
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase stability, redox-behavior and carbon-tolerance of Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ with exsolved nanoparticles†
Alexander
Schwiers
 *ab,
Daniel
Röhrer‡
*a,
Christian
Lenser
a,
Benjamin
Steinrücken
c,
Doris
Sebold
a,
Hartmut
Spliethoff
c,
Olivier
Guillon
abd and
Norbert H.
Menzler
ab
*ab,
Daniel
Röhrer‡
*a,
Christian
Lenser
a,
Benjamin
Steinrücken
c,
Doris
Sebold
a,
Hartmut
Spliethoff
c,
Olivier
Guillon
abd and
Norbert H.
Menzler
ab
aForschungszentrum Jülich GmbH, Institute of Energy and Climate Research (IEK), Materials Synthesis and Processing (IEK-1), Wilhelm-Johnen-Straße, Juelich 52428, Germany. E-mail: a.schwiers@fz-juelich.de
bRWTH Aachen University, Institute of Mineral Engineering (GHI), Aachen, Germany
cTechnical University of Munich, Chair of Energy Systems, Garching 85748, Germany
dJARA-Energy, Juelich, Germany
First published on 11th March 2024
Abstract
Ni-exsolution from perovskites has gained significant attention in the area of electrode materials for fuel and electrolysis cells due to their potential for high catalytic activity, tolerance to degradation, improved redox-cycling stability, and regeneration of the nanoparticles. We investigated the exsolution behavior and phase stability of bulk Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ ceramics at various temperatures and atmospheres. To assess its tolerance to carbon deposition, Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ was compared with commonly used Ni-YSZ materials in the presence of tar-containing biogas. Nanoparticle exsolution was observed in the entire investigated temperature range of 710–900 °C. Above a reduction temperature of 740 °C, Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ partially transforms to a Ruddlesden–Popper phase, while reverting back to the perovskite phase during reoxidation. This phase transition resulted in material expansion and the formation of cracks. Moreover, exposure of reduced Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ to ambient air or H2O led to the formation of secondary phases and fragmentation of sintered pellets. Remarkably, when compared to Ni-YSZ, these materials exhibited no degradation in terms of carbon deposition or Ni dusting after exposure to tar-containing biogas.
1. Introduction
Exsolution is defined as the phase separation of a solid solution due to a miscibility gap under specific conditions.1 In the context of catalysis, exsolution refers to the reduction of metal cations to the metallic state, forming nanosized particles on the surface.2 This process is initiated by exposing the material to reducing conditions, resulting in the release of oxygen from the host lattice and thus providing the driving force to exsolve certain cations.3 Perovskite materials are very popular in this field due to the possibility to be doped with various exsolvable cations, since adjusting their crystal structure is undemanding while preserving stability.4For electrochemical applications such as solid oxide cells (SOCs), the general approach is to exsolve a catalytically active metal from its host lattice to achieve enhanced catalytic activity and high resistance to degradation due to the higher surface-to-volume ratio and socketing of the nanoparticles in the host lattice.
State-of-the-art fuel electrode cermets in SOCs, which consist of an oxygen ion conductor (e.g., yttria-stabilized zirconia or gadolinia-doped ceria) and Ni, suffer from significant degradation when exposed to fuels containing higher hydrocarbons and sulfur species.5–7 The Ni surface can be poisoned, leading to a loss of catalytic activity, or carbon deposition can result in metal dusting. Another concern is the migration of Ni, especially during electrolysis.8 Exsolved and socketed nanoparticles might be more stable against migration and redox cycling compared to Ni particles distributed in an ion conducting ceramic (although they may suffer from Ostwald ripening and agglomeration).9–11 Redox cycling of exsolved nanoparticles might be an option to self-regenerate degraded catalyst particles.12,13
In the case of SOCs, a mixed ionic and electronic conductor (MIEC) is doped with a metal that shows activity towards hydrogen oxidation or water splitting. Using a single material instead of a cermet increases the reaction zone as the material forms a double phase boundary with the gas molecules (compared to the active triple phase boundaries – pores, oxygen-ion conducting phase, electron conducting phase – in cermets). However, despite possessing a certain catalytic activity, they often cannot match the performance of a typical Ni-containing cermet.14 Furthermore, their electronic conductivity may not be sufficient for practical operation.15
In this context, the properties of the oxide host lattice after exsolution are insufficiently investigated. Since the exsolved nanoparticles are not percolated, their function is very different from that of a percolated Ni network and the electrode performance will strongly depend on the properties of the oxide backbone. The phase stability of the oxide during the exsolution process and its influence on mechanical stability of the perovskite backbone is not well investigated. It is therefore important to understand how the loss of a significant amount of B-site cations influences the physical properties of the oxide.
In this study, we selected Sr1−x(Ti0.3Fe0.7−yNiy)O3−δ as a SOC electrode material to investigate its overall phase stability and exsolution behavior, as well as its tolerance to degradation from carbon deposition in tar-containing biogas. To enhance catalytic activity, Ni was added to the B-site to induce exsolution. To potentially address the problem of limiting electronic conductivity, particularly in the case of Sr(Ti1−yFey)O3−δ, the Fe to Ti ratio needs to be high.16,17
Based on these considerations, we utilized the stoichiometry SrTi0.3Fe0.7O3−δ (STF) and the Ni co-doped stoichiometries SrTi0.3Fe0.63Ni0.07O3−δ (STFN) and A-site substoichiometric Sr0.95Ti0.3Fe0.63Ni0.07O3−δ (sSTFN). These were also investigated by e.g. Zhu et al.18 and Santaya et al.,19 respectively, reporting promising electrochemical properties.
Our focus was on characterizing the phase formation in STF, STFN and sSTFN and its influence on mechanical stability during reduction in the temperature range of 710 °C to 900 °C. Additionally, we compared the carbon deposition tolerance in tar-containing biogas to a Ni-yttria stabilized zirconia (Ni-YSZ) cermet.
2. Materials & methods
2.1 Materials synthesis
Commercially available powders purchased from Sigma-Aldrich (St. Louis, USA) were used as precursors: SrCO3, TiO2, Fe2O3 and NiO. The purity of the powders was measured by inductively coupled plasma optical emission spectrometry (ICP-OES). On average the powders show only minor impurities <∼0.05 wt% (see Table 1, ESI†). Notable is a 2.78 ± 0.15 wt% Si contamination in NiO and a 0.67 ± 0.03 wt% Mn contamination in Fe2O3.STF, STFN and sSTFN were synthesized using the solid–state reaction route. STF was employed as a reference to compare the behavior to non Ni-doped material.
The precursors were mixed, and ball milled in ethanol with 3 mm zirconia milling balls in a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 for 24 h. The powders were dried at 80 °C for 24 h and subsequently calcined at 1200 °C for 8 h. Since the resulting powders were not phase pure, they were ball milled again for 24 h. A second calcination step at 1300 °C for 8 h was performed, resulting in a monomodal particle size distribution (see Table 1). Particle sizes were measured in ethanol with a laser scattering particle size distribution analyzer (Partica LA-950V2, Retsch Technology). An ultrasonic treatment was used to break loose agglomerates.
:1:1 for 24 h. The powders were dried at 80 °C for 24 h and subsequently calcined at 1200 °C for 8 h. Since the resulting powders were not phase pure, they were ball milled again for 24 h. A second calcination step at 1300 °C for 8 h was performed, resulting in a monomodal particle size distribution (see Table 1). Particle sizes were measured in ethanol with a laser scattering particle size distribution analyzer (Partica LA-950V2, Retsch Technology). An ultrasonic treatment was used to break loose agglomerates.
| Material | d 10 (μm) | d 50 (μm) | d 90 (μm) |
|---|---|---|---|
| STF | 0.60 | 1.10 | 3.12 |
| STFN | 0.69 | 1.41 | 5.02 |
| sSTFN | 0.55 | 1.15 | 3.33 |
2.2 Microstructure & exsolution analysis
The microstructure and exsolution behavior of the three materials was analyzed by scanning electron microscopy (SEM) using a GeminiSEM 450 (Carl Zeiss AG, Oberkochen, Germany) equipped with an energy-dispersive X-ray spectrometer (EDS) ULTIMAX 170 (Oxford Instruments, Abingdon, Great Britain). Powder samples were investigated in their as prepared (ap), reduced (red) and reoxidized states. The reduction process was carried out at 900 °C in 50% Ar/50% H2 atmosphere for 8 h. Reoxidation was performed at 900 °C in ambient air. Additionally, polished cross-sections of pellets reduced at 900 °C were investigated. To investigate the carbon degradation behavior, the microstructure of polished cross-sections of pellets reduced at 710 °C for 3 h in 100% H2 and exposed to tar-containing biogas were analyzed (see Section 2.5). Furthermore, cross-sections of these pellets reduced at 710 °C were investigated in terms of their exsolution behavior.2.3 Thermogravimetric analysis
Thermogravimetric analysis (TGA) of the materials was conducted in a NETZSCH STA 449 F1 Jupiter (Erich Netzsch GmbH & Co. Holding KG, Selb, Deutschland). Redox cycling experiments were performed at 900 °C. The samples were initially heated up in air with a heating rate of 5 K min−1. The samples were kept at 900 °C for 4 h to remove organics and reach an equilibrium state. The respective mass plateau was defined as a relative mass of 100%. Afterwards, the samples were reduced in Ar/3% H2 for 12 h. This cycle was repeated three times to study the redox behavior of the materials.Additionally, the materials were reduced in Ar/3% H2 at temperatures of 710 °C for 3 h and 900 °C for 30 h. The samples reduced at 710 °C have been reoxidized and cooled down slowly. Beforehand, the samples were reduced and reoxidized for 3 h each to remove organic impurities.
