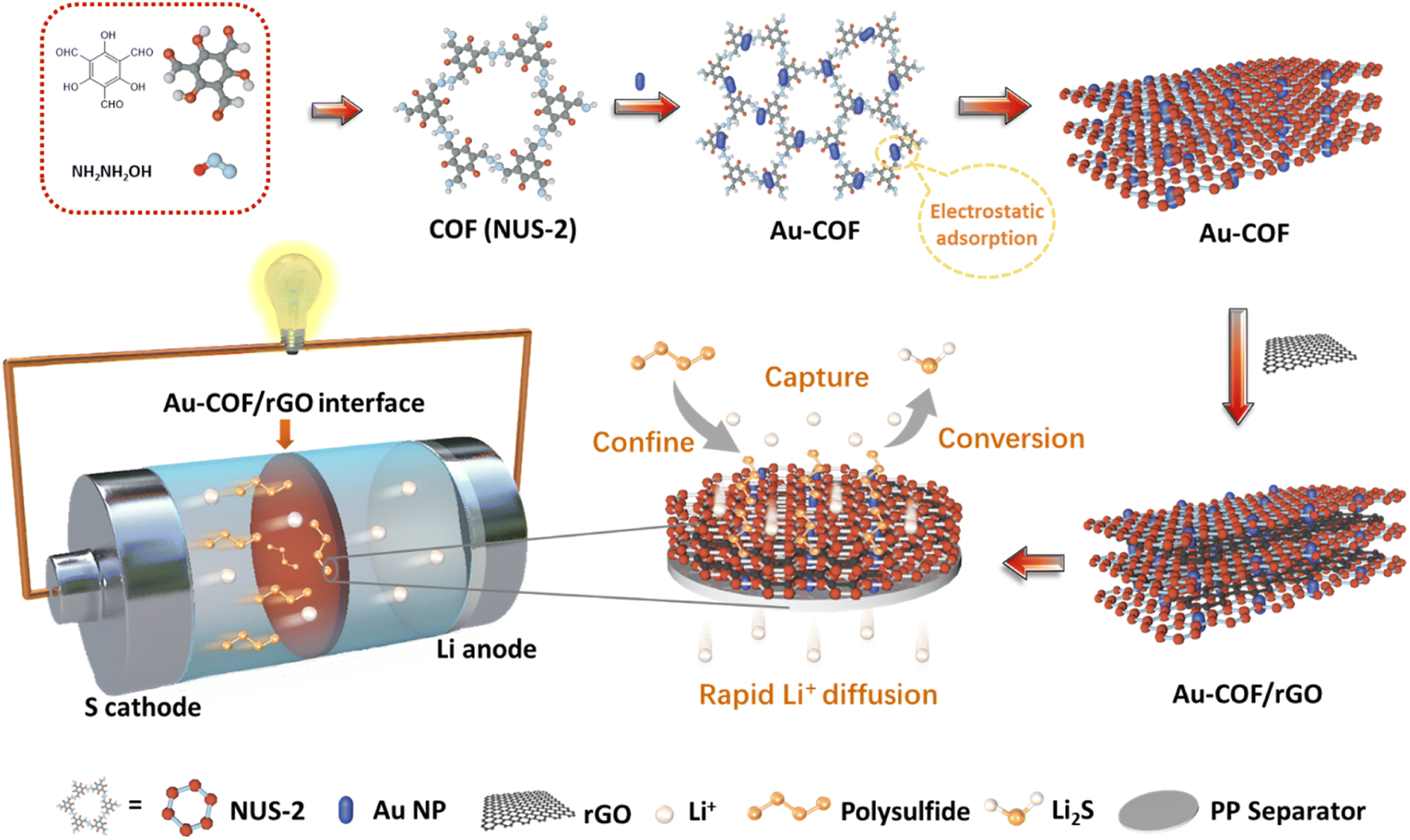
Regulating the kinetic behaviours of polysulfides by designing an Au–COF interface in lithium–sulfur batteries†
Received
26th September 2023
, Accepted 17th November 2023
First published on 21st November 2023
Abstract
The kinetic behaviors of polysulfides related to sluggish/incomplete conversion and repeated shuttles are the key factors behind the unsatisfactory performance of Li–S batteries. Herein, an Au-decorated covalent organic framework/graphene (Au–COF/rGO) is employed to construct a multifunctional interface to solve such issues. The narrow porous structure of COF can confine the diffused polysulfides via a physical barrier, and Au can further anchor and facilitate the catalytic conversion of intercepted polysulfides. Meanwhile, the well-conducting rGO nanosheets are also able to act as an upper current collector to activate polysulfides to quickly participate in the next electrochemical reaction. Thus, an orderly connection between the confinement–capture–conversion of polysulfides is formed. Additionally, the good lithium affinity of Au and imine linkages on COF can promote Li+ diffusion, achieving a fast charge of batteries. Attributed to the multi merits of the Au–COF/rGO interface, the assembled Li–S batteries not only deliver an ultra-high specific capacity (1525 mA h g−1 at 0.1C) with a prominent rate performance (568 mA h g−1 at 4C), but also exhibit outstanding cycling performance in both the cases of conventional and high sulfur loadings. The rational design of the Au–COF/rGO interface provides a new insight into developing advanced Li–S batteries.
Introduction
With the advent of a sustainable and green energy era, the requirements for reliable high-energy-density rechargeable battery systems have increased rapidly.1,2 However, the limited energy density of battery systems currently under development, such as lithium-ion batteries (≈387 W h kg−1), contradicts this growing demand.3–5 Owing to the low cost, natural abundance, and high theoretical energy density (≈2600 W h kg−1), lithium–sulfur (Li–S) batteries are considered one of the most promising high-performance energy storage systems in recent years.6–9 Unfortunately, there is still a big gap between the actual and theoretical energy densities of Li–S batteries due to some serious issues, among which the sluggish/incomplete conversion kinetic and shuttle effect of polysulfides results in the attenuation of capacity, severe self-discharge, and loss of active substances.10–13
Many studies have been devoted to overcoming this issue. Attributed to the design of functional separators, the electrochemical performance and cycle stability of Li–S batteries have been effectively improved.14–18 An ideal functional separator requires great synergy between the various components of separators. Specifically, at first, the homogeneous pores in the separators/interlayer can physically defend against polysulfides but allow the passage of lithium ions. Meanwhile, the manufacture of a functional separator with “chemical traps” can chemically capture polysulfides to restrain the shuttle effect.19–21 To date, various materials, such as metal oxides/carbides/nitrides,22–24 carbon-based materials,25 organic polymers26 and metal–organic/covalent organic framework materials,27 have been successfully used as functional separators, effectively improving the comprehensive electrochemical performance of Li–S batteries. Especially, more and more attention has been paid to covalent organic framework (COF) materials owing to their characteristics of functional diversity and controllable pore size.28 By selecting the COF material in a suitable size during preparation, the COF-interlayer could physically inhibit the shuttle of polysulfides but allow the transmission of smaller-sized lithium ions.29–31 In addition, COF materials can also be compounded with carbon/metal-based materials or modified with functional groups so that the COF interlayer allows the chemical capture of polysulfides and facilitates the transport of lithium ions, offering the possibility of constructing long-life and highly stable Li–S batteries.32,33 These works effectively improve the electrochemical performance of Li–S batteries, promoting the development of COF-based separators. Nevertheless, the sluggish/incomplete conversion kinetic of polysulfides is still a bottleneck to the manufacture of practical Li–S batteries. Additionally, the intrinsic electric insulativity of COF materials increases the internal impedance of the battery to a certain extent causing mass transfer difficulties.34,35 In this case, combining COF with a well-conducting catalyst that could facilitate the catalytic conversion of polysulfides to construct a functional separator may be more suitable for Li–S batteries.
Herein, club-shaped Au nanoparticles (NPs) were prepared to decorate the COF (NUS-2), which was then employed to construct a multifunctional interface between the cathode and the common separator to address the issues related to the kinetic behaviors of polysulfides. Reduced graphene oxide (rGO) nanosheets were introduced to assist the Au–COF to form a uniform interlayer on the surface of the PP membrane. Attributed to the rational design of the Au–COF/rGO interface, the Li–S battery not only obtained an ultra-high specific capacity with a prominent rate performance, but also achieved good cycling performance in both the cases of conventional and high sulfur loadings.
Experimental
Materials
Reduced graphene oxide (rGO) was purchased from Jiangsu Xianfeng Nanomaterials Technology Co., Ltd. (China). Silver nitrate solution (AgNO3, 0.1 N) was supplied by Shanghai Maclean Biochemical Technology Co., Ltd. (China). Hexadecyl trimethyl ammonium bromide (CTAB, 99%) was obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd. (China). 1,3,5-Trimethylresorcinol (TFP), 1,4-dioxane (C4H8O2, 99.8%), tetrachloroauric(III) acid trihydrate (99.9%, Au 50%), ascorbic acid (C6H8O6, >99%), N-methylpyrrolidone (NMP, AR), mesitylene (C9H12, 98%), and tetrahydrofuran (C4H8O, AR) were purchased from Beijing InnoChem Science & Technology Co., Ltd. (China). Other chemical reagents, including hydrazine (H4N2·H2O, 85%), hydrochloric acid (HCl, AR), glacial acetic acid (C2H4O2, GR), acetone (CH3COCH3, AR), absolute ethanol (C2H6O, AR), and sodium borohydride (NaBH4, AR), were supplied by Sinopharm Chemical Reagents Co., Ltd. (China).
Preparation of COF
COF was prepared according to the method reported in the literature. Briefly, 1,3,5-trimethylresorcinol (TFP, 42 mg, 0.2 mmol), hydrazine hydrate (15 μL, 0.3 mmol), mesitylene (1 mL), dioxane (1 mL), and aqueous acetic acid (6 M, 0.2 mL) were added to 10 mL Pyrex container in the same sequence. The mixture was degassed by liquid nitrogen freeze–vacuum–thaw. The process was repeated thrice and the contents were sealed under vacuum. The ampoules were then placed in a constant temperature oven at 120 °C for 72 h after the reaction, yielding a red solid. The obtained products were washed with acetone, anhydrous tetrahydrofuran, absolute ethanol, and methanol (thrice) in sequence. The powder was dried at 120 °C in a vacuum oven for 12 h to obtain the final product which was a deep red powder (35 mg, 77% yield).
Preparation of Au nanoparticles (NPs)
The preparation of Au NPs was adapted from the literature.37,38 Firstly, the seed solution was prepared. HAuCl4·3H2O (0.25 mL, 0.01 mol) was mixed with CTAB (9.75 mL, 0.01 mol) to prepare a gold solution. A fresh cold NaBH4 (0.6 mL, 0.01 mol) solution was quickly added to the above gold solution and stirred for 10 min; the solution was allowed to stand still at room temperature (27 °C) for 1.5 h. The growth solution was prepared by adding HAuCl4·3H2O (2.5 mL, 0.01 mol) to the stirred CTAB (40 mL, 0.10 mol) solution. Subsequently, fresh AgNO3 (150 μL, 0.01 mol), HCl (1 mL, 1.0 mol), and ascorbic acid (400 μL, 0.10 mol) were added to the mixed solution. After the growth solution was inverted and kept stationary at 27 °C for 20 h, Au NPs were obtained by centrifugation and dispersed into deionized water. The final product was a purple dispersion.
Construction of Au–COF/rGO interface
The Au–COF/rGO interface was constructed by a self-assembly method via vacuum filtration. Specifically, 1.5 mg COF and 1.5 mg Au NPs were mixed evenly in ethanol solution (10 mL) by ultrasonic stirring to form a homogeneous dispersion. Then, 1.5 mg of rGO was added to the mixture and sonicated for 30 min. The complex was finally deposited on the PP surface through vacuum filtration. Thus, the Au–COF/rGO/PP separator with a mass ratio of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :COF:rGO = 1:1:1 was obtained after drying at 60 °C for 12 h in a vacuum oven. For comparison, the rGO/PP and COF/rGO/PP separators were also fabricated in the same way. In addition, the amount of Au NPs was adjusted to 0.75, 2.25, and 3.0 mg to investigate the optimal proportion of Au NPs, which are marked 0.5Au–COF/rGO, 1.5Au–COF/rGO, and 2.0Au–COF/rGO, respectively. Furthermore, based on the mass ratio of Au:COF:rGO = 1:1:1, the Au–COF/rGO layer with different thicknesses was constructed by adjusting the total loading amount of the three components to 3.0, 4.5, 6.0, and 7.5 mg, which are abbreviated as L-1.0, L-1.5, L-2.0, and L-2.5, respectively.
:COF:rGO = 1:1:1 was obtained after drying at 60 °C for 12 h in a vacuum oven. For comparison, the rGO/PP and COF/rGO/PP separators were also fabricated in the same way. In addition, the amount of Au NPs was adjusted to 0.75, 2.25, and 3.0 mg to investigate the optimal proportion of Au NPs, which are marked 0.5Au–COF/rGO, 1.5Au–COF/rGO, and 2.0Au–COF/rGO, respectively. Furthermore, based on the mass ratio of Au:COF:rGO = 1:1:1, the Au–COF/rGO layer with different thicknesses was constructed by adjusting the total loading amount of the three components to 3.0, 4.5, 6.0, and 7.5 mg, which are abbreviated as L-1.0, L-1.5, L-2.0, and L-2.5, respectively.
Materials characterization
The FTIR spectra were observed at Tensor II (BRUKER Vertex 70). The morphology and chemical element composition of the materials were observed by scanning electron microscopy (SEM) and energy dispersive X-ray spectrometry (EDS), respectively, which were performed on the FEI Helios G4 CX scanning electron microscope (FEI, USA). The chemical binding states of various materials were analyzed by X-ray photoelectron spectroscopy (XPS), which was employed on a PHI-5000 VersaProbe III spectrometer (ULVAC-PHI, Japan). The crystal phases of the materials were characterized by XRD tests obtained on the D8 Advance Powder diffractometer (BRUKER, German, using Cu-Kα radiation, λ = 1.5418 Å).
Assembly of Li–S batteries
Cathode materials were fabricated by grinding 70 wt% sublimed sulfur and 30 wt% commercial conductive porous carbon (KJC) together. After being heated at 155 °C for 6 h, the composite was pestled again and heated at 200 °C for 2 h to obtain the KJC/S composite. The cathode was prepared by mixing KJC/S composite, Super P, and PVDF with a mass ratio of 7:2:1 in N-methyl-2-pyrrolidone (NMP) and grinding until a homogeneous sulfur slurry was formed. The slurry was spread flat on an aluminum foil and put into a 60 °C vacuum oven for 12 h, and finally cut into wafers with a diameter of 10 mm as cathodes. The anodes were prepared by directly pressing Li metal foils on stainless steel foils. The electrolyte was 1 M LiTFSI in DOL/DME (v/v = 1:1) with 2 wt% LiNO3 as an additive. The ratio between electrolyte volume and sulfur loading was 10. The separators were PP separators with various interlayers. It should be noted that the interlayers were faced with sulfur cathode. All coin cells were assembled in an argon-filled glove box with moisture and oxygen contents below 1 ppm.
Electrochemical measurements
Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted on the EC-Lab electrochemical workstation. CV curves of Li–S cells and galvanostatic charge–discharge profiles were collected from 1.7–2.8 V versus Li/Li+. The EIS was measured in a frequency range of 100 kHz to 0.01 Hz with an amplitude of 5 mV. The galvanostatic charge/discharge was measured on the NEWARE cell test system (BTS8-5 V 10 mA) at a constant temperature of 26 °C. Cycling stability was tested at various charge/discharge rates within a potential window of 1.7–2.8 V versus Li/Li+. The mass-specific capacities were calculated based on the total mass of sulfur in this work.
