
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
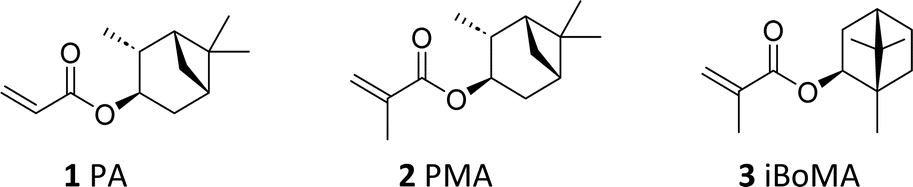
Sustainable, upscaled synthesis of pinene-derived (meth)acrylates and their application as high Tg monomers in styrene/acrylic-based bioderived copolymer coatings†
María
Pin-Nó
ac,
Philippa L.
Jacob
 a,
Vincenzo
Taresco
a,
Maud
Kastelijn
b,
Tijs
Nabuurs
b,
Chandres
Surti
c,
John
Bilney
c,
John
Daly
c,
Daniel J.
Keddie
a,
Steven M.
Howdle
*a and
Robert A.
Stockman
*a
a,
Vincenzo
Taresco
a,
Maud
Kastelijn
b,
Tijs
Nabuurs
b,
Chandres
Surti
c,
John
Bilney
c,
John
Daly
c,
Daniel J.
Keddie
a,
Steven M.
Howdle
*a and
Robert A.
Stockman
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: robert.stockman@nottingham.ac.uk; steve.howdle@nottingham.ac.uk
bCovestro (Netherlands) BV, Sluisweg 12, 5145PE, Waalwijk, The Netherlands
cCornelius Specialties Limited, 5c Rookwood Way, Haverhill, Suffolk CB9 8PB, UK
First published on 22nd July 2024
Abstract
An improved synthesis of the pinene-derived monomers (3-pinanyl acrylate 1 and 3-pinanyl methacrylate 2), replacing hazardous and/or expensive reagents from established methods with cheaper, more innocuous and sustainable reagents, is reported; the monomers of high purity are obtained at up to 160 g scale, without the need for chromatographic separation. Subsequently, these monomers (1 and 2) were successfully copolymerized with n-butyl acrylate/methacrylic acid or styrene/methacrylic acid using a radical semi-batch emulsion copolymerization process. For comparison, materials incorporating the more established terpene-derived monomer iso-bornyl methacrylate 3 were also prepared in an analogous fashion. The obtained polymer latexes had particle sizes between 65 and 90 nm and very low polydispersities (<0.08) and were stable for several years without any coagulum formation. Gradient liquid chromatography indicated that all copolymers had relatively uniform chemical composition distributions. The n-butyl acrylate containing copolymers (P1–P3) were obtained with high molar masses (Mn > 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and Mw > 400000), very high dispersities (Ð > 9.5), and low glass transition temperatures (Tg < −5 °C). The styrene-based copolymers (P4–P6) had slightly lower molar masses (Mn > 40000 and Mw > 150000), lower dispersities (Ð > 3) and high glass transition temperatures (95 °C < Tg < 120 °C). Preliminary testing of the n-butyl acrylate-based materials demonstrated the potential of these copolymers for use in coating applications. The poly(n-butyl acrylate)/pinanyl methacrylate copolymer P2 was found to be harder (König hardness) and had better stain resistance properties towards water-based substances than the analogous n-butyl acrylate-based copolymers containing 3-pinanyl acrylate (P1) or iso-bornyl methacrylate (P3). Through further refinement of the copolymerization process we expect that the properties of these polymers may be further tailored towards a range of coating applications.
000 and Mw > 400000), very high dispersities (Ð > 9.5), and low glass transition temperatures (Tg < −5 °C). The styrene-based copolymers (P4–P6) had slightly lower molar masses (Mn > 40000 and Mw > 150000), lower dispersities (Ð > 3) and high glass transition temperatures (95 °C < Tg < 120 °C). Preliminary testing of the n-butyl acrylate-based materials demonstrated the potential of these copolymers for use in coating applications. The poly(n-butyl acrylate)/pinanyl methacrylate copolymer P2 was found to be harder (König hardness) and had better stain resistance properties towards water-based substances than the analogous n-butyl acrylate-based copolymers containing 3-pinanyl acrylate (P1) or iso-bornyl methacrylate (P3). Through further refinement of the copolymerization process we expect that the properties of these polymers may be further tailored towards a range of coating applications.
Sustainability spotlightThis study investigates the scale up of the synthesis of α-pinene-derived monomers and their subsequent application as high Tg components in copolymer coatings. The newly developed, upscaled syntheses of these terpene-derived monomers employ less hazardous and/or toxic reagents and lead to new materials and coatings that have a very high biobased content. This work aligns with UN Sustainable Development Goal 12 (Responsible Consumption and Production). |
Introduction
Water-based polymer coating formulations are routinely prepared as acrylic-based copolymer resins, typically via emulsion polymerization. Incorporation of different monomers, at specifically defined feed ratios, is well known to influence polymer properties, i.e. aspects such as hardness and flexibility can be easily tailored.1Currently, most acrylic-based commercial resins are prepared from petrochemical sources.2 There is a growing global concern about carbon emissions linked to fossil fuel consumption.3 Alarmingly, it has been predicted that polymer production will account for 20% of global fossil fuel consumption by 2050.4 In response, the chemical industry is intensifying its efforts to find more sustainable, biobased chemicals to produce high quality synthetic materials.5 Clearly, this motivation needs to be balanced against the often-competing pressures of cost and performance. However, it has been shown that market penetration of bioderived products is possible where the product brings specific advantages, such as avoiding toxicity and facilitating degradability (e.g. epoxidized soybean oil (ESO), isosorbide and poly(lactic acid) (PLA)).4,6–8
One strategy used to enhance the green credentials of polymers for coating technology focusses on the use of plant-based building blocks containing a reactive double bond that can be exploited via radical polymerization.9–12 There are numerous examples of the application of biobased monomers in radical polymerization13–16 and of these, terpenes appear as a versatile pool of molecules whose production and extraction do not compete with food production.17 In this regard, we have recently focused on developing biobased radically polymerizable monomers derived from terpenes.18–22
Terpenes and terpenoids are found abundantly in plant oils and many of these molecules are considered to be industrial by-products from processes including citrus processing as well as the pulp and paper industry.23,24 Turpentine, a by-product of the paper and pulp industry is produced on a scale of 330000 tons per year and is composed of mostly α- and β-pinene.25
From a coating standpoint, monomers derived from α-pinene, namely 3-pinanyl acrylate (PA, 1) and 3-pinanyl methacrylate (PMA, 2), appear particularly promising, due to the bicyclic ring present as a pendant moiety. (Meth)acrylic polymers derived from these compounds have relatively high glass transition temperatures (Tg),19 suggesting a potential role as ‘hard’ components in copolymer formulations.26 Previous work has demonstrated their compatibility with ‘soft’ monomers such as limonene acrylate in the synthesis of ABA block copolymers.19,26
Herein, we investigate the utility of the α-pinene derived monomers PA 1 and PMA 2 in this context. We describe both their improved, scaled-up synthesis and their use as a component in binder formulations prepared by semi-batch aqueous emulsion copolymerization. Importantly, we directly examine the effect of incorporation of the pinene-based monomers (PA 1 and PMA 2) into the final materials, comparing them to analogous polymers derived from the more established high Tg monomer, isobornyl methacrylate (iBoMA, 3).
Experimental
Materials
All reagents and solvents were purchased from commercial suppliers (i.e. Sigma Aldrich, Fisher Scientific etc.) unless otherwise stated.Characterisation
Gas chromatography (GC) was performed on a Shimadzu GC-2010 Pro, using a Shimadzu HS-20 headspace sampler, a SGE SilFlow GC 3 port splitter, a retention gap (30 cm × 0.32 mm i. d. polar; CP nr 4083 or similar), and a combination of SGE BP-5 30 m × 0.25 mm i. d. df 1 μm (SGE 054203 or similar) and SGE BP-20 30 m × 0.25 mm i. d. df 1 μm (SGE 054439 or similar) columns, operating with flame ionization detection (with H2, air and N2 as make up gas). The carrier gas was helium. Conditions involved a 120 °C sample incubation temperature, 150 °C sample and transfer line temperatures, a 150 kPa gas pressure, and a temperature ramp from 40 °C (2 min hold time) to 130 °C (at 15 °C min−1) and then to 240 °C (at 25 °C min−1). Gradient-liquid chromatography (GLC) was performed on a Waters uPLC H-Class system using a Waters HSS T3, 2.1 × 100 mm, 1.8 μm column and a solvent flowrate of 0.5 mL min−1. Analysis was performed using a three solvent system: water (with 0.1% aq. trifluoroacetic acid (TFA)), acetonitrile (MeCN), and tetrahydrofuran (THF). The gradient program was as follows: t = 0 to 0.2 min (95% H2O/5% MeCN); t = 0.2 to 8 min (ramp from 95% H2O/5% MeCN to 100% MeCN); t = 8 to 8.2 min (ramp from 100% MeCN to 100% THF); t = 8.2 to 8.4 min (100% THF); t = 8.4 to 8.6 min (ramp from 100% THF to 50% H2O/50% MeCN); t = 8.6 to 8.8 min (ramp from 50% H2O/50% MeCN to 95% H2O/5% MeCN); t = 8.8 to 12 min (95% H2O/5% MeCN). Samples were injected as filtered methanol (with 0.5% TFA) solutions. Size exclusion chromatography (SEC) was conducted on a Waters Alliance e2695 LC system with a Waters 2414 DRI detector and a Waters 2996 PDA detector, using three PLgel 10 μm Mixed-B columns. N-Methylpyrrolidone (NMP) containing 10 mM lithium bromide (LiBr) was used as eluent with a flow of 1 mL min−1 THF at 70 °C and polystyrene standards were used for the calibration. DSC was performed on a TA Instruments DSC250 using Tzero aluminium pans. Indium was used for the enthalpy and temperature calibration of the instrument and an empty pan was used as the reference. Prior to analysis all samples were dried overnight at 120 °C, in air. DSC samples (∼5 mg) were subjected to a cool–heat–cool–heat protocol; all cooling temperature increments were 20 °C min−1, while all heating temperature increments were 10 °C min−1. The samples were first cooled to −85 °C, then heated to 160 °C and then re-cooled to −85 °C before being reheated to 160 °C. Glass transition temperature (Tg) measurements were performed from analysis of the second heating scan. Particle size analysis was performed on a Malvern Zetasizer. 1H and 13C NMR spectra were obtained using a Bruker DPX 400 MHz spectrometer. COSY, HSQC and HMBC were used to facilitate spectral assignments. Deuterated chloroform (CDCl3) was used as solvent, and chemical shifts were assigned in parts per million (ppm) and referenced to the (residual) solvent.27 All spectra were obtained at ambient temperature (22 ± 1 °C).Scaled-up pinene-derived monomer synthesis
The synthesis of the pinene-derived monomers, 3-pinanyl acrylate (PA 1) and 3-pinanyl methacrylate (PMA 2), and their precursor 3-pinanol was adapted from previously reported procedures.18,28,29 Reaction products of high purity were obtained and characterized by comparison of gas chromatography elution times to those of previously reported authentic samples.18Synthesis of (1R,2R,3R,5S)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-ol (3-pinanol)
This synthesis was adapted from previously reported methods.18,29 To a solution of α-pinene (50 g, 367 mmol) in THF (250 mL) NaBH4 (11.1 g, 294 mmol, 0.8 equiv.) was added. To the resulting suspension acetic acid (17 mL, 294 mmol, 0.8 equiv.) was added dropwise. The mixture was left to stir for 1 h at room temperature and then heated at 50 °C for 3 h. The reaction was cooled to room temperature and was basified with 3 M aq. NaOH solution (50 mL, 150 mmol, 0.4 equiv.) which was added dropwise, followed by dropwise addition of 30% aq. H2O2 (45 mL, 440 mmol, 1.2 equiv.) whilst maintaining the temperature below 25 °C. The reaction was subsequently quenched with brine and extracted with petroleum ether. The combined organics were dried (MgSO4), and solvent was removed under reduced pressure. The crude product was purified by vacuum distillation (bp 68–70 °C, 1.5 mbar) to give 3-pinanol as a colourless oil that solidifies upon standing (33.1 g, 214 mmol, 58%). The sample was of >99% purity, with GC retention time matching that of previously reported authentic samples.18Synthesis of (1R,2R,3R,5S)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl acrylate (3-pinanyl acrylate, 1)
This synthesis was adapted from previously reported methods.18 3-Pinanol (140 g, 907.6 mmol) was dissolved in dichloromethane (DCM) (320 mL) and the solution was cooled to 0 °C. Triethylamine (139 mL, 998.4 mmol, 1.1 equiv.) was added in one portion. Subsequently, acryloyl chloride (81.1 mL, 998.4 mmol, 1.1 equiv.) dissolved in DCM (160 mL) was added dropwise keeping the temperature <10 °C. The reaction mixture was stirred at <10 °C for 7 h and left to stir at room temperature for 16 h. Afterwards, 4-methoxyphenol (MEHQ) (132 mg, 1.06 mmol, 0.12 mol%) and water (200 mL) were added and the resulting layers separated. The aqueous phase was extracted once with DCM and the combined organics were washed (aq. NaHCO3 and then brine) and dried (MgSO4) and the solvent was removed under reduced pressure. The crude product was purified by vacuum distillation (bp ∼65 °C, 0.6 mbar) to give 3-pinanyl acrylate 1 as a colourless liquid (164.0 g, 787.9 mmol, 87%). The sample was of >95% purity, with GC retention time matching that of previously reported authentic samples.18Synthesis of (1R,2R,3R,5S)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl methacrylate (3-pinanyl methacrylate, 2)
This synthesis was adapted from previously reported methods.18,28 3-Pinanol (50 g, 324.1 mmol) was melted (∼60 °C) after which copper(II) tetrafluoroborate (Cu(BF4)2) (1.12 g, 3.24 mmol, 1 mol%), methacrylic anhydride (77 mL, 486 mmol, 1.5 equiv.) and phenothiazine (183 mg, 0.82 mmol, 0.25 mol%) were added and the resultant mixture was heated at 80 °C for 5 h. The reaction mixture was stirred at room temperature for 18 h after which water was added (100 mL) and the layers separated. The aqueous phase was extracted three times with petroleum ether. The combined organics were washed with 2% aq. NaOH, dried (MgSO4) and the solvent was removed under reduced pressure. The crude product was purified by fractional vacuum distillation (Vigreux) (bp 70–74 °C, 0.9 mbar) to give 3-pinanyl methacrylate 2 as a colourless liquid (64.9 g, 291.9 mmol, 90%). The sample was of >95% purity, with GC retention time matching that of previously reported authentic samples.18Polymer synthesis
All emulsion copolymerization experiments were performed at 80 °C, using ammonium persulfate (9.5 mM) as an initiator and sodium lauryl sulfate (18.8 mM) as a surfactant. At the end of the feed process, a ‘burn-up’ reaction was performed to further reduce residual monomer levels, using t-butyl hydroperoxide in combination with isoascorbic acid. This synthesis of poly[(n-butyl acrylate)-co-(3-pinanyl acrylate)-co-(methacrylic acid)] (P1) is shown as an example, below. Modifications from this procedure for the preparation of the other butyl acrylate-based polymers (P2 and P3) and styrene-based copolymers (P4–P6) were achieved by substitution of the monomers used as per the details given in Table 1 (and Table S1, ESI†).| Entry | Sample label | Copolymera | M n (g mol−1) | M w (g mol−1) | Đ | Biobased contentc (wt%) | Particle sized (nm) | PDId | Solids contente (%) | T g (°C) | T g, predicted (°C) | MFFTh (°C) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Monomer feed ratios (wt%) are given as subscripts in the name abbreviations. b From SEC data (NMP eluent). c For total solid content from radiocarbon analysis.30 d From DLS analysis. e Calculated from gravimetry. f From DSC analysis, the Tg is reported as the onset temperature. g Calculated from the Fox equation31 (using the following homopolymer Tg: PBA = −54 °C,32 PSt = 100 °C,32 PMAA = 228 °C,32 PiBoMA = 150 °C,33 PPA = 84 °C,19 and PPMA = 168 °C19) h The minimum film forming temperatures (MFFTs) are outside the temperature range of our tester (0–60 °C) and are hence presented as estimates only. | ||||||||||||
| 1 | P1 | P(BA63%-co-PA35%-co-MAA2%) | 43100 |
417700 |
9.69 | 27 | 67 | 0.06 | 44.6 | −13.5 | −19 | <0 |
| 2 | P2 | P(BA63%-co-PMA35%-co-MAA2%) | 52400 |
1077000 |
20.55 | 26 | 87 | 0.03 | 43.4 | −2.0 | −7 | <0 |
| 3 | P3 | P(BA63%-co-iBoMA35%-co-MAA2%) | 45900 |
494300 |
10.77 | 25 | 88 | 0.01 | 44.1 | 4.0 | −9 | <0 |
| 4 | P4 | P(St63%-co-PA35%-co-MAA2%) | 47200 |
186000 |
3.94 | 27 | 87 | 0.03 | 44.1 | 97.5 | 96 | >60 |
| 5 | P5 | P(St63%-co-PMA35%-co-MAA2%) | 50300 |
210900 |
4.19 | 26 | 87 | 0.03 | 44.3 | 114.0 | 121 | >60 |
| 6 | P6 | P(St63%-co-iBoMA35%-co-MAA2%) | 47600 |
158000 |
3.31 | 25 | 94 | 0.08 | 44.4 | 113.5 | 116 | >60 |
Residual monomer concentrations were determined using head-space GC for butyl acrylate (BA), styrene (St) and isobornyl methacrylate (iBoMA, 3). High-performance liquid chromatography (HPLC) was used to determine residual monomer concentrations for methacrylic acid, PA 1 and PMA 2.