2.4 X-ray diffraction and phase analysis of redox and exsolution stability
The phase formation of the materials was analyzed. Powders were reduced at temperatures of 710–900 °C in 50% Ar/50% H2 atmosphere for 8 h. A subset of the samples reduced at 900 °C was subsequently reoxidized at 900 °C for 8 h in ambient air to assess their redox stability. X-ray diffraction (XRD) analysis of the samples was performed using a Bruker D4 endeavor X-ray diffractometer. The phase composition was analyzed using a polynomial fit to determine the position of the diffraction lines, and a subsequent Rietveld refinement method. During certain reduction experiments, Ruddlesden–Popper (RP) phases were formed. Since the exact stoichiometries of the RP-phases are unknown due to broad reflexes and material instabilities in ambient air, the stoichiometries were assumed to be Sr4Ti3O10, Sr2TiO4 (ST-RP) and Sr3Fe2O6 (SF-RP) for the purpose of Rietveld refinement to estimate the quantities of the original perovskite, the formed RP-, and nanoparticle phases.For analysis of the secondary phase formation, upon exposing the materials to ambient air or H2O, pieces of pellets that fragmented were used for the XRD analysis. These pellets were prepared analogously to the pellets prepared for carbon degradation experiments (see Section 2.5) but reduced at 900 °C in 50% Ar/50% H2 atmosphere.
2.5 Carbon degradation experiments & Raman analysis
For carbon degradation experiments, the materials were pressed into pellets with 10 wt% starch and a drop of glycerine as a binder. A uniaxial press was used to apply a pressure of 60 MPa for 120 s, resulting in pellets with a diameter of 25 mm. The pellets were sintered at 1300 °C for 4 h, yielding samples with a diameter of ∼20 mm. The heating rate was initially set to 2 K min−1 up to 500 °C to remove the binder. Subsequently, the heating rate was increased to 5 K min−1.The porosity of the pellets was investigated using the Archimedes principle according to DIN 66137-3 and by image analysis of cross-sections using the software Fiji.
The carbon degradation behavior was investigated in a self-built tube reactor invented by the Technical University of Munich. The samples were placed on a Ni-mesh on top of a heated double tube. The gas flow originates from the bottom of the outer tube, flows through the sample, and leaves the reactor through the inner tube. The gas composition is controlled by mass flow controllers (EL-Flow Prestige, Bronkhorst Deutschland GmbH). For the H2O fraction a liquid flow controller (Liqui-Flow, Bronkhorst Deutschland GmbH) was used, since it is vaporized shortly before mixing with the other components.
The pellets were reduced in situ at 710 °C (the maximum temperature reached by the test rig at that time) for 3 h in 100% H2 atmosphere, since pellets pre-reduced at 900 °C fragmented during transport. 18 g N m−3 of toluene as a model tar was used to assess the degradation behavior of the materials. The tar was dosed to an artificial bio-syngas for 3 h. The artificial biogas contained 25 vol% H2/10 vol% CO2/10 vol% CO/5 vol% CH4/50 vol% H2O. Between changing the gas atmospheres, the sample chamber was purged with N2.
After carbon degradation experiments, Raman analysis was conducted on all materials using an inVia Qontor distributed by Renishaw. Several point measurements were performed with a 532 nm laser. Data was collected in a wavenumber range of ∼70–800 cm−1 and ∼1260–2300 cm−1.
2.6 Conductivity measurements
The ohmic resistances of the materials have been estimated using electrochemical impedance spectroscopy (EIS). The high-frequency intercept was evaluated to estimate the total ohmic resistance of the sample. Compacted samples were used, similar to the ones used for degradation experiments (see Section 2.5). Impedance measurements were conducted in Ar/3% H2 at temperatures between 600 and 900 °C. Frequencies between 106–10−1 Hz were applied with a Novocontrol Alpha-A frequency analyzer in a tube furnace. A Pt layer was applied on top and bottom of the pellets to improve contacting between the samples and the Pt-mesh electrodes of the potentiostat.3. Results
3.1 Powder characterization
After one calcination step, the powders contained some secondary phases identical to the precursor materials. After a second ball milling and calcination step, STF and STFN powders were phase pure and showed no additional reflexes according to XRD. For sSTFN minor reflexes are present indicating NiO (see Fig. 1). Broad reflexes around ∼12.5–20° are due to adhesive tape used for sample preparation. | ||
| Fig. 1 Diffractograms of the as prepared STF, STFN and sSTFN powders. Yellow diamonds denote NiO reflexes. | ||
The stoichiometry and purity of the synthesized powders were investigated by ICP-OES. The nominal stoichiometries according to ICP-OES are given in Table 2. The synthesized powders showed some minor impurities (Si, Co, Mn and main group II elements), most likely due to impurities in the precursor materials.
| Material | Sr (mol%) | Ti (mol%) | Fe (mol%) | Ni (mol%) | Impurities (mol%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Target value | Actual value | Target value | Actual value | Target value | Actual value | Target value | Actual value | ||
| STF | 50.0 | 50.5 ± 1.5 | 15.0 | 14.9 ± 0.3 | 35.0 | 34.0 ± 0.9 | — | — | ∼0.6 |
| STFN | 50.0 | 50.4 ± 1.3 | 15.0 | 15.1 ± 0.4 | 31.5 | 30.7 ± 0.9 | 3.5 | 3.3 ± 0.2 | ∼0.5 |
| sSTFN | 48.7 | 49.1 ± 1.4 | 15.4 | 15.4 ± 0.4 | 32.3 | 31.6 ± 1.1 | 3.6 | 3.4 ± 0.1 | ∼0.5 |
The synthesized and ball milled STF powder (as prepared) consists of square shaped, sharp-edged particles. Furthermore, some relatively small primary particles <50–100 nm are present (see Fig. 2).
 | ||
| Fig. 2 SEM micrographs for STF, STFN and sSTFN in (a, d, g) as prepared, (b, e, h) reduced at 900 °C in 50% Ar/50% H2 and (c, f, i) reoxidized state. | ||
The as prepared STFN and sSTFN powders exhibit a more rounded shape compared to STF. Particle size distribution is comparable to STF (see Table 1). Similar to STF, some small primary particles are visible. Additionally, if Ni is present in the perovskite some lichen-like, melted-looking phases form on the facet of the particles and at the grain boundaries (see Fig. 2(d); additional SEM micrographs are shown in Fig. 1, ESI†). Theses lichen-like structures show similar material contrast to the bulk in backscattered electron (BSE) micrographs (see Fig. 1(b), ESI†).
Some minor grains are observed which do not seem to belong to the perovskite phase. We assume these grains belong to leftovers of unreacted NiO precursors since these particles do not exsolve nanoparticles upon reduction and due to the indication of NiO reflexes for as prepared sSTFN.
3.2 Microstructure and exsolution analysis
SEM analysis was performed on powder samples reduced and reoxidized at 900 °C. The SEM micrographs are shown in Fig. 2. Initially, it was intended to use pellets pre-reduced at 900 °C for carbon degradation experiments. The pre-reduced pellets fractured overnight and during transport after being exposed to ambient air (further discussed in Section 3.5). Cross-sections of pieces of these fractured pellets were investigated (see Fig. 3). Therefore, pellets were reduced in situ at 710 °C (maximum temperature of the test rig), where they remained stable. After carbon degradation experiments, cross-sections of these pellets were examined (for an example of STFN, see Fig. 4). Due to material instabilities, low accelerating voltages had to be chosen, which resulted in low signal-to-noise ratios. In addition, lower accelerating voltages improve lateral resolution. Therefore, the accelerating voltage was adapted according to the samples' properties and desired resolution. In case the accelerating voltage was insufficient to resolve the respective spectral line, the signals were omitted from the analysis. | ||
| Fig. 3 Cross-sections of (a) STF reduced at 900 °C in 50% Ar/50% H2, (b) STFN reduced at 900 °C in 50% Ar/50% H2 and (c) STFN reduced at 900 °C in 50% Ar/50% H2 and reoxidized at 900 °C in air, analyzed with EDS. | ||
 | ||
| Fig. 4 EDS analysis of a cross-section of STFN reduced at 710 °C in 100% H2, analyzed with EDS (black spot belongs to a pore). | ||
STF powder forms Fe nanoparticles with a diameter of ∼50–130 nm upon reduction at 900 °C. They seem to be randomly distributed on the facets of the grains. The number of particles tends to decrease after reoxidation. A fraction of the particles remains stable but some agglomerate. In contrast to XRD analysis, nanoparticles are still present after reoxidation (the fraction might just be too low to be resolved with XRD). During reduction and reoxidation, parts of the grains start to form small cracks in investigated powder.
SEM and EDS analyses of cross-sections (of a fractured pellet) reduced at 900 °C show severe crack formation in the structure (see Fig. 3). It is visible that exsolution is not just limited to the surface, but also bulk exsolution takes place. Furthermore, some larger Fe particles are present. There seem to be spots where the number of nanoparticles increase (as in STFN) but this needs more detailed investigation.
A pellet reduced at 710 °C shows no cracks (see Fig. 2, ESI†). It is not clear whether Fe nanoparticles exsolve at 710 °C since the as prepared material contains small primary particles. Exsolution takes place sporadically at best.
STFN powder forms NiFe nanoparticles with a diameter of ∼30–130 nm upon reduction at 900 °C. They uniformly cover the whole area of the grains. Exceptions are the unreacted NiO precursor particles which do not exsolve nanoparticles (see Fig. 2(e)). Additionally, the average size of the nanoparticles in STFN appears to be smaller when compared to STF. After reduction, the material forms cracks similar to STF at 900 °C. The lichen-like phases on the surface vanish.
The material cracks further during reoxidation. The nanoparticles partially agglomerate, resulting in the formation of larger particles with a diameter of up to approximately 850 nm (see Fig. 2(f)). In the vicinity of the agglomerated particles, the surface shows a depletion of nanoparticles.
Cross-sections of a fractured STFN pellet reduced at 900 °C show bulk exsolution (see Fig. 3(b)). For better visibility of the nanoparticles backscattered electron micrographs with a higher magnification are shown in the ESI,†Fig. 3. The structure shows prominent cracking after reduction and reoxidation. The reason why the cross-sections of the pellets reduced at 900 °C are not flat and look aged will be discussed in Section 3.5.
Powder and pellets reduced or reduced and reoxidized at 900 °C exhibit larger particles (∼500–1500 nm). These particles primarily consist of an Ni/Fe alloy, while some of the larger particles solely consist of Ni (see Fig. 3(c)).
EDS mapping of the cross-section of a pellet reduced at 710 °C reveals that Ni nanoparticles nucleate at the grain boundaries (see Fig. 4). It is not clear if Ni also segregates at grain boundaries of the as prepared material or if this accumulation is solely due to exsolution. Compared to pellets or powder reduced at 900 °C no or just some minor cracks between the grains form after reduction and reoxidation.
sSTFN powder and pellets behave similarly after reduction and reoxidation compared to STFN (see Fig. 2(g–i)).
EDS analysis of a cross-section of the pellet reduced at 900 °C reveals bulk exsolution (see Fig. 4, ESI†). Similarly, the pellet reduced at 710 °C shows preferable nucleation of Ni at the grain boundaries (see Fig. 5(a), ESI†).
All materials exhibit a notable increase in crack formation and Fe exsolution when reduced at 900 °C which is not observable after reduction at 710 °C.
3.3 Thermogravimetric analysis
The thermal decomposition behavior of the materials was investigated by thermogravimetric analysis in air and Ar/3% H2. The results of the cycling experiments are shown in Fig. 5(a). As can be seen in Fig. 5(a), some loss of mass typically occurs during the first heating ramp, which is likely due to the evaporation of contaminations like humidity or carbonate species, and the loss of oxygen during heating. In order to unambiguously correlate mass loss to oxygen loss in the isothermal measurements, samples were exposed to cycling experiments, where the materials were reduced for 12 h in Ar/3% H2 followed by reoxidation for 4 h in air, repeated three times consecutively. The reversible mass loss in Ar/3% H2 and mass gain in air is clearly due to reduction and oxidation, therefore the reversible mass loss in the TGA experiments can be clearly assigned to oxygen loss. | ||
| Fig. 5 TGA analysis of STF, STFN and sSTFN. Blue regions represent air atmosphere, while white regions represent Ar/3% H2 atmosphere. (a) Cycling experiments conducted at 900 °C, during which the materials underwent a reduction phase for 12 h followed by a subsequent reoxidation period for 4 h. This cyclic process was repeated three times. (b) Reduction of the materials at 900 °C for 30 h. (c) Reduction of the materials at 710 °C for 3 h, followed by 1 h of reoxidation and subsequent cooling down to room temperature (25 °C). | ||
At a reduction temperature of 900 °C, none of the materials reach an equilibrium oxygen non-stoichiometry δ after 12 h. At the beginning of the reduction, an abrupt initial mass loss in the first ∼5–10 min is observable, followed by a constant but more moderate mass loss. The initial mass loss is ∼1.2% for STF, ∼1.6% for STFN and ∼1.7% for sSTFN. After 12 h reduction, STFN exhibits a higher mass loss compared to STF (∼0.3%) while sSTFN loses about 0.6% more mass than STF. After reoxidation, the materials tend to reach their initial mass again.
Since an equilibrium was not reached after 12 h at 900 °C in the cycling experiments, the materials were further reduced for 30 h at 900 °C in Ar/3% H2 (see Fig. 5(b)). Again, an abrupt initial mass loss occurred, but even after 30 h, no equilibrium was reached at 900 °C. Initially, STFN exhibited a faster mass loss compared to STF, but both materials ended up with a mass loss of approximately 8% after 30 h of reduction. sSTFN experienced a mass loss of around 8.5% after 30 h of reduction. The initial mass loss (pre-reduction) is assumed to be due to the reduction and oxygen release of the materials without metal exsolution. The subsequent constant mass loss, which is likely kinetically limited compared to the materials' pre-reduction, is attributed to further oxygen release accompanied by metal exsolution. The exsolution process is initiated after a critical amount of oxygen is released from the lattice, resulting in the reduction of the cations.3 The results indicate that the materials constantly release oxygen and potentially exsolve nanoparticles while not reaching an equilibrium at 900 °C after 30 h of reduction.
When the materials are reduced at 710 °C for 3 h (see Fig. 5(c)), the initial abrupt mass loss was still observed and is comparable to the reduction at 900 °C. The subsequent mass loss is significantly lower compared to reduction of the materials at 900 °C (approximately 2.5% for STFN and sSTFN and 2.1% for STF). The samples tend to almost reach an equilibrium at 710 °C but due to the short reduction time the mass loss did not fully equilibrate. The initial oxygen release provides the driving force (critical oxygen deficiency) for the exsolution of metallic cations.20 However, the equilibrium of the oxygen release accompanied by exsolution is likely temperature-dependent.
In the upcoming Section 3.4 we will also provide data that reveal a phase transition if the materials are reduced above a critical temperature. We occasionally observe drifts in the mass signal during the experiments, such as mass gain after reduction and during exsolution. Currently, we attribute these drifts to measurement artifacts like buoyancy or an unsteady gas atmosphere.
The equilibrium oxygen non-stoichiometry δ of the materials was determined based on the observed mass plateaus. To calculate δ, the thermodynamic model of Wagner21 was employed, which was adapted by Nenning et al.22 Therefore, some assumptions were made to calculate δ. The differential 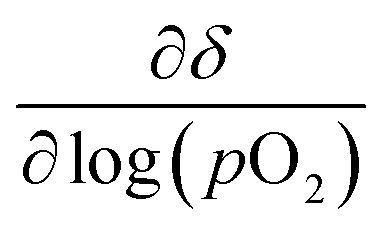 is minimized when the respective cations are present in their oxidation states of Sr2+, Ti4+ and Fe3+. The minimized value corresponds to the measured mass plateaus or an equilibrium oxygen stoichiometry under the given measurement conditions. In our case, this leads to a δ of 0.350, 0.455 and 0.505 for STF, STFN and sSTFN, respectively. That the cations Sr and Fe in fact prefer the above-mentioned oxidation states was confirmed by Mössbauer spectroscopy.23,24 Although we were not able to obtain p(O2) values during our measurements, it can be assumed that
is minimized when the respective cations are present in their oxidation states of Sr2+, Ti4+ and Fe3+. The minimized value corresponds to the measured mass plateaus or an equilibrium oxygen stoichiometry under the given measurement conditions. In our case, this leads to a δ of 0.350, 0.455 and 0.505 for STF, STFN and sSTFN, respectively. That the cations Sr and Fe in fact prefer the above-mentioned oxidation states was confirmed by Mössbauer spectroscopy.23,24 Although we were not able to obtain p(O2) values during our measurements, it can be assumed that  is sufficiently p(O2)-independent at 710 °C.25,26 According to these assumptions, we calculated the oxygen stoichiometries for the samples close to equilibrium in air at 710 °C and after cooling down in air to 25 °C (see Table 3).
is sufficiently p(O2)-independent at 710 °C.25,26 According to these assumptions, we calculated the oxygen stoichiometries for the samples close to equilibrium in air at 710 °C and after cooling down in air to 25 °C (see Table 3).
| Material | Reduced 3 − δ (710 °C) | Oxidized 3 − δ (710 °C) | Oxidized 3 − δ (25 °C) |
|---|---|---|---|
| STF | 2.650 | 2.872 | 2.965 |
| STFN | 2.545 | 2.821 | 2.918 |
| sSTFN | 2.495 | 2.768 | 2.852 |
To calculate the δ-values for the Ni-doped compositions STFN and sSTFN, we assume that Ni is fully exsolved at the equilibrium state under low p(O2) (which is not the case according to XRD analysis presented in Section 3.4 and indicated by EDS analysis in Fig. 4).
In its fully oxidized state, Fe is conventionally presumed to be present as Fe4+. However, due to suboptimal cooling conditions, the average oxidation state of Fe might diverge from Fe4+.
3.4 Redox and exsolution stability
The phase stability during reduction was investigated using XRD analysis. The diffractograms are shown in Fig. 6. The calculated relative phase distributions, according to Rietveld refinement, are shown in Fig. 7 (the exact values are shown in Table 2; ESI†). Upon reduction, all materials exhibited broadening and a shift towards lower 2θ angles of the perovskite reflexes, indicating increased lattice disorder and chemical expansion, respectively, due to the release of oxygen from the lattice. At a temperature of 780 °C, the emergence of metallic Ni/Fe reflexes was observed for STF and STFN, indicating the process of exsolution. In the case of sSTFN, metallic Ni/Fe reflexes were observable starting at 710 °C. Since Ni and Fe reflexes share similar 2θ angles and the corresponding peaks in this analysis are broadened, presumably due to the exsolution of either Ni, Fe or their alloys in different compositions, the fraction of metallic reflexes was summed up for the purpose of the analysis.27,28 The amount of exsolved nanoparticles is higher for sSTFN. In SEM micrographs of pellets used for carbon degradation experiments, the exsolution of nanoparticles is visible starting at 710 °C for STFN and sSTFN (see Fig. 4). For STF exsolution appears at best sporadically at 710 °C. Due to the detection limit of XRD being approximately 1–2 vol%, the method is not suited to resolve the nanoparticle fraction at reduction temperatures below 780 °C. For sSTFN the exsolution process is even detectable at 710 °C with XRD, indicating that exsolution is thermodynamically favored compared to the stoichiometric material. The nanoparticle reflexes shift with rising temperature, presumably due to the higher iron content in the Ni/Fe nanoparticle alloys. These reflexes become more prominent with rising temperature. Furthermore, with rising temperature the perovskite reflexes broaden, and at 780 °C, reflexes related to a Ruddlesden–Popper (RP) phase appear for STFN and sSTFN. In the case of STF, a sudden transition to the RP phase at a higher temperature of 860 °C occurs, coupled with a rapid increase in Fe nanoparticle exsolution. The slower exsolution response observed in STF, compared to STFN and sSTFN, suggests that the RP phase transition occurs after a certain amount of oxygen release and cation exsolution has taken place. For all materials the RP-reflex is split up again at 860 °C, indicating the formation of two RP phases. Interestingly, at 900 °C the amounts of RP-phases and nanoparticles decrease. As the measurements were not conducted in situ but rather with similar but separate samples of the same material sintered at different temperatures sequentially, it is possible that the observed effect could be related to the samples themselves. Since samples of the three different stoichiometries were sintered together at the respective temperatures in the same furnace, it is conceivable that issues related to furnace temperature gradients or the Ar/H2 gas atmosphere heterogeneities may have arisen. This issue will require further investigation. In general, a temperature dependency of the exsolution behavior and the phase transition is evident.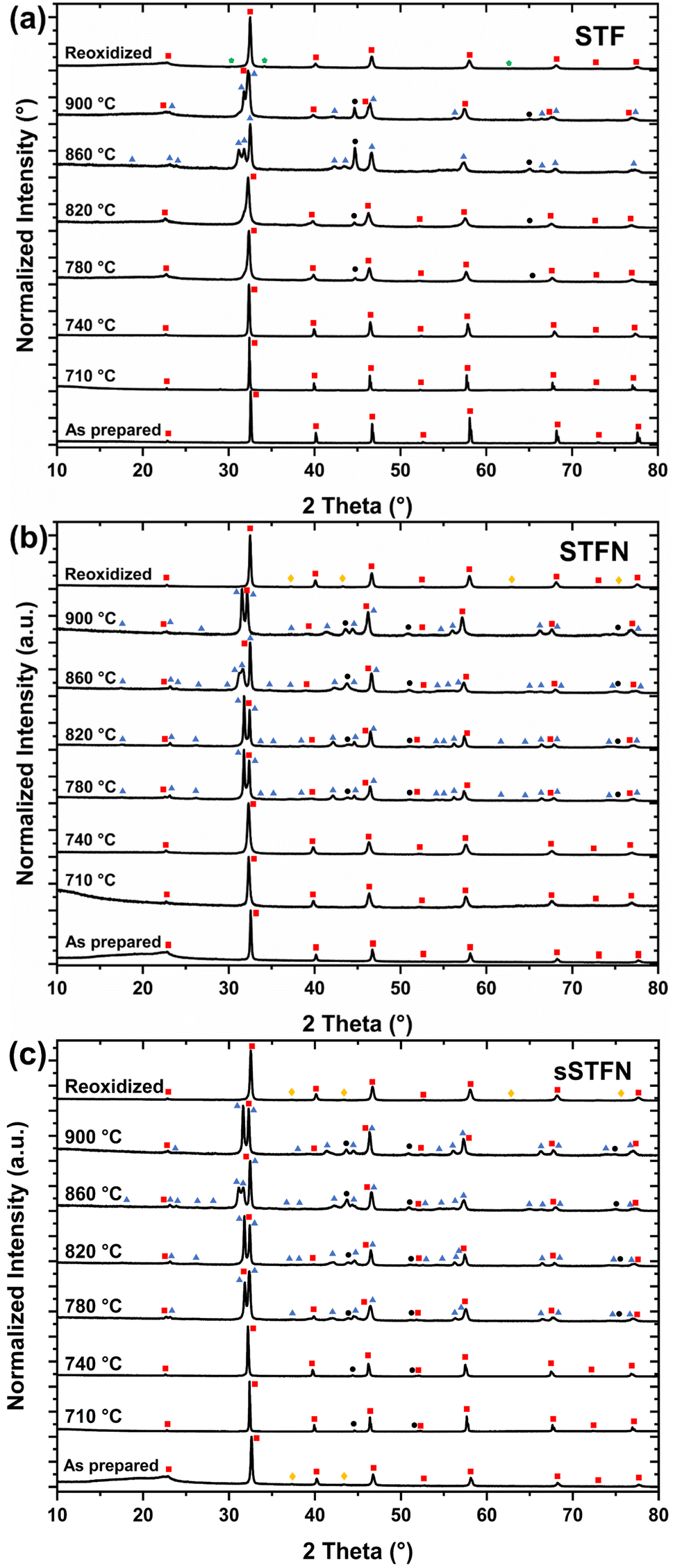 | ||