Ionic conductivity
Coin cells with the PP separator with various interlayers sandwiched between two stainless steel electrodes were calculated to show ionic conductivity using the EIS (0.1–100 kHz) according to the following equation:
where σ is the ionic conductivity (S cm−1); L stands for the thickness of the membrane (cm); Rb represents the bulk resistance (Ω); and A is the area of the stainless-steel electrode (cm2).
Lithium ions transference number
The PP separator with various interlayers was sandwiched between two lithium metal electrodes in coin cells. The lithium ions transference number was calculated by chronoamperometry using an electrochemical working station. The constant step potential was 20 mV, and the lithium ions transference number was defined as the ratio of steady-state current to initial-state current according to the following equation:
| tLi+ = Is(ΔV − I0R0)/I0(ΔV − IsRs) |
where tLi+ is the lithium ions transference number; Is and I0 represent the current at the steady state and initial state (mA); R0 and Rs represent the resistance before and after the potentiostatic polarization (Ω); and V stands for the potentiostatic potential (V).
Lithium ions diffusion coefficient
CV curves with different scan rates were calculated to compare the lithium ions diffusion coefficient according to the Randles–Sevcik equation:
| Ip = 2.69 × 105n1.5ADLi+0.5CLiv0.5 |
where Ip is the peak current (A); n represents the number of electrons involved in the reaction (n = 2 for Li–S battery); A is the area of the electrode (cm2); DLi+ is the lithium ions diffusion coefficient; CLi indicates the lithium ions concentration (mol L−1); and v refers to the scan rate (V s−1).
CV measurement of symmetrical cells
The cathode material was prepared by dispersing the material (Au–COF/rGO, COF/rGO, and rGO) and PVDF binder in NMP in a 9:1 mass ratio. Carbon fiber paper (CP) was cut to a diameter of 12.0 mm to serve as electrode discs. The resulting slurry was dried by dropping it onto a carbon fiber paper (CP). These discs were used as identical working and counter electrodes. The mass loading of the active material was ∼0.5 mg cm−2. The electrolyte was a mixture of Li2S6 (0.5 mol L−1) and LiTFSI (1 mol L−1) in DOL/DME. The CV measurement voltage window for the symmetric cell was between −1.5 and 1.5 V at a scan rate of 10 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were performed at an open circuit voltage with a sinusoidal voltage amplitude of 10 mV and a frequency range of 106 to 0.01 Hz.
Nucleation test of Li2S
The electrode materials (Au–COF/rGO, COF/rGO, and rGO) and PVDF binder were dispersed in NMP in a 9:1 mass ratio. The slurry was scraped onto the aluminum foil, dried, and punched out into electrode discs of 12.0 mm diameter. The mass loading of the active material was 0.5 mg cm−2. Twenty microliters of Li2S8 cathode solution was added dropwise to one side of the Au–COF/rGO, COF/rGO, or rGO working electrode, while 20 μL of Li2S8-free electrolyte was added to the counter electrode side. The assembled cell was discharged at 112 μA at a constant current of 2.06 V and then at a constant voltage of 2.05 V for Li2S nucleation.
Results and discussion
Design principle and material characterization
The fabricating process of the Au–COF/rGO interlayer is shown in Fig. 1. COF was firstly synthesized by using 1,3,5-trimethylresorcinol (TFP) as a monomer (Fig. S1†), which was crystallized in a flower-like morphology composed of each “petal” with a length of 1–2 μm and width of 100–300 nm (Fig. S2–S4†). Subsequently, the club-shaped Au nanoparticles (NPs) with an average width of 10 nm and length of 20 nm were introduced to decorate the COF (Fig. S5 and S6†). Zeta potential analysis revealed that the electropositive Au NPs were anchored on the electronegative COF through electrostatic adsorption (Fig. S7†). Lastly, the rGO nanosheets were employed as a film-forming agent to assist Au–COF in forming a uniform film on the PP membrane via a facile and simple vacuum filtration strategy. The even and tight incorporation of COF with rGO was attributed to the π–π bonds, and the as-prepared Au–COF/rGO composite layer was abbreviated as Au–COF/rGO, which still maintained the flower-like morphology (Fig. S8 and S9†). Such designed interlayer applied between the sulfur cathode and separator in Li–S batteries would offer multiple advantages: (i) the narrow and ordered porous structure of COF can confine the diffusion of dissolved polysulfides to the anodic side via physical barrier; (ii) the Au NPs can further anchor the intercepted polysulfides by chemical adsorption and act as an electrocatalyst to facilitate their conversion; (iii) the introduction of rGO nanosheets with excellent conductivity not only can improve the interface stability but can also act as an upper current collector to activate the anchored polysulfides to quickly participate in the next electrochemical reaction. Thus, an orderly connection between confinement–capture–conversion for polysulfides will be formed at the Au–COF/rGO interface. Moreover, the good lithium affinity of Au and imine linkages on COF can promote the diffusion of Li+, which is beneficial for attaining a higher state of charge at high rates, achieving a fast charge of the battery.
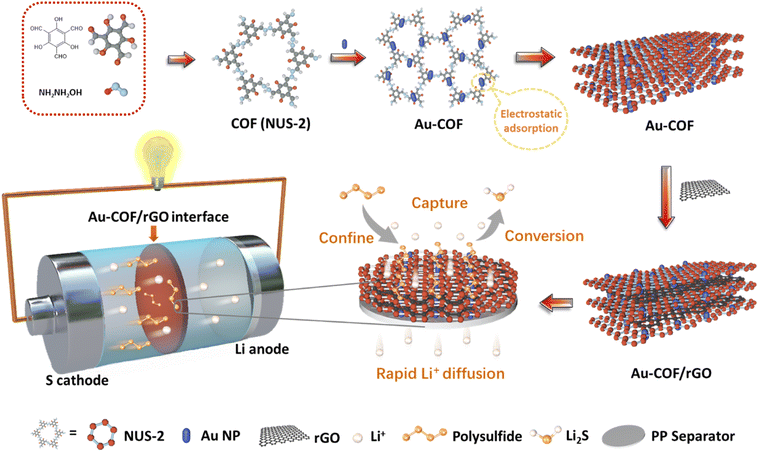 |
| | Fig. 1 Schematic illustration of the preparation process of an Au–COF/rGO interlayer, which enables to form an orderly connection between confinement–capture–conversion of polysulfides in Li–S batteries. | |
The crystalline structure of the as-prepared materials was investigated by PXRD (Fig. 2a). The broad XRD peaks of COF observed at ∼22–28° may arise from defects in the π–π stacking between successive COF layers.39 For the XRD pattern of rGO, a broad peak could be observed at 22.5°, which corresponded to the (002) plane. Meanwhile, a small diffraction peak appeared at 26.6°, which could be attributed to the π–π stacking between neighboring rGO layers.40,41 After combing the COF or Au–COF with rGO nanosheets, the small peak at 26.6° disappeared, which was probably induced by the strong π–π interactions between COF and rGO sheets.41–43 In addition, comparing the characteristic diffraction peaks with the original COF, the excess four peaks of the Au–COF/rGO at 2θ = 38.2°, 44.4°, 64.6°, and 77.6° were assigned to the (111), (200), (220), and (311) planes of Au NPs. The chemical structure of various materials was verified by Fourier transform infrared (FT-IR). As illustrated in Fig. 2b, the characteristic infrared peaks at 1280, 1447, and 1594 cm−1 of Au–COF/rGO and COF/rGO were ascribed to C–N, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO stretching vibration bands,36 which was consistent with primitive COF, indicating that the dopant in both Au NPs and rGO does not affect the functional group structure of COF. The N2 sorption isotherm shows that the COF (Fig. 2c) gives rise to an isotherm with type I and type IV features, suggesting a typical characteristic of microporous and mesoporous structure, which is favorable to restrain polysulfides. In addition, the top-view SEM image (Fig. 2d) shows that Au–COF/rGO is uniformly coated on the surface of the PP separator; meanwhile, the related elemental mapping images exhibit that Au NPs are evenly distributed in the functional separator. In addition, the cross-sectional SEM image (Fig. 2e) shows that the thickness of the coating Au–COF/rGO layer is only ∼5 μm.