Synthesis of poly[(n-butyl acrylate)-co-(3-pinanyl acrylate)-co-(methacrylic acid)]
To a round-bottomed flask equipped with a condenser, thermometer and mechanical stirrer demineralized water (351.4 mL) and 30% aq. sodium lauryl sulfate solution (13.8 mL) were added. The contents of the reactor were heated to 80 °C. At 80 °C a monomer feed consisting of demineralized water (222.0 mL), 30% aq. sodium lauryl sulfate solution (6.9 mL), n-butyl acrylate (326.3 g; 63 wt% of total monomers), 3-pinanyl acrylate (181.2 g; 35 wt% of total monomers) and methacrylic acid (10.4 g; 2 wt% of total monomers) was added over a period of 180 minutes. An initiator feed, comprising ammonium persulfate (2.6 g) in demineralized water (49.2 mL) was fed to the reactor in parallel over a period of 180 minutes. At the end of the addition of the monomer feed, demineralized water (7.4 mL) was used to rinse the feed tank and the washings were added to the reactor. A temperature of 80 °C was maintained for a further 60 minutes. Subsequently, a slurry comprising demineralized water (0.7 mL), 30% aq. sodium lauryl sulfate solution (0.14 mL), and t-butylhydroperoxide (0.46 g) was added, followed by a solution of isoascorbic acid (0.32 g) in demineralized water (5.8 mL) and the temperature was maintained at 80 °C for another 30 minutes. At 80 °C a neutralizing solution consisting of demineralized water (3 mL) and 25% aq. ammonium (3 mL) was fed in for 15 minutes. The reactor content was cooled to 30 °C and the preservative proxel ultra 10 (6.1 mL) was added. The solids content of the emulsion was corrected to 44% using demineralized water, resulting in a stable latex of poly[(n-butyl acrylate)-co-(3-pinanyl acrylate)-co-(methacrylic acid)] (P1).Film properties
Results and discussion
Monomer synthesis
The monomers PA 1 and PMA 2 were previously reported by our group.18 On a standard laboratory scale (up to 10 g) PA 1 or PMA 2 are easily accessible by a facile two-step synthesis from α-pinene via hydroboration/oxidation and subsequent esterification; borane dimethyl sulfide (BH3·SMe2) was the hydroboration reagent, and (meth)acrylation was achieved using the relevant acyl chloride.18For scale up for potential commercial use, neither of the above protocols is ideal either from economic or sustainability standpoints.‡ As such, we sought to improve our syntheses in terms of cost and scale to improve industrial feasibility, whilst exploiting sustainable reactions and methods of product isolation and purification, where possible. We discovered that the combination of sodium borohydride/acetic acid mediated hydroboration29 and hydrogen peroxide induced oxidation delivered 3-pinanol in reasonable yield (58%) at increased lab scale following purification by distillation (>50 g) (see Scheme 1, left), directly from α-pinene. We expect that these yields could be improved with further optimisation. Notably, this method provides a more sustainable pathway to the alcohol, at a fraction of the cost of the initial synthesis.
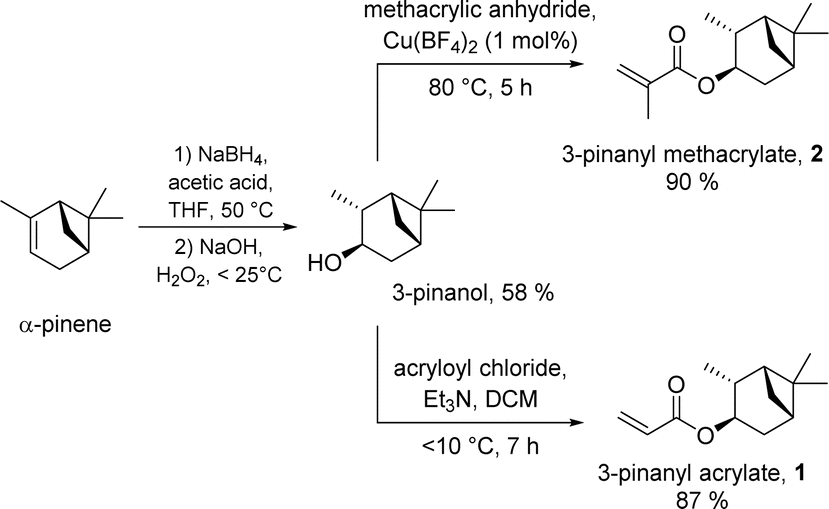 | ||
| Scheme 1 Synthesis of PA 1 and PMA 2 from α-pinene in two-steps via hydroboration/oxidation and (meth)acrylation. | ||
Next, we developed a new approach for the preparation of the methacrylate-based monomer PMA 2 from 3-pinanol, using methacrylic anhydride and catalytic copper(II) tetrafluoroborate,28 to install the ester functionality. This delivered the product 2 (>60 g) in excellent yield (90%) and high purity after distillation (see Scheme 1, right upper).
For the scaled-up synthesis of the acrylate monomer PA 1 from 3-pinanol we used a standard acryloyl chloride method adapted from that reported in our previous investigation,18 due to the lack of commercial availability of acrylic anhydride. This gave the desired monomer PA 1 (>160 g) in high purity (distillation) and high yield (87%) (see Scheme 1, right lower).§
Polymer synthesis
Acrylic and styrene/acrylic dispersions are well established as alternatives to solvent borne resins.34 As such, we selected n-butyl acylate (BA) or styrene (St) as the major components (63 wt%) in the prepared copolymer formulations, due to their wide industrial applicability (see Scheme 2). This allowed us to investigate the effects of PA 1, PMA 2, and iBoMA 3 (incorporated at 35 wt%) on copolymerization performance and the final physical properties. Methacrylic acid (MAA), a latex stabilising monomer, was used in small amounts (2 wt%) in all formulations. | ||
| Scheme 2 Emulsion radical copolymerization of terpene derived monomers PA 1, PMA 2 or iBoMA 3 with BA or St (and MAA). | ||
With reasonable quantities of the monomers PA 1 and PMA 2 in hand, we were able to utilise them in copolymerization of industrial relevance as outlined below.
BA-based copolymers
Initially, we prepared the BA-based materials by semi-batch emulsion copolymerization of BA with PA 1, PMA 2 or iBoMA 3 (and MAA) at 80 °C (see Table 1, Entries 1–3). To determine the copolymerization efficiency, samples were taken during the polymerization process after 15, 30, 60, 90, and 120 minutes during the monomer feed, in the t-butylhydroperoxide/isoascorbic acid promoted ‘burn-up’ reaction (at 150 minutes) and at the end of the process (at 180 minutes).In general, the level of residual BA and terpene monomers (1, 2 or 3) slowly increases across the 120-min feed time, with the ratio between the monomers remaining fairly constant (see Fig. 1a). Following the ‘burn-up’ reaction, quantitative (>99.9%) monomer consumption was achieved for all samples.
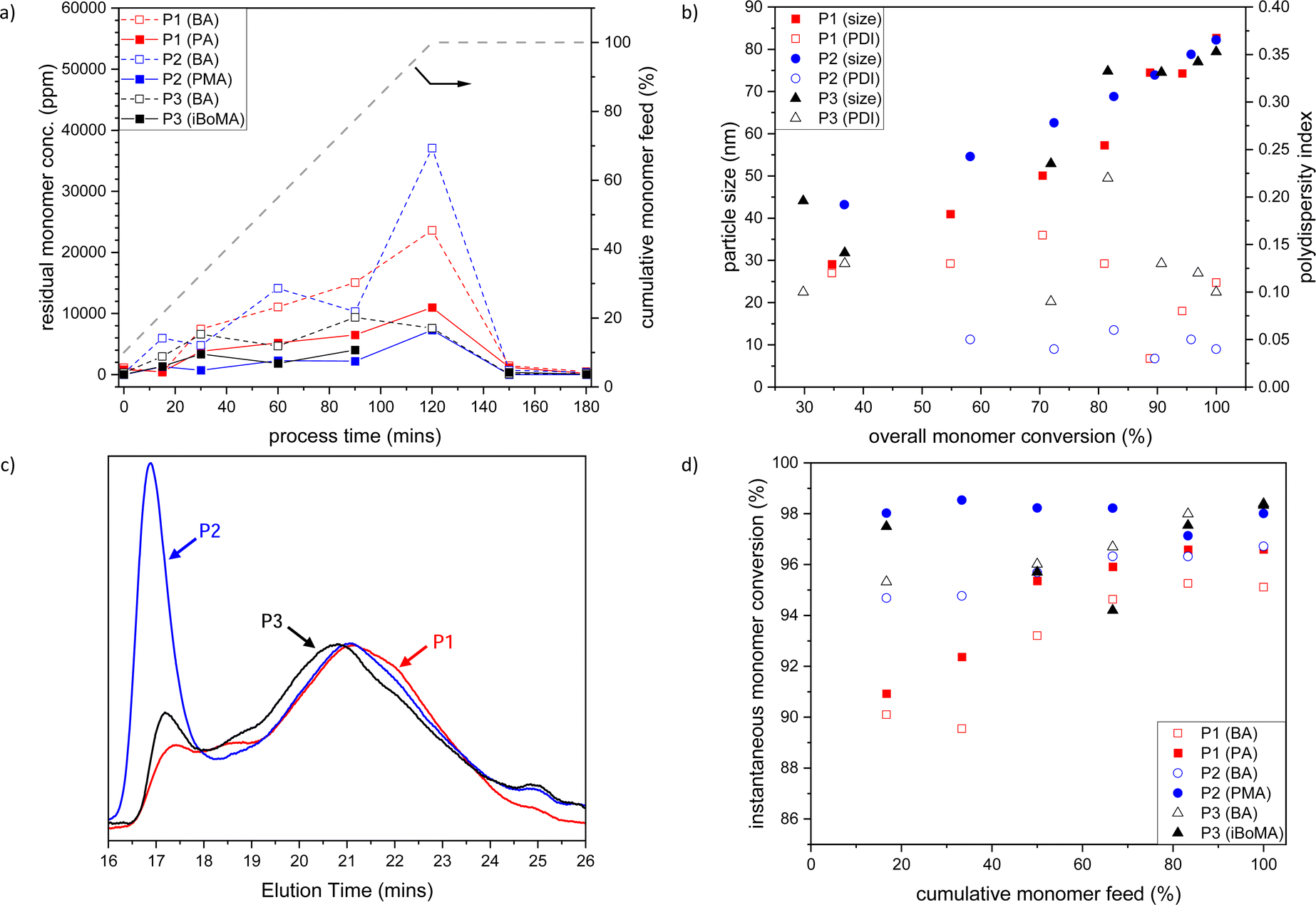 | ||
| Fig. 1 (a) Plot of residual BA (open symbols) and PA 1, PMA 2 or iBoMA 3 (closed symbols) concentrations versus time (the grey dashed line indicates the semi-batch feed profile in cumulative % of the monomers with time); (b) evolution of PBA-based polymer particle size (closed symbols) and polydispersity index (open symbols) with total monomer conversion; (c) SEC traces of BA-based copolymers; and (d) instantaneous monomer conversion for BA (open symbols) and PA, PMA or iBoMA (closed symbols) versus cumulative monomer feed, for BA/PA/MAA (P1, red), BA/PMA/MAA (P2, blue) and BA/iBoMA/MAA (P3, black) semi-batch emulsion radical copolymerizations. Note that the data related to MAA are omitted for clarity. P1–P3 refer to the sample labels given in Table 1. | ||
The size of the BA-based copolymer particles increased with cumulative monomer feed (and therefore total monomer conversion), while the particle size polydispersity index (PDI) remained low (see Fig. 1b). The final obtained latexes (P1–P3) showed particle sizes of 65 to 90 nm and a very low PDI (<0.06) (see Table 1). All samples were highly stable with no sign of coagulum over several years.
In each case, the final polymers (P1–P3) have high molar mass (Mn > 40000, Mw > 400000) and very high dispersity (Ð > 9.5) (see Table 1, entries 1–3). Additionally, bimodality is observed in each of the SEC traces (see Fig. 1c), with the high molar mass fraction being most prominent for the polymer (P2) made with the methacrylate co-monomer, PMA 1. The ‘burn up’ of the residual monomers present after the 120 min feed profile gives rise to the high molar mass peak in the molar mass distributions; the build-up of a higher concentration of residual BA in formation of P2 (see Fig. 1a, blue dashed line) than that observed for the P1 or P3 reactions results in a more prominent high molar mass fraction in the P2 sample.
Early in the reaction (i.e. feed < 40%), lower overall monomer conversion for the PA 1 system (P1) is observed than that for either of the reactions incorporating the other co-monomers (see Fig. 1d). This is likely caused by differences in the copolymerization kinetics between the acrylate/methacrylate systems (P2 and P3) and the acrylate/acrylate system (P1).¶ The broad molar mass distributions observed in each case are attributed to changes in polymerization reaction kinetics over the course of the reaction due to the starved feed nature of the process, rather than any drastic composition drift caused by differences in comonomer reactivity. The relative chemical uniformity of the obtained materials is confirmed by GLC data, where each polymer (P1–P3) is observed as a single narrow peak (see Fig. S1, ESI†). Notably, both 3-pinanyl-containing polymers (P1 and P2) remained completely soluble, as indicated by the GLC (see Fig. S1, ESI†) and solution NMR analysis following freeze drying of the latexes (see Fig. S2–S5, ESI†). This confirmed that no significant crosslinking of the copolymers occurs under the current reaction conditions. This contrasts with our prior observations from solution polymerization, where polymers of PMA 2 were found to crosslink via H-abstraction from the tertiary carbon of the pinanyl moiety.35
All PBA-based emulsion polymers (P1–P3) had low Tgs (i.e. ∼−5 to −20 °C), which were very close to that predicted using the Fox equation31 (see Table 1, entries 1–3). This demonstrates that materials with predictable thermal properties can be readily prepared through this copolymerization process. As expected, based on the monomer feed compositions all BA-based polymer emulsions had ∼25% bioderived content based upon radiocarbon analysis of total solids (see Table 1).
St-based copolymers
To further investigate the copolymerization performance of the terpene-derived monomers (1–3) in industrially relevant systems, we prepared St-based copolymers via analogous methods to that discussed above. Again, samples were taken across the experiment: during the monomer feed regime (at 15, 30, 60, 90, and 120 minutes), during the ‘burn-up’ reaction (at 150 minutes) and at the end of the process (at 180 minutes). The amount of residual monomer St and either 1, 2 or 3 remains relatively low across the reaction (see Fig. 2a), indicating acceptable rates of copolymerization throughout the process. The final St-based polymers were obtained with high molar mass (Mn > 40000 and Mw > 150000) and reasonably high dispersity (Ð > 3) (see Table 1, entries 4–6). All molar mass distributions for the St samples were unimodal (see Fig. 2b), in contrast to the BA-based samples.|| Particle sizes were all close to 90 nm, with a very low PDI (<0.08).
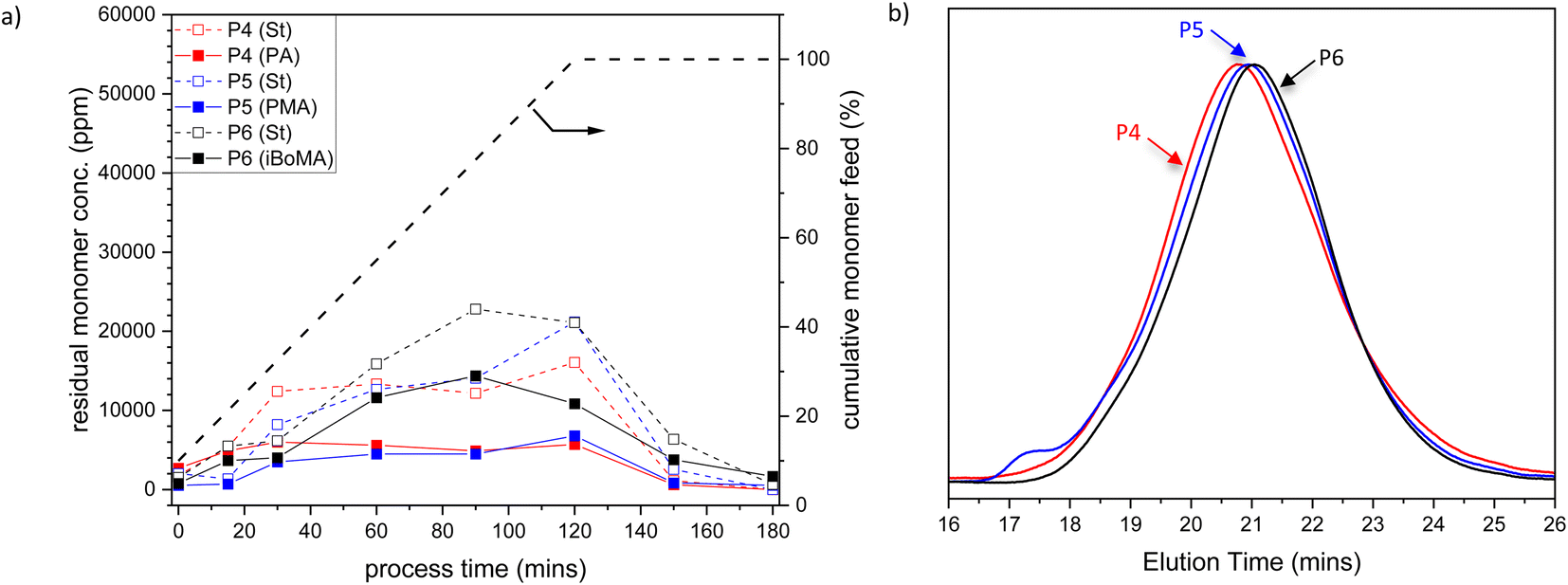 | ||
| Fig. 2 (a) Plot of residual St (open symbols) and PA 1, PMA 2 or iBoMA 3 (closed symbols) concentrations versus time (the grey dashed line indicates the semi-batch feed profile in cumulative % of the monomers with time), and (b) SEC traces of St-based copolymers, for St/PA/MAA (P4, red), BSt/PMA/MAA (P5, blue) and St/iBoMA/MAA (P6, black) semi-batch emulsion radical copolymerizations. Note that the data related to MAA are omitted for clarity. P4–P6 refer to the sample labels given in Table 1. | ||
The Tgs of the St-based polymers (P4–P6) were relatively high (i.e. between 95 and 120 °C) (see Table 1, entries 4–6) Again, the Tgs were in agreement with those predicted using the Fox equation.31
As with the BA-copolymers, the St-based polymer emulsions had ∼25% bioderived content based upon total solids (see Table 1), in line with the monomer feed compositions.
Preliminary coating application testing for poly(n-butyl acrylate)-based formulations
To investigate the potential of the copolymers incorporating the pinene-based high Tg monomers (PA 1 and PMA 2) towards coating applications we conducted preliminary, industry standard hardness (on glass) and stain resistance (on Leneta cards) tests for the low Tg PBA-based formulations. For comparison, data for the iBoMA 3 containing PBA-copolymer P3 were also obtained (see Table 1). The minimum film forming temperatures (MFFTs) for the St-containing copolymers were too high to allow good film formation and, hence, relevant testing.Due to the presence of the acrylate co-monomer PA 1, the PBA-based polymer P1 had by far the lowest hardness on the König scale of the samples tested (7 s, see Table 2, entry 1); in view of their higher Tgs the PBA polymers (P2 and P3) containing methacrylate-based comonomers had much higher König hardness values as expected. Interestingly, polymer P2 which contains the pinene-based monomer (2) has the highest König value (22 s, see Table 1, entry 2), with the iBoMA (3) containing polymer P3 being slightly softer (14 s, see Table 1, entry 3). Hardness values can be further improved via the Tg by optimising the BA/P(M)A ratio in the copolymer.
| Entry | Sample label | Copolymer | König hardnessa (s) | Stain resistanceb | |||
|---|---|---|---|---|---|---|---|
| Water (24 h) | 50% aq. ethanol (1 h) | Coffee | |||||
| (1 h) | (16 h) | ||||||
| a König hardness of copolymer films prepared on glass, and the values are an average of duplicate analyses, DFT = 40 μm. b Stain resistance of films prepared on cards, ranked by visual inspection on a scale from 0 to 5 (0 = no stain resistance; 5 total stain resistance), and the values in parentheses indicate exposure time. | |||||||
| 1 | P1 | P(BA63%-co-PA35%-co-MAA2%) | 7.0 ± 0.3 | 3 | 3 | 4 | 2 |
| 2 | P2 | P(BA63%-co-PMA35%-co-MAA2%) | 22.0 ± 0.9 | 4 | 2 | 4 | 3 |
| 3 | P3 | P(BA63%-co-iBoMA35%-co-MAA2%) | 14.0 ± 0.6 | 3 | 3 | 4 | 3 |
All three coating formulations had good to very good water stain resistance over a 24 h period, with the PMA 2 containing polymer P2 visually outperforming the other two formulations (P1 and P3) (see Table 2). In contrast, when exposed to 50% aqueous ethanol solution, P2 performed marginally poorer than P1 and P3 (see Table 2). All three polymer coatings (P1–P3) had excellent coffee stain resistance at 1 h, with performance decreasing as expected when exposed for 24 h (see Table 1). Notably, the polymers P2 and P3 which each contain a methacrylate-based comonomer behaved similarly, outperforming the acrylate-comonomer containing polymer P1.
Conclusions
We have described the improved synthesis and scale up of the pinene-derived monomers 3-pinanyl acrylate (PA, 1) and 3-pinanyl methacrylate (PMA, 2). Additionally, we have reported their copolymerization performance in the preparation of copolymer emulsions based on BA or St and examined the preliminary performance of the BA-based copolymer formulations through hardness and stain resistance testing, following industry norms.Key developments in monomer syntheses include the replacement of hazardous and expensive reagents for established transformations. The one-pot hydroboration/oxidation of α-pinene was readily achieved using NaBH4 and AcOH in THF, followed by H2O2 and NaOH, to give 3-pinanol in moderate yield (58%), while the reaction of 3-pinanol with methacrylic anhydride and catalytic Cu(BF4)2) provided PMA 2 in high yield (90%). Importantly, both of these syntheses are sustainable, cost efficient and scalable. Additionally, PA 1 was readily prepared at a >100 g scale through the reaction of 3-pinanol with acryloyl chloride in high yield (87%), demonstrating the ease of access to larger volumes of these materials.
Copolymerization performance of PA 1 and PMA 2 was assessed by preparing copolymers with BA or St (and MAA) by semi-batch emulsion copolymerization. Analogous materials from the more established monomer iBoMA 3 were prepared as a reference. Successful copolymerization of the monomers 1–3 with BA (and MAA) gave latexes (P1–P3) with particle sizes between 65 and 90 nm and with a very low PDI (<0.06). The BA-based copolymers (P1–P3) had high molar mass (Mn > 40000, Mw > 400000) and very high dispersity (Ð > 9.5). The BA-based materials showed bimodal molar mass distributions and low Tgs (∼−5 to −20 °C). Throughout the reactions the monomer feed ratios remained relatively constant, particle size was found to increase with monomer conversion, and particle size polydispersity index (PDI) remained low (<0.2). Copolymerization of the monomers 1–3 with St (and MAA) behaved similarly, giving latexes (P4–P6) with particle sizes close to 90 nm and with a very low PDI (<0.08). The St-based copolymers (P4–P6) had high molar mass (Mn > 40000 and Mw > 150000) and reasonably high molar mass dispersity (Ð > 3). In contrast to the BA-cases, the St-based copolymers had monomodal molar mass distributions and relatively high Tgs (∼95 to 120 °C). Importantly, GLC indicated that all the copolymers (P1–P6) had relatively uniform chemical composition distributions.
Preliminary coating application testing was performed on the low Tg BA-based materials (P1–P3). Overall, the PMA-containing copolymer (P2) appeared to be the best performing coating material; in comparison to the iBoMA-containing copolymer (P3), P2 was harder and had slightly better stain resistance. The PA-containing copolymer (P1) was the softest coating, which is in line with it having the lowest Tg, and displayed similar stain resistance to the iBOMA-containing material (P3).
In summary, we have demonstrated the industrially relevant scale up of the monomers PA 1 and PMA 2 using sustainable and cost-efficient processes, successfully incorporated them into copolymers via emulsion polymerization, and finally demonstrated the potential of these new polymers as coating materials. We expect that through refinement of the copolymerization process the properties of the polymers derived from these renewable monomers may be improved further. Companies that are interested in steps to bring these pinene (meth)acrylates to the market are invited to contact the authors.
Data availability
Further data is available upon request to the authors.Author contributions
Maria Pin-No – investigation, methodology, formal analysis, writing-original draft; Philippa L. Jacob – writing-original draft, writing-review & editing; Vincenzo Taresco – writing-review & editing; Maud Kastelijn – investigation, methodology, formal analysis, writing-original draft, writing-review & editing; Tijs Nabuurs – conceptualisation, methodology, writing-original draft; writing-review & editing; Chandres Surti – supervision, project administration; John Bilney – supervision, project administration; John Daly – supervision, project administration; Daniel J. Keddie – formal analysis, visualization, writing-original draft, writing-review & editing; Steven M. Howdle – conceptualisation, funding acquisition, resources, supervision, project administration, writing-review & editing; Robert A. Stockman – conceptualisation, funding acquisition, resources, supervision, project administration, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from Innovate UK (KTP10677) (MPN). VT would like to thank the University of Nottingham for his Nottingham Research Fellowship. We also acknowledge the EPSRC for funding an EPSRC Doctoral Prize [EP/W524402/1] for PLJ and the Engineering and Physical Sciences Research Council and SFI Centre for Doctoral Training in Sustainable Chemistry [EP/S022236/1]. The authors would also like to thank Cornelius and Covestro for technical and financial support.Notes and references
- R. Janani, D. Majumder, A. Scrimshire, A. Stone, E. Wakelin, A. H. Jones, N. V. Wheeler, W. Brooks and P. A. Bingham, Prog. Org. Coat., 2023, 180, 107557 CrossRef CAS.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Prog. Polym. Sci., 2021, 113, 101353 CrossRef CAS.
- P. Achakulwisut, P. Erickson, C. Guivarch, R. Schaeffer, E. Brutschin and S. Pye, Nat. Commun., 2023, 14, 5425 CrossRef CAS PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y. X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- C. Zhang, J. Xue, X. Yang, Y. Ke, R. Ou, Y. Wang, S. A. Madbouly and Q. Wang, Prog. Polym. Sci., 2022, 125, 101473 CrossRef CAS.
- A. Scott, C&EN Archives, 2012, 90, 16–17 Search PubMed.
- J. Thomas and R. Patil, Ind. Eng. Chem. Res., 2023, 62, 1725–1735 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- P. Sahu and A. K. Bhowmick, Ind. Eng. Chem. Res., 2019, 58, 20946–20960 CrossRef CAS.
- J. Zhang, C. Aydogan, G. Patias, T. Smith, L. Al-Shok, H. Liu, A. M. Eissa and D. M. Haddleton, ACS Sustain. Chem. Eng., 2022, 10, 9654–9664 CrossRef CAS PubMed.
- C. Veith, F. Diot-Néant, S. A. Miller and F. Allais, Polym. Chem., 2020, 11, 7452–7470 RSC.
- K. Satoh, Polym. J., 2015, 47, 527–536 CrossRef CAS.
- M. Mousa, H. Bergenudd, A. L. Kron and E. Malmström, Macromolecules, 2021, 54, 6127–6134 CrossRef CAS.
- Q. Hua, L.-Y. Liu, M. Cho, M. A. Karaaslan, H. Zhang, C. S. Kim and S. Renneckar, Biomacromolecules, 2023, 24, 592–603 CrossRef CAS PubMed.
- M. Decostanzi, J. Lomège, Y. Ecochard, A.-S. Mora, C. Negrell and S. Caillol, Prog. Org. Coat., 2018, 124, 147–157 CrossRef CAS.
- S. Molina-Gutiérrez, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Green Chem., 2019, 21, 36–53 RSC.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC.
- R. L. Atkinson, O. R. Monaghan, M. T. Elsmore, P. D. Topham, D. T. W. Toolan, M. J. Derry, V. Taresco, R. A. Stockman, D. S. A. De Focatiis, D. J. Irvine and S. M. Howdle, Polym. Chem., 2021, 12, 3177–3189 RSC.
- M. Cutajar, F. Andriulo, M. Thomsett, J. C. Moore, B. Couturaud, S. M. Howdle, R. A. Stockman and S. E. Harding, Sci. Rep., 2021, 11, 7343 CrossRef CAS PubMed.
- T. M. Bennett, J. Portal, V. Jeanne-Rose, S. Taupin, A. Ilchev, D. J. Irvine and S. M. Howdle, Eur. Polym. J., 2021, 157, 110621 CrossRef CAS.
- U. Montanari, V. Taresco, A. Liguori, C. Gualandi and S. M. Howdle, Polym. Int., 2020, 70, 499–505 CrossRef.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC.
- M. Gscheidmeier and H. Fleig, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a27_267.
- R. L. Atkinson, M. Elsmore, S. Smith, M. Reynolds-Green, P. D. Topham, D. T. W. Toolan, M. J. Derry, O. Monaghan, V. Taresco, D. J. Irvine, R. A. Stockman, D. S. A. De Focatiis and S. M. Howdle, Eur. Polym. J., 2022, 179, 111567 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- A. K. Chakraborti, R. Gulhane and Shivani, Synthesis, 2004, 2004, 111–115 CrossRef.
- V. Hach, Synthesis, 1974, 1974, 340–342 CrossRef.
- ASTM D6866-18: Standard Test Methods for Determining the Biobased Content of Solid, Liquid, and Gaseous Samples Using Radiocarbon Analysis (Method B), 2018 Search PubMed.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, John Wiley & Sons, 2005 Search PubMed.
- F. Alvarez, J. Colmenero, C. H. Wang, J. L. Xia and G. Fytas, Macromolecules, 1995, 28, 6488–6493 CrossRef CAS.
- P. Holub, Double Liaison--Phys., Chim. Econ. Peint. Adhes., 2004, 539, 24–30 CAS.
- O. R. Monaghan, S. T. Skowron, J. C. Moore, M. Pin-Nó, K. Kortsen, R. L. Atkinson, E. Krumins, J. C. Lentz, F. Machado, Z. Onat, A. Brookfield, D. Collison, A. N. Khlobystov, D. De Focatiis, D. J. Irvine, V. Taresco, R. A. Stockman and S. M. Howdle, Polym. Chem., 2022, 13, 5557–5567 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for polymer syntheses, gradient liquid chromatographs of all copolymers, and 1H and 13C NMR spectra of polymers P1 and P2. See DOI: https://doi.org/10.1039/d4su00210e |
| ‡ BH3·SMe2 is toxic, flammable, and preventatively expensive. It also has limited commercial availability for use on larger scales. Both methacryloyl chloride and acryloyl chloride are also toxic highly reactive reagents, and thus would be best avoided from a sustainability standpoint. |
| § Previously we also reported this synthesis using the reagent propanephosphonic acid anhydride (T3P) and acrylic acid (see ref. 18). While this approach is more sustainable than the route reported here, it is currently not economically (and industrially) viable at the reported scale because of the current cost of T3P. |
| ¶ Clearly the MAA presence in each of these systems will also play a role in the copolymerization kinetics. Due to its low feed ratio MAA is omitted from the discussion here to aid in the direct comparison across the copolymerizations using the acrylate (PA) and methacrylate (PMA & iBoMA) terpene-derived comonomers. |
| || This is attributed to the differences in polymerization rates for the ‘burn up’ reactions of the two different copolymerization systems, (i.e. the BA-based high kp systems and the St-based low kp systems). Note that as a fresh initiator is added in the ‘burn-up’ step of the reactions, the kinetics differ from that of the main polymerization reactions. |
| This journal is © The Royal Society of Chemistry 2024 |