
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cleavage of β-O-4 bonds in lignin model compounds with polymer-supported Ni–Salen catalysts†
Qiongli
Liu‡
,
Dianyong
Yang‡
,
Xiuge
Zhao
,
Zhiwei
Xu
,
Ji
Ding
,
Danqi
Wu
,
Ning
An
,
Huiying
Liao
and
Zhenshan
Hou
 *
*
State Key Laboratory of Green Chemical Engineering and Industrial Catalysis, Research Institute of Industrial Catalysis, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: houzhenshan@ecust.edu.cn
First published on 19th September 2024
Abstract
Transition metal-catalyzed lignin oxidative cleavage reactions have attracted considerable attention. In this work, polymerized ionic liquid-tagged Salen ligands have been initially synthesized, followed by anion exchange, and then coordination with Ni(II) via a –N2O2– tetradentate structure. Finally, the as-obtained Ni–Salen complexes were polymerized to give a Ni–Salen polymer catalyst (poly-Ni-[Salen-Vim][OAc]2). The resulting catalyst showed 99% conversion and 88% selectivity to oxidative cleavage products for the oxidative cleavage of a lignin model compound (2-phenoxy-1-phenylethanone) without any base additive at 110 °C. The polymeric ionic liquid-tagged Salen(Ni) catalysts can be separated easily by centrifugation after the reaction and recycled for five runs with a slight loss of activity. Additionally, studies on birch lignin depolymerization indicated that polymer-supported Ni Salen catalysts were able to cleave β-O-4 linkages to produce dimeric products. Further investigation suggests that the oxidative cleavage reaction was proceeded via a radical pathway.
Sustainability spotlightLignocellulose, as the most abundant renewable biomass, could be employed to obtain various high-value added platform chemicals through physical separation and chemical transformation, which represents a promising approach for sustainable development. Due to the complex structure of natural lignin, β-O-4 model compounds like 2-phenoxy-1-phenylethanone (PP-one) have been utilized as materials for catalytic valorization to form highly valued aromatic compounds through different pathways. In this work, we have developed a mild catalytic system for selective oxidative cleavage of β-O-4 lignin models to aromatic products using a recoverable polymeric Salen-Ni(II) catalyst (poly-Ni-[Salen-Vim][OAc]2), which was constructed via the polymeric Salen ligand coordinating with Ni(OAc)2. The as-resulting catalyst can show 99% conversion of (2-phenoxy-1-phenylethanone) and 88% selectivity to the oxidative cleavage products for the oxidative cleavage of the lignin model compound without any base additive at 110 °C. The polymeric Salen-Ni(II) catalysts can be separated easily by centrifugation after the reaction and can be recycled for five runs with a slight loss of activity. Further investigation indicates that the catalyst was highly selective for oxidative cleavage of β-O-4 bonds and the oxidative cleavage reaction proceeded via a radical pathway. |
1 Introduction
With the continuous consumption of fossil energy sources, it has become particularly important to find and utilize renewable resources efficiently. Lignin, as an abundant, easily available and renewable biomass resource, is considered a promising alternative to fossil energy due to its renewability, and thus selective and efficient conversion of lignin into value-added chemicals is of great significance for achieving industrial applications of renewable biomass resources.1,2 It is important to note that its complexity and the diversity of bonds in its structure make the conversion of lignin very challenging. Typically, effective methods have been reported for selective depolymerization of lignin to produce aromatic monomers, including reduction,3,4 oxidation,5–7 pyrolysis,8 acid catalysis, base catalysis,9,10 photocatalytic and electrocatalytic depolymerization.11 Among them, the oxidative cleavage of lignin is a promising method. Especially, specific aromatic chemicals with multifunctional groups such as aromatic alcohols, aldehydes, ketones, acids, and phenolic compounds are currently obtainable by the selective oxidative cleavage of lignin.12,13The C–O and C–C bonds in lignin exist mainly in the forms of β-O-4, 4-O-5, α-O-4, 5-5, and β-5 bonds,14 of which the β-O-4 bond is an important connecting structure of lignin. The higher dissociation energy of C–C relative to those in the lignin β-O-4 structure makes the oxidative cleavage of lignin more favorable by pre-oxidation of Cα–OH to Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ketones. Most oxidation approaches have focused on the activation and cleavage of β-O-4 bonds in the lignin structure because of its relatively low bond dissociation energy (60 kcal mol−1).15,16 However, selective β-O-4 bond cleavage reactions usually involve harsh acidic or basic as well as high temperature conditions. Highly reactive carbon positive ions and other intermediates are inevitably produced during β-O-4 bond cleavage. The reactive species are highly unstable, leading to the production of a large number of condensation products. As a result, the monomers or low molecular products directly arising from lignin oxidation normally tend to be poor.
O ketones. Most oxidation approaches have focused on the activation and cleavage of β-O-4 bonds in the lignin structure because of its relatively low bond dissociation energy (60 kcal mol−1).15,16 However, selective β-O-4 bond cleavage reactions usually involve harsh acidic or basic as well as high temperature conditions. Highly reactive carbon positive ions and other intermediates are inevitably produced during β-O-4 bond cleavage. The reactive species are highly unstable, leading to the production of a large number of condensation products. As a result, the monomers or low molecular products directly arising from lignin oxidation normally tend to be poor.
Different catalytic systems have been studied for lignin oxidation.17–19 It has been found that inexpensive commercially available copper salts (CuCl, CuCl2, and Cu(OAc)2) and bases (NaOH) are efficient catalysts for the selective cleavage of C–C bonds of β-O-4 lignin model substrates under mild conditions.17 It has been demonstrated that the addition of a base has a facilitating effect on the breaking of the β-O-4 bond, in which the base is able to elongate the Cα–H bond of the lignin model substrate so that hydrogen can be easily dissociated. For instance, the covalent triazine skeletons, a metal-free catalyst, showed good catalytic activity for the C–C bond cleavage of PP-one due to the presence of strongly basic sites, which helped to activate the Cβ–H bond.20 Besides, it was found that potassium tert-butoxide with strong basicity exhibited high catalytic activity for the oxidative cleavage of PP-ol.9 Moreover, a catalytic amount of solid base (K7HNb6O39), coupled with the Cu/C3N4 catalyst, showed enhanced catalytic performance for the oxidative cleavage of PP-one.21 However, the above reaction system could require severe reaction conditions or addition of a base, which might cause corrosion and difficulties in separation and recycling. In view of these considerations, our group has developed protonated ionic liquid [Bim][Pic]-stabilizing vanadium–oxygen clusters, which exhibited high catalytic activity for selective aerobic oxidation to cleave β-O-4 linkages into phenols, esters and acids. It was found that the highly reversible interconversion of V4+ and V5+ species in vanadium oxo-clusters allowed the coexistence of mixed valence vanadium species, which was responsible for oxidizing PP-ol to PP-one.22 However, it is still difficult to separate the catalyst from the reaction system.
Salen complexes have the advantages of being inexpensive, easy to synthesize and relatively stable.23 In addition, it has been found that the catalytic activity of metal-Salen catalysts can be tuned by modulating the moieties of Salen ligands.24 V, Cu, Co, and Mn Salen based catalysts have been reported to catalyze the oxidation of lignin and model compounds.25–27 For example, Co Salen catalysts have been most frequently investigated in the catalytic oxidation of lignin and model compounds because of their compatibility with aqueous reaction media. Exposure of Co complexes to oxygen has been reported to form a CoIII-superoxide adduct and CoIII-hydroperoxide adduct.28,29 Although homogeneous metal-Salen complexes have shown high catalytic properties, their cost-effectiveness, poor stability and separation difficulties would still hinder their further application. There have been more studies on the design and application of heterogeneous Salen complex catalysts,30 such as covalent grafting of a Salen complex on graphene,31 covalent grafting on silica32 and constant potential condition deposition methods,33 which might improve stability and recyclability. Especially, the entrainment of am ionic liquid (IL) moiety on a polymeric Salen complex catalyst can facilitate the isolation of products from the catalyst, and also the counter anions of the IL have the ability to regulate the oxidative activity of the catalyst.34–36 Furthermore, divinylbenzene has been widely used as a cross-linker in polymeric catalysts since the copolymerization between divinylbenzene and 1-vinylimidazolium can form highly thermally stable and porous polymers, being favorable for catalytic performance.37–39
Inspired by other and our group's previous studies on oxidative cleavage of β-O-4 bonds in lignin model compounds, in this work we reported polymeric ionic liquid-tagged Salen ligands, coordinating with a Ni(II) via the –N2O2– tetradentate structure to give a polymeric Ni–Salen catalyst (poly-Ni-[Salen-Vim][OAc]2), and the polymeric ionic liquid-tagged Salen(Ni) catalysts acted as highly efficient catalysts for oxidative cleavage of β-O-4 lignin model compounds under mild and non-alkaline/acidic reaction conditions.
2 Experimental section
2.1 Chemicals and materials
High-purity O2 (99.5%) was obtained from Shanghai Shangnong Gas Co., Ltd. K2CO3, CH3COOK, C4H6NiO4·4H2O, C4H6CoO4·4H2O and butylated hydroxytoluene were obtained from Sinopharm Chemical Reagent Co., Ltd. 1-Vinylimidazole (98%), divinylbenzene, azobisisobutyronitrile, acetylacetone, ethylenediamine, N-bromosuccinimide, methanol, ethanol, acetonitrile, toluene, N,N-dimethylformamide, dichloromethane, dimethyl sulfoxide, anhydrous ethyl ether, tert-butanol, anhydrous sodium sulfate, 2-bromoacetophenone, α-bromo-4-methoxyacetophenone, bromo-3,4-dimethoxy-acetophenone, diphenylethanone, 1,4-benzoquinone, benzoic acid, 1-phenylethanol and 5,5-dimethyl-1-pyrroline-N-oxide were obtained from Shanghai Titan Technology Co., Ltd. Phenol, p-methoxyphenol, phenylglyoxylic acid, methyl benzoylformate, and methyl benzoate were purchased from Shanghai McLean Biochemical Technology Co., Ltd. DMSO-d6 was obtained from Shanghai Bailing Wei Chemical Technology Co., Ltd and CDCl3 from Shanghai Haohong Biomedical Technology Co., Ltd.Birch lignin was derived from birch sawdust according to a reported method.4 Briefly, 10 g of wood chips and 100 mL of methanol containing 3% (w/w) hydrogen chloride were carefully added to a 500 mL round-bottomed flask with a condenser. The mixture was refluxed under vigorous stirring for 70 h. The reaction was stopped and the reaction mixture was cooled to room temperature, and then the mixture was filtered to remove residual solids and washed several times with a small amount of methanol. The collected filtrate was concentrated by rotary evaporation to less than 50 mL and transferred to a 250 mL beaker containing ice water with vigorous stirring. Finally, the birch lignin was collected by filtration and dried under vacuum at 60 °C.
2.2 Synthesis and characterization of lignin model compounds and lignin preparation
The synthetic route of β-O-4 lignin model compounds is shown in Fig. S1.† Taking PP-one as an example, 6.9 g (73 mmol) of phenol was dissolved in a Schlenk bottle containing 100 mL of acetone, followed by addition of K2CO3 (10.4 g, 75 mmol), and the mixture was stirred at room temperature for 0.5 h. Next, 2-bromoacetophenone (14.0 g, 70 mmol) was dissolved in 50 mL of acetone and then was added slowly into the Schlenk bottle. Then the resulting mixture was refluxed at 60 °C for 12 h. After the reaction, K2CO3 was separated by filtration and the solvent was removed under reduced pressure. The resulting solid was then dissolved in ethyl acetate and washed with water. Anhydrous MgSO4 was used to remove the residual water in the organic phase. The crude product of PP-one was recrystallized from ethanol three times. Other substrates were prepared using a similar method. The as-synthesized substrates were all characterized by 1H NMR and 13C NMR (Fig. S2–S6†).2.3 Catalyst preparation
2.4 Typical reaction procedure
Lignin model compound (LMC) oxidation was performed in a stainless steel high-pressure batch autoclave with a 25 mL PTFE inlet. The autoclave was equipped with a gas supply system and a magnetic stirrer. Typically, 0.25 mmol of PP-one and 50 mg catalyst were added to 3 mL MeOH and the autoclave was purged with 0.5 MPa oxygen pressure three times. Then the autoclave was charged with molecular oxygen and then the reaction temperature was increased to the desired level. After the reaction, the reactor was quenched in an ice-water bath. Then the reaction solution was collected and the catalyst was separated by centrifugation. A 100 μL reaction solution was diluted in a 5 mL volumetric flask with a mobile phase (acetonitrile/water = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The solution was filtered with a 0.22 mm membrane filter prior to injection. The reaction substrates and products were analyzed by using HPLC [a LC-100 Liquid Chromatograph with a GH0515046C18A column (150 mm × 4.6 mm), a UV detector at a column temperature of 30 °C and a flow rate of 0.5 mL min−1 (270 nm for phenol, PP-one, MB and MP detection) and 1.0 mL min−1 (230 nm for BA detection)]. The conversion of lignin models, the yields of aromatic products and the selectivity of oxidative cleavage products are given as follows:
:3). The solution was filtered with a 0.22 mm membrane filter prior to injection. The reaction substrates and products were analyzed by using HPLC [a LC-100 Liquid Chromatograph with a GH0515046C18A column (150 mm × 4.6 mm), a UV detector at a column temperature of 30 °C and a flow rate of 0.5 mL min−1 (270 nm for phenol, PP-one, MB and MP detection) and 1.0 mL min−1 (230 nm for BA detection)]. The conversion of lignin models, the yields of aromatic products and the selectivity of oxidative cleavage products are given as follows: | (1) |
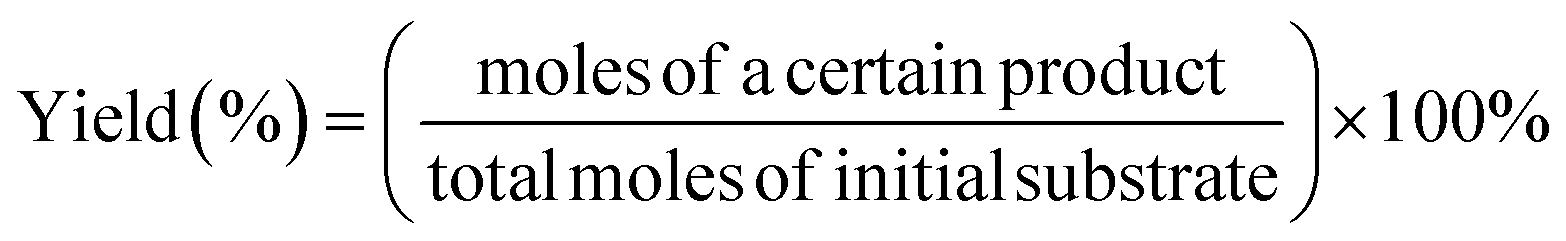 | (2) |
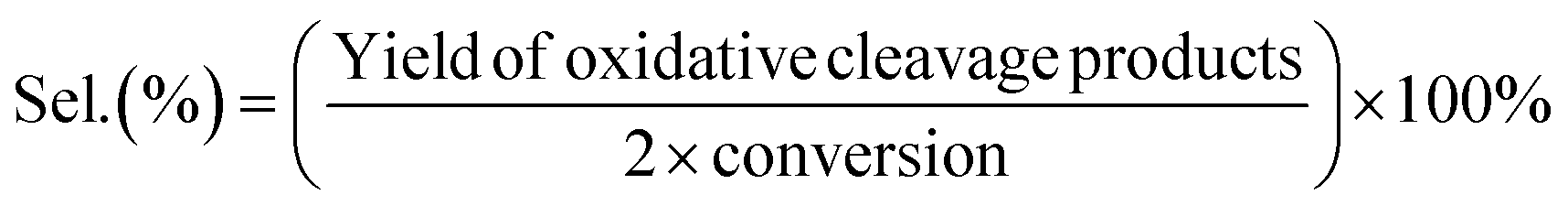 | (3) |
For oxidative degradation of birch lignin, after the oxidative degradation reaction, the autoclave was cooled to room temperature and depressurized. Then the reaction mixture was centrifuged, and the insoluble fraction was washed with methanol. The total solution was combined and transferred to a round-bottom flask and the methanol was removed under vacuum. The resulting oil was dissolved in CH3CN and was analyzed by HPLC-MS (Q Exactive Orbitrap LC-MS/MS system, Thermo Fisher Scientific) equipped with a chromatographic column (Thermo Hypersil GOLD C18, 3 μm, 2.1 mm × 100 mm) in secondary mass spectrometry mode (HCD) and an electrospray ion source (ESI). The small molecular weight substances derived from birch lignin oxidative degradation were determined quantitatively by the external standard method according to previous reports.43,44 The dimers were analyzed by using dimeric β-O-4 lignin model compounds as external standards.
2.5 Catalyst characterization
The samples were characterized by various techniques such as Fourier transform infrared spectroscopy (FT-IR), elemental analysis, X-ray photoelectron spectroscopy (XPS), inductively coupled plasma optical emission spectroscopy (ICP-OES), thermogravimetric analysis (TGA), and 1H NMR and 13C NMR spectroscopy. The FT-IR spectra were recorded on a Nicolet Magna 550 FT-IR spectrometer. XPS was performed using a Thermo Scientific K-Alpha with C 1s (284.8 eV) as a reference to correct the binding energy. The elemental analysis of C, H, and N was performed using an Elementar Vario EL III C H N O S elemental analyzer and the ICP-OES analysis of Ni on a Varian 710 instrument, respectively. The BET and pore analyses were performed using a 3Flex 3500. A PerkinElmer Pyris Diamond was used for the TGA measurements to test the thermal stability of catalysts. All samples were vacuumed at 100 °C for 3 h to remove the solvent molecules before TGA. The samples were heated from 40 °C to 800 °C (heating rate: 10 °C min−1) under a flow of anhydrous air (flow rate: 20 mL min−1). 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 400 MHz instrument (400 MHz 1H NMR and 100 MHz 13C NMR) using CDCl3 and DMSO-d6 as solvents and TMS as the reference, respectively.3 Results and discussion
3.1 Synthesis and characterization of the polymeric Ni–Salen catalyst
The preparation route of the polymeric Ni–Salen catalyst is shown in Scheme 1. In the initial step, the Salen ligand was obtained from the condensation between acetylacetone and ethylenediamine, and then the Salen ligand was bromized with NBS to afford the Salen-Br ligand, which was further processed with 1-vinylimidazole by quaternization to afford [Salen-Vim][Br]2. Then the monomer was co-polymerized with divinylbenzene with azobisisobutyronitrile as a free radical initiator. Finally, the polymeric ligand was subjected to anion exchange with KOAc, followed by coordination with Ni(OAc)2 to afford the poly-Ni-[Salen-Vim][OAc]2 catalyst. | ||
| Scheme 1 The preparation route of the polymeric supported-Ni Salen catalyst. | ||
The FT-IR spectra of the ligands, the polymeric Salen ligand and the poly-Ni-[Salen-Vim][OAc]2 catalyst are all displayed in Fig. 1. The characteristic peaks around 1672 cm−1 and 1613 cm−1 belong to the stretching vibration of CN in the imidazolium and Salen backbone, respectively.45 In addition, the bands in the ranges of 1600–1450 cm−1 and 900–650 cm−1 belong to the benzene skeleton stretching and C–H out-of-plane bending vibrations of the benzene ring.46,47 Especially, the characteristic peak of CC stretching vibration was not discerned due to the weak signal, but the characteristic band at 2926 cm−1 attributed to the stretching vibration of –CH2– was observed in Fig. 2b and c, confirming that the [Salen-Vim][Br]2 ligand has been incorporated into the polymeric networks. In Fig. 1b–e, the formation of multi-substituted benzene was further verified by the pan-frequency peaks of mono-substituted benzene in the range of 1739–1947 cm−1.48 In brief, FT-IR characterization along with 1H NMR (Fig. S7–S9†) revealed that the imidazolium and benzene skeletons were introduced into the polymeric Salen ligand.
 | ||
| Fig. 1 FT-IR spectra of (a) [Salen-Vim][Br]2, (b) poly-Ni-[Salen-Vim][Br]2, (c) poly-Ni-[Salen-Vim][OAc]2, (d) poly-Ni-[Salen-Vim][Br]2 and (e) poly-Ni-[Salen-Vim][OAc]2 after 5 runs. | ||
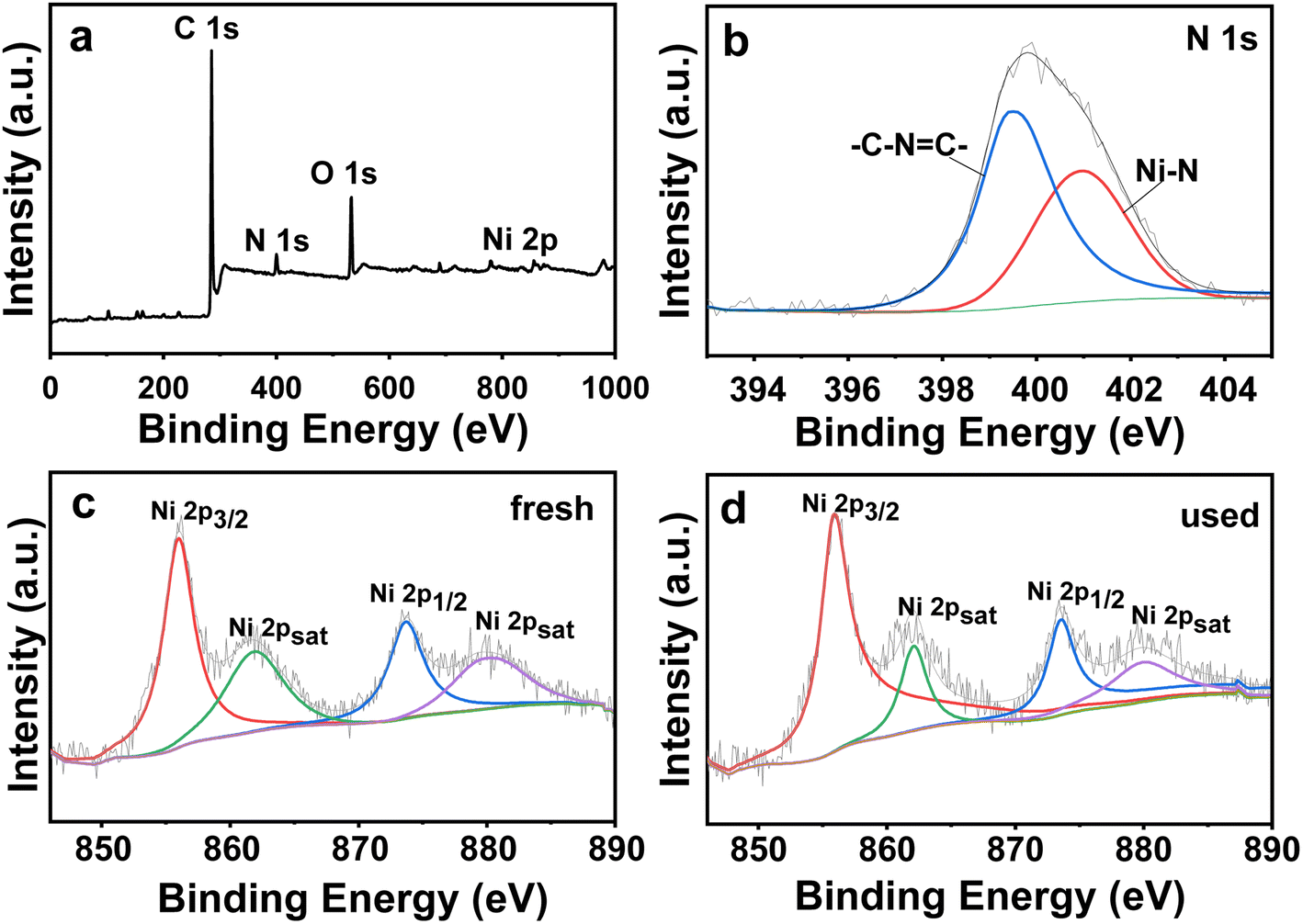 | ||
| Fig. 2 XPS (a) survey spectra, (b) N 1s spectra, and Ni 2p spectra of (c) fresh poly-Ni-[Salen-Vim][OAc]2 and (d) poly-Ni-[Salen-Vim][OAc]2 after five reaction cycles. | ||
X-ray photoelectron spectroscopy was used to investigate the elemental composition and the chemical states of the Ni species in the catalyst. As shown in Fig. 2a, the catalyst surface contains the elements C, N, O, and Ni. The N 1s spectrum (Fig. 2b) can be deconvoluted into two peaks at 400.9 eV and 399.4 eV, which are associated with the N atoms coordinated to Ni sites and N atoms in imidazolium (–C–NC–), respectively.45,49 In addition, two peaks at 856.0 eV and 873.7 eV, matching the Ni 2p3/2 and Ni 2p1/2 binding energies, represented the valence state of active NI(II) species (Fig. 2c and d).48 For the poly-Ni-[Salen-Vim][OAc]2 catalyst, the binding energy of Ni(II) species (856.0 eV) is lower than that of Ni(OAc)2 (856.3 eV),50,51 indicating that significant electron transfer from Salen ligands to Ni(II) sites occurred. As such, the results demonstrated clearly that Ni(II) sites have been coordinated with the polymeric Salen ligand.
The porous structure of the catalyst was determined from nitrogen adsorption–desorption isotherms. As shown in Fig. S10,† the catalyst had a rapidly increasing nitrogen adsorption capacity at low relative pressure (P/P0 < 0.001), and the N2 adsorption–desorption isotherm was a type IV isotherm, with H4 hysteresis loops at higher pressures, which corresponded to the presence of mesoporous and microporous structures in the polymer framework. The pore size distribution confirmed the presence of micropores and abundant mesopores between 1 and 6 nm (Fig. S10b†). Besides, the poly-Ni-[Salen-Vim][OAc]2 catalyst has a high BET surface area of 211 m2 g−1, a mesoporous pore volume of 0.3 cm3 g−1 and an average pore size of 6.5 nm. The high BET surface and porous structures might be conducive to mass transfer and facilitate catalytic reactions.
SEM images showed that the catalyst had a large flaky irregular shape, as seen in Fig. 3a. The voids generated between neighbored sheets correspond to the mesoporous detected from nitrogen sorption isotherms (Fig. S10†). The SEM image confirmed the cross-linked porous structure, indicating that the thin-layer nanostructures could be formed due to the steric effect of long carbon chains. The flaky structure and hierarchical pores are favorable to the mass transfer of reactants as well as full exposure of active sites for efficient oxidative cleavage of PP-one. EDS elemental mapping images showed that the C, N, O and Ni elements were evenly distributed in the polymer framework of poly-Ni-[Salen-Vim][OAc]2.
 | ||
| Fig. 3 (a) SEM image of poly-Ni-[Salen-Vim][OAc]2 and (b) elemental mapping of C, O, N, and Ni in poly-Ni-[Salen-Vim][OAc]2. | ||
Thermogravimetric analysis (TGA) was performed to investigate the thermal stability of the catalyst from 40 °C to 800 °C under a N2 atmosphere. As shown in Fig. S11,† the decrease in mass (6%) was related to the removal of water molecules absorbed by the catalyst below 200 °C, and the main reason for the 52% decrease in mass in the temperature range of 200–500 °C was related to the removal of the ionic liquid groups from the catalyst.52 The further weight loss at higher temperature (>500 °C) could be attributed to the decomposition of the polymeric backbone.37 The above results indicated that the catalyst is thermally stable at the reaction temperature.
3.2 Catalytic performance
PP-one was used as a β-O-4 lignin model compound for oxidative cleavage of the β-O-4 linkage. The catalytic reaction was carried out in CH3OH with O2 as the oxidant. The main products included phenol, benzoic acid (BA), methyl benzoate (MB) and methyl phenylglyoxylate (MP) as detected by HPLC under the reaction conditions. As shown in Table 1, PP-one showed very low reactivity and selectivity to cleavage products in the absence of any catalyst (entry 1), reflecting that side reactions like condensation of active intermediates could significantly occur under the reaction conditions. Additionally, the polymerized Salen ligand also afforded low conversion of PP-one (56%), along with a low yield of phenol (28%) and BA (23%), respectively (entry 2), which was actually close to that without the catalyst. This confirmed that the sole Salen ligand had no obvious catalytic role in the cleavage of β-O-4. Comparatively, the simple inorganic nickel salts showed a significant increase in conversion and product yields compared to those without the catalyst (Table 1, entries 3 and 4), which indicated that the Ni(II) active site favors the oxidative cleavage of PP-one. However, these nickel salts were easily dissolved in MeOH, making it difficult to separate from the reaction system.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Product yields (%) | Sel. (%) | |||
| Phenol | BA | MB | MP | ||||
| a Reaction conditions: 0.25 mmol PP-one, 50 mg catalyst, 3 mL MeOH, 0.5 MPa O2, 110 °C, and 6 h. n.d. = not detected. b 0.06 mmol. c 12 h. d 0.5 MPa N2. | |||||||
| 1 | None | 45 | 29 | 23 | n.d. | n.d. | 58 |
| 2 | Poly[Salen-Vim][OAc]2 | 56 | 28 | 23 | n.d. | n.d. | 46 |
| 3b | NiCl2·6H2O | 66 | 36 | 65 | 1 | n.d. | 77 |
| 4b | Ni(OAc)2·4H2O | 87 | 82 | 76 | 5 | 6 | 97 |
| 5 | Poly-Ni-[Salen-Vim][OAc]2 | 76 | 61 | 71 | n.d. | 3 | 89 |
| 6c | Poly-Co-[Salen-Vim][OAc]2 | 99 | 55 | 88 | 6 | 6 | 78 |
| 7c | Poly-Ni-[Salen-Vim][OAc]2 | 99 | 80 | 81 | 9 | 4 | 88 |
| 8d | Poly-Ni-[Salen-Vim][OAc]2 | 34 | 7 | 2 | n.d. | n.d. | 13 |
| 9 | Poly-Ni-[Salen-Vim][Br]2 | 78 | 52 | 31 | 12 | 30 | 80 |

As shown in Table 1, it was observed that both polymeric Co and Ni Salen catalysts showed high conversion, but the poly-Ni-[Salen-Vim] [OAc]2 catalyst afforded higher selectivity towards oxidative products than the poly-Co-[Salen-Vim] [OAc]2 catalyst (entries 5–7). The low selectivity of the oxidative cleavage products could be attributed to the occurrence of a condensation reaction. Especially, the conversion of PP-one was very poor and the condensation products were formed under the conditions of a N2 atmosphere, which revealed that molecular oxygen plays an essential role in the oxidative cleavage reaction (Table 1, entry 8). Nevertheless, it can be seen that poly-Ni-[Salen-Vim][Br]2 showed a much lower conversion and product yield than the Ni-[Salen-Vim][OAc]2 catalyst (Table 1, entry 9), reflecting that the OAc− anions in the ionic porous polymer exerted a positive impact on the oxidative cleavage likely due to their basicity.53,54 In view of the high catalytic performance of the poly-Ni-[Salen-Vim] [OAc]2 catalyst, further investigation was carried out to gain deeper insight into the role of the poly-Ni-[Salen-Vim][OAc]2 catalyst in the oxidative cleavage of PP-one.
First, the effect of the catalyst dosage on the catalytic performance was studied. As shown in Fig. 4a, the conversion of PP-one and the yields of products obviously increased with the dosage but the catalytic activity remained almost unchangeable as the dosage reached as high as 50 mg. This indicated that polymeric Salen ligand-coordinated Ni(II) sites were actually catalytic sites and more active sites were favorable for the oxidation reaction. Furthermore, the conversion of PP-one increased gradually with the reaction temperature ranging from 80 to 110 °C, but the conversion leveled off at a slightly higher reaction temperature (Fig. 4b). Meanwhile, the yields towards phenol and BA can reach a maximum around 110 °C. Especially, as the temperature was further increased to 130 °C, the yield of MP was slightly reduced along with an increase of the yield of BA, strongly suggesting that MP might be the intermediate product of the reaction. As shown in Fig. 4c, the conversion of PP-one reached 99%, and the highest yields of phenol and MB were 80% and 81% as the time was increased to 12 h. Moreover, if the reaction time was further prolonged to 14 h or 16 h, the conversion and yields of phenol and BA remained almost constant, reflecting that the catalyst can maintain the high selectivity towards oxidative cleavage of β-O-4 bonds even at a longer reaction time. Interestingly, MP reached its maximum yield at 8 h but reduced with reaction time, along with an obvious rise of MB yield, indicating that MP was indeed the intermediate product of the oxidative cleavage reaction in line with that shown in Fig. 4b.
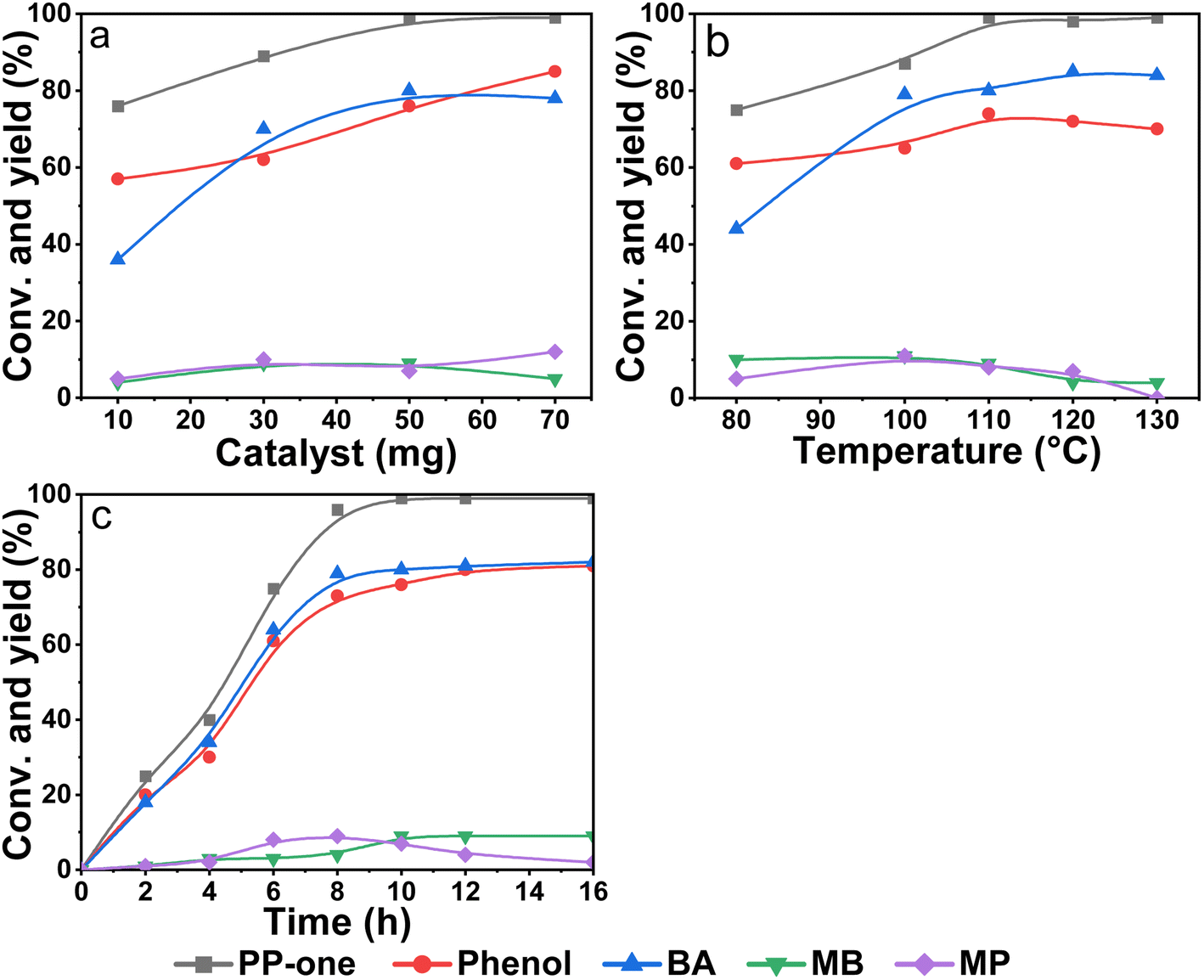 | ||
| Fig. 4 Catalytic oxidation cleavage of PP-one with (a) different amounts of catalyst, (b) different reaction temperatures and (c) different reaction times. Reaction conditions: 0.25 mmol PP-one, 3 mL MeOH, and 0.5 MPa O2. (a) 110 °C and 10 h. (b) 50 mg poly-Ni-[Salen-Vim] [OAc]2 and 10 h and (c) poly-Ni-[Salen-Vim][OAc]2 and 110 °C. | ||
The reusability of the poly-Ni-[Salen-Vim] [OAc]2 catalyst was investigated, as catalyst reusability is one of the most valuable characteristics of the catalyst. After reaction, the poly-Ni-[Salen-Vim][OAc]2 catalyst can be easily separated by simple centrifugation and washed with ethanol. As shown in Fig. 5a, after five recycles, the conversion of PP-one showed a decrease, but the selectivity of oxidative cleavage products maintained a high level (around 90%). Especially, after the five cycles, the spent catalyst showed basically the same characteristic peaks of FT-IR spectra as the fresh catalyst (Fig. 5b), indicating that there was no significant change in the main functional group of the poly-Ni-[Salen-Vim][OAc]2 catalyst. However, the loss of catalyst mass was observed after five cycles, indicating that the polymeric catalyst may undergo degradation under the reaction conditions.55,56 Besides, it was demonstrated by hot filtration experiments (Fig. S12†) that the conversion of PP-one increased by 10% when the catalyst was removed from the reaction system and the reaction time continued to extend from 4 h to 12 h, indicating a slight leaching of Ni species into the effluent.
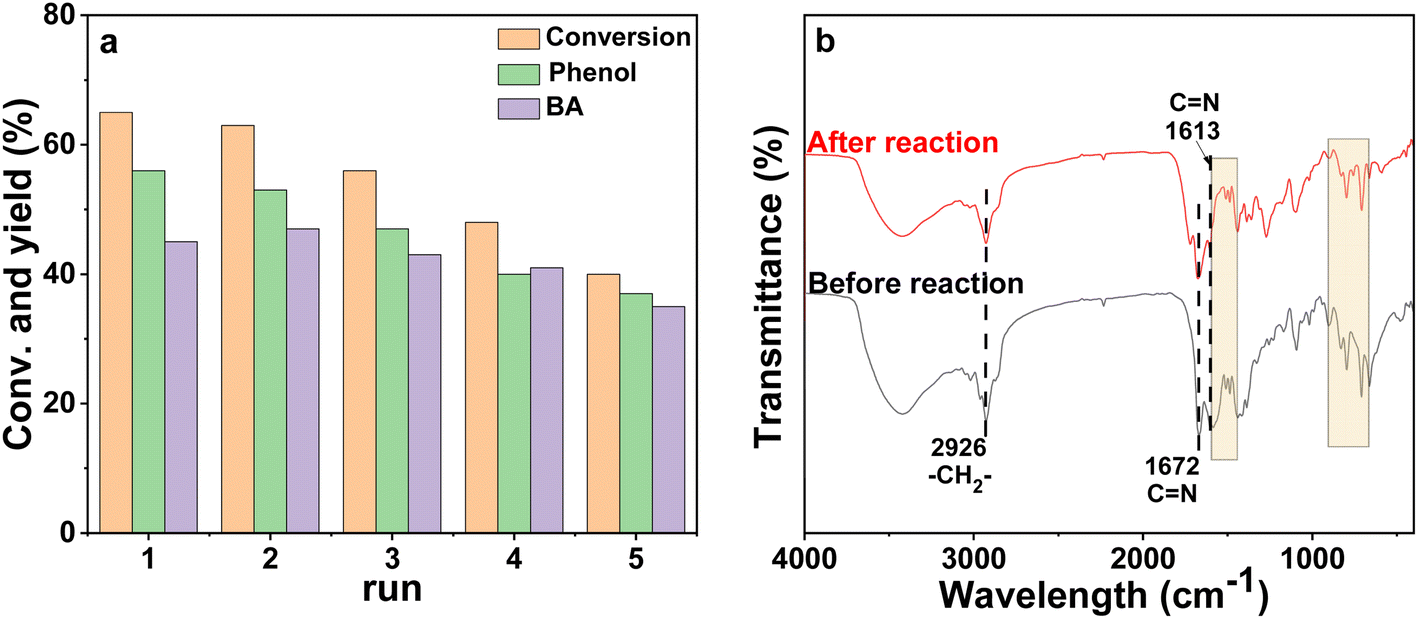 | ||
| Fig. 5 (a) The catalytic recyclability of poly-Ni-[Salen-Vim] [OAc]2 and (b) the FT-TR spectra of the fresh catalyst (upper) and the spent catalyst after five runs (bottom). Reaction conditions: 0.25 mmol PP-one, 50 mg poly-Ni-[Salen-Vim] [OAc]2, 3 mL MeOH, 0.5 MPa O2, 110 °C, and 5 h. | ||
The β-O-4 lignin model compounds contain methoxy groups on the aromatic ring, which are structurally similar to natural lignin. Therefore, the suitability of the poly-Ni-[Salen-Vim][OAc]2 catalyst for the oxidative cleavage of a range of methoxy-containing β-O-4 lignin model compounds other than PP-one was also examined. As shown in Table 2, the catalytic cleavage of the β-O-4 lignin model compounds with the methoxy group can be effectively converted to phenols, aromatic acids and corresponding aromatic esters under the same reaction conditions. These results confirmed clearly that the poly-Ni-[Salen-Vim][OAc]2 exhibits good catalytic performance in the oxidative cleavage of β-O-4 lignin model compounds with the methoxy group.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Conv. (%) | Yields (%) | |||
| Phenol | Acid | Ester 1 | Ester 2 | |||
| a Reaction conditions: 0.25 mmol substrate, 50 mg poly-Ni-[Salen-Vim] [OAc]2, 0.5 MPa O2, 3 mL MeOH, 110 °C, and 12 h. | ||||||
| 1 |

|
92 | 83 | 70 | 16 | 2 |
| 2 |

|
93 | 84 | 69 | 15 | 2 |
| 3 |

|
85 | 50 | 80 | n.d. | 5 |
| 4 |
|
99 | 64 | 24 | 62 | 12 |

Based on the above results, the present poly-Ni-[Salen-Vim][OAc]2 catalyst was compared with reported catalysts for the oxidative cleavage of PP-one using O2 as the oxidant. As shown in Table S1,† the conventional C–O and C–C bond breaking systems for β-O-4 lignin model compounds still require the addition of bases, which are not friendly from the green development point of view or require more severe reaction temperatures to achieve PP-one oxidative cleavage (Table S2,† entries 1–7). Comparatively, the present poly-Ni-[Salen-Vim][OAc]2 catalyst can be used for oxidative cleavage of lignin model compounds under much mild reaction conditions, and furthermore it acted as a heterogeneous catalyst without the addition of a base, facilitating the separation of the catalyst. Moreover, its catalytic activity is comparable with or better than those of reported systems (Table S2,† entry 8).
Finally, to evaluate the applicability of the catalytic system, the oxidative depolymerization of birch lignin was carried out under the same reaction conditions. After the reaction, the conversion of birch lignin was ca. 74%. The liquid fraction was then analyzed by HPLC-MS and HPLC. It was observed that the liquid fraction contained three dimeric products, which are shown in Fig. S13 and S14,† respectively. The results indicated that the present catalyst indeed exhibited activity for the oxidative depolymerization of birch lignin, producing some small molecular products as well as methanol-soluble low-molecular products. After the reaction, only a small amount of birch lignin (around 20%) cannot be dissolved in methanol, indicating that most of the birch lignin has been depolymerized into low-molecular products. However, the corresponding monomers were not detected under the present conditions likely due to the relatively low reaction temperature in methanol media.
3.3 Reaction pathway
For reactions in which molecular oxygen is used as an oxidant, the reaction might proceed through a radical pathway. As shown in Fig. 6, radical inhibition experiments with different scavengers were performed to elucidate the mechanism. When 2,6-dimethyl-p-cresol (BHT) acting as a wide range of radical scavenger, was added to the reaction system, the reaction conversion showed a sharp decrease from 76% to 33%, indicating that the reaction may proceed via the radical pathway. After 1,4-benzoquinone (p-BQ) was added as a superoxide radical quenching agent in the reaction system, the conversion of PP-one remained almost constant. However, after tert-butanol was added in the reaction system as a hydroxyl radical quenching agent, the conversion slightly decreased to 68%. On the basis of the above results, it was indicated that the superoxide radical (O2˙−) and hydroxyl radical were not the main active species in the reaction. Actually, it has been demonstrated that metal Salen complexes usually form mononuclear superoxo− or dinuclear peroxo type complexes with dioxygen, which may be the active species according to previous reports.48,57 | ||
| Fig. 6 Control experiments with different radical scavengers. | ||
In order to further explore the reaction pathways of the oxidative cleavage process, controlled experiments were carried out. The depolymerization of the β-O-4 lignin model compounds can be achieved by either Cα–Cβ bond or Cβ–O bond breakage. C–C bond breakage of PP-one produces BA and phenyl formate, which is subsequently decarboxylated to produce phenol, while C–O bond breakage produces phenol, BA and phenylglyoxylic acid, which is further converted to MP, BA or MB. In order to get deeper insight into the main pathways, the reaction was carried out under the same reaction conditions using possible intermediates as substrates. As shown in Scheme 2 (eqn (4)), when phenylglyoxylic acid, an intermediate product of C–O bond cleavage, was used as a substrate, the conversion was 65%, and the yields to BA, MP, and MB were 52%, 9%, and 1%, respectively, while BA can be converted to MB with a yield of 10% under identical reaction conditions (eqn (5)). Furthermore, if phenyl formate, the intermediate product of C–C bond cleavage, was used as the substrate, it was almost fully converted to phenol (eqn (6)). However, 2-phenylacetophenone gave poor conversion (36%) via the C–C cleavage route (eqn (7)), which indicated that the catalyst is indeed less active for C–C bond cleavage. Actually, the reaction of PP-one catalyzed by the poly-Ni-[Salen-Vim][OAc]2 catalyst produced a small amount of MP (Fig. 5c), which would be produced via the C–O cleavage pathway. As a result, the β-O-4 lignin model compound PP-one is mainly oxidatively cleaved via the C–O cleavage pathway.
 | ||
| Scheme 2 Control experiments with poly-Ni-[Salen-Vim][OAc]2. | ||
Based on the results of radical inhibition experiments, control experiments and previous investigation,58 a plausible catalytic reaction pathway is proposed in Scheme 3. Firstly, molecular oxygen reversibly binds to Ni(II) to generate Ni(III)-superoxide complexes,48,59 and PP-one binds to the Ni(III)-superoxide complexes to form active intermediates. Next, there are two possible paths for the catalytic oxidative cleavage reaction. Path A generates phenol and phenylglyoxylic acid by cleavage of the C–O bond of the intermediate, and phenylglyoxylic acid can be further converted to BA, MB, and MP with the participation of molecular oxygen and methanol by the action of Ni sites. Path B generates phenyl formate and BA via C–C bond cleavage, wherein phenyl formate is easily converted to phenol in the presence of methanol and polymeric Salen Ni catalysts. According to our present investigation as shown in Scheme 2 and Fig. 4, path A should be dominant for the oxidative cleavage reaction.
 | ||
| Scheme 3 Proposed possible cleavage route of PP-one with the poly-Ni-[Salen-Vim][OAc]2 catalyst. | ||
4 Conclusions
Here, we have synthesized polymeric IL-tagged tetradentate Salen ligands, coordinating with Ni(II) sites to afford porous poly-Ni-[Salen-Vim][OAc]2 catalysts. The β-O-4 linkage in lignin model compounds can be transformed into high value added aromatics by selective cleavage of C–O bonds with molecular oxygen on the present polymer catalyst. The as-obtained catalyst can be separated by simple centrifugation or filtration and can be recycled in consecutive catalytic recycles. Further studies indicated that the reaction involves a Ni(II) site-modulated radical pathway based on radical inhibition experiments. Compared to previously reported catalytic systems using base additives, the present polymer catalysts can selectively cleave the C–O bonds of lignin model compounds without extra bases and complex ligands. This work confirms that polymerized Salen ligand-supported metal active sites can be applicable to the oxidation cleavage of β-O-4 model compounds and birch lignin, offering an insight into designing a recoverable catalytic system for biomass conversions.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the support from the National Key Research and Development Program of China (2022YFA1504903) and the National Natural Science Foundation of China (21978095 and 22478113).References
- W. Deng, Y. Feng and J. Fu, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- O. Y. Abdelaziz, I. Clemmensen and S. Meier, ChemSusChem, 2022, 15, e202201232 CrossRef CAS PubMed.
- M. Cao, Y. Ma and T. Ruan, Chem. Eng. J., 2024, 485, 150020 CrossRef CAS.
- S. Xiang, L. Dong, Z. Wang, X. Han, Y. Guo, X. Liu, X. Gong and Y. Wang, J. Energy Chem., 2023, 77, 191–199 CrossRef CAS.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- S. Guo, M. Wang and X. Tong, ChemistrySelect, 2024, 9, e202304750 CrossRef CAS.
- J. X. Song, H. J. Zhang, M. H. Niu, Y. Z. Guo and H. M. Li, Ind. Crops Prod., 2024, 214, 118443 CrossRef CAS.
- C. Chio, M. Sain and W. Qin, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- L. Wang, M. He and X. Liu, Green Chem., 2023, 25, 550–553 RSC.
- S. Dabral, J. Mottweiler and T. Rinesch, Green Chem., 2015, 17, 4908–4912 RSC.
- D. Aboagye, R. Djellabi and F. Medina, Angew. Chem., Int. Ed., 2023, 62, e202301909 CrossRef CAS PubMed.
- C. Li, X. Zhao and A. Wang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Y. Zhu, Y. Liao and W. Lv, ACS Sustainable Chem. Eng., 2020, 8, 2361–2374 CrossRef CAS.
- R. A. Rafael, R. Wojcieszak and E. Marceau, ChemCatChem, 2023, 15, e202300486 CrossRef CAS.
- Y. Feng, S. Long and X. Tang, Chem. Soc. Rev., 2021, 50, 6042–6093 RSC.
- C. Zhang, X. Shen and Y. Jin, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- Y. Hu, L. Yan and X. Zhao, Green Chem., 2021, 23, 7030–7040 RSC.
- S. A. Kim, S. E. Kim and Y. K. Kim, ACS Omega, 2020, 5, 31684–31691 CrossRef CAS PubMed.
- S. Guo, X. Tong and L. Meng, Catal. Sci. Technol., 2023, 13, 1748–1754 RSC.
- G. Zhu, S. Shi and L. Zhao, ACS Catal., 2020, 10, 7526–7534 CrossRef CAS.
- J. Li, Z. Li and J. Dong, ACS Catal., 2023, 13, 5272–5284 CrossRef CAS.
- Y. Wang, B. Ding and J. He, Energy Fuels, 2023, 37, 5429–5440 CrossRef CAS.
- A. Haikarainen, J. Sipilä and P. Pietikäinen, Dalton Trans., 2001, 991–995 RSC.
- V. Sippola, O. Krause and T. Vuorinen, J. Wood Chem. Technol., 2004, 24, 323–340 CrossRef CAS.
- S. D. Springer, J. He and M. Chui, ACS Sustainable Chem. Eng., 2016, 4, 3212–3219 CrossRef CAS.
- X. F. Zhou, Environ. Prog. Sustainable Energy, 2015, 34, 1120–1128 CrossRef CAS.
- S. Sonar, K. Ambrose and A. D. Hendsbee, Can. J. Chem., 2012, 90, 60–70 CrossRef CAS.
- C. J. Cooper, S. Alam and V. d. P. Nziko, ACS Sustainable Chem. Eng., 2020, 8, 7225–7234 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx and A. L. Jongerius, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- L. Tian, X. Wang and H. Zhang, Polym. Adv. Technol., 2023, 34, 3675–3687 CrossRef CAS.
- X. F. Zhou and X. J. Lu, J. Appl. Polym. Sci., 2016, 133, 44133 CrossRef.
- A. Allahresani, E. Naghdi and M. A. Nasseri, Inorg. Chem. Commun., 2020, 119, 108137 CrossRef CAS.
- M. Mierzejewska, K. Łępicka and J. Kalecki, ACS Appl. Mater. Interfaces, 2022, 14, 33768–33786 CrossRef CAS PubMed.
- X. Yun, X. Hu, Z. Jin, J. Hu, C. Yan, J. Yao and H. Li, J. Mol. Catal. A: Chem., 2010, 327, 25–31 CrossRef CAS.
- K. Ambrose, B. B. Hurisso and R. D. Singer, Can. J. Chem., 2013, 91, 1258–1261 CrossRef CAS.
- J. Hu, Y. Hu, J. Mao, J. Yao, Z. Chen and H. Li, Green Chem., 2012, 14, 2894–2898 RSC.
- Y. Zhou, M. Z. Cai, X. J. Shu, Z. H. Xu, L. S. Zhou and X. Wu, Chem. Eng. J., 2022, 435, 134876 CrossRef CAS.
- K. Huang, F. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 13063–13070 RSC.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. X. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- X. Yun, X. Hu and Z. Jin, J. Mol. Catal. A: Chem., 2010, 327, 25–31 CrossRef CAS.
- L. Yuan, Y. Xu and X. Hu, J. Mol. Catal. A: Chem., 2015, 396, 55–60 CrossRef CAS.
- H. Chen, L. Jia and J. Yao, J. Phys. Org. Chem., 2015, 28, 570–574 CrossRef CAS.
- W. Xu, X. Li and J. Shi, Int. J. Biol. Macromol., 2019, 135, 171–179 CrossRef CAS PubMed.
- Y. Zhang, W. Xu, H. Deng, D. Zhang, X. Li and J. Shi, Ind. Crops Prod., 2023, 205, 117481 CrossRef CAS.
- Y. Tian, C. Xing and W. Wang, New J. Chem., 2022, 46, 8855–8862 RSC.
- Z. Jia, K. Wang and T. Li, Catal. Sci. Technol., 2016, 6, 4345–4355 RSC.
- M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 270, 225–235 CrossRef CAS.
- S. Wan, Q. Zou and J. Zhu, Macromol. Rapid Commun., 2023, 44, 2300340 CrossRef CAS PubMed.
- J. Li, D. Jia and Z. Guo, Green Chem., 2017, 19, 2675–2686 RSC.
- Y. Chen, Q. Zhang and F. Zhang, Appl. Catal., A, 2024, 681, 119775 CrossRef CAS.
- M. C. Biesinger, B. P. Payn and A. P. Grosvenor, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- R. Mohammadian, R. Sandaroos and A. Allahresani, Res. Chem. Intermed., 2024, 50, 1313–1329 CrossRef CAS.
- Y. Yang, H. Fan, Q. Meng, Z. Zhang, G. Yang and B. Han, Chem. Commun., 2017, 53, 8850–8853 RSC.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- K. Parkatzidis, H. S. Wang and A. Anastasaki, Angew. Chem., Int. Ed., 2024, 63, e202402436 CrossRef CAS PubMed.
- E. V. Alekseeva, A. A. Vereshchagin and M. V. Novozhilova, J. Electroanal. Chem., 2023, 935, 117310 CrossRef CAS.
- M. Bazarganipour and M. Salavati-Niasari, Chem. Eng. J., 2016, 286, 259–265 CrossRef CAS.
- H. Liu, M. Wang and H. Li, J. Catal., 2017, 346, 170–179 CrossRef CAS.
- S. Kumari and S. Ray, New J. Chem., 2020, 44, 14953–14963 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00331d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |