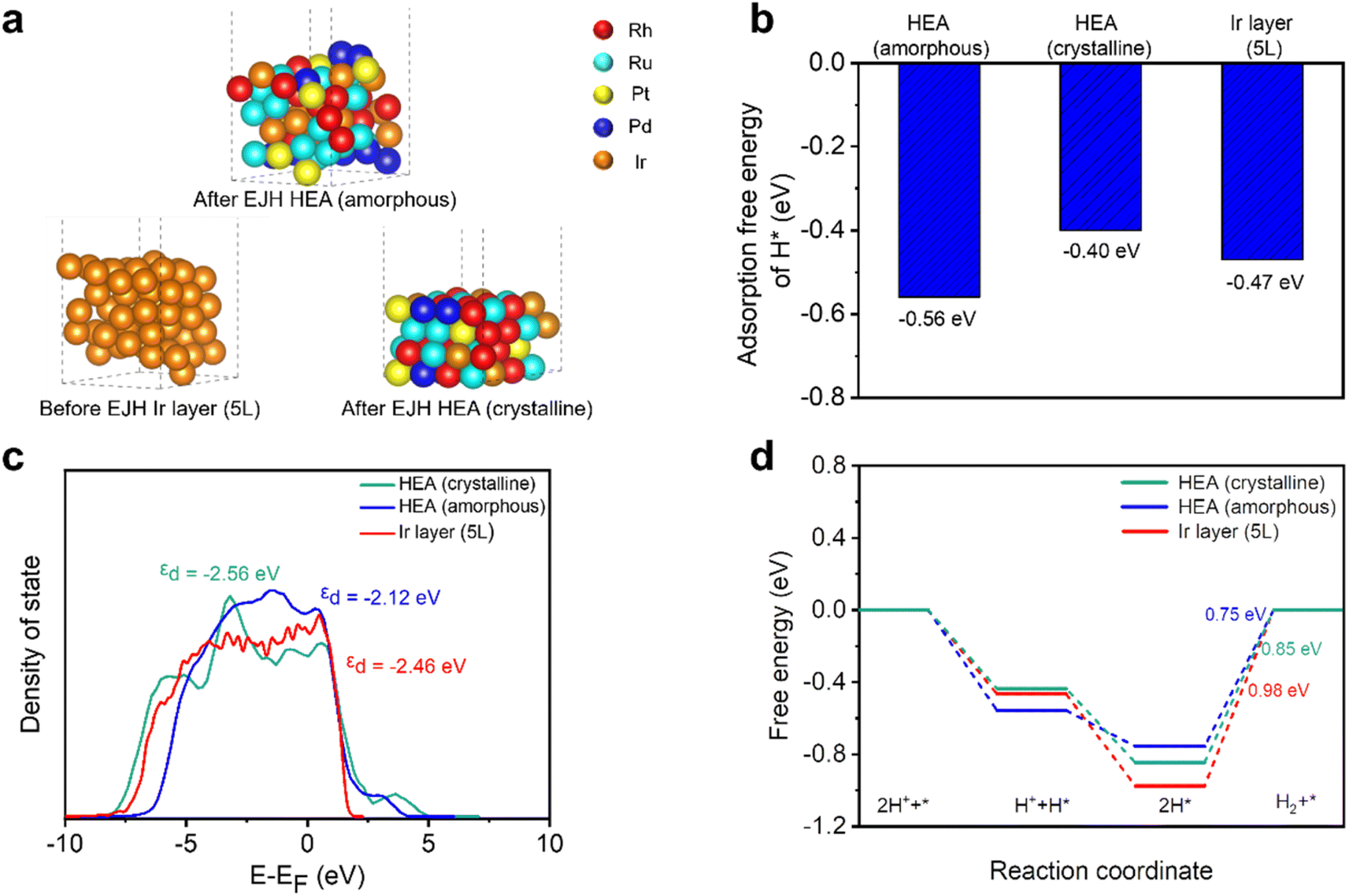 | ||
| Fig. 8 DFT computation results, including (a) calculated structures, (b) average H* adsorption free energy, (c) total DOS, and (d) HER reaction free energy diagram of amorphous HEA, crystalline HEA, and Ir layer (5L). | ||
As shown in Fig. 8b, amorphous HEA has a more negative adsorption free energy of H* (−0.56 eV) than crystalline HEA (−0.40 eV) and the Ir layer (−0.47 eV), which can be attributed to the changes in d-band structure. As depicted by the DOS profiles of the three models in Fig. 8c, amorphous HEA possesses a smoother DOS profile than those of the other two models, reflecting the various atomic configurations and low degeneracy of the energy level.23 The calculated d-band center (εd) of the amorphous HEA, crystalline HEA, and Ir layers are −2.12, −2.56, and −2.46 eV, respectively. The εd value of amorphous HEA approaches that of the Fermi level, indicating the increase in electronic activity and enhancement in the interaction between catalysts and H*.71,72 Additionally, the DOS profile of amorphous HEA (Fig. 8c) shows larger electron density near the Fermi level compared to crystalline HEA and the Ir layer (5L). According to the projected partial density of states (PDOS), as shown in Fig. S8,† the peaks associated with Rh, Ru, Pd, Pt, and Ir in amorphous HEA shift towards the Fermi energy level, suggesting the electronic structure modification of individual metals through amorphization.
Compared with the strong adsorption of H* on the HEA surface, the desorption of H2 was not significantly hindered on surfaces because the adsorption free energy of 2H* was lower than that of two individual H* (Fig. 8d). The Tafel slope of nearly 30 mV dec−1, as depicted in Fig. 5c, suggests that the catalysis predominantly proceeds via the Volmer–Tafel reaction mechanism on both the HEA and Ir layer (5L) catalysts.73 In this pathway, the adsorbed H* combines with the adjacent H* to form H2 molecules. HEA is inclined to have weaker adsorption of 2H* than the Ir layer (5L). Consequently, amorphous (0.75 eV) and crystalline (0.85 eV) HEA have lower thermodynamic barriers than the Ir layer (5L) (0.98 eV) at the subsequent H2 desorption step, which is the limiting step of the entire catalytic reaction.
In summary, even though crystalline HEA has a weaker interaction with hydrogen, than the Ir (5L) surface, the electronic structure can be modified through surface amorphization. Through a DFT study of the d-band structure and adsorption/desorption free energy, the superior HER catalytic activity at the amorphous sites on the HEA surface is demonstrated.
4. Conclusion
A RhRuPtIrPd noble metal-based high entropy alloy (NM-HEA) thin film with surface amorphization was synthesized on glassy carbon using a combined ALD-EJH method. GI-XRD, EDX, TEM, and XPS were employed to characterize the composition, microstructure, and morphology of the HEA/GC catalyst. The ridge-and-valley surface consisting of nanocrystallites embedded in an amorphous structure provided a modified surface for hydrogen adsorption. The as-prepared HEA/GC catalyst demonstrated remarkable activity, exhibiting an overpotential of 13 mV at 10 mA cm−2 and ultra-low Tafel slope of 14 mV dec−1 in an acidic electrolyte, which is primarily due to the intrinsic activity of the HEA material and surface modification. As for the durability, the catalyst showed no current density decline in the 12 h i–t test. In neutral and alkaline solutions, although an enhancement in HER kinetics was observed for the HEA/GC, the HER performance was not as good due to sluggish reactions. This work underscores the significance of the unique properties intrinsic to the NM-HEA, further validating its potential as an exceptional catalyst. This work also introduces a novel method that combines ALD with EJH to synthesize multi-element nanomaterials, significantly expanding the range of materials that can be prepared using ALD and exploring the potential of ALD application in the electrocatalysis field.Author contributions
Yiming Zou: methodology, investigation, formal analysis, data curation, writing – original draft. Lin Jing: investigation, project administration, data curation. Jianghong Zhang: computations, software. Songzhu Luo, Leyan Wang, Yun Li, and Ronn Goei: investigation, data curation. Kwan W. Tan: writing – review and editing. Alfred Iing Yoong Tok: supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agency for Science, Technology, and Research (A*STAR) AME Individual Research Grant (IRG) A1983c0032. K. W. T. and L. W. acknowledge financial support from the Singapore Ministry of Education AcRF Tier 2 grant (MOET2EP 50221-0017). We are thankful for computing resources from the National Supercomputing Centre (NSCC) Singapore.References
- S. Wang, A. Lu and C.-J. Zhong, Nano Convergence, 2021, 8, 1–23, DOI:10.1186/s40580-021-00254-x.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65, DOI:10.1002/anie.201407031.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998, DOI:10.1126/science.aad4998.
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li and H. Fei, Nat. Catal., 2019, 2, 495–503, DOI:10.1038/s41929-019-0279-6.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, Nat. Commun., 2021, 12, 3502, DOI:10.1038/s41467-021-23750-4.
- J. Lu, Z. Tang, L. Luo, S. Yin, P. K. Shen and P. Tsiakaras, Appl. Catal., B, 2019, 255, 117737, DOI:10.1016/j.apcatb.2019.05.039.
- S. L. Zhang, X. F. Lu, Z. P. Wu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 19068–19073, DOI:10.1002/anie.202106547.
- D. Zhang, Z. Wang, X. Wu, Y. Shi, N. Nie, H. Zhao, H. Miao, X. Chen, S. Li and J. Lai, Small, 2022, 18, 2104559, DOI:10.1002/smll.202104559.
- Y.-N. Zhou, X. Liu, C.-J. Yu, B. Dong, G.-Q. Han, H.-J. Liu, R.-Q. Lv, B. Liu and Y.-M. Chai, J. Mater. Chem. A, 2023, 11, 6945–6951, 10.1039/D2TA09784B.
- Y. Xin, S. Li, Y. Qian, W. Zhu, H. Yuan, P. Jiang, R. Guo and L. Wang, ACS Catal., 2020, 10, 11280–11306, DOI:10.1021/acscatal.0c03617.
- Y. Yao, Z. Liu, P. Xie, Z. Huang, T. Li, D. Morris, Z. Finfrock, J. Zhou, M. Jiao and J. Gao, Sci. Adv., 2020, 6, eaaz0510, DOI:10.1126/sciadv.aaz0510.
- X. Huang, G. Yang, S. Li, H. Wang, Y. Cao, F. Peng and H. Yu, J. Energy Chem., 2022, 68, 721–751, DOI:10.1016/j.jechem.2021.12.026.
- Y. Zhai, X. Ren, B. Wang and S. Liu, Adv. Funct. Mater., 2022, 32, 2207536, DOI:10.1002/adfm.202207536.
- M.-H. Tsai and J.-W. Yeh, Mater. Res. Lett., 2014, 2, 107–123, DOI:10.1080/21663831.2014.912690.
- G. Zhang, K. Ming, J. Kang, Q. Huang, Z. Zhang, X. Zheng and X. Bi, Electrochim. Acta, 2018, 279, 19–23, DOI:10.1016/j.electacta.2018.05.035.
- E. P. George, D. Raabe and R. O. Ritchie, Nat. Rev. Mater., 2019, 4, 515–534, DOI:10.1038/s41578-019-0121-4.
- S. Gao, S. Hao, Z. Huang, Y. Yuan, S. Han, L. Lei, X. Zhang, R. Shahbazian-Yassar and J. Lu, Nat. Commun., 2020, 11, 2016, DOI:10.1038/s41467-020-15934-1.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi and O. Sakata, Chem. Sci., 2020, 11, 12731–12736, 10.1039/D0SC02351E.
- L. Jing, Y. Zou, R. Goei, L. Wang, J. A. Ong, A. Kurkin, Y. Li, K. W. Tan and A. I. Y. Tok, Langmuir, 2023, 39, 3142–3150, DOI:10.1021/acs.langmuir.2c03367.
- M. Wei, Y. Sun, Z. Junyu, F. Ai, S. Xi and J. Wang, Energy Environ. Sci., 2023, 16, 4009–4019, 10.1039/D3EE01929B.
- T. A. Batchelor, J. K. Pedersen, S. H. Winther, I. E. Castelli, K. W. Jacobsen and J. Rossmeisl, Joule, 2019, 3, 834–845, DOI:10.1016/j.joule.2018.12.015.
- Y. Maruta, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, S. Yasuno, S. Kawaguchi and O. Sakata, Chem. Commun., 2022, 58, 6421–6424, 10.1039/D2CC01866G.
- D. Wu, K. Kusada, Y. Nanba, M. Koyama, T. Yamamoto, T. Toriyama, S. Matsumura, O. Seo, I. Gueye and J. Kim, J. Am. Chem. Soc., 2022, 144, 3365–3369, DOI:10.1021/jacs.1c13616.
- Z. Jia, K. Nomoto, Q. Wang, C. Kong, L. Sun, L. C. Zhang, S. X. Liang, J. Lu and J. J. Kruzic, Adv. Funct. Mater., 2021, 31, 2101586, DOI:10.1002/adfm.202101586.
- Y. C. Hu, Y. Z. Wang, R. Su, C. R. Cao, F. Li, C. W. Sun, Y. Yang, P. F. Guan, D. W. Ding and Z. L. Wang, Adv. Mater., 2016, 28, 10293–10297, DOI:10.1002/adma.201603880.
- M. W. Glasscott, A. D. Pendergast, S. Goines, A. R. Bishop, A. T. Hoang, C. Renault and J. E. Dick, Nat. Commun., 2019, 10, 2650, DOI:10.1038/s41467-019-10303-z.
- X. Zhang, Y. Yang, Y. Liu, Z. Jia, Q. Wang, L. Sun, L. C. Zhang, J. J. Kruzic, J. Lu and B. Shen, Adv. Mater., 2023, 35, 2303439, DOI:10.1002/adma.202303439.
- Z. Jia, Y. Yang, Q. Wang, C. Kong, Y. Yao, Q. Wang, L. Sun, B. Shen and J. J. Kruzic, ACS Mater. Lett., 2022, 4, 1389–1396, DOI:10.1021/acsmaterialslett.2c00371.
- S. Ju, J. Feng, P. Zou, W. Xu, S. Wang, W. Gao, H.-J. Qiu, J. Huo and J.-Q. Wang, J. Mater. Chem. A, 2020, 8, 3246–3251, 10.1039/C9TA11867E.
- Z. Jin, S. Shin, W.-S. Lee and Y.-S. Min, Nanoscale, 2016, 8, 7180–7188, 10.1039/C5NR09065B.
- Y. Kim, D. H. Jackson, D. Lee, M. Choi, T. W. Kim, S. Y. Jeong, H. J. Chae, H. W. Kim, N. Park and H. Chang, Adv. Funct. Mater., 2017, 27, 1701825, DOI:10.1002/adfm.201701825.
- Y. Cao, Y. Wu, C. m. Badie, S. p. Cadot, C. m. Camp, E. A. Quadrelli and J. Bachmann, ACS Omega, 2019, 4, 8816–8823, DOI:10.1021/acsomega.9b00322.
- S. Jiao, M. Kong, Z. Hu, S. Zhou, X. Xu and L. Liu, Small, 2022, 18, 2105129, DOI:10.1002/smll.202105129.
- W. Xiong, Q. Guo, Z. Guo, H. Li, R. Zhao, Q. Chen, Z. Liu and X. Wang, J. Mater. Chem. A, 2018, 6, 4297–4304, 10.1039/C7TA10202J.
- J. Lu, J. W. Elam and P. C. Stair, Surf. Sci. Rep., 2016, 71, 410–472, DOI:10.1016/j.surfrep.2016.03.003.
- F. Grillo, H. Van Bui, J. A. Moulijn, M. T. Kreutzer and J. R. Van Ommen, J. Phys. Chem. Lett., 2017, 8, 975–983, DOI:10.1021/acs.jpclett.6b02978.
- J. Lu, Acc. Mater. Res., 2022, 3, 358–368, DOI:10.1021/accountsmr.1c00250.
- Y. Yao, B. Chen and F. Zaera, Chem. Mater., 2023, 35, 2155–2164, DOI:10.1021/acs.chemmater.2c03789.
- T. Aaltonen, M. Ritala, T. Sajavaara, J. Keinonen and M. Leskelä, Chem. Mater., 2003, 15, 1924–1928, DOI:10.1021/cm021333t.
- T. Aaltonen, P. Alen, M. Ritala and M. Leskelä, Chem. Vap. Depos., 2003, 9, 45–49, DOI:10.1002/cvde.200290007.
- T. Aaltonen, M. Ritala, Y.-L. Tung, Y. Chi, K. Arstila, K. Meinander and M. Leskelä, J. Mater. Res., 2004, 19, 3353–3358, DOI:10.1557/JMR.2004.0426.
- T. Aaltonen, M. Ritala, V. Sammelselg and M. Leskelä, J. Electrochem. Soc., 2004, 151, G489, DOI:10.1149/1.1761011.
- J. Hämäläinen, M. Kemell, F. Munnik, U. Kreissig, M. Ritala and M. Leskelä, Chem. Mater., 2008, 20, 2903–2907, DOI:10.1021/cm7030224.
- M. J. Weber, A. J. Mackus, M. A. Verheijen, V. Longo, A. A. Bol and W. M. Kessels, J. Phys. Chem. C, 2014, 118, 8702–8711, DOI:10.1021/jp5009412.
- M. Kozodaev, Y. Lebedinskii, A. Chernikova, E. Korostylev, A. Chouprik, R. Khakimov, A. M. Markeev and C. Hwang, J. Chem. Phys., 2019, 151, 204701, DOI:10.1063/1.5107509.
- Y. Zou, C. Cheng, Y. Guo, A. J. Ong, R. Goei, S. Li and A. I. Y. Tok, RSC Adv., 2021, 11, 22773–22779, 10.1039/D1RA03942C.
- Y. Zou, J. Li, C. Cheng, Z. Wang, A. J. Ong, R. Goei, X. Li, S. Li and A. I. Y. Tok, Thin Solid Films, 2021, 738, 138955, DOI:10.1016/j.tsf.2021.138955.
- M. C. Gao, J.-W. Yeh, P. K. Liaw and Y. Zhang, High-Entropy Alloys: Fundamentals and Applications, Springer, 2016 Search PubMed.
- J. Hämäläinen, M. Ritala and M. Leskelä, Chem. Mater., 2014, 26, 786–801, DOI:10.1021/cm402221y.
- Y. Guo, Y. Zou, C. Cheng, L. Wang, R. I. Made, R. Goei, K. W. Tan, S. Li and A. I. Y. Tok, Sci. Rep., 2022, 12, 2522, DOI:10.1038/s41598-022-06595-9.
- Y. Zou, L. Jing, L. Wang, H. T. Tan, R. Goei, S. K. R. Yong, A. J. Ong, K. W. Tan and A. I. Y. Tok, Adv. Eng. Mater., 2023, 25, 2201263, DOI:10.1002/adem.202201263.
- M. O. Thompson, G. Galvin, J. Mayer, P. Peercy, J. Poate, D. Jacobson, A. Cullis and N. Chew, Phys. Rev. Lett., 1984, 52, 2360, DOI:10.1103/PhysRevLett.52.2360.
- G. C. Egan, T. W. Heo, A. Samanta and G. H. Campbell, Acta Mater., 2019, 179, 190–200, DOI:10.1016/j.actamat.2019.08.026.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu and M. Rehwoldt, Science, 2018, 359, 1489–1494, DOI:10.1126/science.aan5412.
- L. Wang, G. L. Seah, Y. Li, W. H. Tu, W. Manalastas Jr, M. J. H. Reavley, E. W. Corcoran Jr, A. K. Usadi, Z. Du and S. Madhavi, Adv. Mater. Interfaces, 2022, 9, 2200151, DOI:10.1002/admi.202200151.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296, DOI:10.1002/adma.201806296.
- S. Anantharaj and S. Noda, J. Mater. Chem. A, 2022, 10, 9348–9354, 10.1039/D2TA01393B.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169, DOI:10.1103/PhysRevB.54.11169.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758, DOI:10.1103/PhysRevB.59.1758.
- H. Idriss, Surf. Sci., 2021, 712, 121894, DOI:10.1016/j.susc.2021.121894.
- J. Landoulsi, M. J. Genet, S. Fleith, Y. Touré, I. Liascukiene, C. Méthivier and P. G. Rouxhet, Appl. Surf. Sci., 2016, 383, 71–83 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801, DOI:10.1038/srep13801.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204, DOI:10.1038/ncomms11204.
- Z. Jin, J. Lv, H. Jia, W. Liu, H. Li, Z. Chen, X. Lin, G. Xie, X. Liu and S. Sun, Small, 2019, 15, 1904180, DOI:10.1002/smll.201904180.
- K. Rui, G. Zhao, M. Lao, P. Cui, X. Zheng, X. Zheng, J. Zhu, W. Huang, S. X. Dou and W. Sun, Nano Lett., 2019, 19, 8447–8453, DOI:10.1021/acs.nanolett.9b02729.
- H. Jin, S. Sultan, M. Ha, J. N. Tiwari, M. G. Kim and K. S. Kim, Adv. Funct. Mater., 2020, 30, 2000531, DOI:10.1002/adfm.202000531.
- Z.-X. Cai, H. Goou, Y. Ito, T. Tokunaga, M. Miyauchi, H. Abe and T. Fujita, Chem. Sci., 2021, 12, 11306–11315, 10.1039/D1SC01981C.
- S. Sultan, M. H. Diorizky, M. Ha, J. N. Tiwari, H. Choi, N. K. Dang, P. Thangavel, J. H. Lee, H. Y. Jeong and H. S. Shin, J. Mater. Chem. A, 2021, 9, 10326–10334, 10.1039/D1TA01067K.
- J. Huang, C. Du, Q. Dai, X. Zhang, J. Tang, B. Wang, H. Zhou, Q. Shen and J. Chen, J. Alloys Compd., 2022, 917, 165447, DOI:10.1016/j.jallcom.2022.165447.
- W. Qiao, S. Yan, D. Jin, X. Xu, W. Mi and D. Wang, J. Phys.: Condens. Matter, 2021, 33, 245201, DOI:10.1088/1361-648X/abe9da.
- J. Ekspong, E. Gracia-Espino and T. Wågberg, J. Phys. Chem. C, 2020, 124, 20911–20921, DOI:10.1021/acs.jpcc.0c05243.
- J. M. Bockris and E. Potter, J. Electrochem. Soc., 1952, 99, 169, DOI:10.1149/1.2779692.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05908a |
| This journal is © The Royal Society of Chemistry 2024 |