C, and CO stretching vibration bands,36 which was consistent with primitive COF, indicating that the dopant in both Au NPs and rGO does not affect the functional group structure of COF. The N2 sorption isotherm shows that the COF (Fig. 2c) gives rise to an isotherm with type I and type IV features, suggesting a typical characteristic of microporous and mesoporous structure, which is favorable to restrain polysulfides. In addition, the top-view SEM image (Fig. 2d) shows that Au–COF/rGO is uniformly coated on the surface of the PP separator; meanwhile, the related elemental mapping images exhibit that Au NPs are evenly distributed in the functional separator. In addition, the cross-sectional SEM image (Fig. 2e) shows that the thickness of the coating Au–COF/rGO layer is only ∼5 μm.
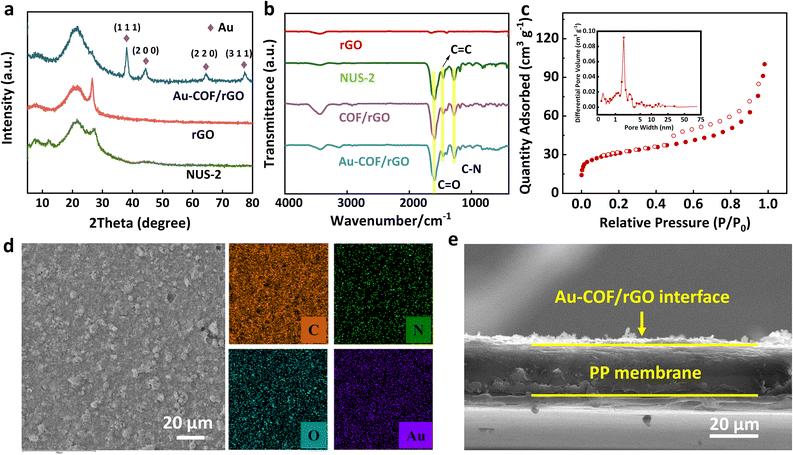 |
| | Fig. 2 Characterization results of Au–COF/rGO interface. (a and b) XRD patterns and FT-IR spectra of various materials. (c) N2 adsorption–desorption curve and the corresponding pore-size distribution curve of Au–COF/rGO. (d) SEM image and the corresponding elemental mapping images of Au–COF/rGO interlayer. (e) Side-view SEM image of Au–COF/rGO interlayer. | |
Interaction between Au–COF/rGO and polysulfides
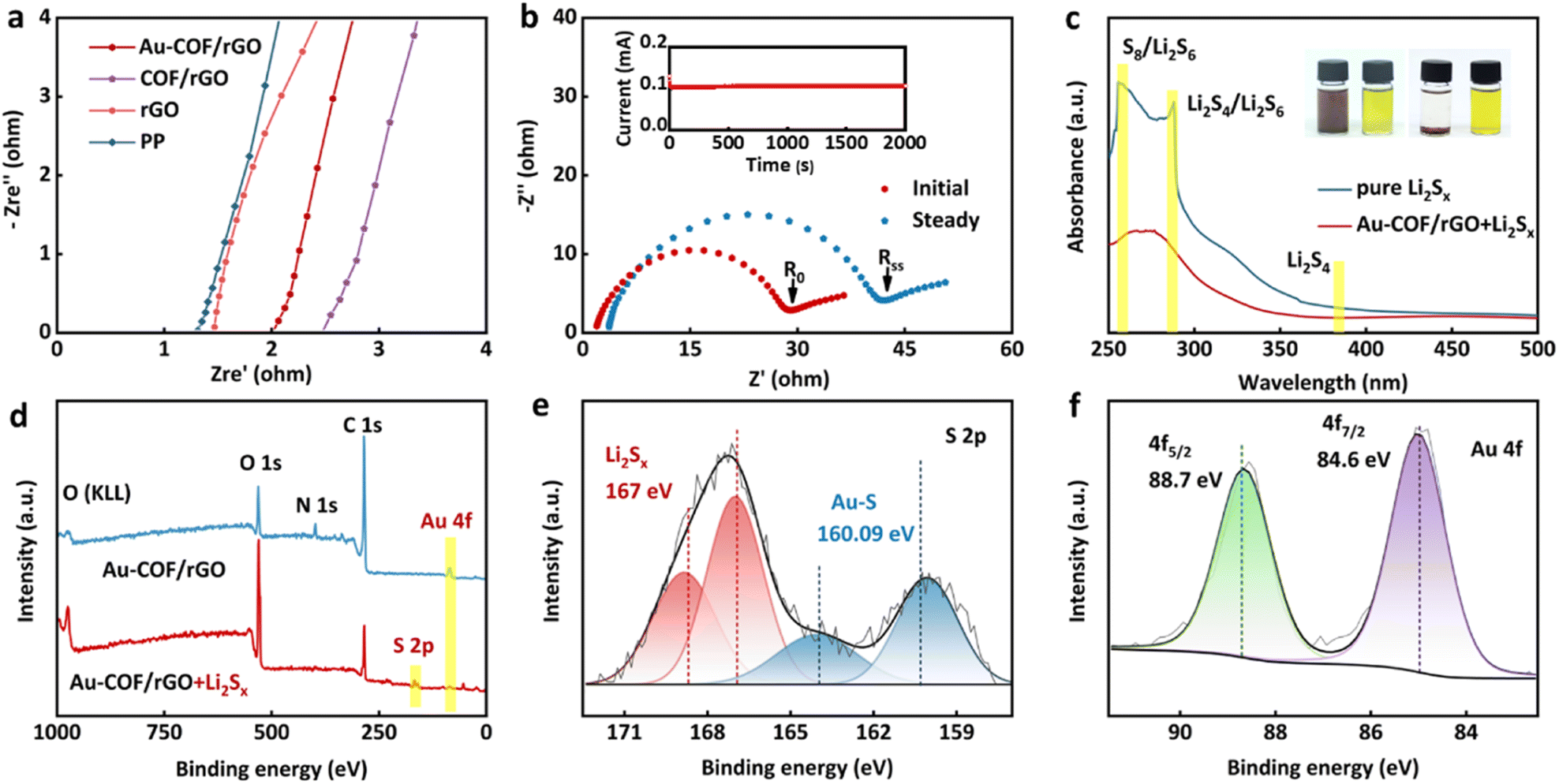
Good electrolyte wettability of the separator is conducive to absorbing and reserving electrolytes to obtain low internal resistance and high Li+ conductivity, which plays an important role in the electrochemical performance of Li–S batteries.44 Herein, the electrolyte wettability of the separator was assessed by contact angle test (Fig. S10†). It could be observed that the initial contact angle between pure PP separator and electrolyte was 50.9°, and there was only a slight decrease even after 10 min. However, after introducing an Au–COF/rGO layer on the surface of the PP separator, super-affinity to electrolyte could be achieved within 1 s, which benefited from the porous structure of COF. The ionic migration resistance and Li+ conductivities of various separators were evaluated by electrochemical impedance spectroscopy (EIS) (Fig. 3a and S11†). It can be seen that the resistance of Au–COF/rGO modified PP separator was slightly larger than that of pure PP rGO/PP separators, while it was smaller than that of COF/PP separators, which may be attributed to the good electrical conductivity of Au NPs. The corresponding Li+ conductivities of various separators showed that the Au–COF/rGO/PP separator possessed a high Li+ conductivity, which was close to others, suggesting that the uniform pore structure of COF substrate combined with the good lipophilicity of Au NPs is beneficial for efficient Li+ conduction. In addition, the Li+ transference number of the Au–COF/rGO/PP separator was measured via a potentiostatic polarization method and calculated using the current and resistance obtained according to the Randles–Sevcik equation.45 Similar to the Li+ conductivity, the Au–COF/rGO modified separator presented a Li+ transference number of 0.82 (Fig. 3b), which was not only higher than that of commercial PP separator (0.3–0.5),46–48 but also superior to that of the most reported COF-based separators (0.5–0.8).49,50 The results indicate that the as-constructed Au–COF/rGO interlayer can work as an ionic sieve to facilitate the diffusion of Li+, while restraining the transmission of polysulfides. This was further demonstrated by the visual permeation experiments (Fig. S12†).
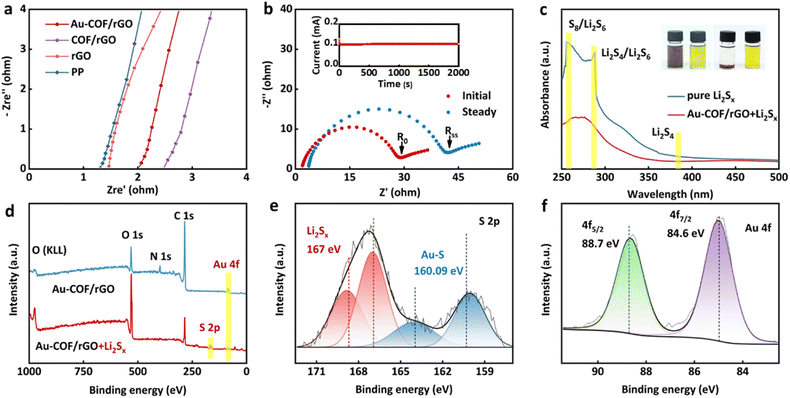 |
| | Fig. 3 Characterization of the principles of Au–COF/rGO. (a) EIS plots of symmetric batteries with various separators. (b) EIS plot before and after polarization (inset: the polarization curve of symmetric cell with an Au–COF/rGO modified separator). (c) UV-Vis spectra of the Li2Sx solution with and without Au–COF/rGO (inset: corresponding electronic photos). (d) Wide-scan survey XPS spectra of Au–COF/rGO before and after treating in Li2Sx solution. High-resolution elemental XPS spectra (e) S 2p and (f) Au 4f from Au–COF/rGO after treating in Li2Sx solution. | |
Visualized adsorption test was carried out by adding the Au–COF/rGO powder into Li2S6 solution to evaluate the special ion selectivity of the constructed interlayer (Fig. 3c). It was observed that the solution with Au–COF/rGO became colorless after 12 h. The supernatant solutions were used for UV-Vis absorption measurement. As a result, the intensity of characteristic absorbance peaks of polysulfides in solutions with Au–COF/rGO was significantly decreased, indicating the strong adsorption effect on polysulfides. X-ray photoelectron spectroscopy (XPS) analysis was employed to explore the interaction between Li2S6 and Au–COF/rGO (Fig. 3d–f). After treatment in Li2S6 solution, the emerged S 2p and S 2s peaks on the survey spectrum of Au–COF/rGO further reflected the effective absorption of Au–COF/rGO towards polysulfides. Additionally, the splitting of S 2p peaks rationally resulted from the varying complex chemical environment of S after being anchored by Au NPs. The binding energy peak at 160.9 eV can be attributed to the S–Au band, in agreement with values for metal sulfides or with the Au–S bond in self-assembled monolayers.51,52 Meanwhile, the splitting peaks of Au 4f located at 88.7 and 84.6 eV further confirmed that a new local environment of Au was created after combining with the polysulfide species. XPS results revealed that the interaction between Au–COF/rGO and polysulfides was attributed to the formation of the n order to explore the effect of the Au–S bond.
Regulation of lithium ions and polysulfides
To explore the effect of the constructed Au–COF/rGO interface on lithium-ion transport, CVs were tested on the cells assembled with four separators at different scan rates from 0.1 to 0.4 mV s−1 to quantitatively analyze the lithium ions diffusion coefficients (DLi+) (Fig. 4a–d and S13†). It can be seen that all CV curves presented two characteristic reduction peaks at ∼2.3 and 2 V (assigned as R1 and R2), which were attributed to the reduction of sulfur to high-order lithium polysulfide species at the higher potential and the reduction of high-order lithium polysulfide species intermediates to insoluble Li2S2/Li2S at the lower potential.42 The peaks at ∼2.5 V (assigned as O1) were contributed by the oxidation of Li2S2/Li2S to elemental sulfur. From the linear fitting of the peak currents, the DLi+ was calculated by the classical Randles–Sevcik equation53 and presented in Table S1.† It was found that the battery with the Au–COF/rGO-based separator showed the highest DLi+ among the four separators, indicating the addition of COF and Au NPs synergistically promotes the transport of lithium ions. In addition, to investigate the contribution of functional separators to the redox kinetics of polysulfides, Li2S precipitation experiments were carried out via constant voltage discharge of asymmetric Li–S cells at 2.05 V. As shown in Fig. 4e–g, the responsive speed of Li2S nucleation in Au–COF/rGO is faster than that in COF/rGO and rGO. Meanwhile, the Li2S precipitation for cells with Au–COF/rGO shows superior capacity at 380.2 mA h g−1 compared to that for cells with COF/rGO (135.4 mA h g−1) and rGO (48.6 mA h g−1). The results showed that the Au–COF/rGO interlayer could significantly promote Li2S nucleation and improve the rapid kinetics of Li2S precipitation. CV tests for symmetrical cells with Li2S6-containing electrolytes were further conducted to verify the catalytic conversion of polysulfides by a functional separator. The CV curves (Fig. 4h) illustrate that the current density of the cells with Au–COF/rGO is much higher than that of the cells with COF/rGO or rGO at the scan rate of 10 mV s−1, indicating the excellent electrocatalytic activity of Au–COF/rGO interlayer. The results may be attributed to the Au NPs which possess the ability to capture polysulfides and catalytic activity. Meanwhile, the EIS curves (Fig. S14†) confirm that Au–COF/rGO interlayer exhibits lower electron transfer resistance than COF/rGO and rGO interlayers, further demonstrating the superior ultrafast conversion of Au–COF/rGO towards polysulfides.
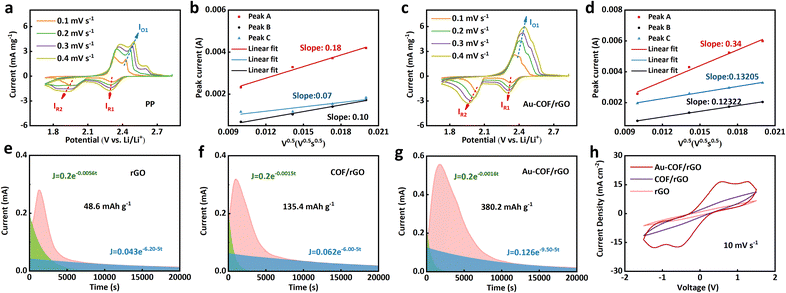 |
| | Fig. 4 CV curves at different scan rates and the corresponding linear fits of the peak current of the cells with the (a and b) PP and (c and d) Au–COF/rGO/PP separators. The Li2S nucleation experiment of (e) Au–COF/rGO, (f) COF/rGO, and (g) rGO. (h) CV curves of symmetric cells with different separators at a scan rate of 10 mV s−1. | |
Electrochemical performance of Li–S batteries
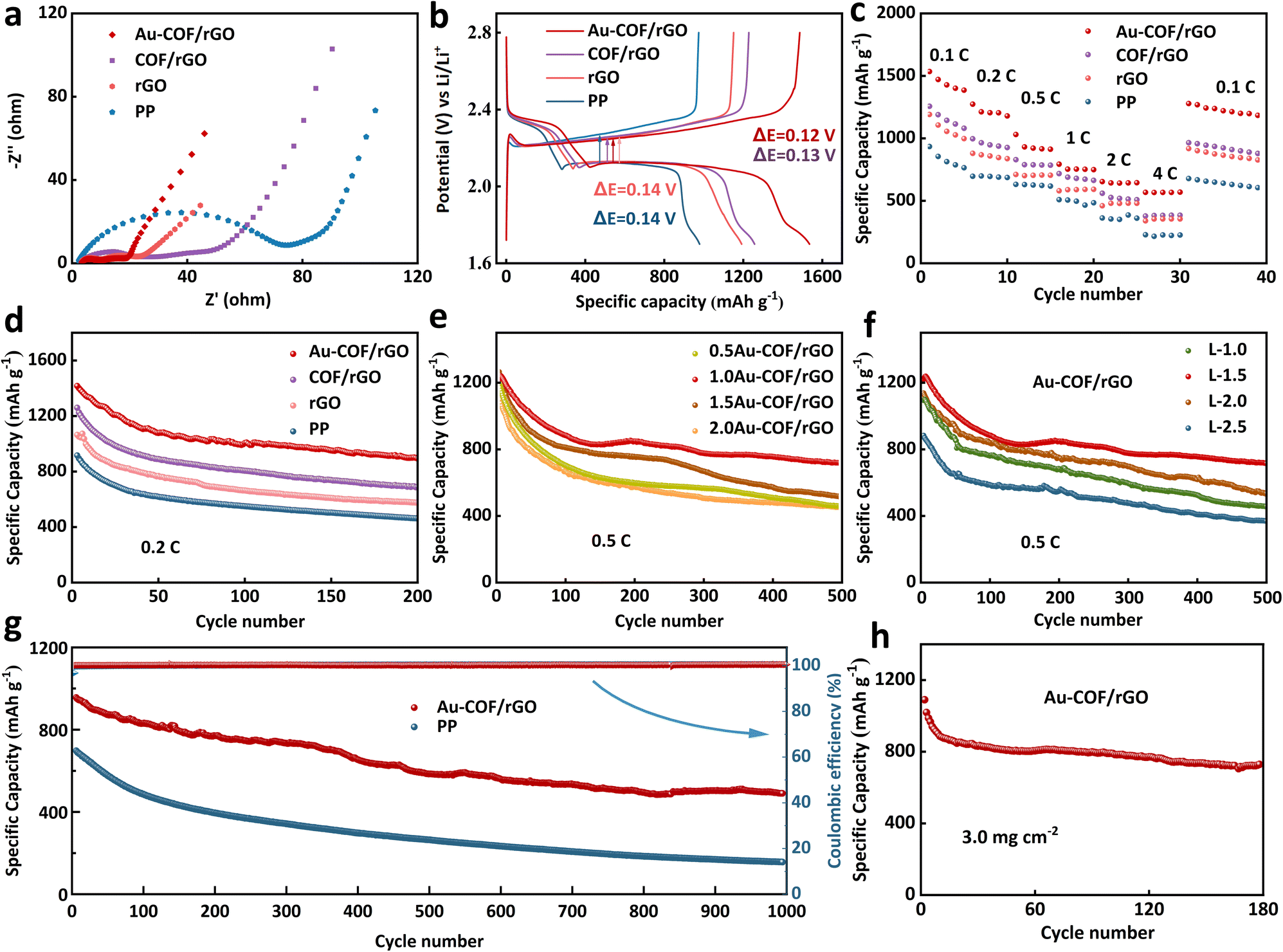
The electrochemical performance of Li–S batteries assembled with various separators was systematically investigated and analyzed. Electrochemical impedance spectroscopy (EIS) for cells with four different separators is illustrated in Fig. 5a. It can be seen that when the separator is incorporated with an Au–COF/rGO layer, the assembled battery shows the lowest electrolyte resistance (Rs) and charge transfer resistance (Rct), which contributes to the synergistic effect between the excellent conductivity of rGO and ordered porous structure of Au–COF. Fig. 5b presents the initial discharge capacity of Li–S batteries with various separators at 0.1C (1C = 1675 mA g−1). It can be seen that all three kinds of interlayers can effectively enhance the discharge capacity of Li–S batteries, while there is a certain gap in the degree of improvement, which is mainly reflected in the second discharge plate at 2.1 V, corresponding to the reduction of higher-order soluble polysulfides to Li2S2/Li2S. The results indicate that the COF/rGO interlayer can effectively reserve the soluble polysulfides in the cathode side, and the Au NPs can further capture them and promote their conversion to lower-order polysulfides, thus achieving a high discharge capacity of 1525 mA h g−1. The rate performance of Li–S batteries at various currents was further tested (Fig. 5c). It was seen that the Li–S batteries with various interlayers not only showed higher initial capacities than that of the pure PP assembled cell, but also delivered superior reversible capacities at high current densities. Especially for the cell with an Au–COF/rGO coated separator, a high capacity of 568 mA h g−1 could still be released even at a high rate of 4C, and a satisfactory capacity of 1279 mA h g−1 was retrieved when the current density was abruptly turned from 4C to 0.1C. The outstanding rate performance and excellent reversibility of Li–S battery further demonstrate the multifunction of Au–COF/rGO-based separator.
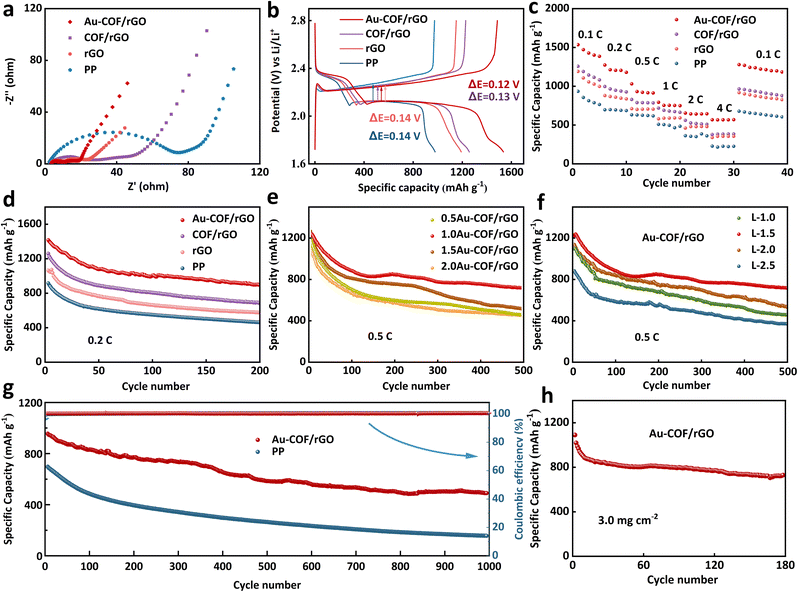 |
| | Fig. 5 The electrochemical performance of Li–S cell with various separators: (a) EIS curves. (b) Charge–discharge voltage profiles at 0.1C. (c) Rate performance. (d) Cycling performance at 0.2C. Cycling performance of Li–S cell assembled with Au–COF/rGO: (e) with different proportions of Au NPs at 0.5C. (f) With different thickness based on the mass ratio of Au:COF:rGO = 1:1:1. (g) Comparison of the cycling performance of Li–S cell with different interfaces at 1.0C. (h) Cycling performance of the Au–COF/rGO assembled Li–S cell with a high sulfur loading of 3.0 mg cm−2 at 0.2C. | |
The cycling performance of the Li–S batteries with various separators was evaluated at 0.2C (Fig. 5d). The corresponding first five charge–discharge curves are presented in Fig. S15.† It can be seen that the modification of separator interface layers has no significant effect on the initial coulomb efficiency of the Li–S battery, while the battery capacity has been greatly improved. For the cell with a pristine PP separator, only a reversible capacity of 462 mA h g−1 was provided after 200 cycles, corresponding to a capacity retention of 45.8%. After introducing a rGO layer or a COF/rGO layer on the PP surface, both the initial capacity and the 200th capacity were increased, attributable to the physical barrier of the rGO layer and COF/rGO towards soluble polysulfides. Surprisingly, when combining the Au with COF, the constructed Au–COF/rGO-based separator enabled the Li–S battery to realize a high reversible capacity of 902 mA h g−1 after 200 cycles, while the corresponding capacity retention increased by 21.4% compared with that of the cell with a pure PP separator. The electrochemical test results show that the introduction of Au in COF greatly improved the electrochemical performance of lithium–sulfur batteries, and thus, the optimal proportion of Au NPs to COF was further explored by cyclic testing at 0.5C. As shown in Fig. 5e and Table S2,† it was found that introducing too little or too much Au in COF degraded the battery performance. This may be because the little doping amount of Au is unable to provide enough active sites for the catalytic conversion of polysulfides, while too much additive amount causes agglomeration of Au NPs. Thus, the optimal proportion of Au and COF was determined to be 1:1, which could provide a high reversible capacity of 719 mA h g−1 even after long-term cycling, with a low capacity decay rate of 0.082%. The thickness of the separator also plays a crucial role in the electrochemical performance of Li–S batteries. Herein, based on the mass ratio of Au:COF:rGO = 1:1:1, an Au–COF/rGO layer with different thicknesses was constructed by adjusting the total loading amount of the three components (Fig. S16†), which were employed to study the effect of the separator thickness on the electrochemical performance of Li–S batteries. The test results are shown in Fig. 5f and Table S3;† it can be seen that the cell with the 4.5 nm thickness of Au–COF/rGO layer exhibits the highest initial/final capacity with the lowest capacity decay rate compared to that of the cells with the thinner and thicker Au–COF/rGO layer. This is mainly because when the loading amount of Au–COF/rGO composites is too small, it cannot completely cover the microporous surface of the PP separator, which can mitigate the shuttle of polysulfides to some degree, but it cannot inhibit the diffusion of polysulfides for a long time. On the other hand, when the separator is too thick, it will cause a high transfer resistance and severe polarization, resulting in degradation of the electrochemical performance of the Li–S cell. What's worse, excessive coating load also will increase the weight of the separator, which may sacrifice the overall power/energy density of the battery.
To further evaluate the applied potential of the Au–COF/rGO modified separator, the long-term cycle life of the Li–S batteries was performed at 1C (Fig. 5g). It can be seen that the cell with a commercial PP separator exhibited a low initial capacity of 695 mA h g−1 with a rapid capacity decay rate of 0.2% per cycle upon 1000 cycles, which is mainly caused by low availability of active materials and serious shuttle effect. By replacing the PP separator with an Au–COF/rGO modified separator, not only did the initial capacity increase by 269 mA h g−1, but the average capacity attenuation rate also decreased to 0.048% per cycle. As a result, the reversible capacity of the Au–COF/rGO-modified Li–S cell was five times higher than that of the ordinary PP separator after 1000 cycles (Fig. S17†). Compared with the charge and discharge curves of the two cells after 1000 cycles, the cell with Au–COF/rGO interlayer still had a relatively smooth discharge platform, indicating that the functionally modified separator reduced the loss of active substances and improved the stability of the battery. The relatively smooth and continuous surface of the lithium metal anode further demonstrated that the Au–COF/rGO layer could effectively restrain the shuttle effect, mitigating the corrosion of lithium metal by polysulfides (Fig. S18†). An interesting phenomenon is that the Au–COF/rGO layer became smoother and denser after circulation compared with the pristine one (Fig. S19†). This was attributable to the reversible interaction between Au–COF and the polysulfides. Lastly, the sulfur loading was augmented to 3.0 mg cm−2 to evaluate the feasibility of the Au–COF/rGO modified separator in practical application. It was observed that the Li–S cell was able to release a high initial capacity of 1162 mA h g−1 at 0.1C (Fig. S20†), and it kept stable cycling at 0.2C (Fig. 5h). The Li–S battery with an Au–COF/rGO interface exhibited superior electrochemical performance than that of most Li–S cells with a functional separator in the case of low and high sulfur loading (Tables S4 and S5†), suggesting the availability and reliability of Au–COF/rGO-demodified separator for practical applications.
Conclusions
In summary, an Au-decorated covalent organic framework (Au–COF) was employed to construct a multifunctional interface in Li–S batteries to address issues related to the kinetic behaviors of polysulfides, including sluggish/incomplete conversion and repeated shuttle. Graphene (rGO) nanosheets were introduced to assist the Au–COF to form a uniform interlayer on the surface of the PP membrane. Thus, COF can confine the diffusion of dissolved polysulfides via a physical barrier, and then Au can further anchor and facilitate the catalytic conversion of the trapped polysulfides. Meanwhile, the well-conducting rGO nanosheets are also able to act as an upper current collector to activate the intercepted polysulfides to quickly participate in the next electrochemical reaction. Consequently, confinement–capture–conversion progress for dissolved polysulfides was performed at the Au–COF/rGO interface. Furthermore, the good lithium affinity of Au and imine linkage on COF can promote the diffusion of Li+, achieving a fast charge of the battery. Due to the multifunctional nature of the Au–COF/rGO interface, the as-assembled Li–S batteries gave an outstanding electrochemical performance in both cases of conventional and high sulfur loadings, presenting the potential for practical applications.
Author contributions
Chan Li: formal analysis, methodology, data curation, writing the original draft. Ke Yang: investigation, formal analysis. Zelin Ma: data curation. Fei Zhao: software. Juan Li: formal analysis. Xinwu Wu: software. Xiaoyu Hao: software. Yibo He: writing-review & editing, conceptualization, supervision.
Conflicts of interest
The authors declare that they have no conflict of interest. They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The financial support from the National Nature Science Foundation of China (22109127), the Chinese Postdoctoral Science Foundation (2021M702666), and the Fundamental Research Funds for the Central Universities (D5000210129) are sincerely appreciated. Yibo He is also very grateful for the support from the Youth Project of “Shaanxi High-level Talents Introduction Plan” and the State Key Laboratory of Solidification Processing.
Notes and references
- S. Xia, Z. Chen, L. Yuan, J. Song, Q. Zhou, X. Yuan, L. Liu, L. Fu, Y. Chen and Y. Wu, J. Mater. Chem. A, 2023, 11, 19870–19876 RSC
 .
.
- N. Yang and X. Jiang, Acc. Chem. Res., 2022, 56, 117–127 CrossRef PubMed .
- H. Duan, L. Liao, R. Bi, Y. Deng and G. Chen, J. Mater. Chem. A, 2023, 11, 19046–19055 RSC .
- X. Han and X. Xu, J. Mater. Chem. A, 2023, 11, 18922–18932 RSC .
- B. Sun, Q. Zhang, W. Xu, R. Zhao, H. Zhu, W. Lv, X. Li and N. Yang, Nano Energy, 2022, 94, 106937 CrossRef CAS .
- L. Zhou, D. L. Danilov, R. A. Eichel and P. H. Notten, Adv. Energy Mater., 2021, 11, 2001304 CrossRef CAS .
- X. Wu, Z. Shen, D. Cai, B. Fei, M. Zhao, J. Fu, Q. Chen and H. Zhan, J. Mater. Chem. A, 2023, 11, 18313–18322 RSC .
- Z. Shi, Y. Ding, Q. Zhang and J. Sun, Adv. Energy Mater., 2022, 12, 2270124 CrossRef .
- R. Deng, H. Yu, J. Liu, F. Chu, J. Lei, L. Yang and F. Wu, J. Mater. Chem. A, 2023, 11, 15769–15777 RSC .
- K. Yang, C. Li, H. Qi, Y. Dai, Y. Cui and Y. He, J. Mater. Chem. A, 2023, 11, 10425–10434 RSC .
- Y. Huang, L. Lin, C. Zhang, L. Liu, Y. Li, Z. Qiao, J. Lin, Q. Wei, L. Wang, Q. Xie and D. L. Peng, Adv. Sci., 2022, 9, e2106004 CrossRef .
- M. Sadd, S. De Angelis, S. Colding-Jørgensen, D. Blanchard, R. E. Johnsen, S. Sanna, E. Borisova, A. Matic and J. R. Bowen, Adv. Energy Mater., 2022, 12, 2103126 CrossRef CAS .
- Y. S. Ye, M. G. Mohamed, W. C. Chen and S. W. Kuo, J. Mater. Chem. A, 2023, 11, 9112–9124 RSC .
- W. Xi, J. Zhang, Y. Zhang, R. Wang, Y. Gong, B. He, H. Wang and J. Jin, J. Mater. Chem. A, 2023, 11, 7679–7689 RSC .
- B. Wang, L. Wang, B. Zhang, S. Zeng, F. Tian, J. Dou, Y. Qian and L. Xu, ACS Nano, 2022, 16, 4947–4960 CrossRef CAS PubMed .
- D. Cao, X. Sun, F. Li, S.-M. Bak, T. Ji, M. Geiwitz, K. S. Burch, Y. Du, G. Yang and H. Zhu, Angew. Chem., Int. Ed., 2023, e202302363 CAS .
- L.-Y. Yao, L.-P. Hou, Y.-W. Song, M. Zhao, J. Xie, B.-Q. Li, Q. Zhang, J.-Q. Huang and X.-Q. Zhang, J. Mater. Chem. A, 2023, 11, 7441–7446 RSC .
- Z. Zhou, Z. Chen, H. Lv, Y. Zhao, H. Wei, G. Huai, R. Xu and Y. Wang, J. Mater. Chem. A, 2023, 11, 5883–5894 RSC .
- Y. Zhang, C. Guo, J. Zhou, X. Yao, J. Li, H. Zhuang, Y. Chen, Y. Chen, S. L. Li and Y. Q. Lan, Small, 2022, 2206616 Search PubMed .
- Z. Zhang, J. N. Wang, A. H. Shao, D. G. Xiong, J. W. Liu, C. Y. Lao, K. Xi, S. Y. Lu, Q. Jiang, J. Yu, H. L. Li, Z. Y. Yang and R. V. Kumar, Sci. China Mater., 2020, 63, 2443–2455 CrossRef CAS .
- B. Patra, K. Kumar, D. Deb, S. Ghosh, G. S. Gautam and P. Senguttuvan, J. Mater. Chem. A, 2023, 11, 8173–8183 RSC .
- T. Zhang, L. Zhang and Y. Hou, eScience, 2022, 2, 164–182 CrossRef .
- W. Yao, W. Zheng, J. Xu, C. Tian, K. Han, W. Sun and S. Xiao, ACS Nano, 2021, 15, 7114–7130 CrossRef CAS PubMed .
- T. Wang, J. He, Z. Zhu, X. B. Cheng, J. Zhu, B. Lu and Y. Wu, Adv. Mater., 2023, 2303520 CrossRef CAS PubMed .
- P. Wang, X. Dai, P. Xu, S. Hu, X. Xiong, K. Zou, S. Guo, J. Sun, C. Zhang, Y. Liu, T. Zhou and Y. Chen, eScience, 2023, 3, 100088 CrossRef .
- X. Wu, C. Zhou, C. Dong, C. Shen, B. Shuai, C. Li, Y. Li, Q. An, X. Xu and L. Mai, Nano Res., 2022, 15, 8048–8055 CrossRef CAS .
- Z. Zhu, Y. Zeng, Z. Pei, D. Luan, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2023, 62, e202305828 CrossRef PubMed .
- M. Wu and Z. Zhou, Interdiscip. Mater., 2023, 2, 231–259 CrossRef .
- Y. Cao, C. Liu, M. Wang, H. Yang, S. Liu, H. Wang, Z. Yang, F. Pan, Z. Jiang and J. Sun, Energy Stor. Mater., 2020, 29, 207–215 Search PubMed .
- Y. Yang, X.-J. Hong, C.-L. Song, G.-H. Li, Y.-X. Zheng, D.-D. Zhou, M. Zhang, Y.-P. Cai and H. Wang, J. Mater. Chem. A, 2019, 7, 16323–16329 RSC .
- J. Xu, S. An, X. Song, Y. Cao, N. Wang, X. Qiu, Y. Zhang, J. Chen, X. Duan and J. Huang, Adv. Mater., 2021, 33, 2105178 CrossRef CAS .
- Y. Ge, J. Li, Y. Meng and D. Xiao, Nano Energy, 2023, 109, 108297 CrossRef CAS .
- D. Chen, S. Huang, L. Zhong, S. Wang, M. Xiao, D. Han and Y. Meng, Adv. Funct. Mater., 2020, 30, 1907717 CrossRef CAS .
- J. Wang, W. Qin, X. Zhu and Y. Teng, Energy, 2020, 199, 117372 CrossRef CAS .
- J. Wang, L. Si, Q. Wei, X. Hong, S. Cai and Y. Cai, ACS Appl. Nano Mater., 2017, 1, 132–138 CrossRef .
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28, 1277–1285 CrossRef CAS .
- H.-H. Chang and C. J. Murphy, Chem. Mater., 2018, 30, 1427–1435 CrossRef CAS PubMed .
- N. S. Abadeer, M. R. Brennan, W. L. Wilson and C. J. Murphy, ACS Nano, 2014, 8, 8392–8406 CrossRef CAS .
- Z. Kang, Y. Peng, Y. Qian, D. Yuan, M. A. Addicoat, T. Heine, Z. Hu, L. Tee, Z. Guo and D. Zhao, Chem. Mater., 2016, 28(5), 1277–1285 CrossRef CAS .
- Z. Qiu, Z. Liu, G. Wang, C. Huangfu, Z. Li, Y. Yan, C. Chi, P. Gao, X. Lu, S. Zhang, T. Wei and Z. Fan, Energy Stor. Mater., 2023, 60, 102824 Search PubMed .
- C. Jiang, M. Tang, S. Zhu, J. Zhang, Y. Wu, Y. Chen, C. Xia, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 16072–16076 CrossRef CAS PubMed .
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228 CrossRef CAS .
- J. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., Int. Ed., 2018, 57, 1034 CrossRef CAS PubMed .
- G. Nunes, Energy Environ. Sci., 2014, 7, 3439–3440 RSC .
- Y. Yang and J. Zhang, Adv. Energy Mater., 2018, 8, 1801778 CrossRef .
- Y. He, Z. Chang, S. Wu, Y. Qiao, S. Bai, K. Jiang, P. He and H. Zhou, Adv. Energy Mater., 2018, 8, 1802130 CrossRef .
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS .
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed .
- J. Liu, J. Zhang, J. Zhu, R. Zhao, Y. Zhang, Y. Ma, C. Li, H. Zhang and Y. Chen, Nano Res., 2023, 265, 1–7 Search PubMed .
- H. X. Yang, Z. K. Liu, Y. Wang, N. W. Li and L. Yu, Adv. Funct. Mater., 2023, 33, 2209837 CrossRef CAS .
- B. Zhang, A. Sels, G. Salassa, S. Pollitt, V. Truttmann, C. Rameshan, J. Llorca, W. Olszewski, G. Rupprechter, T. Burgi and N. Barrabes, ChemCatChem, 2018, 10, 5372–5376 CrossRef CAS PubMed .
- S. Chai, Y. Wang, Y.-n. Zhang, M. Liu, Y. Wang and G. Zhao, Environ. Sci. Technol., 2017, 51, 8067–8076 CrossRef CAS .
- S. Das, S. Majumder and R. Katiyar, J. Power Sources, 2005, 139, 261–268 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*