| Fig. 6 Diffractograms for (a) STF, (b) STFN and (c) sSTFN in their as prepared, reduced (at 710–900 °C in Ar/50% H2) and reoxidized (at 900 °C for 3 h in air) state. Red squares denote the perovskite phase, blue triangles Ruddlesden–Popper phases, black circles metallic Fe/Ni phases, green pentagons denote a strontium ferrite perovskite phase, and yellow diamonds iron or nickel oxide phases. | ||
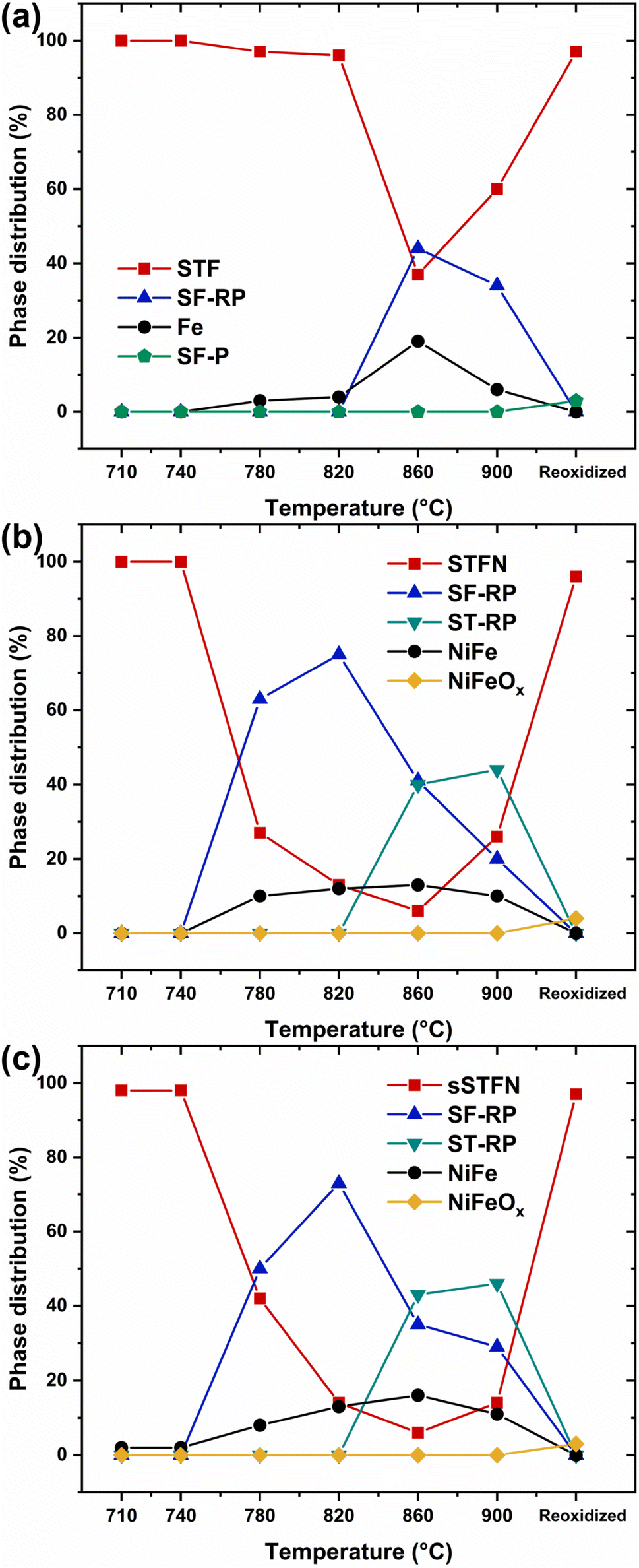 | ||
| Fig. 7 Relative phase distribution for (a) STF, (b) STFN and (c) sSTFN reduced at temperatures of 710–900 °C in 50% Ar/50% H2 for 8 h and reoxidized at 900 °C in air for 8 h. | ||
The samples reduced at 900 °C were subsequently reoxidized. After reoxidation STFN and sSTFN transition back to the perovskite phase, although parts of the exsolved nanoparticles remain on the surface and form oxides. According to XRD the exsolved fractions decrease after reoxidation. For STF, small amounts of a strontium ferrite perovskite phase (SrFe12O19, SF-P) form, but most of the material transitions back to the original perovskite phase, while no metal oxide fraction is detectable. This indicates a partial reintroduction of the exsoluted metals back into the perovskite lattice. It should be noted that, due to the broad nature of the reflexes, the assignment of the phases for the purpose of Rietveld refinement is not distinct. Therefore, the calculated relative phase distributions indicate a broader trend.
3.5 Mechanical and moisture stability
Upon reduction at 900 °C, all materials form cracks in the grains. Furthermore, pellets that were reduced at 900 °C fractured and completely disintegrated during prolonged exposure to ambient air (4 weeks) or immersion in H2O (24 h). Pellets reduced at 710 °C maintained mechanical stability. Diffractograms for sSTFN samples recently reduced, after four weeks of storage in ambient air, and after 24 h exposure to H2O are shown in Fig. 8. Samples stored in ambient air or immersed in H2O show broadened reflexes. Additionally, some new minor reflexes appear. The diffractograms indicate the formation of strontium hydroxides and iron(oxide)hydroxides. However, due to the broadened reflexes, the precise determination of the emerging side phases by XRD alone is not feasible. After embedding, grinding (using water as a coolant), and polishing, the prepared cross-sections exhibit signs of aging, displaying an irregular and non-flat surface, revealed in the subsequent SEM analyses (see Fig. 3, additionally secondary electron (SE) micrographs with topographic information are shown in Fig. 6, ESI†). This further indicates that the materials are not stable in atmospheric conditions or moisture/contact with H2O. These phenomena are not observed for pellets reduced at 710 °C, suggesting that the mechanical instabilities are triggered by pronounced metal exsolution, the accompanied phase transitions and potential secondary phase formation within the materials. | ||
| Fig. 8 Diffractograms for sSTFN recently reduced (at 900 °C in 50% Ar/50% H2), reduced and stored in ambient air for 4 weeks or immersed into H2O. Brown stars denote potential secondary phase reflexes (e.g., strontium hydroxides or iron(oxide)-hydroxides). | ||
3.6 Carbon degradation behavior & Raman analysis
Due to the mechanical instabilities of pellets reduced at 900 °C and heating limitations of the test rig, pellets for carbon degradation experiments were reduced in situ at 710 °C.The fabricated pellets possess a porosity of approximately 25–28 vol%, which is comparable to the reference Ni-YSZ substrate material.
For the reference Ni-YSZ substrate material, severe degradation in terms of carbon deposition and Ni dusting was observed. The whole sample decomposed and turned black. Fig. 9(d) shows a SEM micrograph of the carbon-degraded sample. The Ni grains show carbon deposition in the form of fibers which disintegrate the microstructure. Raman measurements confirmed carbon in the structure due to the presence of the amorphous and graphitic carbon D′ and G′ band signals at ∼1350 and ∼1580 cm−1 (see Fig. 10(h)).29,30
 | ||
| Fig. 9 SEM micrographs of (a and b) reduced (710 °C) and (c) reoxidized STFN and (d) 8YSZ after carbon degradation experiments. (a and d) Show SE and (b and c) show backscattered electron (BSE) micrographs. | ||
 | ||
| Fig. 10 Raman spectra for (a and b) STF, (c and d) STFN, (e and f) sSTFN and (g and h) Ni-8YSZ in reduced state (red) and after carbon degradation experiments (tar). | ||
In contrast to Ni-YSZ, STF as well as STFN and sSTFN show no signs of carbon-related degradation or Ni dusting either in the bulk or the nanoparticles. SEM micrographs show the clean material surfaces without any presence of carbon deposition (see Fig. 9(a) and (b)). Raman spectra taken at different spots of the samples confirm the absence of carbon D′ and G′ band signals. It should be noted that potentially formed carbon species on the materials might have decomposed during cooling.
Moreover, we provide Raman data for the reduced materials stoichiometries, up to a wavenumber of 2300 cm−1 (see Fig. 10). The detection of vibrational modes for the materials provides evidence that they deviate from the ideal cubic perovskite structure.31 Metallic Ni exhibits no allowed Raman mode. Peak broadening may be attributed to the presence of cationic defects and oxygen vacancies. Defects might not be distributed homogeneously throughout the sampling area. The signals in the range of 200–400 cm−1 and 550–800 cm−1 are related to Ti–O bonds. Signals around 200, 300, 600 and 700 cm−1 are related to Fe–O bonds, but they partly overlap with the Ti–O signals.24,32–34 All materials exhibit a broad signal around 1440 cm−1 which could be related to two magnon scattering (observed in SrFeO3−δ at ∼1350–1400 cm−1).24 The obtained spectra exhibit a low signal-to-noise ratio, likely attributed to a certain roughness of the surfaces of the investigated pellets. Furthermore, some signals shift slightly, likely induced by locally varying chemical configurations due to different sampling spots.
Following carbon degradation experiments, transformations of the materials' surfaces were observed that were in contact with Ni. In the tube reactor, Ni meshes are commonly used for sample placement, given the presence of Ni in common electrode materials or substrates for SOCs. The surface of all materials that are in direct contact with the Ni-mesh forms a melted-looking layer after degradation experiments. This layer still consists mainly of bulk perovskite (see Fig. 7, ESI†). After reoxidation, this layer shrinks and forms islands (see Fig. 9(c)).
3.7 Conductivity
STF shows the highest overall conductivity, followed by STFN and sSTFN (see Fig. 11). The higher iron content of STF seems to have a higher impact on the total conductivity compared to further doping with Ni and exsolved Ni/Fe nanoparticles. The obtained values are in the same order of magnitude compared to literature,15,35,36 although the samples have a certain porosity (∼25%).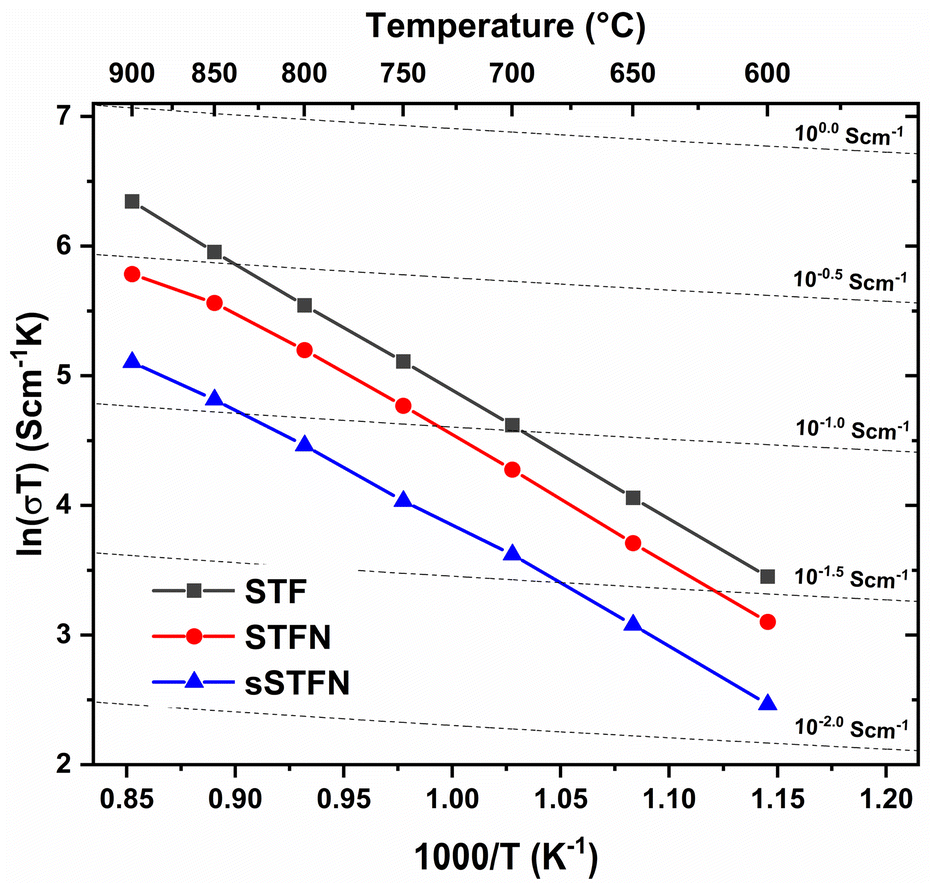 | ||
| Fig. 11 Arrhenius plot of the specific conductivity for STF, STFN and sSTFN, measured at temperatures of 600–900 °C in Ar/3% H2. | ||
4. Discussion
As we have examined the materials as potential candidates for SOC electrodes (or other suitable electrochemical devices) to be used in practical applications, the discussion will primarily focus on the results in this context. When considering a material for a SOC electrode, certain requirements need to be met, with cost aspects being neglected for the time being. These requirements include: (i) sufficient electrical and ionic conductivity, (ii) sufficient catalytic activity, (iii) degradation and carbon deposition tolerance and (iv) electrochemical, chemical and mechanical stability during fabrication and operation conditions.We will discuss the suitability of STFN and sSTFN as SOC electrode materials in the following. In this study, we mainly focused on carbon deposition tolerance and stability-related aspects.
Data for electrochemical evaluation have been reported elsewhere.18,19
Synthesis of a relative phase pure STF, STFN and sSTFN is rather uncomplicated if the precursors are milled down to similar particle sizes and if a calcination temperature of at least 1200 °C is employed. Impurities and secondary phases are below the detection limit of XRD. Some secondary phases and left-over precursors (primarily NiO) are present, potentially due to the solubility limit of Ni. For SrTiO3−δ, the suggested solubility limit is about 5 atom%.37 Minor impurities of NiO are unlikely to significantly impact the electrochemical performance. It has been demonstrated that lower calcination temperatures (<1100 °C) can lead to the formation of Ni perovskite secondary phases and RP phases in Ni-doped SrTiO3−δ.38
Interestingly, the SEM analysis indicates that Ni(O) has an influence on the sintering/calcination behavior and potentially induces the formation of a transient liquid phase with the perovskite. This assumption is based on the presence of the lichen-like structures on the grains of Ni-doped STF after calcination, resembling melted material that solidified upon cooling. Additionally, similar melted-like structures were observed on pellets that were in contact with a Ni-mesh during carbon degradation experiments. The presence of Ni(O) is known to serve as a sintering aid and enhance cation diffusion in perovskite materials, as observed in compounds like Ba(Zr,Ce,Y)O3−δ.39–41 This behavior has also been documented in fluorite-type materials such as yttria-stabilized zirconia42 or gadolinia-doped ceria.43–46
After an initial pre-reduction of the materials, exsolution takes place in reducing conditions starting at 710 °C (for Fe-exsolution in STF probably later around 740–780 °C). At 710 °C the oxygen release and exsolution of metals reach an equilibrium. Reduction at 900 °C leads to an increase in exsolved nanoparticles while no equilibrium is reached within 30 h of reduction, indicating continuous oxygen release and possibly exsolution beyond this point. The exsolution process is triggered by the oxygen loss of the host material. Therefore, after a certain pre-reduction, exsolution begins. It is expected that a higher degree of oxygen non-stoichiometry and faster oxygen release kinetics will enhance the amount of exsolved nanoparticles.20
The exsolution of Ni (and probably Fe) in STFN is thermodynamically favored in comparison to Fe exsolution in STF. Furthermore, the exsolved Fe nanoparticles in STF are fewer by number and larger on average compared to Ni/Fe nanoparticles exsolved in STFN and sSTFN. The doping with Ni reduces the onset temperature for Fe reduction.47 The Sr-substoichiometry further enhances the amount of released oxygen and favors metal exsolution. Due to the high nanoparticle coverage observed on both STFN and sSTFN (particularly after reduction at 900 °C), it is reasonable to assume a certain level of catalytic activity.
All materials undergo a phase transition from the perovskite to a RP phase when the reduction temperature exceeds about 820–860 °C for STF and 740–780 °C for STFN and sSTFN. This phase transition is accompanied or induced by an increase in exsolved nanoparticles. The calculated relative phase distributions indicate that sSTFN is slightly more stable compared to STFN since the formed amounts of RP-phases is lower, while the nanoparticle fraction is even larger. Notably, the investigated pellets reduced at 710 °C, which exhibit preferential Ni exsolution for STFN and sSTFN, do not undergo a phase transition. This suggests that the RP-phase transition may be triggered by the exsolution of a critical amount of Fe. Furthermore, the transition of STF to the RP phase is accompanied by a sharp increase in the content of Fe nanoparticles.
The conductivities of the synthesized materials are in the same order of magnitude compared to literature (see Section 3.7). Although the conductivity is improved by a high Fe content in the perovskite, it still might be insufficient since the common La0.6Sr0.4Co0.2Fe0.8O3−δ SOC air electrode material exhibits approximately two orders of magnitude higher conductivity.17 Therefore, further doping may be required where Co might be a suitable candidate.48
Regarding the carbon degradation tolerance in tar-containing biogas, the materials show promising properties. In fact, none of the materials exhibit any degradation under the same experimental conditions, in contrast to a common Ni-YSZ cermet. The improved degradation tolerance is probably related to socketing of the nanoparticle and a strong host–particle interaction.9 These factors hinder the tip-growth mechanism of carbon, wherein carbon dissolves into the Ni-lattice and grows at the Ni-oxide interface, thereby suppressing carbon deposition.49
Regarding all the above discussed properties STFN and sSTFN might be suitable SOC electrode materials.
One of the major challenges associated with STFN and sSTFN as metal exsolution electrodes, both in practical SOC applications and potentially in other fields of use, is their chemical and mechanical stability. At a certain degree of oxygen non-stoichiometry and accompanied metal exsolution the perovskite transitions to RP-phases. These phase transitions are temperature-dependent and initiated at temperatures around 740–780 °C for STFN and sSTFN (and 860 °C for STF). Importantly, these temperatures fall within the range of typical application temperatures for SOC materials. The Sr-substoichiometric sSTFN seems to be slightly more stable compared to STFN, indicating that the initial A-site deficiency stabilizes the host during exsolution of B-site cations. The phase transition might not be a hindrance in terms of conductivity.50
The essential problem is the mechanical stability of compacted or sintered structures. The processed pellets were not mechanically stable in a sense that they fragmented after reduction at 900 °C, despite having a porosity of approximately 25%. Samples reduced and reoxidized at 900 °C show severe cracking. Furthermore, Fe exsolution tends to increase (compared to pellets reduced at 710 °C). The transition back to the perovskite phase during reoxidation, along with the accompanying volume change, is likely to further worsen the materials' stability over multiple redox cycles. Furthermore, agglomeration of nanoparticles was observed after reoxidation, which is likely to intensify during subsequent redox cycling.
The long-term exposure to ambient air or short-term contact with H2O exacerbates fragmentation of the materials. Considering the operational temperature of a SOC reaching up to 900 °C and the presence of water vapor during SOC applications, either in fuel cell or in electrolysis mode, STFN and sSTFN might lack phase and mechanical stability under operating conditions. At first, the present p(O2) and temperature lead to the desired exsolution but potentially also to further oxygen release and RP-phase transition. Subsequently, when exposed to a certain water vapor partial pressure (p(H2O)), secondary phase formations can take place. In a real electrode structure, this would lead to deterioration of the microstructure, potentially worsening the contact at interfaces between the electrode and electrolyte. Furthermore, the loss of contact between individual grains can significantly impact the ionic and electronic percolation paths. In the worst-case scenario, the entire electrode may be destroyed.
Nevertheless, porous and thin layers on a mechanically stable substrate might be more tolerant against internal stresses as Zhu et al.18 were able to measure electrochemical properties in model cells up to 850 °C. Notably, their Ni-exsolution electrode layers were pre-reduced in situ at 850 °C. Furthermore, under real operating conditions the fuel gas is balanced by steam which might stabilize the materials by equilibrating the oxygen release and thus the nanoparticle exsolution and RP phase transformation. Therefore, further experiments need to be conducted under more realistic operating conditions.
There might be some reasons for the observed mechanical instability and cracking of the materials reduced at 900 °C: (i) the RP phase transition during reduction (and reoxidation) resulting in a volume change of the unit cell which induces strain in the material, (ii) the loss of oxygen during reduction and the possible oxygen uptake in ambient air or water leads to chemical strain, (iii) during exposure to ambient air and moisture (or H2O) segregated Sr(O) (a common phenomenon in perovskites and shown for STFN19 and SOC air electrode like (La,Sr)(Co,Fe)O3−δ51) and exsolved Fe/Ni nanoparticles form secondary phases, e.g. strontiumhydroxides52 and iron(oxide)-hydroxides53 (corrosion). Since nanoparticles have a high surface area and the forming secondary phases might have large volumes, the pellets fragment. For example, Fe(OH)2 possesses a volume 3.5 times higher than Fe, and Fe3O4 has a volume 5.9 times higher than Fe.54 The reactions are favored in H2O compared to ambient air.
It remains unclear which of these factors is the predominant cause of the mechanical instability observed. The observed bulk exsolution, especially of Fe, could be a severe problem regarding mechanical stability, especially if the mentioned secondary phases form.
Pellets of all materials, reduced in situ at 710 °C, seem to be mechanically stable, meaning they did not fragment. They do not reveal pronounced Fe bulk exsolution compared to pellets reduced at 900 °C. This indicates that (bulk) exsolution, phase transitions, and the accompanied effects like secondary phase formation and volume changes are the triggers for crack formation.
This investigation highlights the existence of a temperature and p(O2) range within which STFN and sSTFN can be potentially utilized for SOC operations. It is reasonable to expect that other exsolution materials may also possess such operating windows where the perovskite remains stable while exsolution takes place. Consequently, greater emphasis should be placed on evaluating the (mechanical) stability of potential SOC materials in the context of exsolution. Although the initial properties of STFN and sSTFN appear promising for SOC applications, their practical applicability is potentially compromised by the necessity to operate within a specific temperature range and by the presence of low and high H2O partial pressures. To use STFN and sSTFN in an SOC, either the operation temperature must be lowered or precise control of the exsolution process and phase stability may be required, (potentially by restricting exsolution to the material's surface or a refinement of the microstructure) while maintaining its desirable properties.
5. Conclusion
SrTi0.3Fe0.7O3−δ and its Ni-doped analogues SrTi0.3Fe0.63Ni0.07O3−δ and Sr0.95Ti0.3Fe0.63Ni0.07O3−δ were successfully synthesized as potential SOC exsolution fuel electrode materials. Most of the materials were phase pure, according to XRD, but some minor particles of the precursors were still present, presumably NiO. The total conductivity is comparable to literature values. The exsolution of Ni/Fe nanoparticles was observed at temperatures of 710–900 °C. Samples reduced at 710 °C show preferential Ni nucleation at grain boundaries. Bulk exsolution occurred, particularly at a reduction temperature of 900 °C and pronounced in the case of Fe.TGA analyses under reducing atmosphere reveal an abrupt mass loss in the beginning, attributed to the pre-reduction of the materials, which triggers the exsolution process. The materials tend to reach an equilibrium oxygen non-stoichiometry at 710 °C, while for reduction at 900 °C, no equilibrium was reached, even after 30 h indicating continuous oxygen release, possibly associated with exsolution.
Above a certain threshold temperature (740–780 °C for STFN and sSTFN and 840–860 °C for STF) the perovskites underwent phase transitions to Ruddlesden–Popper phases. These phase transitions were accompanied by mechanical instabilities. Cracking of the materials was observed after reduction at 900 °C. Compacted and sintered pellets fragmented after contact with ambient air and H2O. The formation of strontium hydroxides and iron(oxide)hydroxides is presumed. During reoxidation, the materials transition back to the perovskite phase through oxygen uptake, which could lead to further mechanical degradation during redox cycling. Moreover, the nanoparticles show signs of agglomeration during reoxidation.
Compared to a commonly used Ni-YSZ cermet, the materials show no signs of carbon degradation in tar-containing fuels, as confirmed by microstructural and Raman analysis.
Despite promising initial properties and favorable carbon degradation behavior, the observed mechanical instabilities pose significant challenges for practical application. The general operating conditions of SOCs, including temperature, oxygen partial pressure, and water vapor, are likely to induce mechanical degradation in these materials. Future work should focus on the design of such materials for specific operation conditions, and the stability of porous electrode layers during operation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Deutsche Forschungsgemeinschaft (DFG) within the project SynSOFC II (275388933) which is gratefully acknowledged. We gratefully thank Dr Yoo Jung Sohn for performing Rietveld refinement.References
- C. Noguera and B. Fritz, in Encyclopedia of Engineering Geology, ed. P. Bobrowsky and B. Marker, Springer International Publishing, Cham, 2018, pp. 1–8 Search PubMed.
- J. Mei, T. Liao and Z. Sun, Mater. Today Energy, 2023, 31, 101216 CrossRef CAS.
- O. Kwon, S. Joo, S. Choi, S. Sengodan and G. Kim, J. Phys. Energy, 2020, 2, 032001 CrossRef CAS.
- P. Goel, S. Sundriyal, V. Shrivastav, S. Mishra, D. P. Dubal, K.-H. Kim and A. Deep, Nano Energy, 2021, 80, 105552 CrossRef CAS.
- P. V. Aravind, J. P. Ouweltjes, N. Woudstra and G. Rietveld, Electrochem. Solid-State Lett., 2007, 11, B24 CrossRef.
- H. Jeong, M. Hauser, F. Fischer, M. Hauck, S. Lobe, R. Peters, C. Lenser, N. H. Menzler and O. Guillon, J. Electrochem. Soc., 2019, 166, F137 CrossRef CAS.
- J. F. B. Rasmussen and A. Hagen, J. Power Sources, 2009, 191, 534–541 CrossRef CAS.
- M. B. Mogensen, M. Chen, H. L. Frandsen, C. Graves, A. Hauch, P. V. Hendriksen, T. Jacobsen, S. H. Jensen, T. L. Skafte and X. Sun, Fuel Cells, 2021, 21, 415–429 CrossRef CAS.
- D. Neagu, T.-S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- T. Zhang, Y. Zhao, X. Zhang, H. Zhang, N. Yu, T. Liu and Y. Wang, ACS Sustainable Chem. Eng., 2019, 7, 17834–17844 CrossRef CAS.
- D. Neagu, V. Kyriakou, I.-L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS PubMed.
- P. Steiger, D. Burnat, H. Madi, A. Mai, L. Holzer, J. Van Herle, O. Kröcher, A. Heel and D. Ferri, Chem. Mater., 2019, 31, 748–758 CrossRef CAS.
- K.-Y. Lai and A. Manthiram, Chem. Mater., 2018, 30, 2838–2847 CrossRef CAS.
- D. Udomsilp, C. Lenser, O. Guillon and N. H. Menzler, Energy Technol., 2021, 9, 2001062 CrossRef CAS.
- N. Del
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) baş, S. B. Gharamalek, M. Mansour and A. Nae, Int. Adv. Res. Eng. J., 2022, 6, 56–67 CrossRef.
baş, S. B. Gharamalek, M. Mansour and A. Nae, Int. Adv. Res. Eng. J., 2022, 6, 56–67 CrossRef. - S. Molin, W. Lewandowska-Iwaniak, B. Kusz, M. Gazda and P. Jasinski, J. Electroceram., 2012, 28, 80–87 CrossRef CAS.
- W. Jung and H. L. Tuller, Solid State Ionics, 2009, 180, 843–847 CrossRef CAS.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef CAS.
- M. Santaya, C. E. Jiménez, H. E. Troiani, E. A. Carbonio, M. D. Arce, L. M. Toscani, R. Garcia-Diez, R. G. Wilks, A. Knop-Gericke, M. Bär and L. V. Mogni, J. Mater. Chem. A, 2022, 10, 15554–15568 RSC.
- J. Wang, D. Kalaev, J. Yang, I. Waluyo, A. Hunt, J. T. Sadowski, H. L. Tuller and B. Yildiz, J. Am. Chem. Soc., 2023, 145, 1714–1727 CrossRef CAS PubMed.
- C. Wagner, Prog. Solid State Chem., 1971, 6, 1–15 CrossRef CAS.
- A. Nenning, L. Volgger, E. Miller, L. V. Mogni, S. Barnett and J. Fleig, J. Electrochem. Soc., 2017, 164, F364 CrossRef CAS.
- L. Fournès, Y. Potin, J. C. Grenier, G. Demazeau and M. Pouchard, Solid State Commun., 1987, 62, 239–244 CrossRef.
- O. I. Barkalov, S. V. Zaitsev and V. D. Sedykh, Solid State Commun., 2022, 354, 114912 CrossRef CAS.
- J. Mizusaki, M. Yoshihiro, S. Yamauchi and K. Fueki, J. Solid State Chem., 1985, 58, 257–266 CrossRef CAS.
- M. Kuhn, S. Hashimoto, K. Sato, K. Yashiro and J. Mizusaki, Solid State Ionics, 2011, 195, 7–15 CrossRef CAS.
- K. Sreedeviamma Dijith, R. Aiswarya, M. Praveen, S. Pillai and K. Peethambharan Surendran, Mater. Chem. Front., 2018, 2, 1829–1841 RSC.
- S. H. Khezri, A. Yazdani and R. Khordad, Eur. Phys. J. – Appl. Phys., 2012, 59, 30401 CrossRef.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- H. Jeong, M. Geis, C. Lenser, S. Lobe, S. Herrmann, S. Fendt, N. H. Menzler and O. Guillon, Int. J. Hydrogen Energy, 2018, 43, 20911–20920 CrossRef CAS.
- B. E. Hayden and F. K. Rogers, J. Electroanal. Chem., 2018, 819, 275–282 CrossRef CAS.
- M. Vračar, A. Kuzmin, R. Merkle, J. Purans, E. A. Kotomin, J. Maier and O. Mathon, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 174107 CrossRef.
- W. G. Nilsen and J. G. Skinner, J. Chem. Phys., 2003, 48, 2240–2248 CrossRef.
- V. Damljanovic, doctoral thesis, Universität Stuttgart & Max-Planck-Institut für Festkörperforschung, 2008.
- D. P. Fagg, V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, E. N. Naumovich and J. R. Frade, J. Eur. Ceram. Soc., 2001, 21, 1831–1835 CrossRef CAS.
- A. Mroziński, S. Molin, J. Karczewski, T. Miruszewski and P. Jasiński, Int. J. Hydrogen Energy, 2019, 44, 1827–1838 CrossRef.
- A. M. Beale, M. Paul, G. Sankar, R. J. Oldman, C. R. A. Catlow, S. French and M. Fowles, J. Mater. Chem., 2009, 19, 4391–4400 RSC.
- I. A. Sluchinskaya, A. I. Lebedev and A. Erko, J. Adv. Dielectr., 2013, 03, 1350031 CrossRef.
- S. Nikodemski, J. Tong and R. O'Hayre, Solid State Ionics, 2013, 253, 201–210 CrossRef CAS.
- K.-R. Lee, C.-J. Tseng, S.-C. Jang, J.-C. Lin, K.-W. Wang, J.-K. Chang, T.-C. Chen and S.-W. Lee, Int. J. Hydrogen Energy, 2019, 44, 23784–23792 CrossRef CAS.
- Y. Huang, R. Merkle and J. Maier, J. Mater. Chem. A, 2021, 9, 14775–14785 RSC.
- R. M. Batista and E. N. S. Muccillo, Ceram. Int., 2011, 37, 1047–1053 CrossRef CAS.
- F. Ye, T. Mori, D. R. Ou, J. Zou, J. Drennan, S. Nakayama and M. Miyayama, Solid State Ionics, 2010, 181, 646–652 CrossRef CAS.
- C. Lenser, H. Jeong, Y. J. Sohn, N. Russner, O. Guillon and N. H. Menzler, J. Am. Ceram. Soc., 2018, 101, 739–748 CrossRef CAS.
- D. Xu, K. Li, Y. Zhou, Y. Gao, D. Yan and S. Xu, J. Eur. Ceram. Soc., 2017, 37, 419–425 CrossRef CAS.
- A. Schwiers, C. Lenser, O. Guillon and N. H. Menzler, J. Eur. Ceram. Soc., 2023, 43, 6189–6199 CrossRef CAS.
- M. C. D. Lacharme and A. Donazzi, ECS Trans., 2023, 111, 1867 CrossRef.
- Y. Liu, S. Baumann, F. Schulze-Küppers, D. N. Mueller and O. Guillon, J. Eur. Ceram. Soc., 2018, 38, 5058–5066 CrossRef CAS.
- F. Abild-Pedersen, J. K. Nørskov, J. R. Rostrup-Nielsen, J. Sehested and S. Helveg, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 115419 CrossRef.
- M. V. Patrakeev, I. A. Leonidov, V. L. Kozhevnikov and V. V. Kharton, Solid State Sci., 2004, 6, 907–913 CrossRef CAS.
- X. Yin, L. Bencze, V. Motalov, R. Spatschek and L. Singheiser, Int. J. Appl. Ceram. Technol., 2018, 15, 380–390 CrossRef CAS.
- V. A. Mikhailov, G. Akibo-Betts and A. J. Stace, J. Phys. Chem. A, 2002, 106, 8583–8589 CrossRef CAS.
- P. Sarin, V. L. Snoeyink, D. A. Lytle and W. M. Kriven, J. Environ. Eng., 2004, 130, 364–373 CrossRef CAS.
- S. Caré, R. Crane, P. S. Calabrò, A. Ghauch, E. Temgoua and C. Noubactep, CLEAN – Soil, Air, Water, 2013, 41, 275–282 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05813a |
| ‡ Current address: Technical University of Munich, Chair of Materials Science, Garching 85748, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |